

Abstract
FLT3 ligand (FLT3L) has diverse roles in the hematopoietic system, which include stimulating proliferation of hematopoietic precursors and development of natural killer cells and dendritic cells. FLT3L is initially synthesized as a membrane-bound protein, which must be cleaved to become a soluble growth factor. However, little is known about the enzyme involved in the proteolytic release of FLT3L. In the current study, we show that shedding of FLT3L is metalloprotease-dependent, and that this proteolytic activity was abolished in fibroblasts lacking TNFα converting enzyme (TACE) and could be rescued by reintroducing wildtype TACE in these cells. Moreover, we found that cells derived from the thymus of conditional TACE-deficient mice produce less FLT3L, and that serum FLT3L levels in these TACE mutant mice are significantly lower, both after LPS treatment and in the absence of such a challenge, further corroborating the relevance of TACE as FLT3L sheddase in vivo. Considering the involvements of FLT3 and FLT3L in hematopoietic malignancies and stem cell mobilization, the identification of the enzyme involved in FLT3L shedding may have important clinical implications.
Keywords: TACE / ADAM17, FLT3 ligand, ectodomain shedding
Introduction
The ligand for the receptor tyrosine kinase fms-like tyrosine kinase 3 (FLT3)3 has important roles in hematopoiesis (1-3). Mice with a targeted disruption of FLT3 ligand (FLT3L) are viable and fertile, however they develop several hematopoietic deficiencies, including reduced leukocyte cellularity in the bone marrow, peripheral blood, lymph nodes and spleen, and reduced numbers of myeloid progenitors and B lymphocytes, dendritic cells and natural killer cells (4). While the expression of FLT3 is restricted mainly to early myeloid and lymphoid progenitors, FLT3L has a widespread expression pattern. The protein sequence of FLT3L and the alternative splicing pattern of its mRNA exhibits remarkable similarity to that of CSF-1 (also known as M-CSF) and c-kit ligand (KITL) (also known as stem cell factor or Steel factor), in that all three growth factors are initially produced as membrane-spanning proteins that have to be proteolytically cleaved to become soluble (5-7). There are at least three different protein isoforms for murine FLT3L. The first is the membrane-spanning form, which is efficiently cleaved from cell surface and active as both membrane-bound and soluble growth factor. The second isoform is also membrane tethered, but lacks a cytoplasmic domain due to an alternative splicing of the mRNA and is less efficiently cleaved. The third isoform is generated as a soluble protein without a membrane anchor, and is far less abundant than the other two (1, 4, 8). Intriguingly, the membrane-tethered isoform lacking the cytoplasmic domain, which is one of the two major isoforms in mouse, has not been identified in human (1).
Previous studies have identified the proteases involved in the proteolytic processing of KITL and the membrane-bound isoform of CSF-1 (note that CSF-1 is primarily produced as a soluble growth factor) (9). MMP-9 and Cathepsin K have been implicated in the cleavage of KITL (10, 11), and moreover, the TNFα converting enzyme (TACE or a disintegrin and metalloprotease 17 (ADAM17)) has been shown to be involved in the shedding of both KITL and membrane-bound CSF-1 (12-14). TACE is a type-1 membrane protein and belongs to a large family of transmembrane metalloproteases (ADAM gene family) that have crucial roles in ectodomain shedding (for reviews on ADAMs see (15, 16)). The domain structure of the ADAMs consists of prodomain, a metalloprotease domain, an EGF-like domain, a transmembrane domain, and a cytoplasmic tail. The metalloprotease domain of TACE contains the consensus catalytic sequence, HExxHxxGxxH, which predicts that TACE is an active protease. TACE was originally identified as the enzyme responsible for the cleavage of pro-TNFα (17, 18), but subsequent studies have revealed numerous additional substrates and functions for this molecule, including a critical role in activating the ligands of the epidermal growth factor receptor, and in the modulation of immune reactions (16, 19, 20). In line with these observations, Tace null mice exhibit diverse developmental defects and perinatal lethality (21, 22).
In the present study we evaluated the proteolytic activity of FLT3L shedding and found that TACE is critical for the processing of FLT3L in cell-based assays. Furthermore we found that serum FLT3L levels were much lower in conditional TACE-deficient mice (22) compared with control animals, further corroborating that TACE is the major sheddase for FLT3L in vivo. Taken together with previous studies, in which we showed the involvement of TACE in the shedding of KITL (12) and membrane-bound CSF-1(13), the observations in the current study suggest that TACE plays a central role in the processing of ligands for the type III tyrosine kinase receptors, CSF-1, KITL and FLT3L, thereby regulating the functions of these cytokines in vivo.
Materials and Methods
Mice, cells and reagents
Generation of Taceflox/flox/Mx1-Cre+ (henceforth referred as Tace/Mx1) mice used in the current study had been previously described (22). For temporal deletion of floxed Tace, 6-wk-old Tace/Mx1 mice were injected intraperitoneally with 250 μg of polyinosinic-polycytidylic acid (pIpC, Sigma-Aldrich) three times at 2-day intervals. In all experiments with Tace/Mx1 mice, littermate Taceflox/flox/Mx1-Cre- (henceforth referred as Control) mice served as controls. TACE-deficient and wildtype immortalized embryonic fibroblasts were generated by introducing a vector carrying Large-T antigen as previously described (13). The antiserum directed against the TACE cytoplasmic domain was described previously (23). Recombinant murine IL-7 was from R & D Systems (Minneapolis, MN). Trans-epoxysuccinyl-L-leucylamido-butane (E64), pepstatin A, leupeptin, trypsin inhibitor from Glycine max (STI), and pefabloc SC were from Sigma. GM6001 was from Chemicon International, and recombinant human TIMPs 1 3 were purchased from R&D Systems. U0126 and SB202190 were purchased from Calbiochem. All other reagents were obtained from Sigma-Aldrich unless otherwise indicated.
Cloning of murine FLT3L and generation of expression vectors
A 0.8 kb cDNA fragment including the entire coding sequence of FLT3L was cloned by RT-PCR utilizing the RNA from mouse bone marrow tissue as a template. Sequencing of the PCR product revealed that it encoded a membrane-spanning isoform of FLT3L (6C (8) or T110 (6) form, respectively), which is known to be efficiently cleaved from the cell surface to become a soluble growth factor in vivo (6, 8). The epitope tagged FLT3L was generated by PCR and cloned into pAPtag5 (Genhunter, Nashville, TN) and pcDNA4/Myc-His (Invitrogen) (see Fig.1A for the schema of each vector). FLT3L mutants were generated by a PCR based method using KOD-plus-Mutagenesis Kit (Toyobo, Tokyo, Japan) following the manufacturer’s instruction. All constructs were sequenced to confirm that the desired deletion had been obtained, and to rule out other undesired mutations (see Fig.5A and B for the schema of each mutant FLT3L constructs).
