

Abstract
The objective of this study was to evaluate the effect of vector architecture on DNA condensation, particle stability, and gene transfer efficiency. Two recombinant non-viral vectors with the same amino acid compositions but different architectures, composed of lysine-histidine (KH) repeating units fused to fibroblast growth factor, were genetically engineered. In one vector lysine residues were dispersed (KHKHKHKHKK)6-FGF2, whereas in the other they were in clusters (KKKHHHHKKK)6-FGF2. Organization of lysine residues in this manner was inspired by the sequence of DNA condensing motifs that exist in nature (e.g., histones) where lysine residues are organized in clusters. These two constructs were compared in terms of DNA condensation and gene transfer efficiency. It was observed that the construct with KH units in clusters was able to condense pDNA into more stable particles with sizes <150 nm making them suitable for cellular uptake via receptor mediated endocytosis. This in turn resulted in five times higher transfection efficiency for the cKH-FGF2. This study demonstrates that in targeted non-viral gene transfer, the vector architecture plays as significant a role as its amino acid sequence. Thus, in the design of the non-viral vectors (synthetic or recombinant) this factor should be considered of paramount importance.
Keywords: Lysine-histidine, Non-viral vector, Targeted gene therapy, Vector architecture, Gene delivery
1. Introduction
A major limiting factor to gene therapy is the lack of a suitable gene delivery system to carry the therapeutic genes to the target tissues [1]. Advancements in gene therapy in general depend on the development of novel gene delivery systems (vectors) with high transfection efficiency at the target site and low toxicity.
Based on current understanding of the barriers to systemic gene transfer [2,3], serum nucleases, endosomal entrapment, and the nuclear membrane play significant roles in limiting the number of intact genes that reach the cell nucleus for successful transcription. Viruses have evolved to efficiently overcome these barriers; however, safety and toxicity issues have limited their use for systemic gene delivery [4].
In contrast, non-viral vectors can be utilized to deliver therapeutic genes to target cells without significant toxicity; however, they suffer from low transfection efficiency, thus conferring the need for the design and development of new vectors that are both efficient and safe. For a vector to be maximally effective, it should protect the DNA from serum endonucleases, disrupt the endosome membrane promoting escape of the DNA into cytosol, and facilitate translocation of the genetic material towards the cell nucleus. Positively charged motifs such as adenovirus μ peptide (arginine rich), histones (lysine rich), poly L-lysines, and others have been found to be efficient in condensing pDNA and protecting it from degradation by nucleases [5–7]. After endocytic uptake, plasmid DNA (pDNA) is sequestered in endosomes and trafficked through the cytoplasm for eventual fusion with lysosomal vesicles. To avoid degradation by lysosomal enzymes pDNA must escape from the endosomes prior to lysosomal fusion [8]. It has been shown that histidine residues can be utilized in the vector structure to disrupt endosome membranes thereby promoting endosomal escape [9–12]. This endosomolytic property is thought to arise from the “proton-sponge” effect [13,14]. Although the science of targeted non-viral gene transfer has come a long way, inefficiency still remains a significant challenge due to the many hurdles that must be overcome [15].
The focus of this research is on engineering well-defined non-viral vectors using genetic engineering techniques and evaluating their potential for targeted gene transfer. Recombinant DNA technology has empowered us to exert full control over vector structure at the molecular level and fine tune its physicochemical properties for specific gene delivery needs.
The biosynthesis and characterization of a prototype recombinant vector with the structure (KHKHKHKHKK)6-FGF2, namely dKH-FGF2, which contained 36 lysine residues (K) in the dKH segment to condense pDNA, and 24 histidine residues (H) to promote endosomal escape has been reported previously [16]. At the C-terminus of the dKH segment, FGF2 represents basic fibroblast growth factor, a ligand for the basic fibroblast growth factor receptor (FGFR). This receptor is known to be over-expressed in subpopulations of lung, prostate, and breast cancer, thus conferring the potential for targeted gene delivery via receptor-mediated endocytosis [17]. As a starting point, the arrangement of lysine residues in the dKH tail (i.e., KHKHKHKHKK) was designed as dispersed, while keeping the lysine to histidine ratio constant at 6:4. This ratio was chosen based on previous studies reported by Midoux and Monsigny [18]. Although dKH-FGF2 was able to condense pDNA into nano-size particles (average 231 nm) and transfer genes into target cancer and non-cancer cells [16], the percentage of the transfected cells in the absence of serum was five times higher than in the presence of serum. This prompted us to further characterize dKH-FGF2 and modify its structure to an extent that the transfection efficiency increased in the presence of serum. It was hypothesized that by changing the arrangement of KH residues in the KHKHKHKHKK repeating units and organizing the lysine residues in clusters, the pDNA condensation efficiency will be improved resulting in more compact and stable nanocarriers with higher transfection efficiency. This hypothesis was inspired by motifs that exist in nature (e.g., histones and adenovirus μ peptide) that have lysine and arginine residues arranged in clusters and have been shown to be highly efficient in DNA condensation [19–21]. To test the hypothesis, an analogue of dKH-FGF2 was designed: (KKKHHHHKKK)6–FGF, namely cKH-FGF2.
In this article, the word “architecture” literally refers to the manner in which the components of the vector are organized and integrated.
2. Materials and methods
2.1. Cloning and expression of cKH-FCF2
The gene encoding cKH was designed, expression optimized, and synthesized (Blueheron Biotechnology Inc., Bothel, WA) with N-terminal Ndel and C-terminal EcoRI restriction sites. The synthesized gene was double digested with NdeI and EcoRI (New England Biolabs, lpswich, MA) restriction enzymes and cloned into a pET21b-FGF2 expression vector which contained the FGF2 gene in between EcoRI and HindIII. The parent pET21b vector was purchased from EMD Biosciences (Gibbstown, NJ). The successful cloning of the cKH gene in pET21b-FGF2 vector was verified by DNA sequencing and back translation into the corresponding amino acid sequence.
