

Abstract
Objective
Skin disease is the second most common manifestation in patients with systemic lupus erythematosus (SLE). TNF receptor (TNFR) preligand assembly domain (PLAD) has been found to block the effect of TNF-α and TNFR1 PLAD (P60 PLAD) inhibits inflammatory arthritis. We asked whether TNFR PLAD can limit inflammatory skin injury in SLE.
Methods
Female MRL/lpr mice received P60 PLAD (100 µg/mouse, i.p.) or P80 PLAD (100 µg/mouse, i.p.) or PBS (100 µl/mouse, i.p.) three times a week starting at age of 6 weeks for 26 weeks.
Results
Immunohistochemistry studies demonstrated that TNFR1 but not TNFR2 is dominantly expressed in skin lesions in MRL/lpr mice. We found that TNFR1 PLAD but not TNFR2 PLAD (P80 PLAD) protein significantly inhibited skin injury in lupus MRL/lpr mice. P60 PLAD significantly inhibited NF-κB, MCP-1 and iNOS expression in skin lesions. P60 PLAD reduced lupus serum-induced monocyte differentiation into dendritic cells. P60 PLAD did not reduce IgG deposition in the skin and improve kidney pathology progression in MRL/lpr mice.
Conclusion
Our results indicate that TNFR1 is involved in the expression of skin injury in lupus MRL/lpr mice and P60 PLAD or similar biologics may be of clinical value if applied locally.
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease characterized by high levels of autoantibody and multi-organ tissue damage including kidney and skin (1, 2). Keratinocyte apoptosis represents one of the histological features of skin lesions in SLE patients and may represent a source of autoantigens which in turn may be involved in the production of antibody. Antibody is deposited in the skin of patients with SLE even in non-affected areas (band test) and the presence of circulating anti-Ro (SS-A) antibodies has been associated with skin disease (3). Exposure to ultraviolet light is known to trigger disease (photosensitivity) but the exact mechanisms are not clear (4).
Tumor necrosis factor α (TNF-α) is a proinflammatory cytokine mainly produced by macrophages/monocytes (5). TNFα exerts its effect by binding to extracellular domains of TNFR1 (P60) and TNFR2 (P80). Recently, TNFR PLAD has been found to exert a crucial role in TNFR signaling (6). PLAD protein block effects of TNF-α and P60 PLAD protein inhibits inflammatory arthritis induced by TNF-α, CpG DNA and collagen (7). Given that PLAD has a key role in TNFR signaling, and that TNF-α induces skin inflammation (8), we investigated potential inhibitory role of soluble PLAD proteins in skin inflammation of lupus MRL/lpr mice. We found that P60 not P80 PLAD protein inhibited skin injury in MRL/lpr mice. Our data demonstrates that P60 PLAD protein can ameliorate skin injury in MRL/lpr mice.
MATERIAL AND METHODS
Mice and materials
Female MRL/lpr/2J mice, TNFR1 and TNFR2 gene deficient mice and C57BL/6 mice were purchased from the Jackson Laboratories (Bar Harbor, ME) and housed in the animal facility of Beth Israel Deaconess Medical Center. Double strand (ds) DNA, mouse IgG antibodies were purchased from Sigma-Aldrich. Antibodies to TNFR1, TNFR2, MCP-1, iNOS and NF-κB were purchased from Santa Cruz (CA).
PLAD protein preparation
PLAD60 and PLAD80 proteins were prepared by using GST-fusion protein as described previously (7). LPS was removed from the purified PLAD protein using Detoxi-Gel AffinityPak Columns (pierce) before use.
Treatment of MRL/lpr mice by P60 and P80 PLAD protein
Female MRL/lpr mice received P60 PLAD (100 µg/mouse, i.p. n =8) or P80 PLAD (100 µg/mouse, i.p. n=8) or PBS (100 µl/mouse, i.p. n=8) three times a week starting at age of 6 weeks for 26 weeks. Skin and kidney from sacrificed mice were collected for histological examination and serum was collected for measurement of serum IgG and anti-dsDNA antibody. During the experimental period, urine protein content and mortality were monitored.
Histology of skin and kidney
Histopathological examination of skin and kidney was done after routine fixation and paraffin embedding of the tissue. Tissue sections from skin were cut and stained with hematoxylin and eosin. All slides were coded and evaluated in a ‘blinded’ to sample identity manner. Severity of skin inflammation will be scored 0–4. Grade 0: normal; Grade 1: hyperplasia of epidermis; Grade 2–4: different amounts of infiltrating inflammatory cells in the skin. Renal pathology is graded by glomerular, interstitial and perivascular inflammation. Scores from 0 to 4 are assigned for each of the features. A minimum of 100 glomeruli were assessed to determine the glomerular scoring index in each mouse.
Urine analysis
The mice in each group were placed overnight in a Nalgene metabolic cage to collect urine. Urine was measured with Multistix 10 SG and analyzed by Clinitek Status analyzer (Bayer Healthcare). Proteinuria is expressed as 0–4, 0 (none), 1 (30–100 mg/dl), 2 (100–300 mg/dl), 3 (300–2000 mg/dl), or 4 (> 2000 mg/dl).
Measurement of serum IgG and anti-DNA antibody
Serum IgG and anti-dsDNA antibody were detected by ELISA. 96-well plate was coated with double calf thymus DNA (1.5 mg/ml, Sigma) or mouse IgG antibody (0.1 mg/ml, Sigma) in PBS at 4°C overnight. After blocking with 1% BSA for 2 hours at room temperature (RT), serum samples were added to plate and incubated for 2 hours at RT. After washing 4 times with PBS, HRP-conjugated goat anti-mouse IgG antibody (Sigma) was added to plate and incubated for 1 hour at RT. Antibody binding was visualized using TMB (3’, 3’, 5’, 5’- tetramethylbenzidine, Sigma), and plates were read the optical density at 450 nm.