Figure 1.

A. Schematic presentation of AP-tagged (AP-FLT3L), and HA- and Myc/His-tagged FLT3L (HA-FLT3L). SS, signal sequence; GFD, growth factor domain; TM, transmembrane domain; CT, cytoplasmic tail. B. Western blot analysis of HA-FLT3L expressed in COS-7 cells. (-), non-transfected control. C. In-gel detection of the cleaved AP-FLT3L released in the supernatant. COS-7 cells transfected with AP-FLT3L were incubated with PMA (25 ng/ml) and / or GM6001 (10 μM) for 1 h, and the supernatants and cell lysates were collected for analysis.
Figure 5.
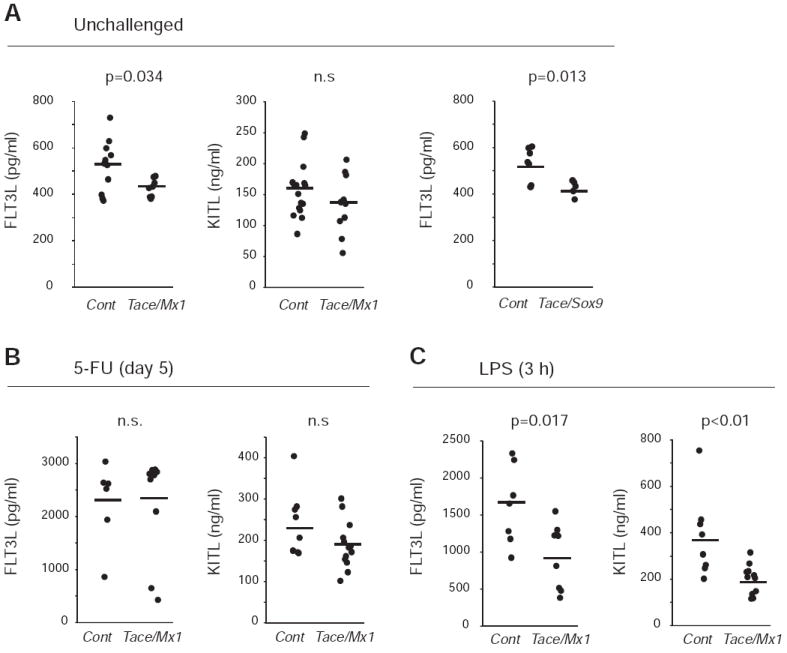
Serum FLT3L and KITL levels are significantly decreased in Tace/mx1 mice and Tace/Sox9 mice. The serum levels of FLT3L and KITL from unchallenged mice (A), mice challenged with 5-FU injection (B) and mice challenged with LPS (C). The serum samples were collected on day 5 (5-FU) or 3 h after (LPS) the treatment. All cytokine levels were analyzed by ELISA. Bars, average. n.s., not significant.
Cell culture, transfection and shedding assay
COS-7 cells and embryonic fibroblasts were grown in DMEM supplemented with 5 % FCS and antibiotics. The cells were transfected with alkaline phosphatase (AP)-tagged FLT3L (AP-FLT3L) using Fugene HD (Roche). Fresh Opti-MEM (Invitrogen) medium with or without the indicated reagents (PMA and / or GM6001) was added 18-24 h after transfection and incubated for an hour. “In-gel” visualization of AP activity in the supernatants and the lysates was done as previously described (24). The supernatants were collected and cleared by centrifugation for 15 min to remove cell debris. The cells were lysed in lysis buffer (1% Triton-X100, protease inhibitor cocktail, 5 mM 1-10-phenanthroline, PBS), and the lysates were collected and cleared by centrifugation. The cleared supernatants were concentrated using ConA-Sepharose lectin beads (Amersham Biosciences) followed by elution with 0.5 M α-methyl-D-mannoside in 50 mM Tris-HCl, pH 7.4 for 2 h at 37°C. The samples were then separated by 10% SDS-PAGE. After electrophoresis, the gels were washed with 2.5% Triton-X100 solution and the AP activity in the gel was visualized by adding detection buffer containing the AP substrate, NBT/BCIP (Sigma-Aldrich). The AP activity in the supernatant was also measured by colorimetry as previously described (13, 24). Cleared cell supernatants applied in 96-well plates were incubated with the same volume of 2 mg/ml 4-nitrophenylphosphate at 37°C for color development. After the incubation, OD405 was measured by a microplate reader (Model 680XR; Bio-Rad). The supernatant from nontransfected cells incubated with the substrate for the same amount of time was used as a spectrophotometric blank to normalize for background AP activity. The average of the constitutive or PMA-stimulated shedding was set to 1, and used as a reference to calculate relative decrease in shedding following inhibition by compounds. All experiments were repeated at least three times with similar results.
Determination of cytokine levels
The serum levels of FLT3L and KITL were measured by sandwich ELISA (Quantikine, R & D Systems) following the manufacturer’s instructions. To enhance serum sFLT3L and sKITL levels in vivo, Tace/Mx1 or littermate Control mice were treated with 100 μg of LPS and 20 mg of D-galactosamine, or 250 mg/kg body weight of fluorouracil (5-FU; Kyowa Hakko, Tokyo, Japan). The mice were sacrificed after 3 h (LPS) or 5 days (5-FU) after injection, respectively. All animal experiments were approved by the Institutional Animal Care and Use Committee of the Keio University School of Medicine (Tokyo, Japan).
Preparation of thymus cells
Single cell suspensions from the thymus were generated by teasing cells isolated from the thymus through a 70 μm strainer (BD Biosciences). RBCs were removed by RBC Lysis Buffer (Roche). The cells were incubated in FCS-free RPMI-1640 medium with or without recombinant IL-7 for 72 h. At the end of incubation, the supernatants were collected, and the levels of soluble FLT3L in the medium were measured by ELISA as described above. The remaining cells were collected and lysed in lysis buffer. The lysates were separated by SDS-PAGE and analyzed by Western blot using anti-TACE antisera and anti-actin antibody.
Statistical analysis
All data are presented as mean ± S.D. Student’s t-test for two samples assuming equal variances were used to calculate the P-values. P-values smaller than 0.05 were considered statistically significant. Statistical analyses were performed by using Excel (Microsoft).