The pET21b-cKH-FGF2 vector was transformed into E. coli BL21 (DE3) (Novagen, San Diego, CA), grown using Barnstead-Labline MAX Q 4000 shaking incubator, and expressed by the addition of IPTG to a final concentration of 0.4 mM at 30 °C. The cells were harvested, lysed, and centrifuged for 40 minutes at 30,000 g (4 °C) to pellet the insoluble fraction. The soluble fraction was removed and loaded onto a Ni-NTA column (Amersham Biosciences, Piscataway, NJ) for purification. The column was washed with 50 volumes of wash buffer (50 mM phosphate buffer, 10 mM Tris, 1 M NaCl, 20 mM immidazole) and eluted with 300 mM immidazole. The purity and expression of the vector were confirmed by SDS-PAGE and western blot analysis, respectively. The vector was dialyzed versus Dulbecco’s phosphate buffered saline (DPBS) and stored at −80°C after addition of 20 mM Tris buffer (pH=7.4), 250 mM NaCl, 50 mM KCl, 2 mM (β–mercaptoethanol and 20% glycerin.
The exact molecular weight and amino acid content of the purified cKH-FGF2 was determined by mass spectroscopy and amino acid content analysis (Commonwealth Biotechnologies Inc., Richmond, VA).
2.2. Particle size and charge analysis
The mean hydrodynamic sizes and surface charges of vector/pDNA complexes were determined using a Malvern zeta/particle sizer and software (Malvern Instruments, UK). Before complexation, the vector solution was dialyzed versus 10 mM phosphate buffer and 5 mM NaCl for 30 minutes. Various amounts of vector were added to 2 μg of pDNA (pEGFP) to form complexes at N/P ratios of 0.5,1, 2, 4, and 6. After 30 minutes of incubation time, the size and zeta potential of the complexes were measured and reported as mean ± SEM (n=3).
The particles size measurements are performed using Dynamic Light Scattering (DLS). The particles are illuminated with a laser and the intensity of scattered light is collected. Due to Brownian motion, particles continue moving resulting in fluctuating intensity of the scattered light. A digital correlator measures the degree of similarity of scattered light intensity at different times and generates a correlation curve that reflects the decay rate. Based on the Stokes–Einstein equation, larger particles move more slowly and therefore the correlation decay rate is slower for larger particles. The correlation function is then used to generate the size distribution of the particles.
2.3. Mitogenic assay
The details of the mitogenic assay for dKH-FGF2 have been reported previously [16]. Briefly, NIH 3T3 cells were grown in F12/DMEM (1:1 ratio) with 10% fetal calf serum (FCS). Cells were washed with a serum-free medium (SFM) and 5×103 cells were seeded in a 96-well dish in 150 μl of SFM. A serial dilution of cKH-FGF2 and native FGF2 (Promega, Madison WI, USA) was prepared across the plate ranging from 0 to 50 ng/ml. The control well with 0 ng/ml concentration received PBS. Cells were incubated for 44 hours and after the incubation time, WST-1 (Roche Applied Science, IN, USA) reagent was added and after 4 hours the absorbance was measured at 440 nm. The data is reported as Mean ± SD, n=6.
2.4. Cell toxicity assay
Cell toxicity assays were performed in DMEM/F12 (90%) supplemented with FCS (10%) as described previously for dKH-FGF2 [16]. NIH3T3 cells (5×103/well) were seeded in a 96-well dish in 150 μl of SFM and incubated overnight. A serial dilution of cKH-FGF2 or cKH-FGF2/pEGFP complexes was prepared across the plate (equivalent of 0 to 80 μg/ml cKH-FGF2). The control well with 0 μg/ml concentration received PBS. Cells were incubated with test groups for 4 hours, washed, and incubated with fresh media overnight. The next day, WST-1 reagent was added, incubated for 4 hours, and the absorbance was measured at 440 nm. The measured absorbance for test groups is expressed as percent of the control (defined as 1). The control cells were treated with PBS. The data is reported as Mean ± SD, n=6.
2.5. Cell culture and transfection
NIH 3T3 cells (mouse embryo fibroblast) and T-47D cells (human breast cancer) were propagated as suggested by the American Type Culture Collection (VA, USA). Cells were seeded in 12-well tissue culture plates (in triplicate) at 5×104 cells per well in 1 ml growth media with 10% heat-inactivated serum (Invitrogen). Cells were approximately 70–80% confluent at the time of transfection. pEGFP under the control of CMV promoter (Clontech, CA) or pRLCMV-luc (Promega, Madison, WI) at a concentration of 3 μg/50 μl was mixed with vector (N/P=1) in 50 μl of 10 mM phosphate buffer and 5 mM NaCl and incubated for 30 min at room temperature for complex formation. The complexes were then added to the growth media supplemented with 10% heat-inactivated serum. This was added to the cells which were then incubated at 37 °C in humidified 5% CO2 atmosphere. After 4 hours, the growth media was removed and replaced with fresh growth media (DMEM 90% and serum 10%). The GFP expression was visualized using a confocal microscope whereas luciferase activity was measured by using Promega’s luciferase assay kit and protocol. Using a previously reported method [16], the percentage of transfected cells was calculated and reported as mean± standard deviation (n=9) for this experiment. When used, 100 μM chloroquine (Sigma) was added to the culture media at the time of cell transfection. Bafilomycin A1 (Sigma) was added to the cell culture media at the time of transfection at a concentration of 100 nM. The data is reported as Mean ± SD, n=3. Lipofectamine 2000 (Invitrogen, Carlsbad, CA) was used as positive control.
2.6. Inhibition assay by FCF2
This method has previously been reported for dKH-FGF2 [16]. In brief, NIH 3T3 cells were seeded in 12-well tissue culture plates at 5×104 cells per well in 1 ml SFM. Cells were approximately 70–80% confluent at the time of transfection. pEGFP (3 μg/50 μl) was mixed with cKH-FGF2 vector at N/P ratio of 1:1 and incubated for 30 minutes at room temperature for complex formation. In one set of wells, FGF2 (1000 ng/ml) was added followed by addition of complexes. In the second set, SFM was added followed by addition of complexes (control). The cells were incubated at 37 °C in humidified 5% CO2 atmosphere. After 4 hours, the growth media was removed and replaced with fresh growth media with serum. The GFP expression was quantified using a previously reported method [15]. The data is reported as Mean ± SD, n=9.
3. Results
3.1. cKH-FGF2 cloning, expression, and purification
Using the cloning strategy shown in Fig. 1a, the gene encoding cKH-FGF2 was cloned into a pET21b expression vector to make pET21b-cKH-FGF2 and sequenced to verify its fidelity to the original design. The results of the DNA sequencing revealed that both sense and antisense strands were free of any mutations and corresponded to the amino acid sequence shown in Fig. 1b.