Immunohistochemistry staining
After routine fixation and paraffin embedding of the tissue, tissue sections from skin were cut. After deparaffinization and antigen retrieval, samples were stained with antibody to TNFR1, TNFR2, NF-κB, MCP-1, iNOS (Santa Cruz, CA) followed by incubation with biotinylated secondary antibodies and avidin-biotin-peroxidase complexes and 3-amino-9-ethylcarbazole containing H2O2. All sections were counterstained with Mayer’s hematoxylin.
Monocyte differentiation into DCs
Spleens were removed from C57BL/6, TNFR1 or TNFR2-deficient mice. Mononuclear cells were isolated from mouse spleens. Mononuclear cells were incubated in 24-well plates at 37°C. Monocytes were obtained after suspending lymphocytes were removed. Sera (40 µl) prior or after IgG depletion from lupus-prone mice (5 month old MRL/lpr mice) or normal C57BL/6 mice, TNF-α (2 ng) and M-CSF (10 ng) were added, as indicated, in plates (9). Differentiation of monocytes into DCs was examined by light microscopy. DCs were fixed and permeabilized and then stained with monoclonal anti-CD40 and CD80 conjugated to PE (Santa Cruz, CA). Expression of CD40, CD80 on DCs was examined using flow cytometry.
Statistics
Statistical evaluations of skin inflammation, renal pathology, serum IgG and anti-dsDNA antibody data were performed using the Student's t-test or the Mann-Whitney test. P ≤ 0.05 was considered statistically significant.
RESULTS
TNFR1 but not TNFR2 is expressed in skin lesions of lupus MRL/lpr mice
MRL/lpr mice, an established lupus animal model, develop spontaneously lupus skin injury (10) (Fig. 1 A). We investigated the expression of TNFR1 and TNFR2 in skin lesions of MRL/lpr mice using immunohistochemistry and found significant expression of TNFR1 and much less TNFR2 expression (Fig. 1B). This data suggests the involvement of TNFR1 in the expression of inflammatory process in the skin of lupus prone mice. Immunofluorescent staining with an anti-CD11c antibody revealed the presence of dendritic cells in the skin lesions (Fig. 1C).
Figure 1.
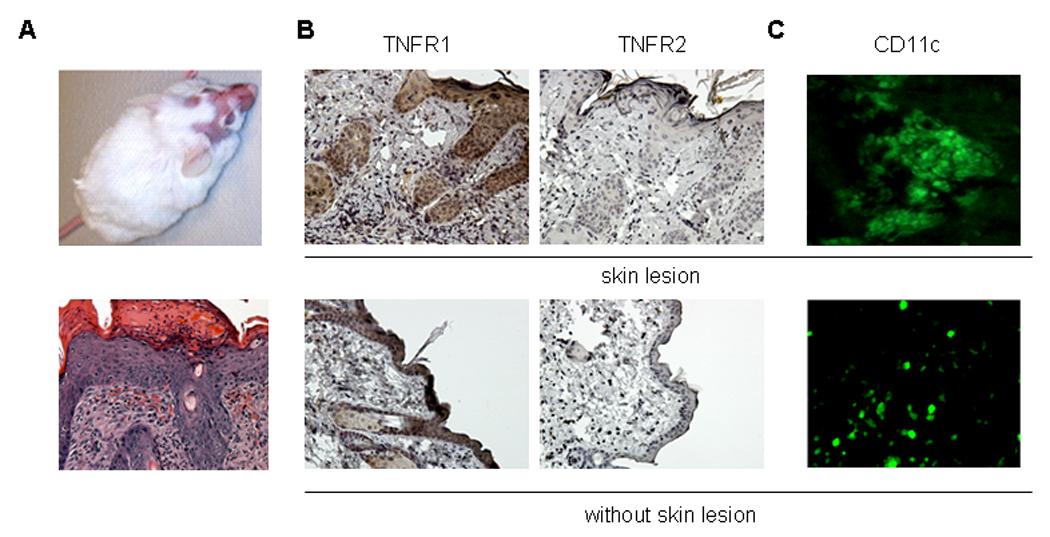
Expression of TNFR and CD11c in skin lesions of MRL/lpr mice. (A) Representative pictures of skin lesions and histopathology in MRL/lpr mice. (B) Representative figures of immunohistochemistry staining of expression of TNFR1, TNFR2 (upper rows). Arrow represents staining of TNFR1 in the skin. Lower rows show TNFR1 and TNFR2 staining in the skin without injury. (C) Representative immunofluorescent staining of CD11c in skin injury of MRL/lpr mice (upper). Lower row shows staining without skin injury.
Treatment of MRL/lpr with P60 PLAD protein limits the appearance of skin lesions
We prepared soluble P60 and P80 PLAD proteins as described previously (7) and injected them in the peritoneum of 6 weeks old MRL/lpr mice for 26 weeks. We assumed that if TNFRs are involved in the expression of skin lesions of these mice then their inhibition should suppress their appearance. During the treatment period, we observed that skin lesions developed on both sides of the face of MRL/lpr mice treated with P80 PLAD or PBS but not in the face of mice treated with P60 PLAD (Fig. 2A–B). The skin lesions on the face appeared earlier and the incidence of skin lesions was higher in mice treated with P80 PLAD than with control PBS (Fig. 2A–B). Histopathologic examination demonstrated a markedly thickened epidermis with inflammatory cell infiltration in the dermis in PBS and P80 PLAD-treated mice but not in mice treated with P60 PLAD (Fig. 2C–D). Also, one mouse treated with P80 PLAD developed skin injury on the back. In a group of mice we discontinued PLAD treatment and we observed the development of cutaneous injury on month later in mice previously treated with P60 PLAD (Fig. 2E). These data indicates that P60 PLAD protein can suppresses skin injury in MRL/lpr mice.
