

Abstract
A correct alignment is an essential requirement in homology modeling. Yet in order to bridge the structural gap between template and target, which may not only involve loop rearrangements, but also shifts of secondary structure elements and repacking of core residues, high-resolution refinement methods with full atomic details are needed. Here we describe four approaches that address this ‘last mile of the protein folding problem’ and have performed well during CASP8, yielding physically realistic models:
YASARA, which runs molecular dynamics simulations of models in explicit solvent, using a new partly knowledge-based all atom force field derived from Amber, whose parameters have been optimized to minimize the damage done to protein crystal structures.
The LEE-SERVER, which makes extensive use of conformational space annealing to create alignments, to help Modeller build physically realistic models while satisfying input restraints from templates and CHARMM stereochemistry, and to remodel the side-chains.
ROSETTA, whose high resolution refinement protocol combines a physically realistic all atom force field with Monte Carlo minimization to allow the large conformational space to be sampled quickly.
And finally UNDERTAKER, which creates a pool of candidate models from various templates and then optimizes them with an adaptive genetic algorithm, using a primarily empirical cost function that does not include bond angle, bond length, or other physics-like terms.
Keywords: high-resolution refinement, template-based modeling, Rosetta, under-taker, Modeller, conformational space annealing, YASARA
INTRODUCTION
The four groups of this paper were selected because their template-based predictions at CASP8 scored especially well on two aspects: First, they showed a good match to the target H-bonds and target side-chain positions and rotamers1. Second, they were physically realistic, with few all-atom clashes and good bond lengths, bond angles and Ramachandran plot. While the latter features are not among the most important ones in structure prediction, getting them right is both essential and difficult: essential because the fine-grained energy functions for high-resolution refinement depend on correct stereochemistry, and difficult because a simple energy minimization not only improves the look of the model, but also tends to move it away from the target2.
The authors’ prediction methods use distinctly different ways to obtain these results, and this article is an attempt to synthesize some commonality from these different approaches. There are two basic approaches to getting the details right: correct by construction and optimizing cost functions (also called energy functions).
The correct-by-construction approach makes sure that all parameters being considered are set correctly in initial models, and that conformation-change operators do not change these parameters. For example, in Rosetta’s initial stages, all backbone bond angles and bond lengths are set to ideal values, and only torsion angles are modified. In undertaker, all backbone fragments and side-chains are copied from PDB files, and bond angles and bond lengths are not changed by conformation-change operators.
The cost function approach requires attention to two issues: the accuracy of the function and the efficiency of the conformational search. Many terms are included in the cost function, and these terms often are in conflict, so setting the weights of the different components is important when optimizing the weighted sum. Rosetta and YASARA have put considerable effort in optimizing their energy functions so that low-energy protein models are much more likely to be correct predictions, and experimental crystal structures have very low energy. Rosetta has also put effort into improving their Monte-Carlo-based search strategy to sample conformation space sufficiently. The Lee method focuses on improved conformational search by conformational space annealing3, using the standard Modeller4 cost function, which includes competing constraints from “many” templates and CHARMM5.
One problem with the cost-function approach is that some terms of the cost function (such as the Lennard-Jones potential for clash detection) are much stiffer than other terms (such as bond length and bond angle), so that poor bond lengths and bond angles are accepted to remove clashes, rather than doing more difficult combinatorial searches that remove the clashes without damaging the bond angles and bond lengths. One solution (used in YASARA and Rosetta) is to keep bond lengths and angles fixed until the worst clashes are gone. Another solution is to perform straightforward (but difficult) global optimization of the function considering all degrees of freedom as done by the LEE server. Undertaker ramps up the weight of the clash terms as the optimization progresses.
All four methods extract considerable information from the templates, using them to provide initial starting models and, for the LEE server and undertaker, constraints for the cost function. None of the four methods use the old “frozen-core” approach, in which portions of the backbone copied from the templates are not allowed to change.
The next sections will describe in more detail the approaches of each of the four methods.
The self-parameterizing knowledge-based YASARA force field
Improving the physical correctness of protein models looks like the ideal task for a widely used physics-based method: all-atom molecular dynamics simulation with explicit solvent. Still, this approach has traditionally had a difficult stand at CASP: computers are too slow to simulate the folding of the CASP targets from a random initial conformation, and empirical force fields are usually too crude to really improve models built with other methods. The latter problem is related to the observation that high resolution X-ray structures ‘jump away’ during the first picoseconds of a simulation, accompanied by a deterioration of knowledge-based indicators like Ramachandran plot quality.
The NMR community, being haunted by the poor quality of structures obtained from molecular dynamics refinement (Ramachandran plot Z-scores of −7 were common6), soon came up with a solution: The force field was augmented with knowledge-based torsional potentials, that were extracted from high-resolution X-ray structures and ensured that the resulting models looked the same7.
Interestingly, the models often looked even better than X-ray structures, raising the question whether these knowledge-based potentials really improved the accuracy, or just created artificially good-looking models.
