

Abstract
Background
Chronic pulmonary infection with Pseudomonas aeruginosa (PA) is responsible for significant morbidity and mortality in cystic fibrosis (CF). Because of the limited studies evaluating early exposure and the progression of genetic variability of PA, our goal was to assess PA in young children with CF followed in two clinic types.
Methods
A total of 39 infants with CF diagnosed through newborn screening were randomly assigned to either a segregated (PA-free) or mixed (PA-positive) clinic at two different CF centers, one of which replaced an older, mixed clinic where nosocomial acquisition was suspected. Oropharyngeal (OP) swab cultures were examined with subsequent genotyping to characterize the strains of PA isolated.
Results
We found that 13/21 segregated clinic patients and 14/18 mixed clinic patients showed positive PA, with median acquisition ages of 3.3 and 2.2 years, respectively (P=0.57). The median time to PA acquisition, however, was significantly longer in the new clinic with proper hygiene precautions compared to an old site (5.0 vs 1.7 years, P<0.001). The majority of subjects isolated a single genotype of PA or AP-PCR types during the study period with 8 subjects clearing the isolate after only one positive culture. The development of chronic colonization yielded the predominance of a single major genotype or AP-PCR type.
Conclusions
Segregation of infants and young children with CF in PA negative or PA positive clinics did not alter the time to first PA isolation in this randomized assessment of facilities with hygienic precautions. During the early infection period where PA is first isolated in young children with CF, patients cleared different PA strains until a predominant strain established permanent colonization.
Keywords: Pseudomonas aeruginosa, cystic fibrosis, newborn screening, arbitrarily primed polymerase chain reaction, genotype, segregated clinic, integrated clinic
INTRODUCTION
Cystic Fibrosis (CF) is an autosomal recessive disorder characterized by chronic pulmonary disease with recurrent respiratory bacterial infections and with malnutrition due to pancreatic insufficiency. (1) The acquisition of Pseudomonas aeruginosa (PA) in children with CF leads to gradual deterioration of lung disease (2) and is responsible for most of the morbidity and mortality associated with this disease. Limited studies exist on examining the microbiology of respiratory secretions in infants and young children, particularly in relationship to care settings. Li and colleagues reported that 16/56 patients (29%) acquired nonmucoid PA in the first six months of life. (3) Nonmucoid and mucoid PA were acquired at median ages of 1.0 and 13.0 years, respectively. Our group also previously reported a higher prevalence of PA colonization at an urban center in CF patients between the ages of 3 and 9 years. (4) Due to this shorter time to acquisition of PA at a very small clinic setting in a 90-year-old hospital facility in Milwaukee (5), the Wisconsin CF Neonatal Screening Research Project studied this further by statistical analyses using survival curves and stepwise regression analysis of life history covariates available. The results demonstrated statistically significant correlations in the center and old hospital (r=0.42), exposure to PA-positive patients (r=0.29), and the population density and urban location (r=0.49). (5) Although our risk factor analyses suggested cross infection (5), there were other predictors such as aerosol exposure; however, due to the study design (3,4,6), it was not possible to evaluate the PA cultures in these groups by molecular methods.
According to the CF Foundation Registry (7,8), up to one-third of CF patients have PA when diagnosed after signs/symptoms, but the ability to diagnose CF through newborn screening makes it possible to determine the temporal sequence of PA isolation. The prevalence (3,7) and morbidity (3,8) of PA in CF have been described in epidemiological studies with associated risk factors for lung infection (4–5,10–12). The microbiology of young children with CF was better understood after Burns et al followed 40 children during the first three years of age and compared serial oropharyngeal (OP) swab and broncheoalveolar lavage (BAL) cultures with antibody responses to PA. (13) By three years of age, 72.5% of these patients were colonized with PA, and 97.5% of patients had evidence of infection if serologic tests were combined with cultures. (13)
Presently, to our knowledge, although several assessments of PA in CF have been published (7,13–16), no studies have evaluated the progression or genetic variation of PA after acquisition beginning with infants diagnosed with CF by newborn screening, nor randomly investigated a segregated follow-up clinic for such patients. Therefore, this project was developed at the two Wisconsin centers in a new cohort of infants diagnosed with CF by newborn screening and then randomized with a goal of determining if our previous observations at a small, old mixed clinic (5) were reproducible when that CF center moved to a new setting where hygienic precautions could be maintained. Our hypothesis was that the new site would be better, but we also aimed to determine if segregation was better. Therefore, the project was designed to address several goals by comparing segregated and mixed clinics and new versus old clinic in Milwaukee while evaluating the timing of PA acquisition, as well as genotyping (17) the PA isolates to examine for patterns that might imply cross- infection.
MATERIALS and METHODS
The study design was a randomized clinical trial of two clinic types with enrollment, longitudinal cohort follow-up observation and data collection during approximately 10 years (1996–2005). Infants diagnosed with CF by newborn screening who were enrolled at two CF centers: University of Wisconsin Hospital and Clinics in Madison, WI and Children’s Hospital of Wisconsin in Milwaukee, WI. At the time of the study, neither center used PA eradication protocols as this issue was still controversial then (15). The study was approved by the institutional review board (IRB) at the University of Wisconsin Hospital and Clinics, the Medical College of Wisconsin, and the Children’s Hospital of Wisconsin. The subjects were enrolled with informed consent from their parents and followed in either segregated (free of patients with PA) or mixed/integrated clinics that included PA positive patients but operated with hygienic precautions. The segregated clinics that excluded CF patients colonized or infected with PA were held on a separate day in the same clinic space used for mixed/integrated clinics that included CF patients with PA. Initially, during 1996, randomization occurred between centers, but due to an IRB requirement during the reapproval processes after one year, the assignments changed to within-center randomization for 1997–2001. Study inclusion criteria were (1) positive newborn screen (highest 4% of the daily immunoreactive trypsinogen levels followed by a gene mutation analysis for ΔF508), (2) elevated sweat chloride at or above 60 mmol/L by quantitative pilocarpine electrophoresis, and (3) informed consent provided by parents or legal guardians. A total of 39 subjects were studied following randomization, including 21 in the segregated group and 18 in the mixed group. Detailed records were kept of all clinic visits for each patient, and these were examined to detect any PA exposures.