Fig. 1.
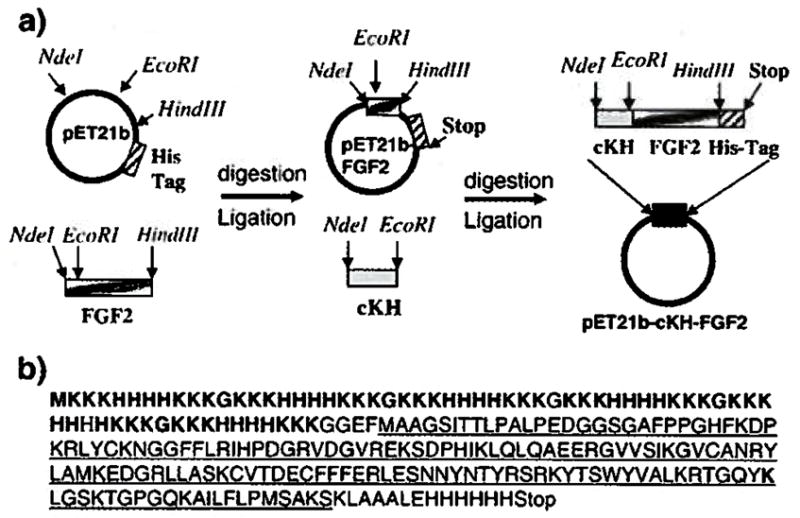
(a) An overview of the cloning strategy used to make a pET21b expression vector containing the gene encoding cKH-FGF2. The FGF2 gene was cloned into pET21b vector in between EcoRI and Hindlll restriction sites to make pET21b-FGF2 vector. The synthesized cKH gene was cloned into pET21b-FGF2 vector in between Ndel and EcoRI restriction sites to make pET21b-cKH-FGF2 expression vector, (b) The corresponding amino acid sequence of the cloned cKH-FGF2 gene. The cKH sequence is shown in bold and the amino acid sequence of FGF2 is underlined.
The expression of the vector was confirmed by western blot analysis and the purity of the vector was determined to be >98% by SDS-PAGE analysis (Fig. 2). The expressed vector was further characterized by mass spectroscopy and amino acid content analysis to determine the exact molecular weight and amino acid composition. The observed molecular weight was determined to be 27,421 Da which was in close agreement with the expected value (i.e., 27,486 Da) (data not shown). The observed amino acid content of the purified vector was also in close agreement with the expected amino acid composition (data not shown).
Fig. 2.
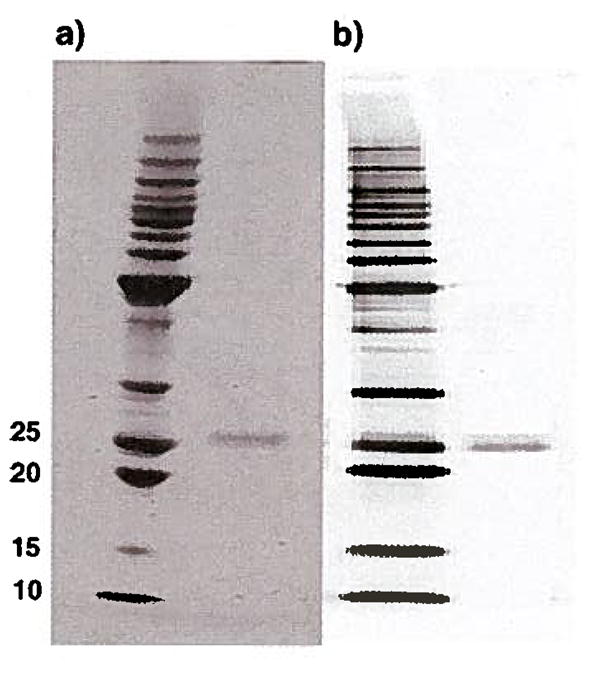
(a) SDS-PAGE analysis of purified cKH-FGF2 with purity higher than 98%. (b) The western blot analysis of the expressed CKH-FGF2 using monoclonal anti-histag antibody. The expected molecular weight of cKH-FGF2 is 27486 Da.
3.2. Particle size, charge, and stability analysis
The ability of both vectors to condense model plasmid DNA (pEGFP) was examined in the presence of 10 mM phosphate buffer and 5 mM NaCl. Various amounts of cKH-FGF2 and dKH-FGF2 were complexed with 2 μg of pEGFP to form complexes. The best results were obtained at N/P ratio of 1 with a resulting average particle size of 115±15 nm for cKH-FGF2 and 205 ± 16 for dKH-FGF2 (Fig. 3a).
Fig. 3.

(a) Particle size analysis of cKH-FGF2/pDNA and dKH-FGF2/pDNA complexes at various N/P ratios in the presence of 10 mM phosphate buffer and 5 mM NaCl. (b) Stability of cKH-FGF2/pDNA and dKH-FGF2/pDNA complexes in the presence of 10 mM phosphate buffer and 150 mM NaCl are demonstrated. Complexes were formed at N/P ratio of 1. For particles formed with cKH-FGF2, the size slightly increased from 115 nm ± 15 to 156 ± 13, but remained stable over 55 min. For particles formed with dKH-FGF2, the size increased steadily from 205±16 nm to 668±24 nm.
The stability of particles under conditions close to physiologic ionic strength (i.e., 10 mM phosphate buffer and 150 mM NaCl) was monitored over 55 minutes. The results showed that CKH-FGF2 was able to form stable particles which only slightly increased in particle size (115 nm → 156 nm) whereas dKH-FGF2 was not able to produce stable particles under the stated conditions (205 nm → 668 nm) (Fig. 3b).
The surface charge of the complexes at N/P ratio of 1 was also determined to be +5±3 for dKH-FGF2 and +7±4 for cKH-FGF2.
3.3. Mitogenic and toxicity assays
Using a WST-1 cell proliferation assay, the bioactivity of the FGF2 segment of cKH-FGF2 was evaluated and compared with native FGF2 in NIH 3T3 fibroblasts known to express the FGFR. The significant mitogenic activity as well as non toxicity of dKH-FGF2 have been reported previously [16]. The FGF2 motif present in cKH-FGF2 was shown to be active in terms of inducing cell proliferation in fibroblasts when they were exposed to concentrations of vector that mimicked physiological FGF2 levels. The results of mitogenic assay showed that the mitogenic activity of cKH-FGF2 was as significant as native FGF2 up to 1 ng/ml; and overall, cKH-FGF2 induced significant cell growth in comparison to PBS control (Fig. 4a).