Figure 2.
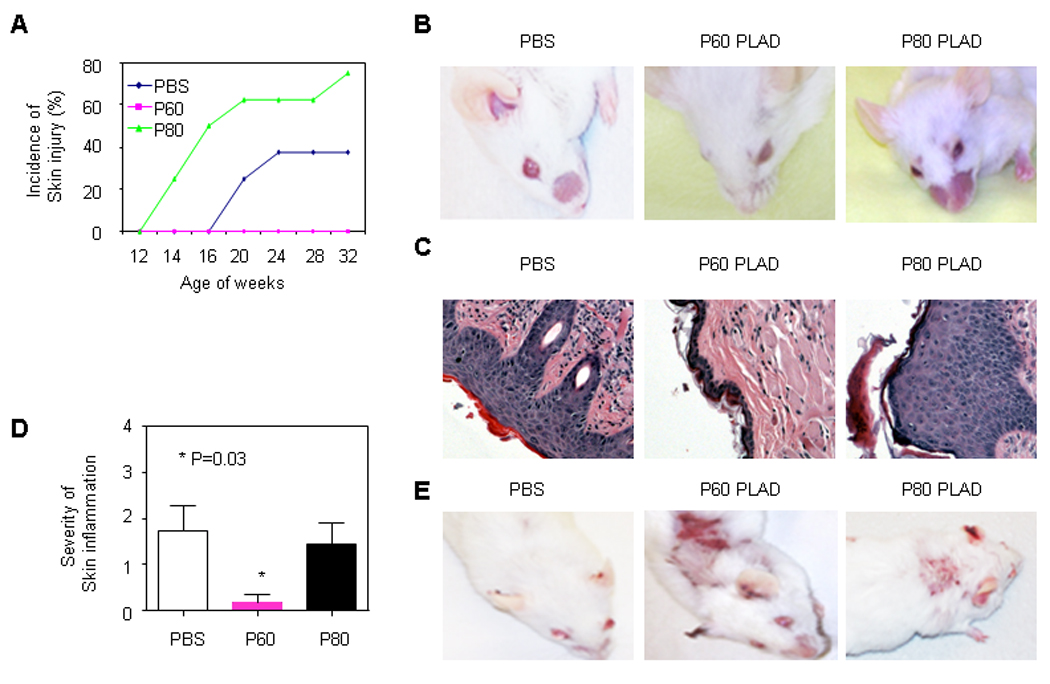
Effect of P60 and P80 PLAD proteins on skin injury of MRL/lpr mice. MRL/lpr mice were treated with P60 PLAD (n= 8, 100 µg/mouse, three times a week, i.p.) or P80 PLAD protein (n= 8, 100 µg/mouse, three times a week, i.p.) or PBS (n= 8, 100 µl/mouse, three times a week, i.p.) starting at 6 week of age for 26 weeks. Skin injury was assessed by incidence and histopathological examination. (A) Incidence of skin injury on the mouse face. (B) Representative picture of MRL/lpr with PBS or P60 or P80 treatment. (C) Representative histopathologic pictures of skin in MRL/lpr mice treated with PBS or P60 or P80 PLAD protein at age of 32 weeks. (D) Severity of skin inflammation. P value= 0.03 (PBS vs P60 PLAD) (E) Pictures of MRL/lpr mice 4 weeks after discontinued treatment.
P60 PLAD protein reduces lupus MRL/lpr serum-induced monocyte differentiation into dendritic cells (DCs)
Monocytes contribute to skin injury in MRL/lpr mice (11). Monocytes not only become macrophages but also represent an important source of DCs during an inflammatory process (12–14). We investigated whether P60 PLAD protein can regulate lupus MRL/lpr serum-induced monocyte differentiation into DCs (9). Serum from 5-month old MRL/lpr mice induced monocyte differentiation into DCs but serum from normal C57BL/6 mice did not (Fig. 3A and supplementary figure 1A). Flow cytometry analysis demonstrated that lupus serum, but normal serum, caused enhanced expression of CD40 and CD80 on the surface membrane of monocytes (Fig. 3B). Depletion of IgG from lupus MRL/lpr serum abolished lupus serum-mediated monocyte differentiation into DCs (Fig. 3C). Immunofluorescent staining demonstrated the cells identified as DCs by light microscopy were CD11c positive cells (Supplementary figure 1B). In addition, we found the accumulated CD11c positive cells in skin sites followed intradermal injection of lupus serum from MRL/lpr mice not from C57BL/6 mice (Fig. 3D).
Figure 3.