A cotton-tipped swab was used to collect the specimen from the oropharynx at each clinic visit using standardized methods previously employed at each CF center (4–6). Protocol visits were scheduled every eight weeks for the first year of life and every three months thereafter. Standard of care at the time of the study was to obtain OP cultures every six months or more frequently if clinically indicated, but the study protocol obtained cultures at each protocol visit. The OP swab cultures were processed at the microbiology laboratory at the two respective centers, University of Wisconsin Hospital and Clinics and Children’s Hospital of Wisconsin. The samples were inoculated directly on eosin-methylene blue (EMB) and oxidative-fermentative base-polymyxin B-bacitracin-lactose (OFPBL) agars. Confirmation of PA was performed by standard biochemical testing. The PA isolates were directly removed from the primary culture plate and initially frozen at −80°C in skim milk broth; prior to molecular analyses, they were thawed and transferred to a solution of glycerol salts for further storage at −80°C.
Arbitrarily primed polymerase chain reaction (AP-PCR) was used for genetic analysis because Kersulyte et al (17) demonstrated using PA isolates from CF patients that AP-PCR was as efficient and sensitive as pulsed field gel electrophoresis (PFGE) for genetically characterizing strains of PA. To prepare DNA, 500 μl of an overnight, stationary-phase, Mueller-Hinton broth culture of each PA isolate was spun for one minute in a microcentrifuge. DNA was extracted as previously described by Kersulyte and colleagues (17). A 2 μl aliquot of the DNA was electrophoresed in a 1X Tris-acetate-buffered 2% agarose gel that stained with ethidium bromide and photographed under ultraviolet light to estimate and verify the integrity of the DNA. AP-PCR was performed in a 50 μl volume containing approximately 20 ng of bacterial DNA; GeneAmp 10X Buffer II (Applied Biosystems, Foster City, CA); 25 mM MgCl2; 2.5 mM (each) of dATP, dCTP, dGTP, and dTTP (Applied Biosystems, Foster City, CA); 1 Unit of AmpliTaq Gold DNA Polymerase (Applied Biosystems, Foster City, CA); 20 pmol of primer (Intregrated DNA Technologies, Coralville, IA); and sterile distilled/deionized water in a MJ Research Peltier Thermal Cycler (PTC)-200 (Global Medical Instrumentation, Inc., Ramsey, MN). The two arbitrary primers used were CF10514 (5′-TGGTGGCCTCGAGCAAGAGAACGGAG) and CF14306 (5′-GGTTGGGTGAGAATTGC). Cycling parameters for the MJ Research PTC-200 after a 5 minute hotstart at 95°C were: 4 cycles of 96°C for 5 minutes, 40°C for 5 minutes, and 72°C for 5 minutes; then 30 cycles of 95°C for 1 minute, 55°C (CF10514) or 61°C (CF14306) for 1 minute, and 72°C for 2 minutes; and then 72°C for 10 minutes. The annealing stage for the AP-PCR was optimized with excellent reproducibility at 55°C for primer CF10514 and 61°C for primer CF14306. After AP-PCR, an 8 μl aliquot was electrophoresed using a 1X Tris-acetate-buffered 2% agarose gel that was stained with ethidium bromide and photographed under ultraviolet light. A 2-log (0.1–10 kb) DNA ladder (New England BioLabs, Beverly, MA) was used as a size marker in all gels. Gel photographs were processed for presentation by scanning with Hewlett-Packard ScanJet 6100 C/T using Adobe Photoshop CS version 8.0 (Adobe Systems, Inc., Mountain View, CA). Major AP-PCR types were defined as differing by two or more prominent bands with further subdivision into minor subtypes if they differed by one or two faint bands. (17)
Statistical Methods
To determine if there were any differences in patterns of PA acquisition between patients followed in either the segregated or mixed clinics, we combined data from the two subgroups and compared the ages of first PA positive culture using a log rank test. In addition, Kaplan-Meier PA-free survival curves were produced to compare segregated versus mixed clinics. Log rank test was also used to compare the time of PA acquisition in patients followed at the large new clinic to the previous, old clinic at the Milwaukee clinic that was previously reported by Farrell et al (4).
RESULTS
Patient Characteristics
Characteristics of the patient cohort were similar between the two respective CF centers as shown in Table 1. The demographics listed in Table 1 were not statistically different and provided a representative cohort for the CF population in Wisconsin with the majority (97%) being Caucasian, slight male predominance (59%), high incidence of ΔF508 genetic mutation (95%), moderate meconium ileus rate (33%), and high prevalence of pancreatic insufficiency (87%). Once randomly assigned, all patients adhered to the protocol for evaluation without any crossover of clinic types.
Table 1.