The YASARA force field described here addresses these issues by combining the AMBER all-atom force field equation8 with multi-dimensional knowledge-based torsional potentials1 (Figure 1) and with a consistent set of force field parameters to maximize the accuracy: this is achieved by making a random change to one or more parameters (e.g. a certain van der Waals radius, a charge, or the weight of a knowledge-based potential, see Table 1), energy-minimizing a training set of 25 high-resolution X-ray structures, measuring the damage done, and rejecting or accepting the new force field based on a Monte Carlo criterion9. To ensure that all forces responsible for the experimentally observed structure are considered, minimizations are done in crystal space, using complete unit cells2. As a result, one obtains a force field that has stable energy minima as close as possible to native structures. And as shown before, this is essentially equivalent to a force field that moves models closer to native structures during a simulation9.
Figure 1.
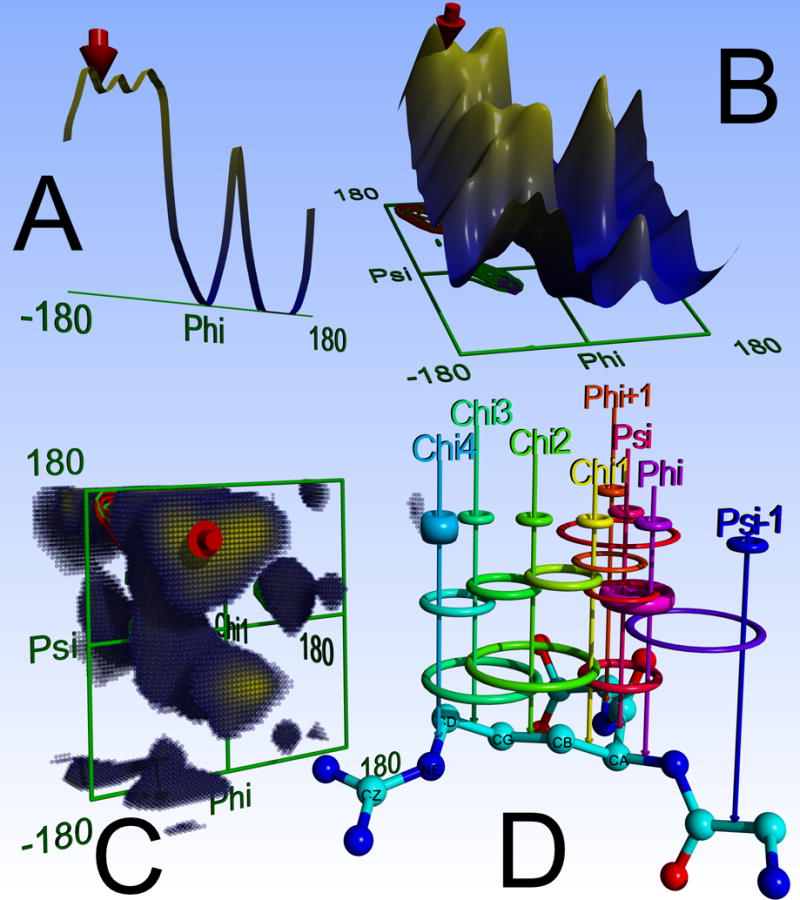
Figure 1A. The 1D knowledge-based potential of the backbone Phi dihedral of Arg. To aid visualization, the energy axis is flipped: favorable low energy regions are at the top and colored yellow, high energy regions are colored blue. The red arrow indicates the current conformation shown in ‘D’. B: The 2D PhiPsi potential, most famous for its relationship with the Ramachandran plot. C: The 3D PhiPsiChi1 potential, the highest energy regions are transparent for clarity. D: Distribution of knowledge-based potentials, using the most complicated case of four Chi dihedrals as an example. A residue like Arg (or Lys) has seven 1D potentials (smallest rings at the top: Omega (not shown), Phi, Psi, Chi1–4), seven 2D potentials (rings in the middle: Psi-1Phi, PhiPsi, PhiChi1, PsiChi1, Chi12, Chi23, Chi34) and three 3D potentials (at the bottom: PhiPsiChi1, Chi123, Chi234). Note that the thickness of the rings symbolizes the weight: The 1D Chi4 potential contributes more because Chi4 is only covered by one 2D and one 3D potential. Likewise, the 2D PhiPsi potential (magenta) accounts for the fact that Phi and Psi are covered by just one 3D potential. The 2D PhiChi1 and PsiChi1 potentials count only half to ensure that Chi1 is not overweighted. Finally, the Psi-1 Phi potential, which spans two subsequent residues, fills up the remaining gaps. Since this potential is special (the two dihedrals are not adjacent but separated by Omega), its weight was optimized independently (parameter 41 in Table 1). Graphics created with YASARA and Po-vRay.
Table 1.
The optimized parameters 38 to 42 of the YASARA force field. The first 37 parameters involve bonds, angles, torsions, VdW radii and point charges, and have been described previously2. ‘Height of the energy barrier’ is a synonym for ‘weight of the potential in the force field’, i.e. parameter optimization was used to determine the optimal weights of 1D, 2D and 3D potentials.
| Par. | Description |
|---|---|
| 38 | Scaling factor for those (Y)AMBER torsional potentials that are also covered by knowledge-based potentials (KBPs). (There are no KBPs involving terminal hydrogens and inside rings). |
| 39 | Height of the average 1D KBP energy barrier. |
| 40 | Height of the average 2D KBP energy barrier. |
| 41 | Ratio of 2D PhiPsi and Psi−1Phi KBPs |
| 42 | Height of the average 3D KBP energy barrier. |
The parameter optimization procedure is computationally intensive and took about half a year using the Models@Home distributed computing system10. After convergence, the contributions of 1D : 2D : 3D potentials (Table 1) were 2.6 : 0.33 : 3.82 kcal/mol. So the highest weight was assigned to the 3D potentials, which makes sense since they contain the most information. A simple explanation for the surprising result that 2D potentials came out last could be that they are (except for Psi−1Phi) fully contained in the 3D potentials, whereas the 1D potentials have a higher resolution (256 instead of 64 bins, see footnote).