Fig. 4.
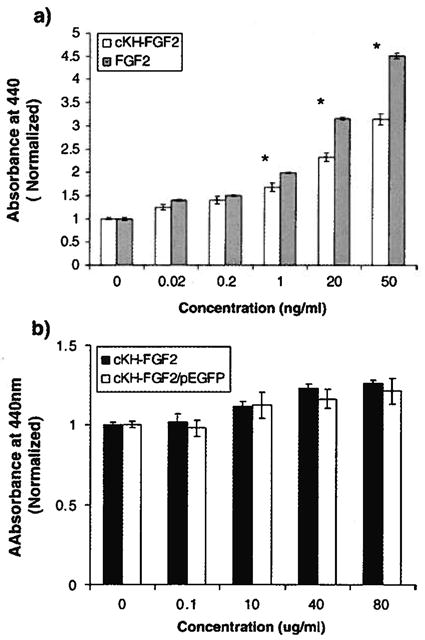
(a) WST-1 cell proliferation assay for NIH 3T3 cells treated with cKH-FGF2 and native FGF2. Cells were treated with various concentrations ranging from 0 (control) to 50 ng/ml and the absorbance of soluble form azan was measured at 440 nm (*t-test, two-tailed, p<0.05). (b) WST-1 cell toxicity assay for NIH 3T3 cells treated with various concentrations of cKH-FGF2 or cKH-FGF2/pEGFP (equivalent of 0 to 80 μg/ml vector). In the tested range, no toxicity was observed with the vector (t-test, two-tailed, p<0.05).
The toxicity of the cKH-FGF2 or cKH-FGF2/pEGFP in NIH3T3 cells was also evaluated by exposing the cells to super-physiological concentrations of the vector ranging from 100 to 80,000 ng/ml. The results demonstrated that the vector does not have any significant effect on cell viability regardless of the dose (Fig. 4b).
3.4. In vitro cell transfection by vector/pEGFP complexes
To evaluate transfection efficiency in terms of percent transfected cells, the pEGFP plasmid was condensed with each vector and functioned as a reporter to monitor the percentage of transfected cells in NIH 3T3 and T-47D cells in the presence of serum. Both vectors were complexed with pEGFP at N/P ratio of 1 and used to transfect NIH3T3 and T47D cells. At N/P ratio of 1, the percent transfected cells with cKH-FGF2 in NIH3T3 and T47D was 41 ±4 and 28±5, respectively (mean ± SD, n=9). The percentages of transfected cells with dKH-FGF2 in NIH3T3 and T47D were 9±3 and 7±2, respectively (Fig. 5).
Fig. 5.
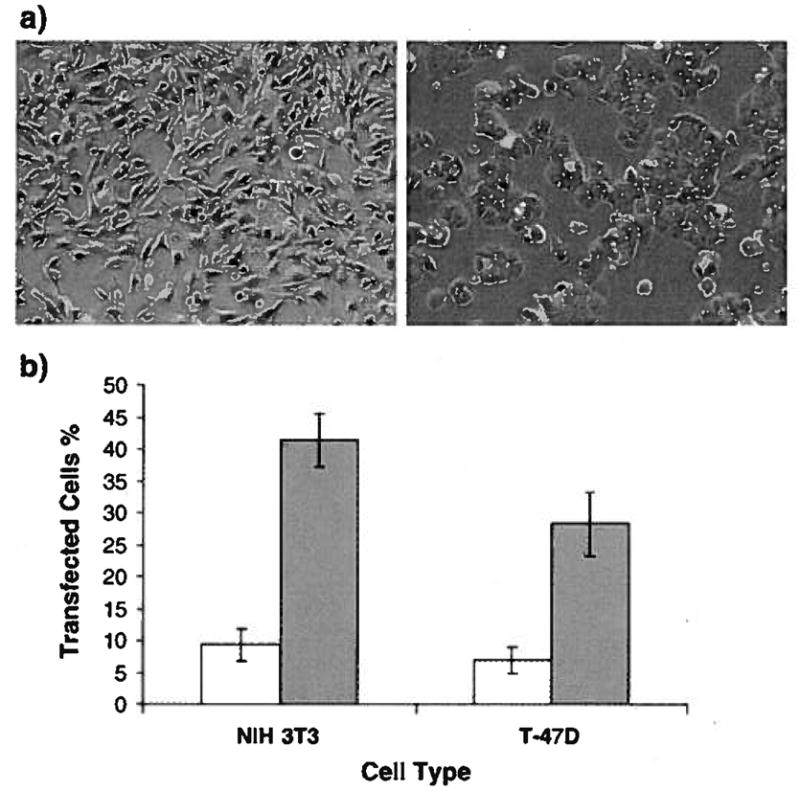
(a) Representative confocal images of NIH3T3 (left) and T47D (right) cells transfected with cKH-FGF2/pEGFP complexes. The green dots are the cells expressing green fluorescent protein (GFP). (b) Percentage of cells transfected with cKH-FGF2/pEGFP (closed bar) and dKH-FGF2/pEGFP (open bar). Cells were transfected with vectors in DMEM supplemented with serum. The percent transfected cells with cKH-FGF2 in NIH3T3 and T47D was 41 ±4 and 28±5, respectively (mean ± SD, n=9). The percentages of transfected cells with dKH-FGF2 in NIH3T3 andT47D were 9±3 and 7±2, respectively. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.5. Inhibition assay
To evaluate whether specific uptake occurred through FGFR, transfection experiments were conducted on NIH 3T3 cells in the presence of 1000 ng/ml free FGF2 (n=3).The results of inhibition assay demonstrated that the presence of FGF2 in the media saturated the FGF2 receptors and significantly inhibited the transfection efficiency of the cKH-FGF2 vector. The transfection efficiency reduced from 49 ± 8% to 6±3% (Fig. 6). This is similar to what we observed for dKH-FGF2 [16].
Fig. 6.