Patient demographics (N = 39) with cystic fibrosis diagnosed through newborn screening.
| Characteristic | Total (N = 39)* | Mixed Clinic, No. (%)(N = 18) | Segregated Clinic, No. (%)(N = 21)* | P value |
|---|---|---|---|---|
| Race | .46 | |||
| Caucasian | 38 (97%) | 17 (94) | 21 (100) | |
| Non-Caucasian | 1 (3%) | 1 (6) | 0 (0) | |
| Sex | .80 | |||
| Male | 23 (59%) | 11 (61) | 12 (57) | |
| Female | 16 (41%) | 7 (39) | 9 (43) | |
| Genotype | .89 | |||
| ΔF508/ΔF508 | 19 (49%) | 8 (44) | 11 (52) | |
| ΔF508/Other | 18 (46%) | 9 (50) | 9 (43) | |
| Other/Other | 2 (5%) | 1 (6) | 1 (5) | |
| Meconium ileus | 1.00 | |||
| Yes | 13 (33%) | 6 (33) | 7 (33) | |
| No | 26 (67%) | 12 (67) | 14 (67) | |
| Pancreatic status | .65 | |||
| Sufficient or probably sufficient | 5 (13%) | 3 (17) | 2 (10) | |
| Insufficient or probably insufficient | 33 (87%) | 15 (83) | 18 (90) | |
Pancreatic status of 1 patient is unknown.
We found that 13 out of 21 segregated patients became PA positive and 14 out of 18 in the mixed/integrated clinics were PA positive. The median ages of PA acquisition were 3.3 and 2.2 years for segregated and mixed clinic, respectively, and there were no statistically significant difference (P=0.57). In addition, Kaplan-Meier PA-free survival curves demonstrated that there were no differences between two clinic types. However, the median time of PA acquisition in patients followed at the new clinic at Milwaukee was significantly greater (P<0.001) at 5.0 years compared to 1.7 years in the previous, old clinic (4,5).
A total of 76 frozen PA isolates were available from 17 patients for molecular studies. Table 2 illustrates patient and PA culture characteristics for individual patients. Eight patients cultured a single PA isolate during the study period. The remaining patients had multiple PA isolates collected with three patients with two isolates, two patients having four isolates, one patient with five isolates, and three patients with more than 10 isolates (13, 14, and 22 isolates). A total of two isolates were not available for analysis – one isolate from patient I and one isolate from patient M. An important point to be aware of is patient I had two older siblings with CF, one of which had mucoid PA during the study period.
Table 2.
Characteristics of patients with positive Pseudomonas aeruginosa cultures.
| Patient | Number of Clinic Visits | Age at 1st Positive Culture (months) | Length of Time of Positive Cultures (months) | Number of Isolates | Number of Major AP-PCR types |
|---|---|---|---|---|---|
| A | 17 | 2.8 | * | 1 | 1 |
| B | 10 | 9.8 | * | 1 | 1 |
| E | 16 | 7 | * | 1 | 1 |
| F | 28 | 3.5 | * | 1 | 1 |
| H | 13 | 13.2 | * | 1 | 1 |
| J | 30 | 42.8 | * | 1 | 1 |
| L | 18 | 23.2 | * | 1 | 1 |
| P | 18 | 5 | * | 1 | 1 |
| D | 23 | 40.5 | 4.5 | 2 | 1 |
| G | 24 | 5.2 | 15 | 2 | 2 |
| K | 22 | 23.5 | 0.5 | 2 | 1 |
| C | 17 | 9.2 | 5.5 | 4 | 4 |
| Q | 26 | 7 | 23 | 4 | 1 |
| N | 25 | 3.2 | 4 | 5 | 1 |
| M† | 19 | 4.5 | 37 | 13 | 1 |
| O | 22 | 10 | 18.2 | 14 | 1 |
| I† | 23 | 1.8 | 43 | 22 | 2 |
One isolate cultured during the study period.
Two isolates were not available for analysis - one isolate from Patient I and one isolate from Patient M.
Patient I was the single patient who developed persistence of a mucoid phenotype at 21 months of age. Although expected to have “mild” CF based on having a class IV allele (R347P) and pancreatic sufficiency, this patient was the youngest sibling of three other children with CF and chronic PA colonization with mucoid phenotype isolated in one of them. In our study population, there was no association with a particular major AP-PCR type and the mucoid phenotypes that were collected. At the conclusion of the observation period, all patients except for two (I and M) had stopped culturing PA, including patient O who had a total of 14 PA isolates collected. Patient O was enrolled four months into the study period before growing PA on OP swab culture for the following 28 months, with negative cultures for the last 26 months of the study period.
Pseudomonas aeruginosa Genotype
The AP-PCR banding profiles of all PA isolates were assessed to determine whether infants are infected with multiple PA genotypes. Figures 1 and 2 illustrate the AP-PCR banding patterns for all major and their minor subtypes with primer CF10514 (Figure 1) and CF14306 (Figure 2). Patients A, B, E, F, H, J, L, and P cultured a single PA isolate that was identified as single distinctive genotypes based on AP-PCR banding patterns with both primers. A single genotype was found in three patients who had more than one isolate, including patients D and K who cultured two isolates and patient Q who cultured four isolates. Patient C was the only patient with more than one genotype identified on the first positive culture. Patient C had two genotypes identified with the first OP swab culture with distinct major AP-PCR banding patterns with both primers. Two more distinct genotypes were identified by AP-PCR banding pattern with both primers one and six months later with no further growth of PA. Three of nine patients (C, G, and I) with multiple PA isolates had more than one genotype identified based upon AP-PCR banding pattern.
Figure 1.
