

Abstract
Epstein–Barr virus (EBV) and the closely related Herpesvirus papio (HVP) are stably replicated as episomes in proliferating latently infected cells. Maintenance and partitioning of these viral plasmids requires a viral sequence in cis, termed the family of repeats (FR), that is bound by a viral protein, Epstein–Barr nuclear antigen 1 (EBNA1). Upon binding FR, EBNA1 maintains viral genomes in proliferating cells and activates transcription from viral promoters required for immortalization. FR from either virus encodes multiple binding sites for the viral maintenance protein, EBNA1, with the FR from the prototypic B95-8 strain of EBV containing 20 binding sites, and FR from HVP containing 8 binding sites. In addition to differences in the number of EBNA1-binding sites, adjacent binding sites in the EBV FR are typically separated by 14 base pairs (bp), but are separated by 10 bp in HVP. We tested whether the number of binding sites, as well as the distance between adjacent binding sites, affects the function of EBNA1 in transcription activation or plasmid maintenance. Our results indicate that EBNA1 activates transcription more efficiently when adjacent binding sites are separated by 10 bp, the spacing observed in HVP. In contrast, using two separate assays, we demonstrate that plasmid maintenance is greatly augmented when adjacent EBNA1-binding sites are separated by 14 bp, and therefore, presumably lie on the same face of the DNA double helix. These results provide indication that the functions of EBNA1 in transcription activation and plasmid maintenance are separable.
Introduction
During latent infection, Epstein–Barr virus (EBV)’s genome is maintained as a 165-kb plasmid in the nuclei of infected cells. This plasmid undergoes DNA synthesis once per cell cycle (Adams, 1987; Yates and Guan, 1991) and partitions faithfully to daughter nuclei at mitoses (Kirchmaier and Sugden, 1995; Sugden and Warren, 1988). Genome maintenance and partitioning requires a single viral protein, Epstein–Barr nuclear antigen 1 (EBNA1), and a viral element in cis, oriP (Lupton and Levine, 1985; Reisman et al., 1985; Yates et al., 1984). There are two clusters of binding sites for EBNA1 within oriP, the dyad symmetry element (DS) that has four sites of lower affinity, and the family of repeats (FR) (Ambinder et al., 1990; Baer et al., 1984; Lupton and Levine, 1985; Reisman et al., 1985). DNA synthesis from oriP initiates at DS (Gahn and Schildkraut, 1989); recent evidence indicates that the cellular origin recognition complex and minichromosome maintenance proteins facilitate that process (Chaudhuri et al., 2001; Schepers et al., 2001). FR functions as a plasmid maintenance and partitioning element. FR from the prototypic B95-8 strain of EBV contains 20 high-affinity binding sites for EBNA1. Each site is 16 bp long, and in all but two instances, adjacent sites are separated by 14 bp (Baer et al., 1984). When bound to FR, EBNA1 prevents the rapid loss of oriP-plasmids (Aiyar et al., 1998). It also activates transcription from at least three viral promoters, the BamH1-C and W promoters (Puglielli et al., 1996), and the promoter for latent membrane protein 1 (LMP1) (Gahn and Sugden, 1995). EBNA1 and FR have also been characterized to activate transcription from heterologous promoters such as the thymidine kinase promoter of herpes simplex virus 1 (HSV-TK) (Reisman and Sugden, 1986; Wysokenski and Yates, 1989), the immediate early promoter of human cytomegalovirus (CMV-IE) (Langle-Rouault et al., 1998), and the long terminal repeat of Rous sarcoma virus (RSV LTR) (A. Aiyar, unpublished data).
While there is no published isolate of Epstein–Barr virus that contains fewer than 15 binding sites for EBNA1 in FR, a virus closely related to EBV, Herpesvirus papio (HVP), contains only eight EBNA1-binding sites in its FR (Loeb et al., 1990). The EBNA1 proteins of EBV and HVP are 56% identical in sequence (Yates et al., 1996). Consistent with the sequence conservation, Yates and co-workers have demonstrated that the EBNA1 protein of each virus supports stable replication and maintenance of plasmids containing the oriP of the other virus. Each protein can also use the heterologous FR to activate transcription (Yates et al., 1996). A striking difference between the FRs of EBV and HVP is that the majority of EBNA1-binding sites in the latter are separated by 10 bp (Loeb et al., 1990). Because the periodicity of a DNA double-helix ranges from 10 to 12 bp (Watson and Crick, 1953), this difference would cause EBNA1 dimers bound to adjacent binding sites in EBV’s FR to lie on the same face of the DNA double-helix, while they would lie on opposite sides in the HVP FR.
Using deletion derivatives of EBV’s FR, Yates and co-workers demonstrated elegantly that stable maintenance of oriP-plasmids, and activation of transcription in Raji cells, required just seven EBNA1-binding sites in FR (Wysokenski and Yates, 1989). In contrast, deletion derivatives with five EBNA1-binding sites supported plasmid maintenance to 1% the level of wild-type FR, and transcription activation to 10% the level of wild-type FR (Wysokenski and Yates, 1989). While this marked decrease is most readily attributed to a difference in the number of EBNA1-binding sites, it remains possible that it reflects differences in the affinity of EBNA1 for individual binding sites. This is because the individual binding sites in FR differ in sequence, and consequently, in the affinity with which they are bound by EBNA1 (Ambinder et al., 1990). Consistent with this latter explanation, these same studies indicated that constructions with 12 low-affinity EBNA1-binding sites from DS were also impaired in their ability to support plasmid maintenance in Raji cells and to activate transcription in Raji as well as Wilson (EBNA1) cells (Wysokenski and Yates, 1989).