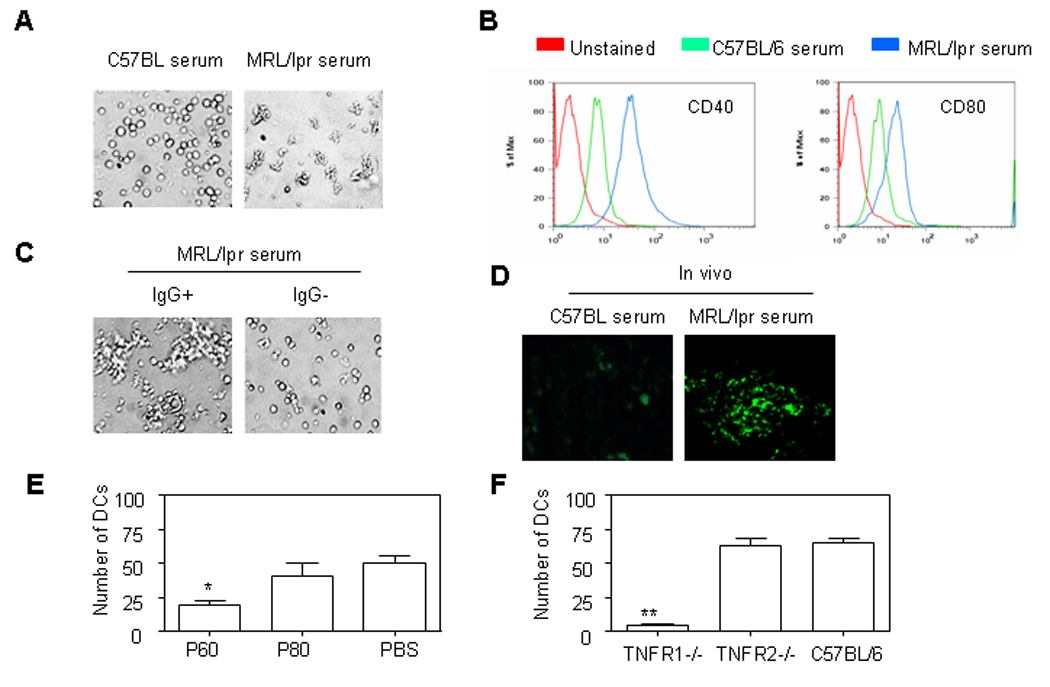
P60 PLAD protein inhibited lupus serum-induced monocyte differentiation into dendritic cells (DCs). (A). Representative pictures of monocytes from C57BL/6 mice cultured with serum from lupus MRL/lpr mice, normal C57BL/6 mice for 40 hours. (B) Expression of CD40 and CD80 on DCs measured by flow cytometry. (C) Representative pictures of monocytes from C57BL/6 mice cultured with serum from lupus MRL/lpr mice with or without IgG depletion for 40 hours. (D) Immunofluorescent staining of DCs in C57BL/6 mice skin with FITC conjugated anti-CD11c Ab 3d after intradermal injection of serum (100 µl) from lupus MRL/lpr or C57BL/6 mice. (E) Cumulative data of DCs differentiated from monocytes of C57BL/6 mouse treated with lupus MRl/lpr serum for 40 hours in the presence of PBS, P60 PLAD (100 µg) or P80 PLAD (100 µg). (F) Cumulative data of DCs differentiated from monocytes of mice with TNFR1 or TNFR2 gene deficiency and wild type mice were cultured with serum from lupus MRL/lpr mice for 40 hours.
To understand the role of TNFR1 in lupus MRL/lpr serum-induced monocyte differentiation into DCs, we investigated whether P60 PLAD regulates lupus serum-induced monocyte differentiation into DCs. We observed that P60 PLAD not P80 PLAD reduced lupus MRL/lpr serum-induced monocyte differentiation into DCs (Fig. 3E). To further confirm the role of TNFR1 in lupus MRL/lpr serum-induced monocyte differentiation into DCs, we repeated experiments using monocytes from TNFR1 or TNFR2 knockout mice. We observed that lupus MRL/lpr serum-induced monocyte differentiation into DCs was markedly limited in TNFR1 but not TNFR2-deficient mice (Fig. 3F). These data indicates that P60 PLAD regulates lupus MRL/lpr serum containing IgG-induced monocyte differentiation into DCs.
P60 PLAD protein inhibited the expression of MCP-1, iNOS, NF-κB in skin lesions of MRL/lpr mice
Immune cells migrate to inflammatory sites from blood vessels in response to increased expression of chemokines (15). MCP-1 has been shown to contribute to the attraction of immune cells to the inflamed skin in MRL/lpr mice (11, 16). MCP-1 expression was significantly reduced in the skin lesions in mice treated with P60 PLAD protein compared to PBS treatment (Fig. 4A).
Figure 4.
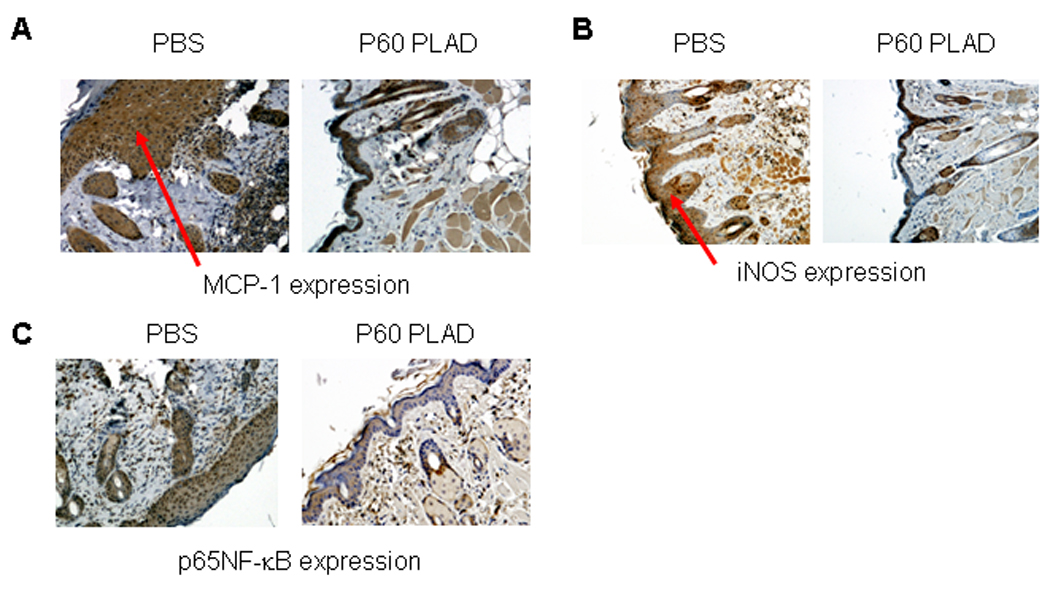
Expression of MCP-1, iNOS and NF-κB in skin lesions of MRL/lpr mice. Expression of MCP-1, iNOS and NF-κB (A–C) in skin lesions of MRL/lpr mice treated with PBS or P60 PLAD at age of 32 weeks. Arrow represents staining of MCP-1, iNOS and NF-κB in the skin.