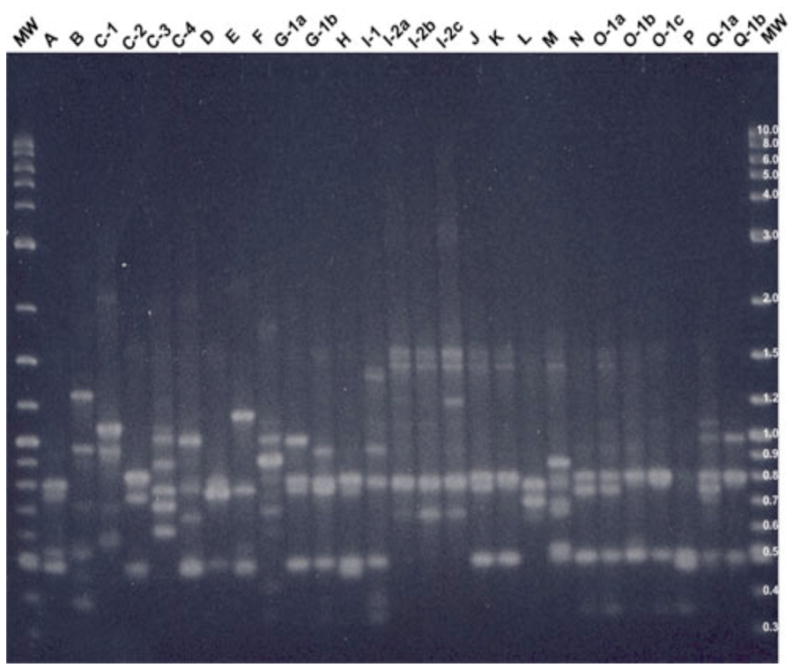
2% agarose gel showing AP-PCR banding pattern for primer CF10514 with all major types and their minor subtypes.
Note: MW is the molecular weight control [2-log (0.1–10 kb) DNA ladder], “A, B, etc.” represent Patient A, B etc., “1, 2, etc.” represent major AP-PCR type, and “a, b, etc.” represent minor subtype. Single letters represent a patient with a single major AP-PCR type.
Figure 2.
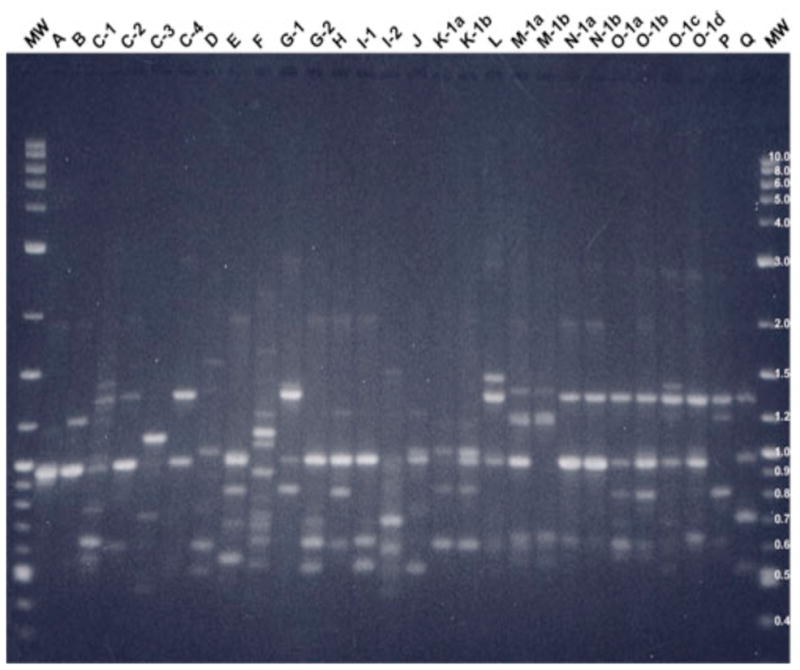
2% agarose gel showing AP-PCR banding pattern for primer CF14306 with all major types and their minor subtypes.
Note: MW is the molecular weight control [2-log (0.1–10 kb) DNA ladder], “A, B, etc.” represent Patient A, B etc., “1, 2, etc.” represent major AP-PCR type, and “a, b, etc.” represent minor subtype. Single letters represent a patient with a single major AP-PCR type.
Pseudomonas aeruginosa Genetic Divergence
The AP-PCR banding profiles were assessed to evaluate if genetic divergence of PA isolates occurs with chronic colonization in those patients with multiple isolates (Figures 1 and 2). Patients M, O, and I were the origin of 12, 14, and 21 isolates, respectively. Patient I had two genotypes identified initially (Figure 3) with both major AP-PCR types cultured 14 months later with no further isolation of the major first major AP-PCR type with subsequent persistence of the second major AP-PCR type. This second major AP-PCR type underwent minor genetic variance with primer CF10514 (Figure 3) and no genetic variance occurred with primer CF14306. Patients M and O underwent minor genetic variation of the initial strain and eventually developed persistence of a single major AP-PCR type. The three patients with more than 10 isolates collected during the study period developed persistence of a single major AP-PCR type after minor genetic variation of the initial strain (patients M and O) or after loss of a major AP-PCR type (patient I).
Figure 3.