To elucidate whether the differences in the space between adjacent binding sites and binding site number within FR affect the functions of EBNA1, we have systematically tested the effects of spacing between adjacent binding sites, and binding site number on the ability of EBNA1 to activate transcription or maintain plasmids in proliferating cells. Our results indicate that EBV’s EBNA1 bound to sites spaced 10 bp apart activate transcription better than binding sites spaced 14 bp apart. Intriguingly, binding sites spaced 14 bp apart support plasmid maintenance far more efficiently than binding sites spaced 10 bp apart.
Results
Design and construction of two sets of synthetic FRs (sFR)
To determine whether the differences in spacing of adjacent EBNA1-binding sites in the EBV and HVP FR affected either plasmid maintenance or transcription activation, we created synthetic FRs with increasing numbers of binding sites that were separated by either 10 or 14 bp. To eliminate the effect of differences in sequence of individual EBNA1-binding sites in the wild-type FR, we chose to construct multimers of the EBNA1-binding site “GGTAGCATATGCTATC” that is present at a higher frequency than any other site in EBV’s FR. Multimers of this site were created as described under Materials and methods and introduced into transcription and replication reporter plasmids diagrammed in Fig. 1. Although sFR contain up to 20 identical copies of the same repeat, we did not observe any change in repeat number or sequence when propagated in recombination-deficient Escherichia coli strain STBL2 (data not shown).
Fig. 1.

Schematic representations of the transcription and replication reporter plasmids used in this study. The name of each plasmid, the number of EBNA1-binding sites within the sFR element in the plasmid, and the space between adjacent EBNA1-binding sites in sFR is indicated. Transcription reporter plasmids contain the firefly luciferase gene under the control of the HSV-1 thymidine kinase promoter. Replication reporter plasmids were constructed using pPUR as a backbone and contain the DS element from EBV, along with EBV sequences between FR and DS. The location of XbaI and BamHI recognition sites used in Southern hybridization experiments is indicated.
The spacing between adjacent EBNA1 binding sites affects transcription activation
EBNA1 activates transcription from at least three EBV promoters used during latency when bound to FR (Gahn and Sugden, 1995; Puglielli et al., 1996). This transcription activation is recapitulated when FR is introduced upstream of many heterologous promoters (Langle-Rouault et al., 1998; Reisman and Sugden, 1986; Wysokenski and Yates, 1989). Yates and co-workers (Wysokenski and Yates, 1989) have previously analyzed the relationship between the number of EBNA1-binding sites and transcription activation of the HSV-1 TK promoter in human and rat cells. Their results indicated that optimal transcription activation required at least seven EBNA1-binding sites in FR, albeit in a manner dependent upon the affinity of EBNA1 for the binding sites. A transcription reporter plasmid with 12 low-affinity binding sites from DS activated transcription to 20–30% the level of wild-type FR (Wysokenski and Yates, 1989). We have extended this analysis in a manner independent of the affinity of individual EBNA1-binding sites by testing transcription reporter plasmids containing increasing numbers of identical high-affinity EBNA1-binding sites. Either the control TK-luciferase plasmid, AGP47, or the derivatives containing 1, 5, 7, 10, or 20 EBNA1-binding sites separated by 10 or 14 bp were introduced into C33A/1553 cells, a derivative of C33A cells that express wild-type EBNA1. An EGFP expression plasmid, 2145, was cotransfected with the luciferase reporter plasmids to normalize for transfection efficiency. Cells were harvested at 48 or 96 h posttransfection and analyzed for luciferase activity as described under Materials and methods. The results of these assays are depicted in fig. 2A and B. By comparing the graphs in Fig. 2A and B, it can be observed that independent of the spacing between adjacent EBNA1-binding sites, the level of luciferase induction at 96 h posttransfection was lower than the level observed at 48 h posttransfection. This decrease can be attributed to plasmid loss as a function of cell division (Aiyar et al., 1998). Forty-eight hours posttransfection, we observed an increase in luciferase induction with increasing numbers of EBNA1-binding sites, reaching a maximum of 110-to 230-fold over the backbone plasmid that lacked any EBNA1-binding sites for plasmids that contained 20 EBNA1-binding sites. However, as can be observed by comparing the solid bars to the hatched bars in Fig. 2, sFRs wherein adjacent EBNA1-binding sites were separated by 10 bp activated transcription to a greater level than the corresponding sFR with a 14-bp spacer between adjacent binding sites. This correlation was significant, with a P (two-sided) of 0.005 by the Kendall rank correlation test, for constructions that contained between 7 and 20 EBNA1-binding sites. Plasmid 985, wild-type FR-TK luciferase, was also tested in this assay. Results from experiments using this plasmid are labeled “wt FR” in Fig. 2 and indicate that EBNA1 equivalently activates transcription from a plasmid containing the wild-type FR, or a plasmid containing an sFR with 20 EBNA1-binding sites spaced 14 bp apart. Results similar to these were obtained from transcription reporter assays performed in 293/EBNA1 cells (data not shown) and when luciferase activity was measured 96 h posttransfection (Fig. 2B). Overall, three conclusions can be drawn from these experiments. First, consistent with the observations of Yates and co-workers (Wysokenski and Yates, 1989), an increasing number of EBNA1-binding sites cause increased transcription activation. Second, because we have used multimers of identical EBNA1-binding sites, this increase can be attributed to the presence of additional EBNA1-binding sites on the transcription template, rather than differences in affinity between individual sites. Finally, the space between adjacent EBNA1-binding sites influences transcription activation. EBNA1-binding sites spaced 10 bp apart were more effective at activating transcription than EBNA1-binding sites spaced 14 bp apart from each other.