Nitric oxide is involved in tissue inflammation (17). We investigated whether P60 PLAD regulates iNOS expression in skin lesions in MRL/lpr mice. iNOS expression was markedly decreased in skin lesions in mice treated with P60 PLAD protein (Fig. 4B).
NF-κB is an important intracellular regulator of genes involved in the inflammatory response (7, 18, 19), we investigated whether NF-κB is downregulated in the skin lesions of MRL/lpr mice treated with P60 PLAD protein. NF-κB expression was markedly decreased in inflammatory lesions of MRL/lpr mice treated with P60 PLAD protein compared to the MRL/lpr mice treated with PBS (Fig. 4 C).
Effect of TNFR PLAD protein on autoantibody production and skin deposition in MRL/lpr mice
We determined the effect of TNFR PLAD protein treatment on the production of autoantibodies in MRL/lpr mice. We measured levels of IgG, anti-dsDNA antibodies in the sera of MRL/lpr mice at age of 16 weeks by ELISA. We noted a tendency for higher IgG levels in mice treated with P60 PLAD protein and also a similar tendency for higher levels of anti-ds-DNA antibody production (Fig. 5A–B) suggesting that blockade of TNFR does not improve systemic autoimmunity. Next we also investigated whether PLAD protein treatment alters IgG deposition in the skin of MRL/lpr mice. Immunohistochemistry staining demonstrated that PLAD protein treatment does not significantly inhibit IgG deposition in the skin of MRL/lpr mice compared to control PBS treatment (Fig. 5C) suggesting again that the effect of PLAD P60 relies on its ability to limit the local skin inflammatory response.
Figure 5.
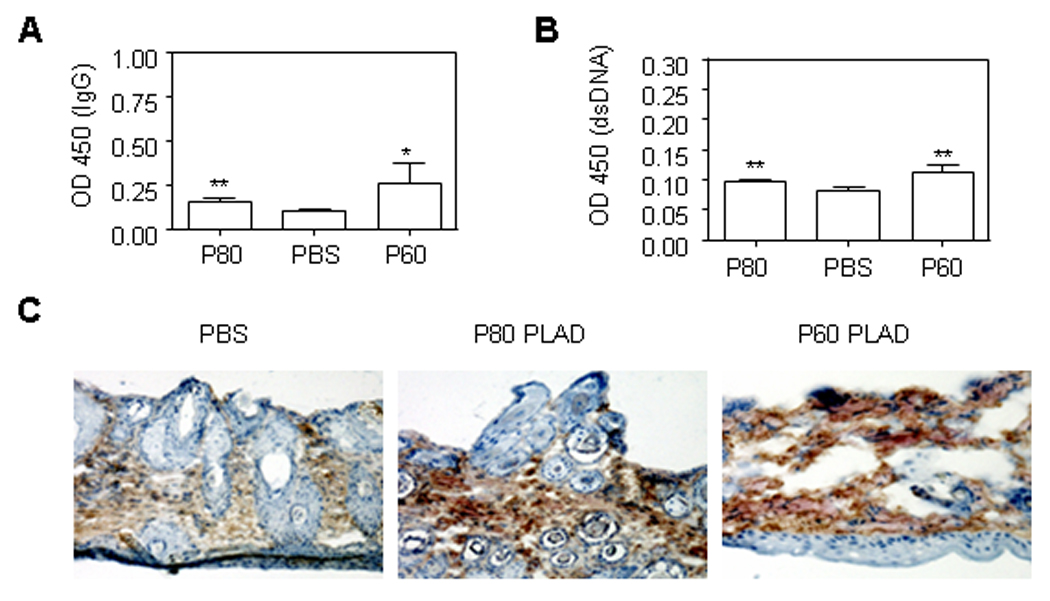
Effects of P60 and P80 PLAD protein on autoantibody production and skin deposition in MRL/lpr mice. At 16 weeks of age, OD450 values of serum IgG (1:4) (A) and serum anti-dsDNA antibody (1:4) in MRL/lpr mice treated with PBS or P60 or P80 PLAD protein. *P < 0.01 and ** P> 0.05 (P value represents P60 or P80 PLAD vs PBS treatment). (C) Representative immunohistochemistry staining of IgG deposition in face skin injury of MRL/lpr mice. Skin from MRL/lpr mice treated with PBS or P80 PLAD or P60 PLAD at the age of 32 weeks.