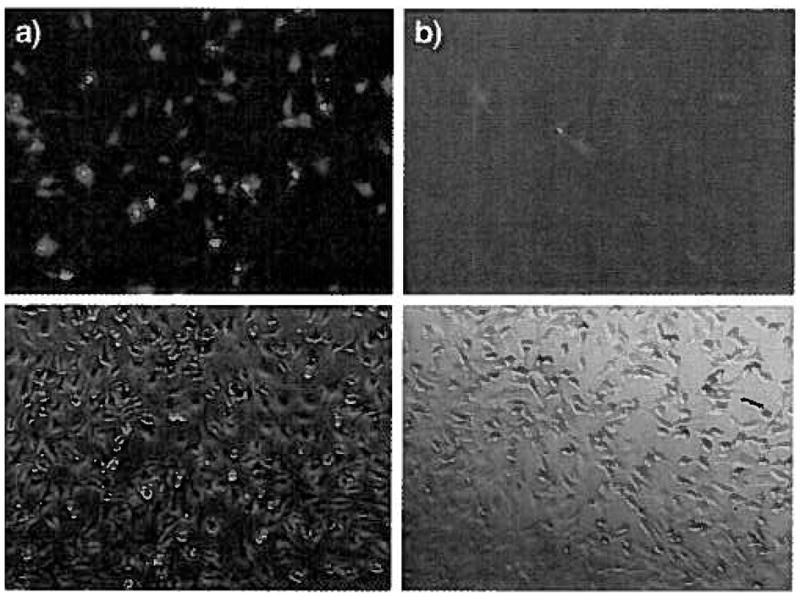
Inhibition assay was performed to demonstrate the transfection of cells via FGF2 receptor-mediated endocytosis. (a) Confocal microscopy image of NIH 3T3 cells transfected with cKH-FGF2/pEGFP in serum free media (SFM); (b) confocal microscopy image of NIH 3T3 cells transfected with cKH-FGF2/pEGFP in SFM with addition of 1000 ng/ml FGF2.
3.6. Influence of histidine on endosomal escape and transfection efficiency
Using plasmid DNA encoding luciferase (pRLCMV-luc), the levels of gene expression by cKH-FGF2 and dKH-FGF2 in NIH3T3 cells were also quantitated. It was observed that CKH-FGF2 was able to transfect NIH3T3 cells approximately five times more than dKH-FGF2. Luciferase activity was determined to be 95 ±5 and 16±4 RLU/μg protein for cKH-FGF2 and dKH-FGF2, respectively (Fig. 7).
Fig. 7.

Comparison of the transfection efficiency between cKH-FGF2 and dKH-FGF2 in NIH3T3 cells. Luciferase activity was measured as described in Materials and methods and determined to be 95±5 and 16±4 for cKH-FGF2 and dKH-FGF2, respectively. Lipofectamine was used as positive control.
The effect of histidine residues in promoting the endosomal escape was also evaluated by complexing the vectors with pRLCMV-luc and transfecting NIH3T3 cells in the presence of 100 μM chloroquine and 100 nM bafilomycin A. In the presence of chloroquine, the transfection efficiency of cKH-FGF2 increased from 95±5 to 142 ±9 and for dKH- FGF2 it increased from 16±4 to 29±6 RLU/μg protein (Fig. 8). In the presence of bafilomycin A, the transfection efficiency of CKH-FGF2 dropped from 95±5 to 15±4, whereas the transfection efficiency of dKH-FGF2 decreased from 16±4 to 6±2 RLU/μg protein.
Fig. 8.
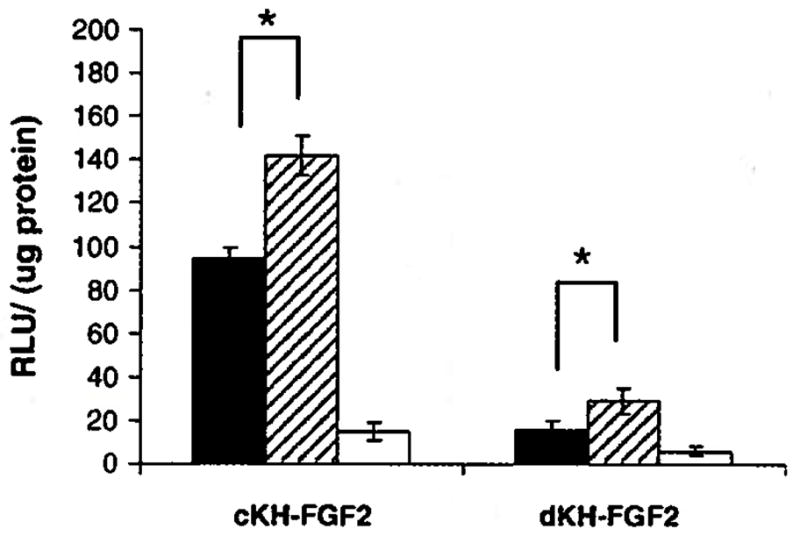
Comparison of transfection efficiency of cKH-FGF2 and dKH-FGF2 in the absence (closed bar) and in the presence of chloroquine (hatched bar) and bafilomycin A (open bar). NIH3T3 cells were transfected with both vector/pDNA complexes and the luciferase activity was measured. The results are shown as mean ± SD (n=3) (*t-test, two-tailed, p<0.05).
4. Discussion
Recombinant DNA technology has allowed the production of amino acid based polymers containing repeating blocks of amino acids with defined compositions, sequences and lengths [22–24]. In this study, the pDNA condensation and gene transfer efficiency of two constructs with similar compositions but different backbone architectures, i.e. dKH-FGF2 and cKH-FGF2, were examined. In the latter, the lysine and histidine residues are arranged in clusters whereas in the former they are dispersed. The biosynthesis and characterization of dKH-FGF2 has been reported previously [16]. The SDS-PAGE and westernblot analysis results demonstrated successful cloning, expression and purification of the cKH-FGF2 vector in E. coli expression system. The molecular weight as well as amino acid content of the expressed vector was also confirmed by mass spectroscopy and amino acid content analysis. Approximately 800 μg of cKH-FGF2 with 98% purity was obtained from one liter of culture media.