Results
Shedding of FLT3L is metalloprotease-dependent and stimulated by PMA
We first cloned an approximately 0.8 kb fragment of FLT3L cDNA and generated HA-tagged (HA-FLT3L) and AP-tagged (AP-FLT3L) vectors as shown in Fig. 1A. When HA-tagged FLT3L was expressed in COS-7 cells, it appeared as several bands (Fig 1B). Treatment with peptide N-glycanase F, which removes all N-linked carbohydrate residues, resulted in disappearance of slower migrating bands, reflecting differential glycosylation during maturation (data not shown). To facilitate the detection of cleaved FLT3L in the supernatant, we took advantage of an AP reporter system (24) (see Materials and Methods for details). As has previously been shown (6), PMA highly stimulated the shedding of AP-FLT3L from transfected COS-7 cells (Fig. 1C). In order to further characterize the enzyme responsible for FLT3L shedding, we examined whether GM6001, a broad spectrum metalloprotease inhibitor, could block this activity. As shown in Fig. 1C, we found that addition of GM6001 effectively inhibited both the constitutive and PMA-stimulated shedding of AP-FLT3L, indicating that the proteolytic activity is potentially dependent of metalloprotease(s).
TACE mediates FLT3L cleavage
To explore possible involvements of other proteolytic enzymes besides metalloproteases in the cleavage of FLT3L, we next screened an array of protease inhibitors. The protease inhibitors used include; serine protease inhibitors, leupeptin, STI and Pefabloc SC; cysteine protease inhibitor, E-64; aspartate protease inhibitor, pepstatin; and GM6001. When the shedding activity of FLT3L was measured by colorimetry in the presence of a given protease inhibitor, only GM6001 showed inhibitory effects on both constitutive and PMA-stimulated shedding, indicating that metalloprotease(s), but not other types of proteases, participates in FLT3L shedding (Fig. 2A and B). To further characterize this proteolytic activity, we examined the inhibitory profile of tissue inhibitor of metalloproteases (TIMPs) in FLT3L shedding. TIMPs are the major cellular inhibitors of metalloproteases and exhibit different efficacies against different metalloprotease subfamily members (25, 26). MMPs are all sensitive to TIMPs 1-3, while MT1-MMP is sensitive to TIMPs 2 and 3. TACE, on the other hand, is sensitive to TIMP3 but resistant to TIMPs 1 and 2. As shown in Fig. 2A and B, both constitutive and PMA-stimulated FLT3L shedding were only sensitive to TIMP3, and neither TIMP1 or TIMP2 showed any impact on this activity, indicating that TACE, but not MMPs or MT1-MMP, is involved in the release of soluble FLT3L. Additionally, MAP kinase inhibitors (U0126 and SB202190), which have been shown to inhibit shedding activity towards several TACE substrates (13, 27, 28), down-regulated both constitutive and PMA-stimulated FLT3L shedding (Fig. 2C and D).
Figure 2.
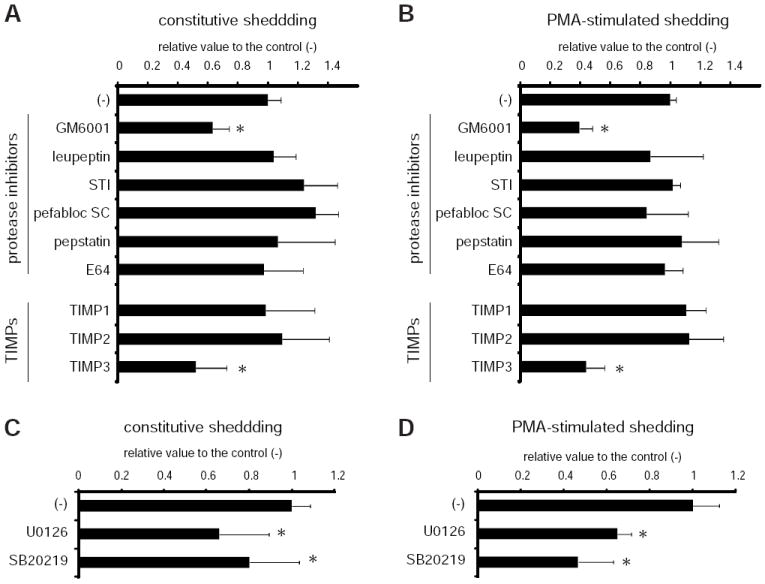
Characterization of FLT3L shedding activity. COS-7 cells transiently expressing AP-FLT3L were incubated with various protease inhibitors (A and B) or MAP kinase inhibitors (C and D) in the presence (B and D) or absence (A and C) of PMA (25 ng/ml). The inhibitors were used at the following concentrations: GM6001 (10 μM), leupeptin (100μM), STI (0.5 mg/ml), pefabloc SC (0.4 mM), E64 (10μM), TIMPs 1-3 (16 nM), U0126 (5 μM) and SB202190 (20 μM). After the incubation, the supernatants were collected and subjected to a colorimetric assay for AP activity as described in Materials and Methods. Each value represents the mean derived from at least three individual experiments; error bars, SD. *, p < 0.05.
To examine if the lack of TACE activity results in a decrease in FLT3L shedding, we next used immortalized mouse embryonic fibroblasts (mEFs) derived from wildtype and Tace-/- embryos (22). When the AP-FLT3L expression vector was introduced into wildtype mEFs, the results were essentially identical to those obtained with COS-7 cells (Fig. 3A). On the other hand, in Tace-/- mEFs, the PMA-induced shedding was almost completely abolished, indicating that TACE is involved in the stimulated processing of FLT3L. To validate this observation, we next attempted a rescue experiment by reintroducing TACE into Tace-/- mEFs. As shown in Fig. 3B, reintroduction of TACE fully recovered the PMA-stimulated shedding activity to a comparable level to that in wildtype mEFs, while the negative control, empty pcDNA vector, did not. Although the remaining constitutive FLT3L shedding from Tace-/- mEFs can be further reduced by the general metalloproteinase inhibitor GM6001 (Fig. 3A) or the ADAM10-selective inhibitor GI254023X (at 0.2 μM, data not shown)(29, 30), these results nevertheless suggest that TACE is a principal enzyme for the cleavage of FLT3L, at least under the condition of the cell-based assays in the current study.
Figure 3.
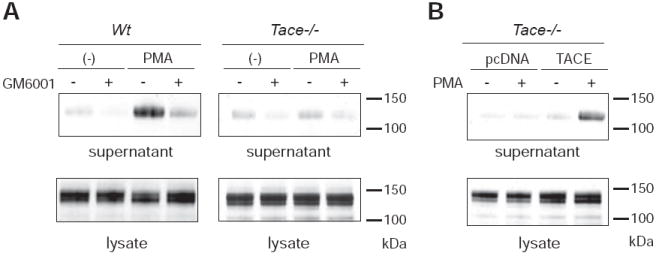
Processing of AP-FLT3L is dependent on TACE activity. A. PMA-induced shedding of AP-FLT3L is present in wildtype (Wt) mEFs but is missing in Tace-/- mEFs. B. Recovery of PMA-induced shedding is observed by the reintroduction of a TACE expression vector but not by an empty vector (pcDNA).