Effect of TNFR PLAD proteins on kidney pathology in MRL/lpr mice
During treatment period, we also investigated effects of PLAD protein on kidney damage. It appeared that mice treated with PLAD proteins had a tendency to develop more proteinuria (Fig. 6A). Kidney pathology worsened in the mice treated with PLAD P60 (Fig. 6B). This data suggest that like systemic autoimmunity, lupus kidney pathology does not benefit from TNFR inhibition.
Figure 6.
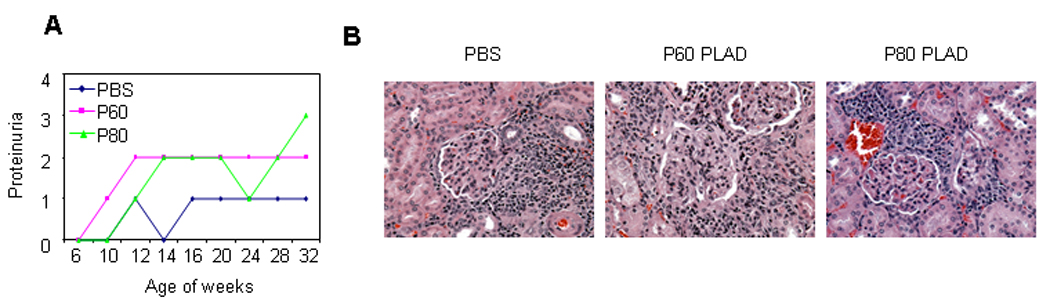
Effect of PLAD proteins on kidney pathology in MRL/lpr mice. MRL/lpr mice were treated with P60 PLAD (n= 8, 100 µg/mouse, three times a week, i.p.) or P80 PLAD protein (n= 8, 100 µg/mouse, three times a week, i.p.) or PBS (n= 8, 100 µl/mouse, three times a week, i.p.) starting at age of 6 weeks for 26 weeks. (A) Proteinuria from MRL/lpr mice treated with PBS or P60 or P80 PLAD protein. (B) Representative images of H&E stained tissue sections of kidney.
Discussion
Our results demonstrate that P60 PLAD but not P80 PLAD protein can suppress skin injury in lupus MRL/lpr mice. The fact that skin lesions express TNFR1 rather than TNFR2 may explain the mitigating effect of the TNFR1 inhibitor P60 PLAD. Our findings are in agreement with previous reports that TNF inhibition does not affect systemic autoimmunity, or it may, as it has been observed in patients receiving anti-TNF treatment (20), instigate systemic autoimmunity manifested by the appearance of anti-nuclear antibodies. Treatment of MRL/lpr mice with P60 or P80 PLAD proteins did not improve systemic autoimmunity, but instead, it appeared to provoke a tendency towards worsening. Treatment with P60 PLAD did not even affect the deposition of immunoglobulin to the skin of MRL/lpr mice despite the fact that the inflammatory process was suppressed. This antithetic effect signifies the independence of local organ injury form systemic autoimmunity and calls for attention to local events which may execute tissue damage and which may become targets of treatment.
Keratinocyte apoptosis is a histologically recognizable feature of skin lesions in patients with lupus. It has been well established that apoptotic material may flood the immune system with autoantigens which may promote the appearance of high levels of autoantibodies (21). On the other hand, since Fas-induced apoptosis is defective in MRL/lpr mice, blocking of TNF-α-induced apoptosis(5) may enhance severity of defective apoptosis in MRL/lpr mice. TNFR1 represents the main receptor in the TNF-α-induced apoptosis (5) and although TNFR2 lacks a death domain it activates NF-κB (5) and is involved in T cell apoptosis, especially CD8+ T cells (22). Thus, blockade of TNFR1 or TNFR2 using P60 PLAD or P80 PLAD may enhance defective apoptosis in T cells and promote the production of autoantibodies in MRL/lpr mice. This data indicates that TNF may protect against nephritis by reducing the production of autoantibodies and by promoting T cell apoptosis in MRL/lpr mice.
Tissue damage in SLE is characterized by autoantibody production and immune complex formation and deposition (2). Antibody-nucleosome complexes and IgG have been found deposited in the renal glomerular basement membrane in lupus nephritis in humans and mice (23–25). These immune complexes activate complement and initiate the glomerulonephritis which has been demonstrated in animal models (26, 27). The anti-dsDNA and anti-nucleosome antibodies cross-react with proteins such as α-actinin in the kidney and may have a direct pathogenic effect on renal cells (28, 29). The absence of TNFR1 in the Fas deficient mouse resulted in greatly accelerated glomerulonephritis and production of IgG and anti-dsDNA antibodies (30). Although the levels of anti-dsDNA antibody in mice treated with P60 or P80 PLAD did not rise significantly more than in mice treated with PBS, both levels of IgG and anti-dsDNA antibody displayed a trend towards higher values (Fig. 5). It is possible that other autoantibodies including anti-histone, anti-phospholipid antibodies might be elevated in MRL/lpr mice treated with P60 or P80 PLAD protein and make contribution to development of proteinuria and kidney damage.
DCs are important resident cells in skin and the first to react to pathogen in the skin inflammation (13). Activated DCs release cytokines and move to the draining lymph nodes where mature DCs activate T cells (14). Monocytes from blood vessels are thought to be an important resource of DCs (15). Serum from lupus patients promotes monocyte differentiation into DCs (9) and serum IgG from MRL/lpr mice was shown here to induce monocyte differentiation into DCs. Although IFN-α has been shown to exert important role in lupus patient serum-induced monocyte differentiation into DCs (9), our results demonstrate that lupus MRL/lpr serum-induced monocyte differentiation into DCs also requires TNFR1 signaling. Blocking TNFR1 signaling by P60 PLAD protein inhibited lupus MRL/lpr serum –induced monocyte differentiation into DCs. Lupus MRL/lpr serum-induced monocyte differentiation into DCs was markedly reduced in monocytes from TNFR1 but not TNFR2 knockout mice. TNFR1 has been shown to be required for differentiation of follicular DCs (31). TNFR1 knockout mice are resistant to TNF-α-induced skin inflammation (32).