The cross-validated results are shown in Table 2. Obviously, the knowledge-based potentials helped a lot: First, the damage done to crystal structures during an energy minimization (RMSD column) is noticeably smaller with the YASARA force field than with YAMBER2 (which used the same parameter optimization approach, but without knowledge-based potentials2) or AMBER. While these RMSD differences look small, they translate to much larger differences during longer simulations2. And second, the old modeler’s rule of thumb to ‘never hurt a protein by energy minimization if it can be avoided’ is no longer an issue: the deterioration of structure validation Z-scores is gone, the minimized structures even look a bit better according to WHAT IF (third column in Table 2). This does not only hold for those checks that are related to the knowledge-based potentials described here (Ramachandran plot, backbone conformation quality), but also for the independent 3D packing quality check (which improves from −0.583 to −0.539).
Table 2.
Simulated annealing minimization of protein crystals using different force fields and a protocol described previously2, the average results for an independent validation set of another 25 proteins are shown. RMSD is the heavy-atom RMSD from the X-ray structure after the minimization converged, Quality Z-Score is the average of the three most sensitive WHAT IF checks28: Ramachandran plot (RAMCHK), backbone conformation (BBCCHK) and 3D packing quality (QUACHK).
| Force field | RMSD | Quality Z-Score |
|---|---|---|
| None | 0.000 | 0.348 |
| AMBER99 (23) | 0.440 | −0.581 |
| AMBER03 (27) | 0.437 | −0.364 |
| YAMBER2 (2) | 0.410 | −0.353 |
| YASARA | 0.379 | 0.616 |
A Z-score is simply the number of standard deviations away from the average, a negative value is assigned to models that are worse than the average high resolution X-ray structure (e.g. have more Ramachandran plot outliers).
Regarding the practical application of the new YASARA force field during CASP8, two results are noteworthy: First, extensive parallel molecular dynamics simulations2 aided by Concoord11 won three of the 12 refinement targets (TR429, TR454 and TR469, based on GDT_TS scores for Model_1). And second, short energy minimizations with a solvent shell helped to improve the physical realism of homology models1 built for the main CASP8 targets. The initial models and thus the starting points for the energy minimizations were obtained using the following protocol: PSI-BLAST12 was run to identify the five closest templates in the PDB, then for each template up to five stochastic alignments were created13 using SSALIGN scoring matrices14. For each of the maximally 25 template/alignment combinations, a 3D model was built using loop conformations extracted from the PDB15 and the SCWRL side-chain placement algorithm16. After the minimization, the models were ranked by quality Z-score (Table 2), and the top five were submitted.
The YASARA force field and the homology modeling protocol have been implemented as part of the molecular modeling program YASARA Structure, available from www.YASARA.org. A web-server can be found at www.YASARA.org/minimizationserver.
Protein 3D modeling by global optimization (LEE)
Resolving two requirements - taking as much information from templates and keeping good stereochemistry is a difficult task to achieve. The LEE and LEE-SERVER models of CASP8 targets are physically realistic while they are in good agreement with native H-bonds and native side-chain conformations. This is achieved firstly by incorporating “many” templates into the standard Modeller4 energy function so that the function contains many competing energy terms and secondly by simply executing rigorous/extensive global optimization to the much frustrated energy function.
An ideal method for logical protein structure modeling would require two conditions. The first one is the availability of an accurate energy function which can identify a good protein model from bad ones solely based on models’ energy values. The other one is the availability of an efficient sampling method which can guarantee to generate a wide spectrum of low-energy conformations including the global minimum energy conformation of a given energy function in a reasonable time scale. Unfortunately the two conditions are not going to be met in near future, and consequently the state of the art protein modeling is carried out with not-so-ideal energy functions and conformational search methods.
Using a given (and preferably more commonly used) energy function, the LEE approach focuses on investigating the effect of utilizing a more rigorous optimization method for the improvement of the protein model quality. Generally, more efficient optimization of an energy function does not necessarily warrant more accurate protein modeling due to the inherent inaccuracy of energy functions currently available. In order to circumvent the situation, we resort to score functions constructed from templates based on consistency, which is shown to be effective for multiple alignment17 and three-dimensional (3D) protein chain building18. In the LEE method, a global optimizer, the conformational space annealing (CSA) method3,19, is applied to the three steps of protein 3D modeling -multiple alignment, 3D chain building, and side-chain re-modeling20.
CSA searches the conformational space of local minima. In addition, CSA combines the general scheme of genetic algorithm with the concept of annealing in the conformational space. CSA maintains a population of conformations and controls its conformational diversity using a distance measure between them. Annealing is achieved by using a large value for the distance measure at the early stages of optimization, and reducing its value in later stages.