Fig. 2.

EBNA1-binding sites spaced 14 bp apart activate transcription more efficiently in C33A/EBNA1 cells than EBNA1-binding sites spaced 14 bp apart. Luciferase assays were performed 2 days (A) or 4 days (B) after introducing the transcription reporter plasmids described in Fig. 1 into C33A/1553 cells. The y-axis represents the fold induction of luciferase activity over that observed with the backbone reporter AGP47 48 h posttransfection, and the number of binding sites in sFR is indicated on the x-axis. The solid bars represent results from reporters in which adjacent EBNA1-binding sites are separated by 14 bp, while hatched bars represent results from reporters in which adjacent EBNA1-binding sites are separated by 10 bp. wtFR refers to use of the plasmid 985, FR-TK-luciferase, as the transcription reporter.
Plasmid maintenance is dramatically influenced by the space between adjacent EBNA1-binding sites
To investigate whether plasmid maintenance in proliferating cells is affected by the distance between adjacent EBNA1-binding sites in FR, the two sets of replication reporter plasmids described in Fig. 1 were individually introduced into 293/EBNA1 cells along with the transfection control, EGFP-expression plasmid 2145. Two days posttransfection, cells were harvested to measure transfection efficiency as a function of EGFP expression and then plated at 2 × 105, 2 × 104, or 2 × 103 cells per 10-cm dish. Two days after plating, cells were subjected to puromycin selection for a total of 2 weeks, following which colonies were stained with methylene blue and counted as described under Materials and methods. As such, this assay tests the ability of EBNA1 bound at sFR to maintain and partition the replication reporter plasmid in proliferating cells and thereby gives rise to a puromycin-resistant colony. Under these conditions, transfection of the parental plasmid, AGP83, that contained DS, but lacked FR, yielded an average of 2.25 colonies per 105 cells plated. Based on Southern hybridization studies (see below and data not shown), this number reflects the efficiency at which plasmid DNA is integrated randomly in transfected cells. Several conclusions can be drawn from the results of this assay that are summarized in Tables 1 and 2.
Table 1.
293/EBNA1 cells transfected with replication reporters in which adjacent EBNA1 binding sites are spaced 14 bp apart form puromycin-resistant colonies with greater efficiency
| Reporter transfected |
EBNA1 binding sites in sFR |
Space between adjacent sites |
Frequency of puromycin resistance in plated cellsa |
|---|---|---|---|
| Mock | — | — | 0 |
| AGP83 | 0 | — | 2.2 × 10−5 |
| AGP72 | 5 | 14 | 1.4 × 10−3 |
| AGP88 | 5 | 10 | 4 × 10−5 |
| AGP100 | 7 | 14 | 1.2 × 10−2 |
| AGP89 | 7 | 10 | 1.1 × 10−3 |
| AGP73 | 10 | 14 | 4.4 × 10−2 |
| AGP90 | 10 | 10 | 3.5 × 10−2 |
| AGP74 | 20 | 14 | 1.3 × 10−2 |
| AGP91 | 20 | 10 | 7.6 × 10−3 |
Data represent the number of puromycin-resistant colonies formed per 105 GFP-positive, live 293/EBNA1 cells plated 48 h posttransfection. Selection was initiated 4 days posttransfection and maintained for 2 weeks. Each replication reporter was transfected independently at least two times. At least two plates were counted from each transfection. Only colonies 2 mm in size and larger were counted.
Table 2.
At least four EBNA1 binding sites spaced 14 bp apart are required for detectable plasmid maintenance in 293/EBNA1 cells
| Replication reporter |
EBNA1 binding sites in sFR |
Frequency of puromycin resistance in plated cellsa |
|---|---|---|
| AGP83 | 0 | 2.2 × 10−5 |
| AGP127 | 3 | 3.5 × 10−5 |
| AGP128 | 4 | 7.9 × 10−4 |
Data represent the number of puromycin-resistant colonies formed, per 105 GFP-positive, live 293/EBNA1 cells plated 48 h posttransfection. Selection was initiated 4 days posttransfection. Each replication reporter was transfected independently at least two times. At least two plates were counted from each transfection. The number of colonies formed for AGP83 are reproduced from Table 1. Transfections testing the efficiency with which AGP83 forms puromycin-resistant colonies were performed concurrently with transfections testing AGP127 and AGP128.
First, as indicated in Table 1, when sFR contained between 5 and 10 EBNA1-binding sites, we observed an increase in colony number as a function of an increasing number of EBNA1-binding sites. This increase mirrors the increase previously observed in Raji cells by Yates and co-workers (Wysokenski and Yates, 1989). However, a difference between our results and those described previously lies in the reproducible observation that replication reporter plasmids with 20 EBNA1-binding sites formed colonies less efficiently than reporter plasmids with 10 EBNA1-binding sites. The observed decrease was statistically significant, with a P value of 0.011 when examined by the Kendall rank correlation test.