Cells derived from the thymus of Tace/Mx1 mice produce less soluble FLT3L
Based on these observations, we next examined whether these findings were applicable to the endogenous FLT3L. FLT3L is, in contrast to its receptor, FLT3, ubiquitously expressed with relatively high expression levels in stromal cells and T lymphocytes (6). We collected cells from the thymus from Tace/Mx1 and Control mice, and incubated these cells with or without recombinant murine IL-7 for 72 h to enhance the expression of FLT3L (31). In line with the results using AP tagged FLT3L, the levels of cleaved FLT3L released into the supernatant were lower in Tace/Mx1 derived cells compared with Control (Fig. 4A). Western blot of thymus cells showed some remaining TACE expression in the thymus in Tace/Mx1mice (Fig. 4B), which is most likely due to incomplete excision by the Mx1-Cre, and provides a likely explanation for the residual shedding activity in the Tace/Mx1 mice derived thymus cells.
Figure 4.
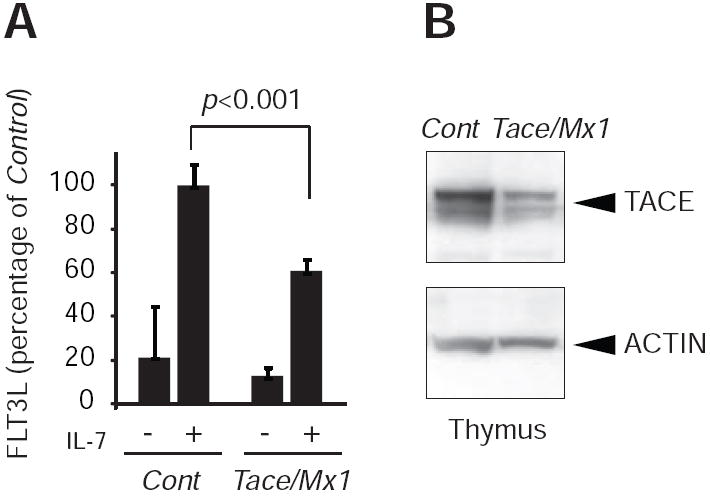
Thymus cells from Tace/Mx1 mice release less soluble FLT3L into the culture supernatant than those of Control mice. (A) Total thymus cells were incubated with (+) or without (-) recombinant IL-7 (15 μg/ml) for 72 h. Soluble FLT3L released in the supernatant was analyzed by ELISA. (B) Western blot analysis shows significant decrease, but not complete disappearance, of TACE in the thymus cells from Tace/Mx1 mice.
Decreased serum FLT3L levels in the conditional TACE-deficient mice
As described above, because FLT3L has a widespread expression pattern, it is not feasible to disrupt Tace in all the cells expressing FLT3L. Moreover, FLT3L can also be produced as a soluble protein via alternative splicing of the transcripts, which could hamper the detection of the soluble FLT3L generated by shedding from its membrane-anchored precursor in vivo. Nevertheless, to confirm the contribution of TACE to the release of soluble FLT3L in vivo, we examined the serum levels of FLT3L and, for comparison, KITL, which had previously been shown to be a TACE substrate in vitro (12). We found that serum FLT3L levels were lower in Tace/Mx1 mice (n=8, 428 ± 36 pg/ml) compared with Control (n=11, 523 ± 111 pg/ml) (Fig. 5A). On the other hand, there was no significant difference in the serum levels of soluble KITL between Control and Tace/Mx1 mice, most likely due to an insufficient excision of floxed Tace in stromal cells and osteoblasts, which represent the major source of KITL. In fact, a significant decrease in soluble serum KITL levels was observed in a different mutant line (Tace/Sox9 mice) in which floxed Tace was removed from stromal cells and osteoblasts but not from blood cells (see (14) for details on Tace/Sox9 mice). Additionally, we also found a comparable decrease in the serum FLT3L levels to that in Tace/Mx1 mice in Tace/Sox9 mice (Fig. 5A, Control (n=7, 517 ± 74.0 pg/ml), Tace/Sox9 (n=6, 409 ± 33.4 pg/ml), p = 0.024). Since it has been shown that both serum FLT3L and KITL are highly upregulated under myelosuppressive conditions (1, 3), we challenged the Control and Tace/Mx1 mice with 5-FU and evaluated the serum levels of both growth factors 5 days after the injection. As shown in Fig. 4B, although there was a trend toward having a lower serum KITL in Tace/Mx1 mice (not statistically significant), the serum FLT3L levels were very similar in Control and Tace/Mx1 mice. The cause for this apparent discrepancy could be a compensatory mechanism, such as an increase in the production of the soluble isoform or upregulation in the production of FLT3L in the remaining TACE-positive cells, although this remains to be established. Consistently, there was no difference in the survival rate between Control and Tace/Mx1 mice when the mice were challenged with successive 5-FU treatments (data not shown).
In a previous study, we showed that Tace/Mx1 mice exhibited lower serum TNFα levels compared with Control when they were challenged with LPS (22), indicating that TACE activity could be upregulated by LPS in vivo. In accordance with these findings, it has also been shown that LPS can stimulate TACE activity and upregulate the shedding of several membrane bound proteins, including the p75 TNF receptor (21, 32), TGFα (33) and CSF-1 receptor (34). Since this upregulation can be induced rapidly (<3 h), we hypothesized that the potential compensatory effects observed with 5-FU treatment might not be seen with a more rapid challenge. We therefore injected the mice with LPS as described previously (22) and collected the serum 3 h after the injection. As shown in Fig. 4C, we found that LPS treatment significantly increased the serum levels of both FLT3L and KITL, and, most importantly, that the increase was much less pronounced in Tace/Mx1 mice (FLT3L, n=8, 936 ± 442 pg/ml; KITL, n=11, 199 ± 64 ng/ml) compared with Control (FLT3L, n=7, 1624 ± 532 pg/ml; KITL, n=10, 382 ± 177 ng/ml), indicating that TACE is required for the LPS-induced release of these growth factors in vivo.
Deletions or mutations in the juxtamembrane domain of FLT3L lead to a decreased shedding efficiency
To gain further insight into the mechanism of FLT3L shedding, we attempted to locate the site(s) essential for proteolytic cleavage of FLT3L. First, we generated mutants with larger deletions (up to 20 amino acid residues) in the juxtamembrane domain (MUT1-3) and tested the shedding profiles of these mutants. As shown in Fig. 6A, constitutive and PMA-stimulated shedding were almost completely abolished in MUT1 and MUT2, whereas the shedding efficiency of MUT3 was comparable to that of the control (AP-FLT3L), indicating that the region deleted in MUT2 (Ile175-Glu184) is required for FLT3L shedding. To narrow-down the region essential for FLTL3 shedding, we then generated MUT4 and MUT5. We found that the AP activity in the supernatant released from MUT4 transfected cells was decreased compared with the wildtype control; however there still was a significant upregulation with PMA stimulation. On the other hand, there was almost no AP activity in the supernatant from the cells transfected with MUT5, which lacked the sequence Ile175-Ala179 in the juxtamembrane region of FLTL3 (see Fig. 6A for details). Furthermore, we generated additional mutants with shorter deletion within this sequence (MUT6 and MUT7) and obtained similar results with those of MUT5.