The ability of the purified cKH-FGF2 vector to condense pDNA into stable nanosize particles was studied in solutions with different ionic strengths. Various amounts of cKH-FGF2 and dKH-FGF2 were complexed with 2 μg of pEGFP (N/P ratios 0.5,1, 2,4, and 6) to form complexes in the presence of 10 mM phosphate buffer and 5 mM NaCl. The best results were obtained when N/P ratio was 1. At higher N/P ratios the average particle size dramatically increased (>300 nm) which could be due to the interaction between the hydrophobic residues in FGF2 sequence resulting in the aggregation of particles. Such particles are too big to fit in clathrin coated vesicles and cannot be endocytosed via receptors [14,25]. However, they could sediment readily and be taken up by the cells via phagocytosis. Apropos, they were eliminated from the pool of data and not reported in transfection studies. This study shows that both vectors were able to condense pDNA into nanosize particles, but cKH-FGF2 was able to interact more efficiently with pDNA producing smaller particles. To examine the particle stability under conditions close to physiologic ionic strength, complexes were formed at N/P of 1, the concentration of NaCl was increased to physiologic levels (i.e., 150 mM), and the particle size was monitored over 55 minutes. The results showed that cKH-FGF2 was able to form stable particles with slight increase in particle size (115 nm→ 156 nm), whereas dKH-FGF2 was not able to produce stable particles under stated conditions (205 nm→668 nm) (Fig. 3b). This difference in particle stability could be the result of more efficient interaction between cationic residues in cKH-FGF2 with negatively charged phosphate groups in pDNA. Once the electrostatic charges are neutralized, vector/pDNA complex collapses into a condensed particle stabilized by hydrophobic pockets. These hydrophobic pockets are created at the points of interaction between neutralized amine groups in lysine residues and phosphate groups in pDNA. Such pockets could inhibit diffusion of water molecules into the core and minimize the possibility of buffer and salt molecules to interfere with the electrostatic interactions; hence, formation of more stable particles. More studies with other KH-FGF2 analogues could provide a clearer picture to better understand the observed differences. Although the histidine residues are positively charged at physiologic pH (pKa=6.0) and participate in pDNA condensation, lysine residues (pKa=10.5) are the major players in this event. Therefore, in comparison to dKH, cKH with blocks of K residues could have produced dominant hydrophobic pockets resulting in more stabilized particles.
As expected, the surface charges of the complexes formed with both vectors were determined to be close to zero. In targeted gene transfer, particle surface charge does not play as significant a role as in non-targeted vectors because particles are internalized via receptors.
Before utilizing the cKH-FGF2 vector for in vitro cell transfection, it was further characterized in terms of mitogenicity and toxicity in NIH3T3 (fibroblast) cells by using WST-1 cell proliferation and toxicity assays. The mitogenicity assay was performed to confirm the activity of the FGF2 motif in cKH-FGF2, whereas the cell toxicity assay was carried out to evaluate the toxicity of the vector in normal fibroblast cells known to over-express FGF2 receptors [26]. The results showed that in comparison to PBS control, cKH-FGF2 induced significant cell growth (Fig. 4a). This indicates that presence of C-terminal histag or N-terminal cKH did not have any significant effect on the ability of FGF2 to bind to FGFR. We have previously reported similar observations with dKH-FGF2 and have also demonstrated that the lysine-histidine repeating units by themselves do not have mitogenic activity [16]. Therefore, the observed mitogenicity is likely due to the presence of FGF2 segment in cKH-FGF2.
Although the treatment of cells with peptide based vectors could have altered the cellular signaling pathways, these changes did not have any deleterious effect on cell growth/viability (Fig. 4b). This could be due to the susceptibility of KH repeating units to serine proteases inside endosomes degrading them into smaller fragments with less toxicity. This concept has been demonstrated by other groups elsewhere [27].
So far, it was demonstrated that the vector is able to condense pDNA efficiently, target FGFR and internalize, while showing no detectable toxicity. The next logical step is to evaluate the ability of cKH-FGF2 vector in terms of gene transfer efficiency in cells over-expressing FGFR such as NIH3T3 (fibroblasts) and T47D (breast cancer) cells [28]. To compare the transfection efficiency of cKH-FGF2 with dKH-FGF2, they were complexed with pEGFP at N/P ratio of 1 and used to transfect both NIH3T3 and T47D cells in the presence of heat-inactivated serum. In both cell lines, the highest number of transfected cells was observed with cKH-FGF2 in comparison to dKH-FGF2 (Fig. 5). These results were expected because the size of the particles that were formed with cKH-FGF2 at N/P of 1 were not only stable (Fig. 3) but in the optimum range for receptor mediated endocytosis (<200 nm) [25]. We used two different cell lines (cancer and non-cancer) to examine whether the cell type had any effect on the observed differences between the two vectors. The vector cKH-FGF2 was more efficient in gene transfer than dKH-FGF2 in both cell lines. In addition, higher numbers of NIH3T3 cells were transfected in comparison to T47D. This could be due to the presence of higher levels of FGFR on the NIH3T3 cell membranes. Here, all the variables were kept constant except the vector architecture (i.e., cKH versus dKH) which influenced the particle size and stability. Thus, it was deduced that the observed difference in transfection efficiency between the two vectors was directly influenced by the manner the KH repeating units were arranged. In subsequent studies we used NIH3T3 cells in testing the hypotheses, because NIH3T3 cells responded to the treatments with higher sensitivity (Fig. 5).
To investigate whether the targeted cKH-FGF2 vector delivered the pEGFP to the NIH3T3 cells via FGFR, an inhibition assay was performed and cells were transfected in SFM in the presence and absence of native FGF2. The results demonstrated that the presence of FGF2 in the media saturated the FGF2 receptors and significantly inhibited the transfection efficiency of the vector (Fig. 6). This observation reveals that internalization of complexes was facilitated by FGFR, although some non-specific uptake was also observed.
The levels of gene expression were also measured quantitatively by transfecting the NIH3T3 cells with vector/pRLCMV-luc complexes (Fig. 7). It was observed that cKH-FGF2 was able to transfect cells approximately five times more than dKH-FGF2. This could be the result of higher numbers of cKH-FGF2/pDNA complexes that were internalized by the cells which correlates directly to the particle size. In this set of experiments, due to its high efficiency, lipofectamine® was used as positive control to validate the transfection efficiency process. Polymers such as poly L-Lysine are not suitable positive controls because of their inability to escape from endosomes and very low transfection efficiency. The transfection efficiency of dKH has been reported previously [16]. It is noteworthy that commercially available gene transferring agents (e.g., lipofectamine, DOSPER, Poly Lysine, PEI) are not targeted vectors and internalize mostly via phagocytosis or pinocytosis, while the targeted vectors in this study internalize via receptor mediated endocytosis. Therefore, the efficiency of targeted vectors is controlled by the number of receptors available on the surface of the target cells. This concept where the internalization of targeting motifs is dependent upon the availability of the receptors on the surface of cells has been demonstrated elsewhere [29]. Thus, cKH-FGF2 could be highly efficient in transfecting cells that over-express FGFR but very poor in transfecting cells that express low levels of FGFR. Hence, lipofectamine (non-targeted) was used merely as a positive control not as a point of reference to be compared with targeted vectors.