In conclusion, this study shows that TNFR1 is important for the expression of skin lesion in the lupus prone MRL/lpr mouse. Blockade of TNFR1 signaling mitigates skin lesions despite the fact that it does not affect skin immunoglobulin deposition. In agreement with previous information and clinical experience, TNF inhibition with PLAD proteins displayed a tendency towards aggravated systemic autoimmunity and kidney pathology. The value of our results lies with the establishment of the importance of local factors in the expression of local organ injury and the possibility of considering the application of local TNFR1 inhibitors in the treatment of lupus skin lesions.
Supplementary Material
Acknowledgements
Work was supported by PHS NIH Grant RO1 AI 42269 and the Mary Kirkland Center for Lupus Research.
Footnotes
Author Contributions
Dr. Deng and Tsokos had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis
Study design. Guo-Min Deng, George C. Tsokos
Acquisition of data. Guo-Min Deng, Lena Liu
Analysis and interpretation of data. Guo-Min Deng, George C. Tsokos
Manuscript preparation. Guo-Min Deng, George C. Tsokos
Statistical analysis. Guo-Min Deng
REFERENCES
- 1.Fu SM, Castillo JD, Deshmukh US, Lewis JE, Waters ST, Gaskin F. Autoantibodies and glomerulonephritis in systemic lupus erythematosus. Lupus. 2003;12(3):175–180. doi: 10.1191/0961203303lu352xx. [DOI] [PubMed] [Google Scholar]
- 2.Rahman A, Isenberg DA. Systemic lupus erythematosus. N Engl J Med. 2008;358(9):929–939. doi: 10.1056/NEJMra071297. [DOI] [PubMed] [Google Scholar]
- 3.Fukuda MV, Lo SC, de Almeida CS, Shinjo SK. Anti-Ro antibody and cutaneous vasculitis in systemic lupus erythematosus. Clin Rheumatol. 2009;28(3):301–304. doi: 10.1007/s10067-008-1043-5. [DOI] [PubMed] [Google Scholar]
- 4.Norris DA. Pathomechanisms of photosensitive lupus erythematosus. J Invest Dermatol. 1993;100(1):58S–68S. doi: 10.1111/1523-1747.ep12355599. [DOI] [PubMed] [Google Scholar]
- 5.Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell. 2001;104(4):487–501. doi: 10.1016/s0092-8674(01)00237-9. [DOI] [PubMed] [Google Scholar]
- 6.Chan FK, Chun HJ, Zheng L, Siegel RM, Bui KL, Lenardo MJ. A domain in TNF receptors that mediates ligand-independent receptor assembly and signaling. Science. 2000;288(5475):2351–2354. doi: 10.1126/science.288.5475.2351. [DOI] [PubMed] [Google Scholar]
- 7.Deng GM, Zheng L, Chan FK, Lenardo M. Amelioration of inflammatory arthritis by targeting the pre-ligand assembly domain of tumor necrosis factor receptors. Nat Med. 2005;11(10):1066–1072. doi: 10.1038/nm1304. [DOI] [PubMed] [Google Scholar]
- 8.Kondo S, Sauder DN. Tumor necrosis factor (TNF) receptor type 1 (p55) is a main mediator for TNF-alpha-induced skin inflammation. Eur J Immunol. 1997;27(7):1713–1718. doi: 10.1002/eji.1830270718. [DOI] [PubMed] [Google Scholar]
- 9.Blanco P, Palucka AK, Gill M, Pascual V, Banchereau J. Induction of dendritic cell differentiation by IFN-alpha in systemic lupus erythematosus. Science. 2001;294(5546):1540–1543. doi: 10.1126/science.1064890. [DOI] [PubMed] [Google Scholar]
- 10.Peng SL, Moslehi J, Craft J. Roles of interferon-gamma and interleukin-4 in murine lupus. J Clin Invest. 1997;99(8):1936–1946. doi: 10.1172/JCI119361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Menke J, Hsu MY, Byrne KT, Lucas JA, Rabacal WA, Croker BP, et al. Sunlight triggers cutaneous lupus through a CSF-1-dependent mechanism in MRL-Fas(lpr) mice. J Immunol. 2008;181(10):7367–7379. doi: 10.4049/jimmunol.181.10.7367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ginhoux F, Tacke F, Angeli V, Bogunovic M, Loubeau M, Dai XM, et al. Langerhans cells arise from monocytes in vivo. Nat Immunol. 2006;7(3):265–273. doi: 10.1038/ni1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leon B, Lopez-Bravo M, Ardavin C. Monocyte-derived dendritic cells formed at the infection site control the induction of protective T helper 1 responses against Leishmania. Immunity. 2007;26(4):519–531. doi: 10.1016/j.immuni.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 14.Shortman K, Naik SH. Steady-state and inflammatory dendritic-cell development. Nat Rev Immunol. 2007;7(1):19–30. doi: 10.1038/nri1996. [DOI] [PubMed] [Google Scholar]
- 15.Auffray C, Sieweke MH, Geissmann F. Blood Monocytes: Development, Heterogeneity, and Relationship with Dendritic Cells. Annu Rev Immunol. 2009 doi: 10.1146/annurev.immunol.021908.132557. [DOI] [PubMed] [Google Scholar]