Figure 6.
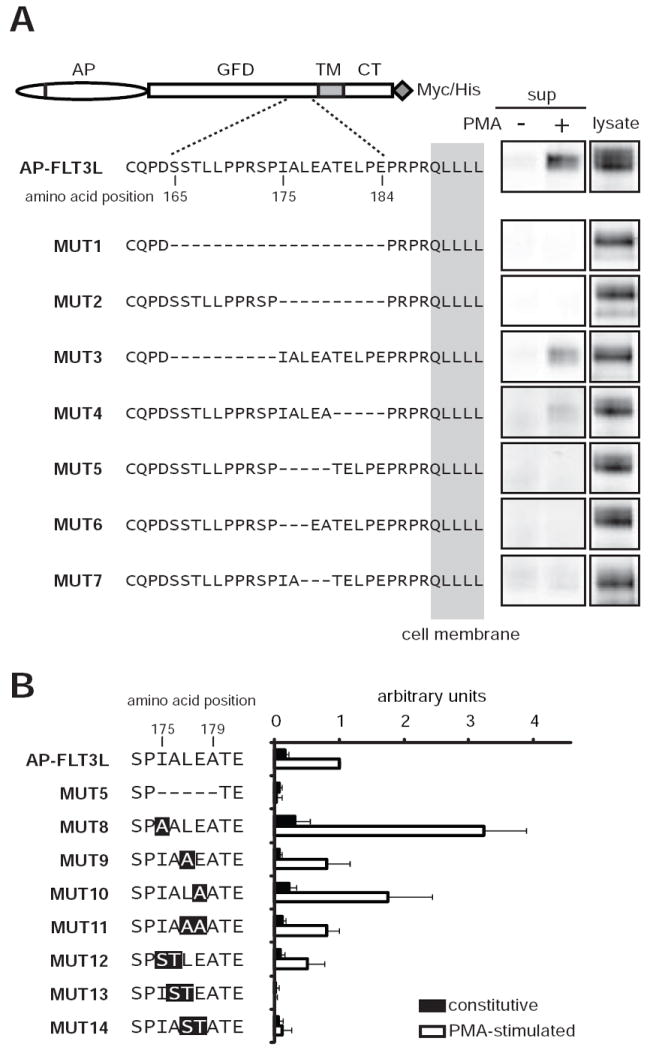
Evaluation of the shedding efficiency of FLT3L deletion mutants (MUT1-7) and substitution mutants (MUT8-14). A. AP-FLT3L mutants were transfected in COS-7 cells and the AP activity released in the supernatant (sup) and that in the cell lysates was visualized by “in-gel” staining as described in Materials and Methods. GFD, growth factor domain; TM, transmembrane domain; CT, cytoplasmic tail. B. Evaluation of shedding efficiency of the substitution mutants. Shedding activity is presented as a ratio of the AP activity in the supernatant and the cell lysate as previously described (29, 35). Boxed letter(s) denotes the mutated amino acids.
To further evaluate how the mutations in this amino acid sequence would affect shedding efficiency, we next generated several constructs with one or two amino acids substitution (MUT8-14, see Fig. 6B for details). AP activity was evaluated by colorimetry, and in order to semi-quantify and normalize the shedding efficiency of each construct, the ratio between AP activity in the supernatant and the cell lysate was calculated (29, 35). As shown in 6B, the shedding efficiency of MUT5 was significantly lower compared to that of the control (AP-FLT3L), consistent with the results with in-gel staining (Fig. 6A). Whereas, the mutant constructs with one or two alanine substitution (MUT8-11) were cleaved with a similar or even higher efficiency compared with the control. Since an uncleavable mutant of heparin-binding EGF like growth factor was successfully generated by introducing a substitution mutation with serine and threonine (36), we undertook a similar strategy and generated three mutant constructs with a serine and threonine substitution (MUT12-14). Although MUT12 (I175S/A176T) showed a comparable shedding efficiency with that of the control, MUT13 (A176S/L177T) and MUT14 (L177S/E178T) were as resistant to shedding as MUT5, indicating that the amino acid sequence Ala176-Leu177-Glu178 in the juxtamembrane domain is important for efficient shedding of FLT3L. The cell surface expression of the constructs, and therefore the maturation of these proteins, was confirmed by immunostaining and confocal microscopic analysis (data not shown).
Discussion
The observations in the current study clearly demonstrate for the first time that TACE has a crucial role in the ectodomain shedding of FLT3L, and, additionally, further corroborate our previous observations that TACE is also involved in the processing of KITL (12). The participation of TACE in FLT3L shedding was identified by 1) cell-based assays using activators and inhibitors of shedding, and TACE-deficient mEFs, 2) an ex vivo assay using cells derived from TACE conditional knockout-mice (Tace/Mx1), 3) and in vivo experiments using Tace/Mx1 mice. Furthermore we also found that FLT3L serum levels were significantly lower in another Tace mutant line, Tace/Sox9 mice, in which Tace is disrupted under the control of a Sox9 promoter (14). Since SOX9 is not expressed in blood cells in Tace/Sox9 mice, and therefore TACE is not inactivated in these cells, the finding also indicates that non-hematopoietic cells are also a major source of soluble FLT3L in vivo. This observation is also in accordance with a wide expression pattern of both FLT3 and TACE in vivo. Taken together, the current study makes a strong case that TACE is a principal sheddase for FLT3L.
By utilizing several FLT3L deletion and substitution mutants, we found Ala176-Leu177-Glu178 in the juxtamembrane domain to be essential for efficient shedding of FLT3L in vitro. Additionally, we also found that the cleavage efficiency is not absolutely dependent on this sequence per se, since substitution of Leu177 and/or Glu178 with alanine (Fig. 6B, MUT9-11) did not suppressed shedding. The precise mechanisms on how TACE recognizes the cleavage site of its substrates are not fully understood, but several determinants affecting the cleavage efficiency have been reported. These include the amino acid sequence of the cleavage site, the distance of the cleavage site from the cell surface and the secondary structure of the extracellular domain of the substrate; on the other hand, no consensus amino acid sequence for cleavage site has so far been identified (35, 37-39). TACE usually releases membrane-bound molecules through cleavage within the stalk region proximal to the transmembrane domain; for example, in the case of EGFR ligands whose shedding is dependent on TACE (TGFα, amphiregulin, heparin biding EGF-like growth factor and epiregulin) (40), the cleavage site is located at 12-16 amino acids from the transmembrane domain (41). Ala176-Leu177-Glu178 in the juxtamembrane domain of FLT3L is located 11-13 amino acids from the transmembrane domain, and therefore this location (Ala176-Leu177 and/or Leu177-Glu178) may be a good candidate for the cleavage site for FLT3L. While this hypothesis will have to be further confirmed by examining the amino acid sequence of the C-terminal end of the cleaved soluble FLT3L, the data presented in the current study may aid in understanding the regulation of the proteolytic processing of membrane bound FLT3L.