For a given set of templates3, we perform multiple alignment between query and templates by generating a pair-wise restraint library from pro le-pro le alignment between query and templates and structure-structure alignment between templates. Unlike the other heuristic (progressive) alignment methods popular in the literature, we apply a more thorough global optimization to a consistency-based score function by using CSA17. The more consistent an alignment is to the restraint library, the higher its score is evaluated. Typically, CSA provides 100 alternative solutions, from which a few good alignments are selected by screening. To evaluate an alignment, we have generated 25 3D models using the standard Modeller package and these models are used to measure the quality of the alignment. For the quality assessment of a 3D model, we used the support vector regression machine trained on decoy structures generated by LEE method in the CASP7 experiments.
Using the alignment selected and templates’ 3D structures as input, we generate 3D structures of the query protein by straightforward optimization of the standard Modeller energy function using CSA, which we call Modeller-CSA18. To apply CSA to the MODELLER energy function, one should be equipped with the following three ingredients: (a) a local minimizer for a given input structure, (b) a distance measure between two given energy-minimized structures, and (c) ways to combine two parent structures to generate a daughter structure which will be energy-minimized subsequently.
For local energy minimization, we have used what is already implemented in the MODELLER package. Root-mean-square-deviation between Cα atoms of two structures are used as the distance measure. To explore the conformational space of the neighborhood of a parent structure P1, we generate a daughter structure by replacing a part of P1 by the corresponding part of another parent structure P2. The actual replacement is performed using internal variables such as bond angles, bond lengths and dihedral angles. As a result, daughter structures partially inherit bond angles, bond lengths, and backbone and side-chain dihedral angles of their parents. To generate the initial population of conformations, Cartesian coordinates of a trial MODELLER structure are randomly perturbed within 2 angstroms, and subsequently energy-minimized using MODELLER version 8v2.
The Modeller energy function has 35 restraint energy terms for 12 features such as distances between atoms, bond angles, dihedral angles, etc. Without additional restraints provided by users, the number of non-zero energy components is typically 15. Besides the spatial restraints, CHARMM energy terms are included in Modeller to enforce proper stereochemistry, which in general creates additional frustration to the energy function. Therefore, models with lower Modeller energies can be considered to satisfy more restraints than those with higher energies. In other words, the lower a model’s Modeller energy is, the more information from template structures and their alignment is accordingly utilized in the model while satisfying as much stereochemistry as allowed by the Modeller energy. For this reason, as much steric clashes are removed accordingly. When we compare the accuracy of the standard Modeller models with that of Modeller-CSA models for 140 TBM domains, the latter is improved by 2.9 % (backbone by GDTHA), 11.0 % (χ1), 18.3 % (χ2) and 7.3 % (H-bond), demonstrating the positive effect of rigorous optimization of the standard Modeller energy function for protein 3D modeling.
The Modeller energy function is a collection of many competing/contradicting restraints arising partly from an alignment containing more than one template and partly from the protein stereochemistry. Generally, it is not possible to satisfy all restraints, naturally setting up a combinatorial optimization problem. The LEE approach intends to include many more (up to 20) templates than a typical template-based protein modeling method, consequently generating a much frustrated energy function for optimization. To handle this kind of problem, a powerful optimizer such as the CSA is shown to be appropriate by providing physically reliable models while efficiently incorporating maximal information from many templates.
Comparison of 3D modeling accuracy between various methods is a tricky procedure since identifying proper template(s) can have a more significant effect than the rest of the modeling procedure. This is more so for medium to more difficult template based modeling (TBM) targets, and less for high accuracy (HA) TBM targets. When we compare the model accuracies for HA-TBM targets, LEE models are especially excellent for all aspects of model quality.
Finally, we remark that additional side-chain improvement (10.8 % for chi_1 and 20.5 % for chi_2) is achieved by side-chain remodeling, for which a rotamer library is constructed based on the consistency of the side chains from the Modeller-CSA models. Into this library, we have added a backbone dependent and sequence-specific rotamer library similar to the SCWRL3.016. Again using the CSA, we have optimized a scoring function containing energy terms from SCWRL and DFIRE21.
Rosetta all-atom refinement
The Rosetta CASP8 models are physically realistic because they were all refined in the physically realistic all atom Rosetta force field22. Lennard Jones repulsive interactions are not damped and all backbone and side-chain atoms are modeled explicitly, so refined models have essentially no steric clashes. Explicit orientation dependent hydrogen bonding potentials derived from high level quantum chemistry calculations are used to refine interactions between hydrogen bond donors and acceptors; this is a considerable improvement over the purely electrostatic treatment of polar interactions in molecular mechanics force fields such as CHARMM5 and AMBER23. The hydrophobic effect and electrostatic desolvation are treated using an implicit solvation model which captures both entropic and enthalpic contributions. Protein structure derived torsional potentials restrict backbone torsion angles to populated regions of the Ramachandran plot and side-chain chi angles to within a standard deviation of significantly populated “rotameric” values. Bond lengths and angles are restrained to close to ideal values. The net result of the above is that Rosetta refined models are physically realistic both in terms of local geometry and global structural properties.
Because the Rosetta force field is reasonably accurate, the native structure almost always has lower (free) energy than non native models. The structure prediction problem in Rosetta is thus the problem of searching for the lowest energy structure for a given amino acid sequence. This is carried out using the Rosetta high resolution sampling/refinement methodology.