Second, the data summarized in Table 1 also indicate that the distance between adjacent EBNA1-binding sites reproducibly and dramatically influences plasmid partitioning and maintenance. The efficiency of colony formation is considerably higher when adjacent EBNA1-binding sites in sFR are separated by 14 bp than when they are separated by 10 bp. This phenomenon is most apparent when the replication reporter plasmids contained either 5 or 7 EBNA1-binding sites in sFR. A plasmid with five binding sites in sFR spaced 14 bp apart, AGP72, gave rise to colonies with a 35-fold higher efficiency than AGP88, a plasmid with five binding sites spaced 10 bp apart in sFR. Similarly, AGP100, a plasmid whose sFR contains seven binding sites spaced 14 bp apart, gave rise to colonies approximately 10-fold more efficiently than the corresponding plasmid with seven binding sites spaced 10 bp apart (AGP89). When examined by the Wilcoxon rank-sum test (Hollander and Wolfe, 1973), both of these differences were found significant, with P values less than 0.05. An effect of binding site spacing was also observed when the replication reporter plasmids contained 10 or 20 EBNA1-binding sites in sFR, although this difference was not as statistically significant as that observed with five or seven EBNA1-binding sites in sFR (P value = 0.06).
Colony formation requires reporter plasmids to undergo DNA synthesis and to be partitioned at consecutive mitoses. The failure of a plasmid to support efficient colony formation can result from a deficiency in DNA synthesis, or from a defect in maintenance and partitioning. Although none of the reporter plasmids tested in this study contained differences in DS, the replication origin within oriP, it is possible that alterations in FR affect the efficiency with which DNA synthesis initiates at DS. We therefore examined the efficiency with which reporter plasmids were replicated 4 days after their introduction into 293/EBNA1 cells, as well as 3 weeks after transfection. If the permutation of EBNA1-binding sites in sFR primarily affects DNA synthesis, differences would be observed at both early and late time points. In contrast, if the spacing between adjacent EBNA1-binding sites only affected the levels of replicated reporter DNA at the later time point, it would indicate that the permutation of EBNA1-binding sites strongly affects plasmid maintenance and partitioning, but not DNA synthesis. These possibilities were distinguished as described below.
Plasmids AGP83, AGP72, AGP100, AGP73, AGP74, AGP88, AGP89, AGP90, and AGP91 were introduced into 293/EBNA1 cells. Episomal DNAs were extracted 4 days posttransfection and then digested with the restriction endonuclease DpnI to selectively digest the input unreplicated DNA as well as the hemimethylated products from the first round of DNA replication in 293/EBNA1 cells. DpnI-resistant DNAs were digested with XbaI and BamHI, to release two fragments, labeled A and B, in the representative analysis shown in Fig. 3. These enzymes were chosen because the size and sequence of fragment A would be the same for all the replication reporter plasmids tested. The representative analysis shown in Fig. 3 indicates that by 4 days posttransfection all the replication reporters have undergone at least two rounds of DNA synthesis. The band labeled D represents the largest fragment released after digestion of the input bacterially dam-methylated replication reporter plasmids with DpnI. From two independent Southern analyses performed using episomal plasmids extracted from transfected 293/EBNA1 cells, we estimate that there are between 5 and 90 DpnI-resistant newly replicated reporter plasmids present per cell after correcting for transfection efficiency and are represented in Table 3. A similar range was observed from one experiment performed using episomal DNAs extracted from C33A/1553 cells 4 days posttransfection. In all three experiments, AGP73 was incompletely digested by BamHI, resulting in a third DpnI-resistant fragment in addition to A and B. This band is labeled as an asterisk in Figs. 3 and 4. It is unclear why this digestion is incomplete; we have determined that the input bacterially synthesized AGP73 DNA is completely digested by BamHI. In addition, plasmid DNAs recovered from bacteria transformed with DpnI-resistant AGP73 DNA extracted from transfected 293/EBNA1 and C33A/1553 cells are completely susceptible to BamHI digestion and fail to show any rearrangement in AGP73. Finally, sequence analysis of AGP73 prior to or after transfection indicates the BamHI site is intact and is in an identical sequence context in all the replication reporters tested in this study. Because this digestion is incomplete, we have quantified the number of replicated molecules in AGP73 transfected cells using the sum of the amount of DNA in fragments A and *. It is emphasized that this incomplete digestion does not affect the conclusions drawn from this study.
Fig. 3.
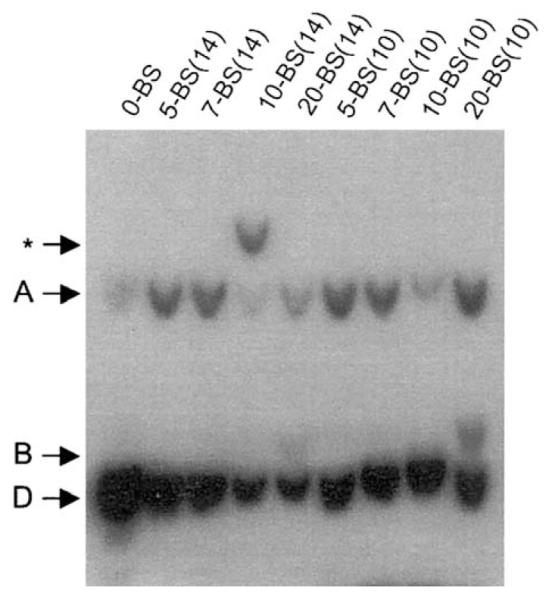
The distance between adjacent EBNA1-binding sites in sFR and the number of EBNA1-binding sites in sFR do not affect synthesis of oriP-plasmids 4 days after their introduction into 293/EBNA1 cells. 293/EBNA1 cells were transfected with the replication reporter plasmids described in Fig. 1. Four days after transfection, episomal DNAs were recovered from transfected cells, digested with DpnI, BamHI, and XbaI, and quantified by Southern hybridization. Digested DNA recovered from 1 × 107 cells was loaded in each lane. The number of EBNA1-binding sites in sFR is indicated above each lane, and the spacer between adjacent EBNA1-binding sites in sFR is indicated in parentheses. DpnI-resistant, BamHI/XbaI-digested fragments are designated as A and B. The intensity of A was used to estimate the copy number of newly synthesized plasmids per cell, by comparison with standards. AGP73 recalcitrant to BamHI-digestion as described in the text is designated as *. D represents the largest fragment created after DpnI-digestion of the input bacterially dam-methylated reporter DNAs.