Despite a considerable amount of studies on FLT3L in the past, the contribution of ectodomain shedding to the function of FLT3L in vivo still remains to be determined. Unlike the ligands for epidermal growth factor receptor, which are produced as membrane-bound proforms and have to be cleaved to be fully active (16, 21, 42), FLT3L, KITL and CSF-1 are all biologically active whether they are expressed on the cell surface or shed to become soluble (1, 7, 9). In the case for KITL, however, it is clear from the analysis of Steel-Dickie (Sld) mutation, that the membrane-bound form has indispensible roles in development, which cannot be compensated by the soluble counterpart (43). Therefore, although membrane-bound and soluble KITL are both biologically active, they also have distinct roles, which are likely regulated through ectodomain shedding in vivo. The contribution of shedding to the function of CSF-1 is less clear, since soluble CSF-1 is primarily produced by alternative splicing and therefore independently from ectodomain shedding (7, 44, 45), and both soluble and membrane-bound forms have significantly overlapping functions in vivo (9, 46-49). Similar to KITL, FLT3L is primarily produced as a membrane-bound protein (1), and thus most soluble FLT3L is generated through ectodomain shedding. Therefore it will be interesting to further dissect the functions of either soluble or membrane-bound FLT3L in vivo. To this end, a generation of knock-in mutant mice exclusively expressing soluble FLT3L or shedding-insensitive mutant FLT3L should be highly informative, and the identification of mutant forms of membrane anchored FLT3L that are resistant to processing by TACE in the current study may be of use for this propose.
In conclusion, our data show that TACE is the major sheddase for FLT3L in both in vitro cell-based assays and in vivo using conditional TACE-deficient mice. The identification of TACE as a principal sheddase for FLT3L in the current study will be of importance for better understanding of the physiological roles of FLT3L and TACE in vivo. Moreover, since FLT3L is involved in stem cell mobilization and it has been shown that FLT3L administration exhibits a potent antitumor activity (via expansion of the dendritic cell population) (1-3), the current study may also have important implications for stem cell modulation and for the treatment of cancer.
Acknowledgments
We thank Ms. Shizue Tomita, Ms. Yuiko Sato and Dr. Takeshi Miyamoto for technical assistance.
Footnotes
Disclosure
The authors have no financial conflict of interest.
This work was supported by The Uehara Memorial Foundation, The Mochida Memorial Foundation, and Grants-in-aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (19591765) to K.H, and NIH GM064750 to C.P.B.
The abbreviations used are: FLT3, fms-like tyrosine kinase 3; FLT3L, FLT3 ligand; TACE, TNFα converting enzyme; KITL, c-kit ligand; AP, alkaline phosphatase; 5-FU, fluorouracil, mEFs, mouse embryonic fibroblasts; E64, Trans-epoxysuccinyl-L-leucylamido-butane; STI, trypsin inhibitor from Glycine max.
References
- 1.Lyman SD, Jacobsen SE. c-kit ligand and Flt3 ligand: stem/progenitor cell factors with overlapping yet distinct activities. Blood. 1998;91:1101–1134. [PubMed] [Google Scholar]
- 2.Stirewalt DL, Radich JP. The role of FLT3 in haematopoietic malignancies. Nat Rev Cancer. 2003;3:650–665. doi: 10.1038/nrc1169. [DOI] [PubMed] [Google Scholar]
- 3.Wodnar-Filipowicz A. Flt3 ligand: role in control of hematopoietic and immune functions of the bone marrow. News Physiol Sci. 2003;18:247–251. doi: 10.1152/nips.01452.2003. [DOI] [PubMed] [Google Scholar]
- 4.McKenna HJ, Stocking KL, Miller RE, Brasel K, De Smedt T, Maraskovsky E, Maliszewski CR, Lynch DH, Smith J, Pulendran B, Roux ER, Teepe M, Lyman SD, Peschon JJ. Mice lacking flt3 ligand have deficient hematopoiesis affecting hematopoietic progenitor cells, dendritic cells, and natural killer cells. Blood. 2000;95:3489–3497. [PubMed] [Google Scholar]
- 5.Savvides SN, Boone T, Andrew Karplus P. Flt3 ligand structure and unexpected commonalities of helical bundles and cystine knots. Nat Struct Biol. 2000;7:486–491. doi: 10.1038/75896. [DOI] [PubMed] [Google Scholar]
- 6.McClanahan T, Culpepper J, Campbell D, Wagner J, Franz-Bacon K, Mattson J, Tsai S, Luh J, Guimaraes MJ, Mattei MG, Rosnet O, Birnbaum D, Hannum CH. Biochemical and genetic characterization of multiple splice variants of the Flt3 ligand. Blood. 1996;88:3371–3382. [PubMed] [Google Scholar]
- 7.Horiuchi K, Toyama Y. Posttranslational regulation of cell-surface colony-stimulating factor-1. Crit Rev Immunol. 2008;28:215–227. doi: 10.1615/critrevimmunol.v28.i3.30. [DOI] [PubMed] [Google Scholar]
- 8.Lyman SD, James L, Escobar S, Downey H, de Vries P, Brasel K, Stocking K, Beckmann MP, Copeland NG, Cleveland LS. Identification of soluble and membrane-bound isoforms of the murine flt3 ligand generated by alternative splicing of mRNAs. Oncogene. 1995;10:149–157. [PubMed] [Google Scholar]
- 9.Pixley FJ, Stanley ER. CSF-1 regulation of the wandering macrophage: complexity in action. Trends Cell Biol. 2004;14:628–638. doi: 10.1016/j.tcb.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 10.Kollet O, Dar A, Shivtiel S, Kalinkovich A, Lapid K, Sztainberg Y, Tesio M, Samstein RM, Goichberg P, Spiegel A, Elson A, Lapidot T. Osteoclasts degrade endosteal components and promote mobilization of hematopoietic progenitor cells. Nat Med. 2006;12:657–664. doi: 10.1038/nm1417. [DOI] [PubMed] [Google Scholar]