The development of the Rosetta sampling methodology has been a continued battle with the challenges posed by refining models in such a high resolution force field. Because the repulsive interactions are not damped and the hydrogen bonding interactions are sensitive to small changes in distance and angles between hydrogen bonding atoms, the total system energy can change drastically with even slight perturbations of the structure, which produces an extremely bumpy free energy landscape riddled with local minima. The key aspects of the Rosetta sampling methodology that allow effective search for the global minimum despite the ruggedness of the landscape are described in the following paragraph.
First, Rosetta uses Monte Carlo Minimization24 rather than traditional Monte Carlo moves to explore backbone conformational space - after a perturbation of the backbone torsion angles, gradient based quasi Newton minimization is used to locate the nearest local minimum, and the decision about whether or not to accept the move is made based on the energy difference between the starting energy and the energy after minimization. Without the minimization step, essentially no moves would be accepted since the chance of introducing a clash is extremely high. Second, side-chain torsional barriers are hopped over by combinatorial optimization of side-chain rotamer conformations after the backbone torsion perturbation but before the minimization (which is carried out with respect to all side-chain and backbone degrees of freedom). This discrete optimization of side-chain conformations provides a big speedup over continuous minimization in for example molecular dynamics simulations in which quite substantial torsional barriers must be overcome. Third, large scale sampling is carried out initially using a low resolution representation in which side-chain degrees of freedom are effectively integrated out; this greatly reduces the computational cost and enables rapid and broad sampling. In the subsequent all-atom refinement stage the goal is to identify the lowest energy local minimum in the vicinity of starting low resolution model, and hence this more computationally intensive refinement is limited to a relatively small region of conformational space. Fourth, the Lennard Jones repulsive interactions in both the continuous quasi-Newton optimization and the discrete side-chain optimization are initially damped considerably and then ramped slowly to full weight—this allows the gradual working out of clashes in starting models generated by the low resolution Rosetta methodology and in general greatly smoothes the landscape which facilitates the early part of the search. Fifth, loop regions and other regions variable in a population of starting models are randomly selected for complete rebuilding by cutting the chain, remodeling the variable region, and then resealing the break to restore near ideal geometry. Following the stochastic rebuilding of randomly selected segments of the chain using the low resolution Rosetta representation, all atom refinement of both the rebuilt region and the remainder of the protein are carried out. The cutting of the chain allows the traversal of huge barriers that would be nearly insurmountable if the chain were kept intact. This “rebuild and refinement” protocol has proven quite effective in refining comparative models and NMR structures25 and was used extensively in CASP8. Sixth, rather than carry out a small number of very long trajectories, the standard approach with Rosetta is to carry out very large numbers of short Monte Carlo Minimization trajectories starting with a diverse collection of starting models generated either from the Rosetta de novo modeling method or from alternative alignments to alternative templates. The advantage of carrying out many short trajectories is that the space of possible structures can be covered much more effectively than in a single long trajectory. While each full atom refinement trajectory typically takes tens of minutes (rather than days or weeks in the case of long time scale MD simulations), because many independent trajectories are carried out Rosetta refinement is quite CPU intensive.
The Rosetta high resolution sampling methodology because of the factors described above is reasonably effective at locating low energy near native structures provided the number of degrees of freedom is not too large. However, because the size of the search space grows exponentially with chain length, for larger proteins (> 200 amino acids) Rosetta rarely happens upon the native free energy minimum unless there is considerable information from homologous structures or from experimental data to guide sampling to this region. The sampling problem—finding the global minimum—remains the most formidable obstacle to consistent high resolution structure prediction with Rosetta.
It is important to recognize that Rosetta models are physically realistic because of the combination of the physically realistic energy function with the powerful sampling methodology. The physically realistic forcefield ensures that predicted structures, which are at energy minima, are physically realistic, but it is not in itself sufficient—without an adequate sampling methodology it would be very difficult to locate deep local minima, let alone the global minimum.
Undertaker keeps the good parts
The SAM-T08-human method at CASP8 builds models using the program undertaker26, developed at University of California, Santa Cruz. Exactly the same protocol is used for template-based and template-free modeling.
First, a random, all-atom conformation is built by joining 1-to-4-residue long fragments from PDB files. This conformation is invariably terrible, but it is complete, and all bond angles, bond lengths, and torsion angles are locally reasonable.
Next, incomplete models are built from alignments to templates. These models copy the backbone from the template in aligned positions. Side-chains are also copied when the residues in the target are identical to the aligned residues in the template. The side-chains for aligned residues that differ are filled in by SCWRL 3.016.
Each of the incomplete models is inserted into the random conformation, to make complete models based on the models from the alignments. These models are scored with a cost function, and the best-scoring one is kept. The incomplete models are then inserted into that model, and the process is repeated a total of nine times. The best model generated from each round of inserting incomplete models is kept to seed a pool for genetic optimization. For targets with good templates, most of the models are very similar and are based on one of the templates, with only small loop regions left from the initial random conformation.
For the SAM-T08-server submissions, these initial models were submitted as the second model and the final optimized models as the first model, so the effect of subsequent optimization can easily be measured. We noticed that essentially all the correct hydrogen bonds are present in the initial models, so undertaker is not creating them during optimization. Indeed for all the measures of accuracy that we have looked at, the difference between the initial and final models is very small (much less than a standard deviation).