Table 3.
Average copy number of replication reporter plasmids during transient and stable replication assays
| Reporter transfected |
EBNA1 binding sites in sFR |
Space between adjacent sites |
Four days post transfectiona |
Three weeks post transfectiona |
|---|---|---|---|---|
| AGP83 | 0 | — | 35 ± 14 | NDb |
| AGP127 | 3 | 14 | 47 ± 34 | ND |
| AGP128 | 4 | 14 | 24 ± 8 | 14 ± 8 |
| AGP72 | 5 | 14 | 72 ± 7 | 22 ± 15 |
| AGP88 | 5 | 10 | 68 ± 31 | 0.9 ± 1 |
| AGP100 | 7 | 14 | 35 ± 16 | 57 ± 16 |
| AGP89 | 7 | 10 | 41 ± 4 | 8 ± 5 |
| AGP73 | 10 | 14 | 43 ± 22 | 87 ± 32 |
| AGP90 | 10 | 10 | 65 ± 17 | 71 ± 14 |
| AGP74 | 20 | 14 | 61 ± 13 | 33 ± 21 |
| AGP91 | 20 | 10 | 73 ± 28 | 28 ± 16 |
Numbers represent the average number of DpnI-resistant episomal plasmid molecules per transfected cell detected in three independent experiments along with the standard deviation.
Not determined.
Fig. 4.
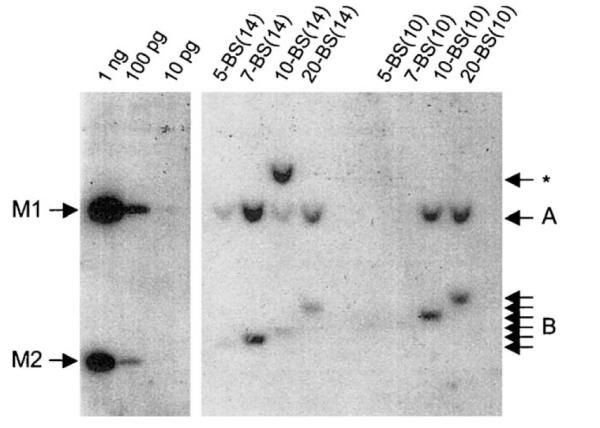
EBNA1-binding sites in FR spaced 14 bp apart support stable replication more effectively than binding sites spaced 10 bp apart. 293/EBNA1 cells were transfected with the replication reporter plasmids described in Fig. 1 and subjected to 3 weeks of puromycin selection. Episomal DNAs were recovered from pools of resistant cells, digested as described in Fig. 3, and quantified by Southern hybridization. Digested DNA recovered from 5 × 106 cells were loaded in each lane. The blot is labeled as in Fig. 3. The number of EBNA1-binding sites in sFR is indicated above each lane, and the spacer between adjacent EBNA1-binding sites in sFR is indicated in parentheses. Measures of 10 pg, 100 pg, or 1 ng of BamHI/XbaI-digested AGP83 were used as standards during quantitation. Probe hybridized to the standards is indicated by M1 and M2.
To determine whether the space between adjacent binding sites in FR affects plasmid maintenance, transfected cells were subjected to puromycin selection for 3 weeks, after which episomal plasmids were recovered from pooled colonies of resistant cells. The recovered plasmids were digested with DpnI, XbaI, and BamHI and examined by Southern analysis. Representative results are shown in Fig. 4, which is labeled similarly to Fig. 3. Because this analysis was performed 3 weeks posttransfection, we do not detect DpnI-sensitive unreplicated plasmids in episomal plasmids extracted from cells. Thus the DpnI-resistant fragments, labeled B in Fig. 4, that vary in size between plasmids, are not obscured as they are in Fig. 3. The analysis shown in Fig. 4 indicates that AGP72, AGP100, AGP73, and AGP74 are maintained as episomal plasmids at copy numbers that vary from approximately 20 (AGP72) to approximately 90 (AGP100, AGP73) copies per puromycin-resistant cell. In contrast, in the analysis shown in Fig. 4 when adjacent binding sites in sFR were separated by 10 bp, only plasmids with 7 (AGP89), 10 (AGP90), or 20 (AGP91) binding sites were detected as episomal DNAs. AGP88, that contains five binding sites spaced 10 bp apart, was present at less than one molecule per cell (0.8 episomal molecules/resistant cell).