One question that still needs to be answered is whether histidine residues in the vector backbone had any effect on endosomal escape and transfection efficiency. Histidine residues can effectively increase the delivery of pDNA into the cytosol via membrane destabilization of acidic endocytotic vesicles containing vector/pDNA complexes following the protonation of the imidazole groups. This was assessed by transfecting NIH3T3 cells in the absence and presence of bafilomycin A1 and chloroquine. Chloroquine is a buffering agent known to disrupt the endosomal membrane by increasing the pH of the endosome environment and facilitating escape of the cargo into cytosol [18]. In contrast, bafilomycin A is an inhibitor of vacuolar ATPase endosomal proton pump which significantly reduces the escape of the cargo into cytosol [30,31].
When NIH3T3 cells were transfected in the presence of chloroquine an approximately 50% increase in transfection efficiency was observed with both vectors (Fig. 8). This indicates that some particles remained trapped inside a subpopulation of endosomes and could not escape into cytoplasm without the help of chloroquine. It was also observed that the transfection efficiency of dKH-FGF2 was not as high as cKH-FGF2 even in the presence of chloroquine suggesting that fewer numbers of particles were internalized in the first place. The fact that chloroquine had a significant positive effect on transfection efficiency implies that addition of more histidine residues to the vector structure could enhance the endosome disrupting efficiency of the vectors. This can be examined by carefully designing and testing other KH-FGF2 analogues with different architecture containing more or less histidine content.
In the presence of bafilomycin, the transfection efficiency of both vectors dropped significantly as a result of the entrapment of vector/pDNA complexes inside endosomes and inability to escape into cytosol. Therefore, it is conceivable that the observed difference between the transfection efficiency of the two vectors was due to the internalization of higher number of cKH-FGF2/pDNA complexes. Consequently, this observation can directly be attributed to the vector architecture highlighting the considerable effect of vector architecture on transfection efficiency.
5. Conclusion
This study demonstrates that besides the amino acid composition, the vector architecture could also play a significant role in targeted gene transfer. This factor becomes more important when using random copolymerization to synthesize polymeric non-viral vectors. Drawing a decisive conclusion at this point seems premature, but the data suggests that more information can be obtained by generating other KH analogues (e.g., KKHHKKHHKK or KKKHHKKKHH) followed by conducting comprehensive physicochemical and biological characterization. Recombinant DNA technology has made it possible to design limitless number of constructs with minute differences in amino acid sequence (up to single amino acid) in order to study the effect of vector structure on transfection efficiency at all levels. By manipulating the vector structure at the molecular level, it can be correlated with DNA condensation and transfection efficiency potentially leading to the creation of vectors with high transfection efficiency and low toxicity. It is expected that the next generations of recombinant non-viral vectors would rival viruses in terms of efficiency without compromising safety.
Acknowledgments
This work was funded in part by the American Cancer Society Institutional Research Grant (1RG-77-003-26).
References
- 1.Louise C. Nonviral vectors. Methods Mol Biol. 2006;333:201–226. doi: 10.1385/1-59745-049-9:201. [DOI] [PubMed] [Google Scholar]
- 2.Rettig GR, Rice KG. Non-viral gene delivery: from the needle to the nucleus. Expert Opin Biol Ther. 2007;7(6):799–808. doi: 10.1517/14712598.7.6.799. [DOI] [PubMed] [Google Scholar]
- 3.Ruponen M, Honkakoski P, Ronkko S, Pelkonen J, Tammi M, Urtti A. Extracellular and intracellular barriers in non-viral gene delivery. J Control Release. 2003;93(2):213–217. doi: 10.1016/j.jconrel.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 4.Thomas CE, Ehrhardt A, Kay MA. Progress and problems with the use of viral vectors for gene therapy. Nat Rev Genet. 2003;4(5):346–358. doi: 10.1038/nrg1066. [DOI] [PubMed] [Google Scholar]
- 5.Li SD, Huang L. Targeted delivery of antisense oligodeoxynucleotide and small interference RNA into lung cancer cells. Mol Pharm. 2006;3(5):579–588. doi: 10.1021/mp060039w. [DOI] [PubMed] [Google Scholar]
- 6.Kaouass M, Beaulieu R, Balicki D. Histonefection: Novel and potent non-viral gene delivery. J Control Release. 2006;113(3):245–254. doi: 10.1016/j.jconrel.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 7.Yamagata M, Kawano T, Shiba K, Mori T, Katayama Y, Niidome T. Structural advantage of dendritic poly(L-lysine) for gene delivery into cells. Bioorg Med Chem. 2007;15(1):526–532. doi: 10.1016/j.bmc.2006.09.033. [DOI] [PubMed] [Google Scholar]
- 8.Vaughan EE, DeGiulio JV, Dean DA. Intracellular trafficking of plasmids for gene therapy: Mechanisms of cytoplasmic movement and nuclear import. Curr Gene Ther. 2006;6(6):671–681. doi: 10.2174/156652306779010688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leng Q, Kahn J, Zhu J, Scaria P, Mixson J. Needle-like morphology of H2K4b polyplexes associated with increases in transfection in vitro. Cancer Ther. 2007;5B:193–202. [PMC free article] [PubMed] [Google Scholar]