Undertaker does optimization using an adaptive genetic algorithm that has 39 different conformation-change operators. Initially, operators that remove clashes and close chain breaks tend to get most heavily applied, while towards the end of the optimization operators that make small tweaks to torsion angles tend to be more successful. For example, in the last 300 generations of the optimizations for the server-only targets, the TweakPsiSegment, which adjusts one psi angle by a small amount, succeeds on average 26% of the time (Tables 3 and 4).
Table 3.
The undertaker conformation-change operators that had the highest probability of success (improving the cost function when applied) for the last 300 generations of the optimization by undertaker for the SAM-T08 server on the server-only models. The “tweak” operators make tiny changes to one or two torsion angles; the “heal” operators replace a peptide plane with a canonical one; and the Backrub operators29 rotate a piece of the backbone a small amount about the axis between two CA atoms (2 apart for Backrub, further for BigBackrub) and make compensating rotations for the peptide planes at either end of rotated piece.
| Operator | % successful |
|---|---|
| TweakPsiSegment | 26.3 |
| TweakPeptide | 25.4 |
| TweakPsiSubtree | 20.2 |
| Backrub | 14.5 |
| HealPeptide | 14.3 |
| TweakPhiSubtree | 12.0 |
| TweakPhiSegment | 11.4 |
| TweakPsiPhiSegment | 7.9 |
| HealGap | 7.9 |
| BigBackrub | 7.6 |
| TweakPsiPhiSubtree | 7.6 |
Table 4.
The undertaker conformation-change operators that had the highest expected value for the change to the cost function (counting the change as 0 when the operator made the cost worse) for the last 300 generations of the optimization by undertaker for the SAM-T08 server on the server-only models. InsertAligment and InsertSpecificFragment are most useful earlier in the optimization, as they make very large changes to the conformation. They rarely improved the best model - most often they were successful when repairing a damaged model that was in the pool for the genetic optimization. CloseGap does a short fragment insertion immediately adjacent to a chain break. FixOmega adjusts the peptide bond to be 180 degrees and does a compensating adjustment to the adjacent phi or psi angle. OneRotamer replaces one side-chain with a side-chain from a PDB file, and TweakHbondSubtree changes the distance and direction of a hydrogen bond by a small amount.
| Operator | avg improvement |
|---|---|
| InsertAlignment | 0.0071242 |
| InsertSpecificFragment | 0.00385188 |
| CloseGap | 0.0033171 |
| FixOmega | 0.00286841 |
| OneRotamer | 0.00276826 |
| TweakPeptide | 0.00268681 |
| BigBackrub | 0.00261841 |
| Backrub | 0.00258232 |
| HealPeptide | 0.00199681 |
| TweakHbondSubtree | 0.00183261 |
Fairly large improvements in the physics-like terms of the cost function are made during optimization, particularly removing clashes and breaks, but also improving the N-CA-C bond angle, hydrogen-bond geometry, and denseness of packing (measured with several different terms). These changes do not improve the measures of correctness of the model, but produce less ugly models of about the same accuracy.
The conformation-change operators are designed to preserve bond lengths and bond angles (or to change them to values copied from an example in PDB), except at breaks in the chain. One special case is that several operators modify peptide planes, either by rotating them around the CA-CA axis or by inserting a canonical peptide plane between the two CA atoms. Both of these types of operations can modify the N-CA-C bond angle, and so the cost function includes explicit scoring of the N-CA-C bond angles---the only bond angle included in the cost function.
The presence of explicit chain breaks means that the cost function needs to include a penalty for chain breaks, so that the optimization process will attempt to close the breaks. The cost function does not increase quadratically with the size of the gap (as is common in physics-based cost functions), but linearly. This ensures that the optimizer does not try spreading a large gap into many smaller gaps, but leaves it in one place, so that combinatorial optimization can try to close the gap with bigger movements of the backbone.
One of the most important tasks for optimizers like undertaker is to remove steric clashes from the conformation. Undertaker’s clash cost function does not use Lennard-Jones potentials, but a simple “softstep” function that is 0 when the atoms are far enough apart and rapidly rises to 1 as the atoms get too close. A table of minimum acceptable differences is used, derived from a training set of PDB files. By not having enormous costs for bad clashes, the optimizer does not destroy everything in a neighborhood to remove a clash. Clash removal is only done when it is possible to do so without damaging the good aspects of the conformation.
During the optimization process the weight of the break and clash costs is increased, so that later stages of optimization try harder to remove the breaks and clashes.
Another component of undertaker’s cost function that is very useful for polishing almost-right models is the hydrogen-bond cost function. Undertaker implements several different H-bond cost functions of differing complexity, from a very simple one that just counts how many donors are close enough to acceptors to potentially form H-bonds, to one that takes into account several different geometric features of the H-bond. For CASP8, the most sophisticated of these H-bond cost functions was used, so that optimization could improve the quality of the modeled H-bonds, and not just the number of them. Somewhat unusually, undertaker’s H-bond scoring is done without explicitly representing the hydrogens---all the geometric terms are parameterized to use just the heavy atoms that are included in X-ray models. Although undertaker does introduce some H-bonds and improve others during optimization, it does not appear to be increasing the overall number or quality of correct H-bonds.