Plasmid maintenance in 293/EBNA1 cells requires at least four EBNA1-binding sites that are spaced 14 bp apart
Because the periodicity of the DNA double-helix varies between 10 and 12 bp, AGP88 and AGP89 are predicted to maximally contain three or four EBNA1-binding sites, respectively, on one face of the DNA double-helix. Colony formation assays and Southern blot analyses for episomal DNA indicate that AGP88 is not stably maintained. AGP88 is maintained poorly in both these assays, such that in Southern analysis it is detected at less than one episomal molecule per transfected cell. In contrast, AGP72, which contains five EBNA1-binding sites separated by 14 bp, functions substantially better than AGP88 and slightly better than AGP89 (Table 1 and Fig. 4). Therefore two replication reporters, AGP127 and AGP128, that contain three or four EBNA1-binding sites separated by 14 bp were constructed and used to test the hypothesis that maintenance in 293/EBNA1 cells requires at least four EBNA1-binding sites on one face of DNA.
AGP127 and AGP128 were tested in colony formation assays in 293/EBNA1 cells as described previously. The results of this assay, summarized in Table 2, clearly indicate that similar to AGP88, AGP127 is not maintained. This replication reporter cannot be distinguished statistically from AGP83, a control reporter that contains DS, but not FR. In contrast, cells transfected with AGP128 formed colonies approximately 20–30 times more efficiently. This efficiency was approximately that observed with AGP89 (compare Tables 1 and 2).
To confirm these results, Southern blot analyses were performed on episomal plasmids extracted from cells transfected with AGP127 or AGP128. As described earlier, the copy number of episomal plasmids was determined 4 days posttransfection, and after 3 weeks of puromycin selection. These results are shown in Fig. 5 and indicate that episomal, newly replicated AGP127 and AGP128 can be detected readily 4 days posttransfection. However, episomal AGP127 was not detected in extracts from puromycin-resistant colonies obtained 3 weeks posttransfection. In contrast, episomal AGP128 was readily detected at about 14 episomal molecules per transfected cell.
Fig. 5.
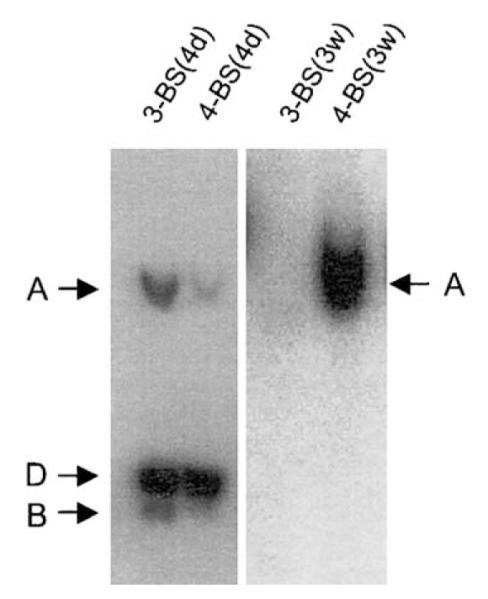
Maintenance of replicated oriP-plasmids in 293/EBNA1 cells requires at least four EBNA1-binding sites spaced 14 bp apart in sFR. Replication reporters that contain three binding sites in sFR, AGP127, or four binding sites, AGP128, were introduced into 293/EBNA1 cells. Episomal DNAs were recovered either 4 days (4d) posttransfection, or from pooled colonies after 3 weeks (3w) of puromycin selection. The number of binding sites in sFR is indicated above each lane, and the time posttransfection is indicated in parentheses. Each lane was loaded with DpnI/BamHI/XbaI-digested DNAs recovered from 1 × 107 cells (4d) or 2 × 107 cells (3w).
AGP89 and AGP128 behave similarly in two independent measures of plasmid maintenance, indicating that although it is inefficient, maintenance of episomal oriP plasmids in 293/EBNA1 cells requires a minimum of four adjacent EBNA1-binding sites on one face of the DNA helix. Plasmids with three such sites (AGP88, AGP127) fail to be maintained. Conversely, a plasmid with five such sites (AGP72) is maintained more efficiently, and the maintenance of a plasmid with seven sites (AGP100) cannot be distinguished from that of a plasmid with 20 binding sites (AGP74) either by colony formation assays or by a Southern analysis quantifying replicated DNAs.
Discussion
The genomes of EBV and HVP contain a cis-acting sequence, termed oriP, that facilitates DNA synthesis and maintenance during latent infection. OriP consists of two regions, one of which, FR, is required for plasmid maintenance and partitioning. While FR of EBV and HVP both contain multiple EBNA1-binding sites, they differ in their organization. EBV’s FR contains 20 EBNA1-binding sites, the majority of which are separated by 14 bp (Baer et al., 1984). Because each EBNA1-binding site is 16 bp long, and the periodicity of the DNA double-helix ranges between 10 and 12 bp, this arrangement is predicted to cause EBNA1 dimers bound to adjacent binding sites to lie on the same face of the DNA double-helix. The EBNA1-binding sites in EBV’s FR are not identical in sequence, although they are all predicted to be bound by EBNA1 with higher affinity that the sites in EBV’s DS (Ambinder et al., 1990). In contrast, FR of HVP contains only eight EBNA1-binding sites, six of which are separated from each other by 10 bp (Loeb et al., 1990). EBNA1 dimers bound to adjacent sites in the HVP FR are predicted to lie on approximately opposite faces of the DNA double-helix. Yates and co-workers have demonstrated that EBNA1 from either HVP or EBV can function to stably replicate plasmids that contain oriP from the other virus (Yates et al., 1996). Separately, they have used deletion mutants to define the minimal sequence within EBV’s FR that would function to stably replicate plasmids in cells. Their results indicate that a minimum of seven EBNA1-binding sites in FR is required for stable plasmid replication in cells (Wysokenski and Yates, 1989). We have determined whether EBNA1’s functions are affected by binding site number or the space between adjacent binding sites by using two sets of constructions with increasing numbers of identical binding sites separated by 10 or 14 bp. Our studies have confirmed the observations of Yates and co-workers and have extended them in an unexpected manner.