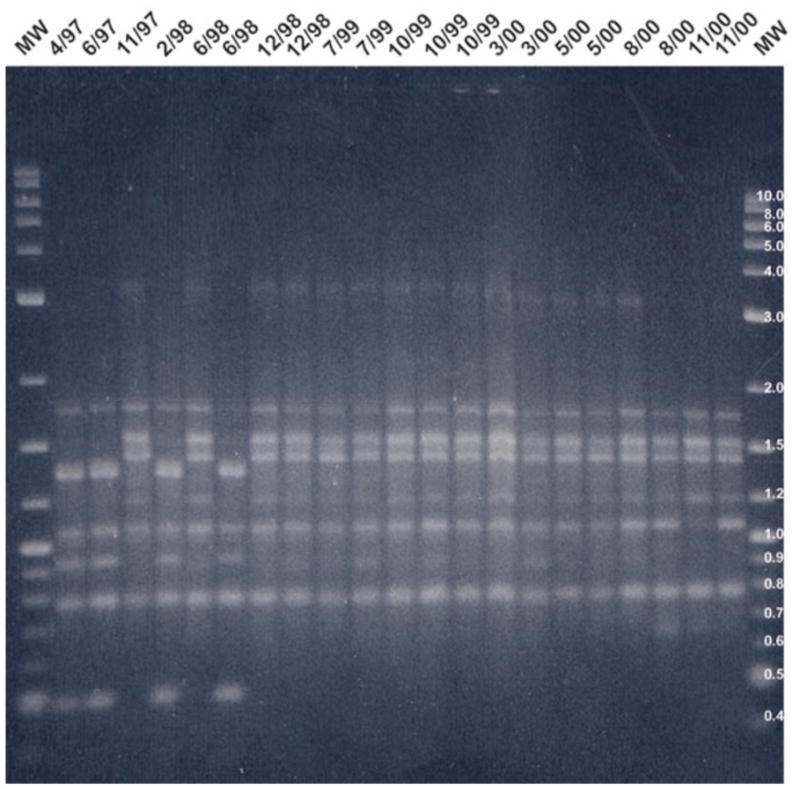
2% agarose gel showing AP-PCR banding pattern for patient I using primer CF10514.
Note: MW is the molecular weight control [2-log (0.1–10 kb) DNA ladder], Positive culture dates are listed along the top.
Pseudomonas aeruginosa Genetic Homogeneity
The AP-PCR banding profiles were assessed to evaluate if genetic homogenity or sharing of a common PA genotype occurred between patients in the cohort (Figures 1 and 2). Common major and minor bands were identified between patients using an individual primer with commonality of AP-PCR banding patterns identified using both primers only in patients N and O who had identical banding patterns with primer CF14306 and a two minor band difference with primer CF10514 as illustrated in Figures 1 and 2. These two particular patients lived in different areas of the state and had no mutual clinic visits or other social interaction during the study period. There were common clinic visit dates for these two patients 15 months after these initial isolates were cultured.
DISCUSSION
Due to limited prospective, serial studies of PA in young children with CF and the opportunity for pre-symptomatic diagnosis through newborn screening, we developed a longitudinal study to evaluate PA acquisition during infancy and early childhood in patients randomly assigned to either a traditional mixed clinic or a segregated ambulatory care setting. The latter objective was considered imperative by the Wisconsin CF centers because of the alarming results in our previous project implicating cross-infection (4,5) and “acceleration of lung disease” (2) and the lack of a randomized clinical trial with newborn screening-diagnosed patients. Due to the previous use of mixed clinics in an old hospital facility demonstrating much earlier acquisition of PA, we felt that person-to-person transmission of PA was likely the underlying problem (4,5). More specifically, out of necessity, patients at the Milwaukee Center received care in a very small clinic with a 110 square foot waiting room where proper hygiene precautions were impossible. (5) Subsequently, a new hospital with larger waiting and clinic rooms became available, and proper hygiene precautions were then able to be instituted. With this dramatic change in the clinic environment, our results in this study did not reproduce previous observations (4), and there was a change in median time to initial PA acquisition from 1.7 years to 5.0 years. The significant difference is likely due to a major reduction in patient-to-patient interactions. Unfortunately, however, the power for this assessment of the age of PA acquisition was only 0.21, and 77 patients per group or 147 total would be needed for 0.80 power. When we recognized during an interim analysis that it would be impossible to accumulate such a group in a reasonable time period and also learned that our IRB were uncomfortable with continuation of mixed clinics for research purposes, we turned our attention to PA genotyping in an effort to discover if there was a specific microorganism responsible for early acquisition.
These CF patients were diagnosed without evidence of lung disease and study enrollment was prior to the first isolation of PA on OP swab culture. With longitudinal observations, this study design would not likely bias data collection as compared to a study design of enrolling patients with a clinical diagnosis of CF. Specimen collection with OP swabs occurred with each clinic visit; although this is a limitation (6), there were no clinical indications arising for lower airway sampling by bronchoscopy with BAL. A total of 11/39 (28.2%) cultured PA by one year of age, and the mean age for the first positive PA culture was 13 months.
The findings of our molecular studies support previous reports by Burns et al (13) and Abman et al (18) that the development of a PA genotype clone occurs in children with CF who develop persistent infection although new PA strains can appear after eradication therapies (14). Restriction fragment length polymorphisms with an exotoxin A gene probe was used by Burns et al (13) and Southern blot analysis with DNA probes derived from the 5′ region of exotoxin A structural gene was used by Abman et al (18). The persistence of a single genotype occurs despite the presence of multiple genotypes initially. In our cohort, the three patients (I, M, and O) who developed persistence of a genotype continued to have genotypic variation until the end of the study period or until PA was no longer isolated in patient O. All three patients acquired PA before one year of age as illustrated in Table 2. There were no distinguishing features among these three patients with two being male, two being pancreatic insufficient, and one having a previous meconium ileus. With regard to CF genotypes, patient M has ΔF508/R553X, and patient O has ΔF508/ΔF508; patient I, a child with pancreatic sufficiency, has a presumptive genotype of R347P (determined in the DNA component of screening) with 2184 insA (based on the determination of the other allele in an older sibling with CF who also isolated PA).