The insignificant gain in accuracy from our initial models to our final optimized models indicates that we need to focus more on generating good alignments to templates and creating the initial models from the alignments---subsequent optimization is making the models prettier without really making them better. The big improvement in CASP8 over older versions of undertaker is that polishing the models no longer decreases their accuracy.
CONCLUSION
In the early days of CASP, the best homology models were built using the ‘frozen core’ approach, keeping the backbone of aligned residues fixed. Optimists who allowed all atoms to move during the refinement usually had to pay the price of reduced accuracy. Unfortunately the ‘frozen core’ approach cannot produce models more accurate than the best available template, so it is good news that all of the four methods described here give the atoms the freedom they deserve.
Cost functions have become accurate enough that they often move models in the right direction during an optimization, particularly when the initial homology model is close to the experimental structure.
One approach to success (Rosetta, undertaker, and YASARA) is multi-level optimization: first address the course-grained features, clipping clash costs and tweaking mainly torsion angles rather than bond lengths and bond angles, so that the latter will not absorb surrounding errors. Fine tuning involving all parameters is done only once the major gaps and clashes have been resolved. An alternative approach is to apply a single straightforward (but difficult) energy optimization to an all-atom cost function as in Modeller-CSA.
All four methods are tied to the template structures and will thus not yield very different solutions: Modeller-CSA and undertaker try to satisfy restraints extracted from the templates, while Rosetta and YASARA don’t use restraints, but will also not move too far away, since the refinement simulation time is too short, or the energy barriers are too high. The differences in accuracy of the models produced by the four methods are probably due mainly to the differences in the initial alignments to templates used by the methods.
The above also implies that the alignment problem has not become obsolete yet. Incorporating structural information from single or multiple templates through accurate alignments is still a very important part of structure prediction. Three-dimensional modeling of apparently unaligned segments is still a challenging problem, though de novo modeling with proper loop closure is showing some promise.
Acknowledgments
Many thanks to the users of YASARA (Yet Another Scientific Artificial Reality Application) for financing this work, and to Bert de Groot, Robbie Joosten and Gert Vriend for their support.
This work was supported by the Korea Science and Engineering Foundation (KOSEF) grant funded by the Korea government (MEST) (No. 2009-0063610).
This work was supported by NIH grant P20 GM076222.
This work was supported in part by NIH grant 1~R01~GM068570-01.
Footnotes
Knowledge-based torsion potentials have been extracted from ~11000 non-redundant PDB files (90% sequence identity cutoff, resolution better than 2.5 Å) using an approach described previously7, with some modifications: to avoid secondary structure bias, only residues outside helices and sheets with an average B-factor<40 were considered, 256 bins were used for 1D potentials, 64*64 bins for 2D potentials and 64*64*64 bins for 3D potentials.
The refinement protocol consisted of 100 MD simulations in explicit solvent (each lasting about 10ps), run in parallel using Models@Home. Then the best model was picked considering YASARA force field energies and WHAT IF validation Z-scores, and subjected to another refinement cycle until the procedure converged. During the first refinement cycles, Concoord was tried as well to quickly sample conformational space just before each MD simulation. The latter then usually managed to restore the model quality scores after the difficult Concoord journey through distance space.
To collect fold candidates of a given query sequence, we considered top scoring templates from the meta-server provided by http://meta.bioinfo.pl, as well as from the in-house fold recognition method called FoldFinder. FoldFinder is a pro le-pro le alignment method utilizing predicted secondary structures. Considered up to 25 top-scoring templates, we performed structural clustering from which typically 5 to 10 sets of templates are generated.
References
- 1.Keedy DA, Williams CJ, Headd JJ, Arendall WB, III, Chen VB, Kapral GJ, Gillespie R, Zemla A, Richardson DC, Richardson JS. The other 90% of the protein: Assessment beyond the Calphas for CASP8 template-based models. Proteins. 2009;77(Suppl 9) doi: 10.1002/prot.22551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Krieger E, Darden T, Nabuurs SB, Finkelstein A, Vriend G. Making optimal use of empirical energy functions: force field parameterization in crystal space. Proteins. 2004;57:678–683. doi: 10.1002/prot.20251. [DOI] [PubMed] [Google Scholar]
- 3.Lee J, Scheraga HA, Rackovsky S. New Optimization Method for Conformational Energy Calculations on Polypeptides: Conformational Space Annealing. JComputChem. 1997;18:1222–1232. [Google Scholar]