- 16.Tesch GH, Maifert S, Schwarting A, Rollins BJ, Kelley VR. Monocyte chemoattractant protein 1-dependent leukocytic infiltrates are responsible for autoimmune disease in MRL-Fas(lpr) mice. J Exp Med. 1999;190(12):1813–1824. doi: 10.1084/jem.190.12.1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Belmont HM, Levartovsky D, Goel A, Amin A, Giorno R, Rediske J, et al. Increased nitric oxide production accompanied by the up-regulation of inducible nitric oxide synthase in vascular endothelium from patients with systemic lupus erythematosus. Arthritis Rheum. 1997;40(10):1810–1816. doi: 10.1002/art.1780401013. [DOI] [PubMed] [Google Scholar]
- 18.Deng GM, Verdrengh M, Liu ZQ, Tarkowski A. The major role of macrophages and their product tumor necrosis factor alpha in the induction of arthritis triggered by bacterial DNA containing CpG motifs. Arthritis Rheum. 2000;43(10):2283–2289. doi: 10.1002/1529-0131(200010)43:10<2283::AID-ANR16>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 19.Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5(10):749–759. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- 20.Williams EL, Gadola S, Edwards CJ. Anti-TNF-induced lupus. Rheumatology (Oxford) 2009;48(7):716–720. doi: 10.1093/rheumatology/kep080. [DOI] [PubMed] [Google Scholar]
- 21.Lorenz HM, Herrmann M, Winkler T, Gaipl U, Kalden JR. Role of apoptosis in autoimmunity. Apoptosis. 2000;5(5):443–449. doi: 10.1023/a:1009692902805. [DOI] [PubMed] [Google Scholar]
- 22.Zheng L, Fisher G, Miller RE, Peschon J, Lynch DH, Lenardo MJ. Induction of apoptosis in mature T cells by tumour necrosis factor. Nature. 1995;377(6547):348–351. doi: 10.1038/377348a0. [DOI] [PubMed] [Google Scholar]
- 23.Berden JH, Licht R, van Bruggen MC, Tax WJ. Role of nucleosomes for induction and glomerular binding of autoantibodies in lupus nephritis. Curr Opin Nephrol Hypertens. 1999;8(3):299–306. doi: 10.1097/00041552-199905000-00005. [DOI] [PubMed] [Google Scholar]
- 24.Kalaaji M, Fenton KA, Mortensen ES, Olsen R, Sturfelt G, Alm P, et al. Glomerular apoptotic nucleosomes are central target structures for nephritogenic antibodies in human SLE nephritis. Kidney Int. 2007;71(7):664–672. doi: 10.1038/sj.ki.5002133. [DOI] [PubMed] [Google Scholar]
- 25.Kalaaji M, Mortensen E, Jorgensen L, Olsen R, Rekvig OP. Nephritogenic lupus antibodies recognize glomerular basement membrane-associated chromatin fragments released from apoptotic intraglomerular cells. Am J Pathol. 2006;168(6):1779–1792. doi: 10.2353/ajpath.2006.051329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kramers C, Hylkema MN, van Bruggen MC, van de Lagemaat R, Dijkman HB, Assmann KJ, et al. Anti-nucleosome antibodies complexed to nucleosomal antigens show anti-DNA reactivity and bind to rat glomerular basement membrane in vivo. J Clin Invest. 1994;94(2):568–577. doi: 10.1172/JCI117371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Bruggen MC, Walgreen B, Rijke TP, Tamboer W, Kramers K, Smeenk RJ, et al. Antigen specificity of anti-nuclear antibodies complexed to nucleosomes determines glomerular basement membrane binding in vivo. Eur J Immunol. 1997;27(6):1564–1569. doi: 10.1002/eji.1830270636. [DOI] [PubMed] [Google Scholar]
- 28.Mostoslavsky G, Fischel R, Yachimovich N, Yarkoni Y, Rosenmann E, Monestier M, et al. Lupus anti-DNA autoantibodies cross-react with a glomerular structural protein: a case for tissue injury by molecular mimicry. Eur J Immunol. 2001;31(4):1221–1227. doi: 10.1002/1521-4141(200104)31:4<1221::aid-immu1221>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 29.Deocharan B, Qing X, Lichauco J, Putterman C. Alpha-actinin is a cross-reactive renal target for pathogenic anti-DNA antibodies. J Immunol. 2002;168(6):3072–3078. doi: 10.4049/jimmunol.168.6.3072. [DOI] [PubMed] [Google Scholar]
- 30.Zhou T, Edwards CK, 3rd, Yang P, Wang Z, Bluethmann H, Mountz JD. Greatly accelerated lymphadenopathy and autoimmune disease in lpr mice lacking tumor necrosis factor receptor I. J Immunol. 1996;156(8):2661–2665. [PubMed] [Google Scholar]
- 31.Le Hir M, Bluethmann H, Kosco-Vilbois MH, Muller M, di Padova F, Moore M, et al. Differentiation of follicular dendritic cells and full antibody responses require tumor necrosis factor receptor-1 signaling. J Exp Med. 1996;183(5):2367–2372. doi: 10.1084/jem.183.5.2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pasparakis M, Courtois G, Hafner M, Schmidt-Supprian M, Nenci A, Toksoy A, et al. TNF-mediated inflammatory skin disease in mice with epidermis-specific deletion of IKK2. Nature. 2002;417(6891):861–866. doi: 10.1038/nature00820. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.