In this cohort, considerable PA phenotypic changes occurred without correlation with genotypic variation, especially among the patients (I, M, and O), who had more than 10 PA isolates. We further substantiate as previously reported by Burns et al (12) and Van Daele et al (15) that phenotypic and genotypic changes lack correlation and are unrelated events. Spencer and colleagues confirmed that most of the lab strain PAO1 genome represents a core PA backbone sequence using three investigative strains (two from CF patients and another from an aquatic environment) possessing additional genetic material that accounted for at least 10% of their genomes. (20) Genomic mosaicism was conserved in PA isolates from the airways of young children with CF. (21) Mucoid phenotype, however, was not commonly seen in our cohort involving infants and young children, which confirms the previous findings of Li et al (3). In fact, only one patient (I) who had a sibling with mucoid PA showed this strain.
Patient-to-patient transmission of PA in CF remains controversial. (22–26) Our only molecular observation that even suggested cross infection involved patients N and O who had similar PA genotypes and four mutual clinic visit dates. The similar clinic visit dates, however, did not occur until 15 months after the initial isolates were cultured for both patients. The last positive PA culture for patient N was 12 months prior to the first mutual clinic visit. This suggests that the isolates were not shared between patients but may have been contracted from a common source in the clinic. In addition, there was no socialization of the two patients outside of the clinic, and they lived 180 miles apart from each other.
In the setting of CF center follow-up care after newborn screening, serious concerns have been expressed about the potential for cross infection because nosocomial acquisition of virulent PA pathogens can negate the value of early diagnosis and possibly lead to more harm than good. (27–28) In fact, there are conflicting reports in the literature regarding the level of contact needed to transmit PA from one patient to another. Transmissible PA strains have been reported to cause cross-infection control problems at large CF centers in Europe and Australia. (27,29–33) Armstrong et al (27) identified a widespread clone of PA in 55% of 118 infected patients in a pediatric CF center in Victoria, Australia. Apparently, close attention to hygienic principles in the absence of patient segregation did not prevent cross-infection of transmissible PA. (23) Successful infection control measures with segregation in a CF clinic provided indirect evidence that person-to-person transmission of an epidemic strain was prevented. (16,24) In direct contrast to other studies, a longitudinal study over two decades with 174 patients demonstrated that the acquisition of PA was primarily environmental with prolonged close contact needed for patient to patient spread, such as siblings. (25)
Collectively, our previous investigations of PA in CF patients diagnosed through newborn screening (2–6) combined with the information generated in this study provide valuable insights on the challenges associated with follow-up care in young children at risk for chronic respiratory infection. In summary, with both Wisconsin CF centers now using modern, large clinics and waiting rooms, as well as hygienic precautions, there were no differences between clinic types (mixed vs segregated) or centers. In addition, we found that children diagnosed through CF newborn screening can be infected initially with more than one PA strain based on genotyping with AP-PCR and that genetic divergence of PA with chronic colonization may occur with major AP-PCR types undergoing minor genotypic changes in some patients with the clearance of single or even persistent genotypes in others. A single major AP-PCR type eventually persists in young children with chronic PA colonization.
From a variety of observations reported herein and elsewhere (16,20), we believe that an appropriate, modern clinic setting with enough space and efficient patient flow to avoid patient-to-patient interactions, combined with compulsive use of recommended hygienic precautions (34), should be sufficient to prevent cross-infection of PA in young children with CF diagnosed through newborn screening. Elimination of waiting rooms is an example of how CF centers can minimize person-to-person interactions. On the other hand, busy clinics may find this strategy impractical and rigorous hygienic precaution (30) impossible to apply religiously. Furthermore, as McKay et. al recently emphasized from their retrospective study, segregation is desirable when a CF clinic has “constraints in the availability of physical space and personnel” (16). This, in fact, was the situation in the old Milwaukee clinic (5) used originally in our RCT of CF newborn screening with a previous cohort of study patients (1). Thus, in such situations, segregated clinics seem warranted after newborn screening, but further epidemiological studies with molecular methods will be important to address this question.
Acknowledgments
The authors want to thank Christopher Green, MD and Anita Laxova, BS as well as other members of the Wisconsin Interdisciplinary Newborn Screening Research Team for assistance in the development of this manuscript.
Financial Support: The National Institutes of Health (grant NIDDK 5 R01 DK34108-17).
Footnotes
This study was approved by the institutional review boards of all participating centers, and informed consent was received from the parents of the participating patients.