As demonstrated previously, transcription activation by EBNA1 increases with an increasing number of binding sites, as does the ability of EBNA1 to maintain oriP plasmids. Surprisingly, both transcription activation and plasmid maintenance are affected by the space between adjacent binding sites. EBNA1 bound to sites spaced 10 bp apart activated transcription more effectively than EBNA1 bound to parallel constructions with sites spaced 14 bp apart. This difference is clearly manifested when the transcription reporters are sensitized by decreasing the number of binding sites to below 10.
Our results indicate that the presence of FR or the number of binding sites in FR does not affect DNA synthesis from replication reporter plasmids when assayed 4 days posttransfection. This observation is similar to recent results published by the groups of Sugden (Leight and Sugden, 2001) and Yates (Yates et al., 2000). Leight and Sugden have observed recently that oriP plasmids are rapidly lost between 5 days and 2 weeks posttransfection, a phenomenon they have attributed to epigenetic events that allow a small subset of oriP plasmids to be replicated stably (Leight and Sugden, 2001). This rapid loss is also evident in results from the colony formation and Southern hybridization experiments reported in this study. While between 20 and 60% of cells are detectably transfected by 2 days posttransfection, less than 5% of these cells proceed to form puromycin-resistant colonies. In addition, although we detect between 5 and 140 newly replicated molecules per transfected cell 4 days posttransfection, this number is substantially decreased after 3 weeks of puromycin selection. While the events that lead to this rapid loss of extrachromosomal plasmids within a week posttransfection are not understood, our results indicate they are not influenced by the number of EBNA1-binding sites on the replication reporter, when the reporter contains anywhere from 5 to 80 EBNA1-binding sites (data not shown). It is therefore unlikely that the cellular epigenetic event that allows a subset of oriP plasmids to be replicated stably involves binding of a cellular protein to EBNA1-binding sites in FR. Although the EBNA1-binding sites in the EBV and HVP FR vary considerably in sequence, for both viruses the sequence of the spacer between adjacent binding sites is highly conserved. Because we have altered the spacer sequence without affecting the ability of EBNA1 to activate transcription, or maintain plasmids in proliferating cells, it is unlikely that epigenetic events leading to stable replication of a subset of plasmids involve the association of cellular proteins with sequences between adjacent EBNA1-binding sites in FR.
Both the number of binding sites and the distance between adjacent sites profoundly impact stable replication and maintenance when assayed 3 weeks posttransfection. While binding sites spaced 10 bp apart activate transcription more efficiently, they are far less efficient in plasmid maintenance as assayed by colony formation or Southern hybridization. This decrease in efficiency is most clearly visible when using replication reporter constructions genetically sensitized by decreasing the number of binding sites to be less than 10. Because binding sites spaced 10 bp apart permit efficient transcription activation by EBNA1, it is unlikely that their failure to support oriP-plasmid maintenance results from an inability to be bound by EBNA1. For both sets of replication reporters, we find plasmid maintenance increases with increasing numbers of binding sites. In 293/EBNA1 cells, stable maintenance requires a minimum of four binding sites, as evidenced by our results with constructions AGP89 and AGP128. This minimal number differs between cell lines and is likely a function of the inherent rate of extrachromosomal plasmid loss for each cell line (Aiyar et al., 1998). Maintenance in C33A/1553 cells requires a minimum of five EBNA1-binding sites spaced 14 bp apart (data not shown). The arrangement of the eight binding sites in the HVP FR predicts that EBNA1 dimers bound to five of these sites will lie on one face of the DNA double-helix, suggesting this may be a general minimal requirement for oriP-plasmid maintenance.
Because the space between adjacent binding sites affects EBNA1’s functions in oriP-plasmid maintenance, and to a smaller extent in transcription activation, it is likely that each of these processes is optimal with a different arrangement of EBNA1 bound to DNA. Schleif and others have shown that binding-site phasing affects the function of many prokaryotic transcription activators (Martin et al., 1986; Schleif, 1996). More recently, studies on the interferon-β enhanceosome by the groups of Thanos and Maniatis demonstrate that phasing affects transcription activation by modulating the ability of proteins within the enhanceosome to interact with components of the basal transcription apparatus (Kim and Maniatis, 1997; Merika et al., 1998). Similar observations have also been made with an enhanceosome that forms at the early promoter of human papillomavirus type 18 (Bouallaga et al., 2000). In light of these results, it is perhaps not surprising that transcription activation by EBNA1 is affected by surface presented by an array of EBNA1 dimers on DNA. We find it more surprising that stable plasmid replication is also affected by phasing. Because the latter requires efficient DNA synthesis as well as plasmid maintenance, it is possible that EBNA1 bound at FR influences the conformation or occupancy of EBNA1 at DS, a model suggested by the observation that EBNA1 at FR can interact with EBNA1 at DS, or affect occupancy of EBNA1 at DS (Su et al., 1991). Consistent with this idea, EBNA1-binding sites are placed differently in the HVP and EBV DS elements, which may explain why these viruses have evolved to contain FR elements wherein EBNA1-binding sites are phased differently. Alternatively, plasmid maintenance by EBNA1 may be optimal when EBNA1 dimers are aligned on the same surface of the DNA double-helix to facilitate interactions with cellular factors required for maintenance. These two possibilities can be distinguished by testing whether the space between adjacent EBNA1-binding sites in FR affects the maintenance of plasmids containing a replication origin other than DS, such as the chromosomal sequences identified by Calos and co-workers to function as replication origins in FR-containing plasmids (Heinzel et al., 1991; Krysan et al., 1989, 1993).