- 11.Heissig B, Hattori K, Dias S, Friedrich M, Ferris B, Hackett NR, Crystal RG, Besmer P, Lyden D, Moore MA, Werb Z, Rafii S. Recruitment of stem and progenitor cells from the bone marrow niche requires MMP-9 mediated release of kit-ligand. Cell. 2002;109:625–637. doi: 10.1016/s0092-8674(02)00754-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kawaguchi N, Horiuchi K, Becherer JD, Toyama Y, Besmer P, Blobel CP. Different ADAMs have distinct influences on Kit ligand processing: phorbol-ester-stimulated ectodomain shedding of Kitl1 by ADAM17 is reduced by ADAM19. J Cell Sci. 2007;120:943–952. doi: 10.1242/jcs.03403. [DOI] [PubMed] [Google Scholar]
- 13.Horiuchi K, Miyamoto T, Takaishi H, Hakozaki A, Kosaki N, Miyauchi Y, Furukawa M, Takito J, Kaneko H, Matsuzaki K, Morioka H, Blobel CP, Toyama Y. Cell surface colony-stimulating factor 1 can be cleaved by TNF-alpha converting enzyme or endocytosed in a clathrin-dependent manner. J Immunol. 2007;179:6715–6724. doi: 10.4049/jimmunol.179.10.6715. [DOI] [PubMed] [Google Scholar]
- 14.Horiuchi K, Kimura T, Miyamoto T, Miyamoto K, Akiyama H, Takaishi H, Morioka H, Nakamura T, Okada H, Blobel CP, Toyama Y. Conditional inactivation of TACE by a Sox9 promoter leads to osteoporosis and increased granulopoiesis via dysregulation of IL-17 and G-CSF. J Immunol. 2009;182:2093–2101. doi: 10.4049/jimmunol.0802491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seals DF, Courtneidge SA. The ADAMs family of metalloproteases: multidomain proteins with multiple functions. Genes Dev. 2003;17:7–30. doi: 10.1101/gad.1039703. [DOI] [PubMed] [Google Scholar]
- 16.Blobel CP. ADAMs: key components in EGFR signalling and development. Nat Rev Mol Cell Biol. 2005;6:32–43. doi: 10.1038/nrm1548. [DOI] [PubMed] [Google Scholar]
- 17.Moss ML, Jin SL, Milla ME, Bickett DM, Burkhart W, Carter HL, Chen WJ, Clay WC, Didsbury JR, Hassler D, Hoffman CR, Kost TA, Lambert MH, Leesnitzer MA, McCauley P, McGeehan G, Mitchell J, Moyer M, Pahel G, Rocque W, Overton LK, Schoenen F, Seaton T, Su JL, Becherer JD. Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-alpha. Nature. 1997;385:733–736. doi: 10.1038/385733a0. [DOI] [PubMed] [Google Scholar]
- 18.Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, Castner BJ, Stocking KL, Reddy P, Srinivasan S, Nelson N, Boiani N, Schooley KA, Gerhart M, Davis R, Fitzner JN, Johnson RS, Paxton RJ, March CJ, Cerretti DP. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature. 1997;385:729–733. doi: 10.1038/385729a0. [DOI] [PubMed] [Google Scholar]
- 19.Murphy G, Murthy A, Khokha R. Clipping, shedding and RIPping keep immunity on cue. Trends Immunol. 2008;29:75–82. doi: 10.1016/j.it.2007.10.009. [DOI] [PubMed] [Google Scholar]
- 20.Levine SJ. Mechanisms of soluble cytokine receptor generation. J Immunol. 2004;173:5343–5348. doi: 10.4049/jimmunol.173.9.5343. [DOI] [PubMed] [Google Scholar]
- 21.Peschon JJ, Slack JL, Reddy P, Stocking KL, Sunnarborg SW, Lee DC, Russell WE, Castner BJ, Johnson RS, Fitzner JN, Boyce RW, Nelson N, Kozlosky CJ, Wolfson MF, Rauch CT, Cerretti DP, Paxton RJ, March CJ, Black RA. An essential role for ectodomain shedding in mammalian development. Science. 1998;282:1281–1284. doi: 10.1126/science.282.5392.1281. [DOI] [PubMed] [Google Scholar]
- 22.Horiuchi K, Kimura T, Miyamoto T, Takaishi H, Okada Y, Toyama Y, Blobel CP. TNFalpha-converting enzyme (TACE/ADAM17) inactivation in mouse myeloid cells prevents lethality from endotoxin shock. J Immunol. 2007;179:2686–2689. doi: 10.4049/jimmunol.179.5.2686. [DOI] [PubMed] [Google Scholar]
- 23.Schlondorff J, Becherer JD, Blobel CP. Intracellular maturation and localization of the tumour necrosis factor alpha convertase (TACE) Biochem J. 2000;347:131–138. [PMC free article] [PubMed] [Google Scholar]
- 24.Sahin U, Weskamp G, Zheng Y, Chesneau V, Horiuchi K, Blobel CP. A sensitive method to monitor ectodomain shedding of ligands of the epidermal growth factor receptor. Methods Mol Biol. 2006;327:99–113. doi: 10.1385/1-59745-012-X:99. [DOI] [PubMed] [Google Scholar]
- 25.Baker AH, Edwards DR, Murphy G. Metalloproteinase inhibitors: biological actions and therapeutic opportunities. J Cell Sci. 2002;115:3719–3727. doi: 10.1242/jcs.00063. [DOI] [PubMed] [Google Scholar]
- 26.Lambert E, Dasse E, Haye B, Petitfrere E. TIMPs as multifacial proteins. Crit Rev Oncol Hematol. 2004;49:187–198. doi: 10.1016/j.critrevonc.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 27.Weskamp G, Schlondorff J, Lum L, Becherer JD, Kim TW, Saftig P, Hartmann D, Murphy G, Blobel CP. Evidence for a critical role of the tumor necrosis factor alpha convertase (TACE) in ectodomain shedding of the p75 neurotrophin receptor (p75NTR) J Biol Chem. 2004;279:4241–4249. doi: 10.1074/jbc.M307974200. [DOI] [PubMed] [Google Scholar]
- 28.Fan H, Derynck R. Ectodomain shedding of TGF-alpha and other transmembrane proteins is induced by receptor tyrosine kinase activation and MAP kinase signaling cascades. Embo J. 1999;18:6962–6972. doi: 10.1093/emboj/18.24.6962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Le Gall SM, Bobe P, Reiss K, Horiuchi K, Niu XD, Lundell D, Gibb DR, Conrad D, Saftig P, Blobel CP. ADAMs 10 and 17 represent differentially regulated components of a general shedding machinery for membrane proteins such as transforming growth factor alpha, L-Selectin, and tumor necrosis factor alpha. Mol Biol Cell. 2009;20:1785–1794. doi: 10.1091/mbc.E08-11-1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weskamp G, Ford JW, Sturgill J, Martin S, Docherty AJ, Swendeman S, Broadway N, Hartmann D, Saftig P, Umland S, Sehara-Fujisawa A, Black RA, Ludwig A, Becherer JD, Conrad DH, Blobel CP. ADAM10 is a principal ’sheddase’ of the low-affinity immunoglobulin E receptor CD23. Nat Immunol. 2006;7:1293–1298. doi: 10.1038/ni1399. [DOI] [PubMed] [Google Scholar]