References
- 1.Farrell PM. Improving the health of patients with cystic fibrosis through newborn screening. Wisconsin Cystic Fibrosis Neonatal Screening Study Group. Adv Pediatr. 2000;47:79–115. [PubMed] [Google Scholar]
- 2.Kosorok MR, Zeng L, West SE, Rock MJ, Splaingard ML, Laxova A, Green CG, Collins J, Farrell PM. Acceleration of lung disease in children with cystic fibrosis after Pseudomonas aeruginosa acquisition. Pediatr Pulmonol. 2001;32:277–87. doi: 10.1002/ppul.2009.abs. [DOI] [PubMed] [Google Scholar]
- 3.Li Z, Kosorok MR, Farrell PM, Laxova A, West SE, Green CG, Collins J, Rock MJ, Splaingard ML. Longitudinal development of mucoid Pseudomonas aeruginosa infection and lung disease progression in children with cystic fibrosis. JAMA. 2005;293:581–8. doi: 10.1001/jama.293.5.581. [DOI] [PubMed] [Google Scholar]
- 4.Farrell PM, Shen G, Splaingard M, Colby CE, Laxova A, Kosorok MR, Rock MJ, Mischler EH. Acquisition of Pseudomonas aeruginosa in children with cystic fibrosis. Pediatrics. 1997;100:E2. doi: 10.1542/peds.100.5.e2. [DOI] [PubMed] [Google Scholar]
- 5.Kosorok MR, Jalaluddin M, Farrell PM, Shen G, Colby CE, Laxova A, Rock MJ, Splaingard M. Comprehensive analysis of risk factors for acquisition of Pseudomonas aeruginosa in young children with cystic fibrosis. Pediatr Pulmonol. 1998;26:81–8. doi: 10.1002/(sici)1099-0496(199808)26:2<81::aid-ppul2>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 6.West SE, Zeng L, Lee BL, Kosorok MR, Laxova A, Rock MJ, Splaingard MJ, Farrell PM. Respiratory infections with Pseudomonas aeruginosa in children with cystic fibrosis: early detection by serology and assessment of risk factors. JAMA. 2002;287:2958–67. doi: 10.1001/jama.287.22.2958. [DOI] [PubMed] [Google Scholar]
- 7.Burns JL, Emerson J, Stapp JR, Yim DL, Krzewinski J, Louden L, Ramsey BW, Clausen CR. Microbiology of sputum from patients at cystic fibrosis centers in the United States. Clin Infect Dis. 1998;27:158–63. doi: 10.1086/514631. [DOI] [PubMed] [Google Scholar]
- 8.FitzSimmons SC. The changing epidemiology of cystic fibrosis. J Pediatr. 1993;122:1–9. doi: 10.1016/s0022-3476(05)83478-x. [DOI] [PubMed] [Google Scholar]
- 9.Emerson J, Rosenfeld M, McNamara S, Ramsey B, Gibson RL. Pseudomonas aeruginosa and other predictors of mortality and morbidity in young children with cystic fibrosis. Pediatr Pulmonol. 2002;34:91–100. doi: 10.1002/ppul.10127. [DOI] [PubMed] [Google Scholar]
- 10.Kerem E, Corey M, Stein R, Gold R, Levison H. Risk factors for Pseudomonas aeruginosa colonization in cystic fibrosis patients. Pediatr Infect Dis J. 1990;9:494–8. doi: 10.1097/00006454-199007000-00008. [DOI] [PubMed] [Google Scholar]
- 11.Demko CA, Byard PJ, Davis PB. Gender differences in cystic fibrosis: Pseudomonas aeruginosa infection. J Clin Epidemiol. 1995;48:1041–9. doi: 10.1016/0895-4356(94)00230-n. [DOI] [PubMed] [Google Scholar]
- 12.Wang SS, FitzSimmons SC, O’Leary LA, Rock MJ, Gwinn ML, Khoury MJ. Early diagnosis of cystic fibrosis in the newborn period and risk of Pseudomonas aeruginosa acquisition in the first 10 years of life: A registry-based longitudinal study. Pediatrics. 2001;107:274–9. doi: 10.1542/peds.107.2.274. [DOI] [PubMed] [Google Scholar]
- 13.Burns JL, Gibson RL, McNamara S, Yim D, Emerson J, Rosenfeld M, Hiatt P, McCoy K, Castile R, Smith AL, Ramsey BW. Longitudinal assessment of Pseudomonas aeruginosa in young children with cystic fibrosis. J Infect Dis. 2001;183:444–52. doi: 10.1086/318075. [DOI] [PubMed] [Google Scholar]
- 14.Munck A, Bonacorsi S, Mariani-Kurkdjian P, Lebourgeois M, Gérardin M, Brahimi N, Navarro J, Bingen E. Genotypic characterization of Pseudomonas aeruginosa strains recovered from patients with cystic fibrosis after initial and subsequent colonization. Pediatr Pulmonol. 2001;32:288–92. doi: 10.1002/ppul.1121. [DOI] [PubMed] [Google Scholar]
- 15.Taccetti G, Campana S, Festini F, Mascherini M, Döring G. Early eradication therapy against Pseudomonas aeruginosa in cystic fibrosis patients. Eur Respir J. 2005;26:458–61. doi: 10.1183/09031936.05.00009605. [DOI] [PubMed] [Google Scholar]
- 16.McKay K, Cooper P, van Asperen P. Segregation of children with CF diagnosed via newborn screening and acquisition of Pseudomonas aeruginosa. J Cystic Fibrosis. 2009;8:400–404. doi: 10.1016/j.jcf.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 17.Kersulyte D, Struelens MJ, Deplano A, Berg DE. Comparison of arbitrarily primed PCR and macrorestriction (pulsed–field gel electrophoresis) typing of Pseudomonas aeruginosa strains from cystic fibrosis patients. J Clin Microbiol. 1995;33:2216–9. doi: 10.1128/jcm.33.8.2216-2219.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abman SH, Ogle JW, Harbeck RJ, Butler-Simon N, Hammond KB, Accurso FJ. Early bacteriologic, immunologic, and clinical courses of young infants with cystic fibrosis identified by neonatal screening. J Pediatr. 1991;119:211–7. doi: 10.1016/s0022-3476(05)80729-2. [DOI] [PubMed] [Google Scholar]