- 10.Leng Q, Goldgeier L, Zhu J, Cambell P, Ambulos N, Mixson AJ. Histidine-lysine peptides as carriers of nucleic acids. Drug News Perspect. 2007;20(2):77–86. doi: 10.1358/dnp.2007.20.2.1083026. [DOI] [PubMed] [Google Scholar]
- 11.Putnam D, Zelikin AN, Izumrudov VA, Langer R. Polyhistidine-PEG: DNA nanocomposites for gene delivery. Biomaterials. 2003;24(24):4425–4433. doi: 10.1016/s0142-9612(03)00341-7. [DOI] [PubMed] [Google Scholar]
- 12.Shigeta K, Kawakami S, Higuchi Y, Okuda T, Yagi H, Yamashita F, Hashida M. Novel histidine-conjugated galactosylated cationic liposomes for efficient hepatocyte-selective gene transfer in human hepatoma HepG2 cells. J Control Release. 2007;118(2):262–270. doi: 10.1016/j.jconrel.2006.12.019. [DOI] [PubMed] [Google Scholar]
- 13.Behr JP. The proton sponge: A trick to enter cells the viruses did not exploit. Chimica. 1997;51:34–36. [Google Scholar]
- 14.Putnam D, Gentry CA, Pack DW, Langer R. Polymer-based gene delivery with low cytotoxicity by a unique balance of side-chain termini. Proc Natl Acad Sci U S A. 2001;98(3):1200–1205. doi: 10.1073/pnas.031577698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haider M, Hatefi A, Ghandehari H. Recombinant polymers for cancer gene therapy: a minireview. J Control Release. 2005;109(1–3):108–119. doi: 10.1016/j.jconrel.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 16.Hatefi A, Megeed Z, Ghandehari H. Recombinant polymer-protein fusion: A promising approach towards efficient and targeted gene delivery. J Gene Med. 2006;8(4):468–476. doi: 10.1002/jgm.872. [DOI] [PubMed] [Google Scholar]
- 17.Blanckaert VD, Hebbar M, Louchez MM, Vilain MO, Schelling ME, Peyrat JP. Basic fibroblast growth factor receptors and their prognostic value in human breast cancer. Clin Cancer Res. 1998;4(12):2939–2947. [PubMed] [Google Scholar]
- 18.Midoux P, Monsigny M. Efficient gene transfer by histidylated polylysine/pDNA complexes. Bioconjug Chem. 1999;10(3):406–411. doi: 10.1021/bc9801070. [DOI] [PubMed] [Google Scholar]
- 19.Rajagopalan R, Xavier J, Rangaraj N, Rao NM, Gopal V. Recombinant fusion proteins TAT-Mu, Mu and Mu-Mu mediate efficient non-viral gene delivery. J Gene Med. 2007;9(4):275–286. doi: 10.1002/jgm.1014. [DOI] [PubMed] [Google Scholar]
- 20.Khadake JR, Rao MR. Condensation of DNA and chromatin by an SPKK-containing octapeptide repeat motif present in the C-terminus of histone H1. Biochemistry. 1997;36(5):1041–1051. doi: 10.1021/bi961617p. [DOI] [PubMed] [Google Scholar]
- 21.Tecle M, Preuss M, Miller AD. Kinetic study of DNA condensation by cationic peptides used in nonviral gene therapy: Analogy of DNA condensation to protein folding. Biochemistry. 2003;42(35):10343–10347. doi: 10.1021/bi034325e. [DOI] [PubMed] [Google Scholar]
- 22.Cappello J, Crissman J, Dorman M, Mikolajczak M, Textor G, Marquet M, Ferrari F. Genetic engineering of structural protein polymers. Biotechnol Prog. 1990;6(3):198–202. doi: 10.1021/bp00003a006. [DOI] [PubMed] [Google Scholar]
- 23.Liu W, Dreher MR, Furgeson DY, Peixoto KV, Yuan H, Zalutsky MR, Chilkoti A. Tumor accumulation, degradation and pharmacokinetics of elastin-like polypeptides in nude mice. J Control Release. 2006;116(2):170–178. doi: 10.1016/j.jconrel.2006.06.026. [DOI] [PubMed] [Google Scholar]
- 24.Urry DW, Harris CM, LCX, LC-H, Gowda DC, Parker TM, Peng SQ, Xu J. Transductional protein-based polymers as new controlled-release vehicles. In: Park K, editor. Controlled drug delivery: Challenges and Strategies. American Chemical Society; Washington, DC: 1997. pp. 405–436. [Google Scholar]
- 25.Rejman J, Oberle V, Zuhorn IS, Hoekstra D. Size-dependent internalization of particles via the pathways of clathrin- and caveolae-mediated endocytosis. Biochem J. 2004;377(Pt 1):159–169. doi: 10.1042/BJ20031253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sosnowski BA, Gonzalez AM, Chandler LA, Buechler YJ, Pierce GF, Baird A. Targeting DNA to cells with basic fibroblast growth factor (FGF2) J Biol Chem. 1996;271(52):33647–33653. doi: 10.1074/jbc.271.52.33647. [DOI] [PubMed] [Google Scholar]
- 27.Manickam DS, Oupicky D. Multiblock reducible copolypeptides containing histidine-rich and nuclear localization sequences for gene delivery. Bioconjug Chem. 2006;17(6):1395–1403. doi: 10.1021/bc060104k. [DOI] [PubMed] [Google Scholar]
- 28.Chandler LA, Sosnowski BA, Greenlees L, Aukerman SL, Baird A, Pierce GF. Prevalent expression of fibroblast growth factor (FGF) receptors and FGF2 in human tumor cell lines. Int J Cancer. 1999;81(3):451–458. doi: 10.1002/(sici)1097-0215(19990505)81:3<451::aid-ijc20>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 29.Oriova A, Magnusson M, Eriksson TL, Nilsson M, Larsson B, Hoiden-Guthenberg I, Widstrom C, Carisson J, Tolmachev V, Stahl S, Nilsson FY. Tumor imaging using a picomolar affinity HER2 binding affibody molecule. Cancer Res. 2006;66(8):4339–4348. doi: 10.1158/0008-5472.CAN-05-3521. [DOI] [PubMed] [Google Scholar]
- 30.Bowman EJ, Siebers A, Altendorf K. Bafilomycins: A class of inhibitors of membrane ATPases from microorganisms, animal cells, and plant cells. Proc Natl Acad Sci U S A. 1988;85(21):7972–7976. doi: 10.1073/pnas.85.21.7972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lampela P, Elomaa M, Ruponen M, Urtti A, Mannisto PT, Raasmaja A. Different synergistic roles of small polyethylenimine and Dosper in gene delivery. J Control Release. 2003;88(1):173–183. doi: 10.1016/s0168-3659(02)00489-3. [DOI] [PubMed] [Google Scholar]