- 4.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. JMolBiol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 5.MacKerell J, AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuczera J, Yin D, Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. JPhysChemB. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 6.Nabuurs SB, Nederveen AJ, Vranken W, Doreleijers JF, Bonvin AM, Vuister GW, Vriend G, Spronk CA. DRESS: a database of refined solution NMR structures. Proteins. 2004;55:483–486. doi: 10.1002/prot.20118. [DOI] [PubMed] [Google Scholar]
- 7.Kuszewski J, Gronenborn AM, Clore GM. Improvements and extensions in the conformational database potential for the refinement of NMR and X-ray structures of proteins and nucleic acids. J Magn Reson. 1997;125:171–177. doi: 10.1006/jmre.1997.1116. [DOI] [PubMed] [Google Scholar]
- 8.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Jr, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. A second generation force field for the simulation of proteins, nucleic acids and organic molecules. JAmChemSoc. 1995;117:5179–5197. [Google Scholar]
- 9.Krieger E, Koraimann G, Vriend G. Increasing the precision of comparative models with YASARA NOVA - a self-parameterizing force field. Proteins. 2002;47:393–402. doi: 10.1002/prot.10104. [DOI] [PubMed] [Google Scholar]
- 10.Krieger E, Vriend G. Models@Home: distributed computing in bioinformatics using a screensaver based approach. Bioinformatics. 2002;18:315–318. doi: 10.1093/bioinformatics/18.2.315. [DOI] [PubMed] [Google Scholar]
- 11.de Groot BL, van Aalten DM, Scheek RM, Amadei A, Vriend G, Berendsen HJ. Prediction of protein conformational freedom from distance constraints. Proteins. 1997;29:240–251. doi: 10.1002/(sici)1097-0134(199710)29:2<240::aid-prot11>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 12.Altschul SF, Madden TL, Schaeffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mueckstein U, Hofacker IL, Stadler PF. Stochastic pairwise alignments. Bioinformatics. 2002;18(Suppl 2):153–160. doi: 10.1093/bioinformatics/18.suppl_2.s153. [DOI] [PubMed] [Google Scholar]
- 14.Qiu J, Elber R. SSALN: An alignment algorithm using structure-dependent substitution matrices and gap penalties learned from structurally aligned protein pairs. Proteins. 2006;62:881–891. doi: 10.1002/prot.20854. [DOI] [PubMed] [Google Scholar]
- 15.Canutescu AA, Dunbrack RLJ. Cyclic coordinate descent: A robotics algorithm for protein loop closure. Protein Sci. 2003;12:963–972. doi: 10.1110/ps.0242703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Canutescu AA, Shelenkov AA, Dunbrack RLJ. A graph-theory algorithm for rapid protein side-chain prediction. Protein Sci. 2003;12:2001–2014. doi: 10.1110/ps.03154503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Joo K, Lee J, Kim I, Lee SJ, Lee J. Multiple Sequence Alignment by Conformational Space Annealing. BiophysJ. 2008;95:4813–4819. doi: 10.1529/biophysj.108.129684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Joo K, Lee J, Seo JH, Lee K, Kim BG, Lee J. All-atom chain-building by optimizing MODELLER energy function using conformational space annealing. Proteins. 2009;75:1010–1023. doi: 10.1002/prot.22312. [DOI] [PubMed] [Google Scholar]
- 19.Lee J, Lee I-H, Lee J. Unbiased global optimization of Lennard Jones clusters for N <= 201 by conformational space annealing method. PhysRevLett. 2003;91:080201. doi: 10.1103/PhysRevLett.91.080201. [DOI] [PubMed] [Google Scholar]
- 20.Joo K, Lee J, Lee S, Seo J-H, Lee SJ, Lee J. High Accuracy Template Based Modeling by Global Optimization. Proteins. 2007;69 (Suppl 8):83–89. doi: 10.1002/prot.21628. [DOI] [PubMed] [Google Scholar]
- 21.Zhou H, Zhou Y. Distance-scaled, finite ideal-gas reference state improves structure-derived potentials of mean force for structure selection and stability prediction. Protein Sci. 2002;11:2714–2726. doi: 10.1110/ps.0217002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Misura KM, Chivian D, Rohl CA, Kim DE, Baker D. Physically realistic homology models built with ROSETTA can be more accurate than their templates. ProcNatlAcadSciUSA. 2006;103:5361–5366. doi: 10.1073/pnas.0509355103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang J, Cieplak P, Kollman PA. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? JCompChem. 2000;21:1049–1074. [Google Scholar]
- 24.Wales DJ, Scheraga HA. Global Optimization of Clusters, Crystals, and Biomolecules. Science. 1999;285:1368–1372. doi: 10.1126/science.285.5432.1368. [DOI] [PubMed] [Google Scholar]
- 25.Meiler J, Baker D. The fumarate sensor DcuS: progress in rapid protein fold elucidation by combining protein structure prediction methods with NMR spectroscopy. JMagnReson. 2005;173:310–316. doi: 10.1016/j.jmr.2004.11.031. [DOI] [PubMed] [Google Scholar]
- 26.Archie J, Karplus K. Proteins. 2009. Applying undertaker cost functions to model quality assessment. In press, published online 30 Sep 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duan Y, Wu C, Chowdhury S, Lee MC, Xiong G, Zhang W, Yang R, Cieplak P, Luo R, Lee T. A Point-Charge Force Field for Molecular Mechanics Simulations of Proteins. JCompChem. 2003;24:1999–2012. doi: 10.1002/jcc.10349. [DOI] [PubMed] [Google Scholar]
- 28.Hooft RWW, Vriend G, Sander C, Abola EE. Errors in protein structures. Nature. 1996;381:272–272. doi: 10.1038/381272a0. [DOI] [PubMed] [Google Scholar]
- 29.Davis IW, Arendall WB, III, Richardson DC, Richardson JS. The backrub motion: how protein backbone shrugs when a sidechain dances. Structure. 2006;14:265–274. doi: 10.1016/j.str.2005.10.007. [DOI] [PubMed] [Google Scholar]