- 19.Van Daele SG, Franckx H, Verhelst R, Schelstraete P, Haerynck F, Van Simaey L, Claeys G, Vaneechoutte M, de Baets F. Epidemiology of Pseudomonas aeruginosa in a cystic fibrosis rehabilitation centre. Eur Respir J. 2005;25:474–81. doi: 10.1183/09031936.05.00050304. [DOI] [PubMed] [Google Scholar]
- 20.Spencer DH, Kas A, Smith EE, Raymond CK, Sims EH, Hastings M, Burns JL, Kaul R, Olson MV. Whole-genome sequence variation among multiple isolates of Pseudomonas aeruginosa. J Bacteriol. 2003;185:1316–25. doi: 10.1128/JB.185.4.1316-1325.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ernst RK, D’Argenio DA, Ichikawa JK, Bangera MG, Selgrade S, Burns JL, Hiatt P, McCoy K, Brittnacher M, Kas A, Spencer DH, Olson MV, Ramsey BW, Lory S, Miller SI. Genome mosaicism is conserved but not unique in Pseudomonas aeruginosa isolates from the airways of young children with cystic fibrosis. Environ Microbiol. 2003;5:1341–9. doi: 10.1111/j.1462-2920.2003.00518.x. [DOI] [PubMed] [Google Scholar]
- 22.Ballman M, Rabsch P, von der Hardt H. Long-term follow up of changes in FEV1 and treatment intensity during Pseudomonas aeruginosa colonisation in patients with cystic fibrosis. Thorax. 1998;53:732–7. doi: 10.1136/thx.53.9.732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jones AM, Dodd ME, Govan JR, Doherty CJ, Smith CM, Isalska BJ, Webb AK. Prospective surveillance for Pseudomonas aeruginosa cross-infection at a cystic fibrosis center. Am J Respir Crit Care Med. 2005;171:257–60. doi: 10.1164/rccm.200404-513OC. [DOI] [PubMed] [Google Scholar]
- 24.Griffiths AL, Jamsen K, Carlin JB, Grimwood K, Carzino R, Robinson PJ, Massie J, Armstrong DS. Effects of segregation on an epidemic Pseudomonas aeruginosa strain in a cystic fibrosis clinic. Am J Respir Crit Care Med. 2005;171:1020–5. doi: 10.1164/rccm.200409-1194OC. [DOI] [PubMed] [Google Scholar]
- 25.Speert DP, Campbell ME, Henry DA, Milner R, Taha F, Gravelle A, Davidson AG, Wong LT, Mahenthiralingam E. Epidemiology of Pseudomonas aeruginosa in cystic fibrosis in British Columbia, Canada. Am J Respir Crit Care Med. 2002;166:988–93. doi: 10.1164/rccm.2203011. [DOI] [PubMed] [Google Scholar]
- 26.Ramsey BW. To cohort or not to cohort: how transmissible is Pseudomonas aeruginosa? Am J Respir Crit Care Med. 2002;166:906–7. doi: 10.1164/rccm.2207005. [DOI] [PubMed] [Google Scholar]
- 27.Armstrong DS, Nixon GM, Carzino R, Bigham A, Carlin JB, Robins-Browne RM, Grimwood K. Detection of a widespread clone of Pseudomonas aeruginosa in a pediatric cystic fibrosis clinic. Am J Respir Crit Care Med. 2002;166:983–7. doi: 10.1164/rccm.200204-269OC. [DOI] [PubMed] [Google Scholar]
- 28.Farrell MH, Farrell PM. Newborn screening for cystic fibrosis: Ensuring more good than harm. J Pediatr. 2003;143:707–12. doi: 10.1016/j.jpeds.2003.09.022. [DOI] [PubMed] [Google Scholar]
- 29.Scott FW, Pitt TL. Identification and characterization of transmissible Pseudomonas aeruginosa strains in cystic fibrosis patients in England and Wales. J Med Microbiol. 2004;53:609–15. doi: 10.1099/jmm.0.45620-0. [DOI] [PubMed] [Google Scholar]
- 30.O’Carroll MR, Syrmis MW, Wainwright CE, Greer RM, Mitchell P, Coulter C, Sloots TP, Nissen MD, Bell SC. Clonal strains of Pseudomonas aeruginosa in paediatric and adult cystic fibrosis units. Eur Respir J. 2004;24:101–6. doi: 10.1183/09031936.04.00122903. [DOI] [PubMed] [Google Scholar]
- 31.McCallum SJ, Corkill J, Gallagher M, Ledson MJ, Hart CA, Walshaw MJ. Superinfection with a transmissible strain of Pseudomonas aeruginosa in adults with cystic fibrosis chronically colonised by P aeruginosa. Lancet. 2001;358:558–60. doi: 10.1016/s0140-6736(01)05715-4. [DOI] [PubMed] [Google Scholar]
- 32.Jones AM, Govan JR, Doherty CJ, Dodd ME, Isalska BJ, Stanbridge TN, Webb AK. Spread of a multiresistant strain of Pseudomonas aeruginosa in an adult cystic fibrosis clinic. Lancet. 2001;358:557–8. doi: 10.1016/s0140-6736(01)05714-2. [DOI] [PubMed] [Google Scholar]
- 33.Cheng K, Smyth RL, Govan JR, Doherty C, Winstanley C, Denning N, Heaf DP, van Saene H, Hart CA. Spread of beta-lactam-resistant Pseudomonas aeruginosa in a cystic fibrosis clinic. Lancet. 1996;348:639–42. doi: 10.1016/S0140-6736(96)05169-0. [DOI] [PubMed] [Google Scholar]
- 34.Saiman L, Siegel J. Infection control in cystic fibrosis. Clin Microbiol Rev. 2004;17:57–71. doi: 10.1128/CMR.17.1.57-71.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]