- 31.Chklovskaia E, Nissen C, Landmann L, Rahner C, Pfister O, Wodnar-Filipowicz A. Cell-surface trafficking and release of flt3 ligand from T lymphocytes is induced by common cytokine receptor gamma-chain signaling and inhibited by cyclosporin A. Blood. 2001;97:1027–1034. doi: 10.1182/blood.v97.4.1027. [DOI] [PubMed] [Google Scholar]
- 32.Zhang Z, Kolls JK, Oliver P, Good D, Schwarzenberger PO, Joshi MS, Ponthier JL, Lancaster JR., Jr Activation of tumor necrosis factor-alpha-converting enzyme-mediated ectodomain shedding by nitric oxide. J Biol Chem. 2000;275:15839–15844. doi: 10.1074/jbc.M000604200. [DOI] [PubMed] [Google Scholar]
- 33.Koff JL, Shao MX, Kim S, Ueki IF, Nadel JA. Pseudomonas lipopolysaccharide accelerates wound repair via activation of a novel epithelial cell signaling cascade. J Immunol. 2006;177:8693–8700. doi: 10.4049/jimmunol.177.12.8693. [DOI] [PubMed] [Google Scholar]
- 34.Rovida E, Paccagnini A, Del Rosso M, Peschon J, Dello Sbarba P. TNF-alpha-converting enzyme cleaves the macrophage colony-stimulating factor receptor in macrophages undergoing activation. J Immunol. 2001;166:1583–1589. doi: 10.4049/jimmunol.166.3.1583. [DOI] [PubMed] [Google Scholar]
- 35.Horiuchi K, Le Gall S, Schulte M, Yamaguchi T, Reiss K, Murphy G, Toyama Y, Hartmann D, Saftig P, Blobel CP. Substrate selectivity of epidermal growth factor-receptor ligand sheddases and their regulation by phorbol esters and calcium influx. Mol Biol Cell. 2007;18:176–188. doi: 10.1091/mbc.E06-01-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hirata M, Umata T, Takahashi T, Ohnuma M, Miura Y, Iwamoto R, Mekada E. Identification of serum factor inducing ectodomain shedding of proHB-EGF and sStudies of noncleavable mutants of proHB-EGF. Biochem Biophys Res Commun. 2001;283:915–922. doi: 10.1006/bbrc.2001.4879. [DOI] [PubMed] [Google Scholar]
- 37.Hinkle CL, Sunnarborg SW, Loiselle D, Parker CE, Stevenson M, Russell WE, Lee DC. Selective roles for tumor necrosis factor alpha-converting enzyme/ADAM17 in the shedding of the epidermal growth factor receptor ligand family: the juxtamembrane stalk determines cleavage efficiency. J Biol Chem. 2004;279:24179–24188. doi: 10.1074/jbc.M312141200. [DOI] [PubMed] [Google Scholar]
- 38.Zheng Y, Saftig P, Hartmann D, Blobel C. Evaluation of the Contribution of Different ADAMs to Tumor Necrosis Factor alpha (TNFalpha) Shedding and of the Function of the TNFalpha Ectodomain in Ensuring Selective Stimulated Shedding by the TNFalpha Convertase (TACE/ADAM17) J Biol Chem. 2004;279:42898–42906. doi: 10.1074/jbc.M403193200. [DOI] [PubMed] [Google Scholar]
- 39.Overall CM, Blobel CP. In search of partners: linking extracellular proteases to substrates. Nat Rev Mol Cell Biol. 2007;8:245–257. doi: 10.1038/nrm2120. [DOI] [PubMed] [Google Scholar]
- 40.Sahin U, Weskamp G, Kelly K, Zhou HM, Higashiyama S, Peschon J, Hartmann D, Saftig P, Blobel CP. Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J Cell Biol. 2004;164:769–779. doi: 10.1083/jcb.200307137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Harris RC, Chung E, Coffey RJ. EGF receptor ligands. Exp Cell Res. 2003;284:2–13. doi: 10.1016/s0014-4827(02)00105-2. [DOI] [PubMed] [Google Scholar]
- 42.Yamazaki S, Iwamoto R, Saeki K, Asakura M, Takashima S, Yamazaki A, Kimura R, Mizushima H, Moribe H, Higashiyama S, Endoh M, Kaneda Y, Takagi S, Itami S, Takeda N, Yamada G, Mekada E. Mice with defects in HB-EGF ectodomain shedding show severe developmental abnormalities. J Cell Biol. 2003;163:469–475. doi: 10.1083/jcb.200307035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brannan CI, Lyman SD, Williams DE, Eisenman J, Anderson DM, Cosman D, Bedell MA, Jenkins NA, Copeland NG. Steel-Dickie mutation encodes a c-kit ligand lacking transmembrane and cytoplasmic domains. Proc Natl Acad Sci U S A. 1991;88:4671–4674. doi: 10.1073/pnas.88.11.4671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wong GG, Temple PA, Leary AC, Witek-Giannotti JS, Yang YC, Ciarletta AB, Chung M, Murtha P, Kriz R, Kaufman RJ, Ferenz CR, Sibley BS, Turner KJ, Hewick RM, Clark SC, Yanai N, Yokota H, Yamada M, Saito M, Totoyoshi K, Takaku F. Human CSF-1: molecular cloning and expression of 4-kb cDNA encoding the human urinary protein. Science. 1987;235:1504–1508. doi: 10.1126/science.3493529. [DOI] [PubMed] [Google Scholar]
- 45.Price LK, Choi HU, Rosenberg L, Stanley ER. The predominant form of secreted colony stimulating factor-1 is a proteoglycan. J Biol Chem. 1992;267:2190–2199. [PubMed] [Google Scholar]
- 46.Chitu V, Stanley ER. Colony-stimulating factor-1 in immunity and inflammation. Curr Opin Immunol. 2006;18:39–48. doi: 10.1016/j.coi.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 47.Yao GQ, Wu JJ, Sun BH, Troiano N, Mitnick MA, Insogna K. The cell surface form of colony-stimulating factor-1 is biologically active in bone in vivo. Endocrinology. 2003;144:3677–3682. doi: 10.1210/en.2002-221071. [DOI] [PubMed] [Google Scholar]
- 48.Nandi S, Akhter MP, Seifert MF, Dai XM, Stanley ER. Developmental and functional significance of the CSF-1 proteoglycan chondroitin sulfate chain. Blood. 2006;107:786–795. doi: 10.1182/blood-2005-05-1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dai XM, Zong XH, Sylvestre V, Stanley ER. Incomplete restoration of colony-stimulating factor 1 (CSF-1) function in CSF-1-deficient Csf1op/Csf1op mice by transgenic expression of cell surface CSF-1. Blood. 2004;103:1114–1123. doi: 10.1182/blood-2003-08-2739. [DOI] [PubMed] [Google Scholar]