Thus far, it has not been possible to distinguish between EBNA1’s functions in transcription activation from its functions in plasmid maintenance. Multiple deletion mutations placed in EBNA1 affect both transcription and plasmid maintenance in proliferating cells to similar extents (Ceccarelli and Frappier, 2000; Mackey and Sugden, 1999), suggesting that both these functions may have a common underlying mechanism. Rittner and co-workers have observed that transcription from oriP-containing luciferase reporter plasmids microinjected into the nuclei of 293/EBNA1 cells is activated to a substantially lower level than transcription from the same plasmids microinjected into the cytoplasm (Langle-Rouault et al., 1998). This result was interpreted to indicate that EBNA1 activates transcription primarily by facilitating nuclear import and retention of plasmids, thereby suggesting that transcription activation of transfected reporter plasmids is a manifestation of EBNA1’s plasmid maintenance function.
If the ability of EBNA1 to activate transcription, and stably maintain oriP plasmids, were both only dependent upon its ability to retain oriP-containing plasmids in the nuclei of dividing cells, differential effects of binding site spacing on transcription activation and stable replication would not have been observed. We interpret the results described in this article to indicate that the transcription activation and plasmid maintenance functions of EBNA1 can be distinguished. This is best illustrated when considering results obtained with transcription and replication reporters that contain seven EBNA1-binding sites spaced either 10 or 14 bp apart. Seven binding sites spaced 14 bp apart activate transcription to approximately 50% the level of seven binding sites spaced 10 bp apart. Results from plasmid replication and maintenance assays indicate that seven binding sites spaced 14 bp apart function 10 times as well as seven binding sites spaced 10 bp apart. This difference in function strongly suggests that EBNA1 uses different mechanisms to activate transcription and to maintain episomal plasmids.
Finally, the binding site constructions described in this article can be exploited to understand the relative contribution of EBNA1 toward transcription activation and genome maintenance during immortalization of primary B cells by EBV. Kieff and co-workers have demonstrated that introduction of a dominant negative form of EBNA1 does not affect the proliferation of IB4, a lymphoblastoid cell line with integrated EBV genomes. This result is interpreted to demonstrate that EBNA1’s primary function during immortalization is to maintain viral genomes (Hung et al., 2001). Previously, Speck and co-workers have shown that EBNA1 bound to oriP activates the viral BamHI-C and BamHI-W promoters that are active during immortalization, suggesting a role for EBNA1’s ability to activate transcription in immortalization (Puglielli et al., 1996). Replacement of the wild-type FR with sFRs that preferentially support transcription activation or plasmid maintenance can be used to discern the contribution of EBNA1 toward each of these processes during immortalization by EBV. Such studies are currently in progress.
Materials and methods
Bacterial strains and plasmid purification
All plasmids were propagated in the E. coli strains DH5α, MC1061/P3, or STBL2 (Invitrogen, Carlsbad, CA). Plasmids used for transfection were purified on isopycnic CsCl gradients (Sambrook et al., 1989).
Oligonucleotides and plasmids
Oligonucleotides EBNA1-BS1 and EBNA1-BS2 contain a single high-affinity binding site for EBNA1 flanked by BsrGI and Acc65I recognition sites as indicated below:
EBNA1-BS1: 5′ GATCCTGTACATTAGGATAGCATATGCTACCCAGAGGTACCG
EBNA1-BS2: 5′ TCGACGGTACCTCTGGGTAGCATATGCTATCCTAATGTACAG
EBNA1-BS1 and EBNA1-BS2 were annealed and cloned into plasmid 2275 (Leight and Sugden, 2001) cut with BamHI and XhoI, to create plasmid AGP6. The complementary 5′ overhangs created after digestion with the endonucleases Acc65I and BsrGI sites flanking the EBNA1-binding site in AGP6 were used to create derivatives of AGP6 that contain increasing numbers of EBNA1-binding sites termed synthetic FR. In the first set of sFRs, adjacent EBNA1-binding sites are separated by a spacer of precisely 14 nucleotides with the sequence “CAGAGGTACATTAG.” A second set of sFRs was created wherein adjacent EBNA1-binding sites were separated by precisely 10 bp. For this, two oligonucleotides AGO31 and AGO32 were synthesized that contain a single high-affinity binding site for EBNA1 flanked by EcoRI and MfeI recognition sites as detailed below:
AGO31: 5′ TCGAGCAATTGAGGATAGCATATGCTACCCAGAATTCG
AGO32: 5′ GATCCGAATTCTGGGTAGCATATGCTATCCTCAATTGC
AGO111 and AGO112 were annealed and cloned into plasmid 2275 cut with BamHI and XhoI to create plasmid AGP56. The EcoRI and MfeI sites flanking the EBNA1-binding site in AGP56 were used to create a series of derivatives of AGP56 that contained an increasing number of EBNA1-binding sites. In this second set of sFRs, adjacent EBNA1-binding sites are separated by a spacer of precisely 10 nucleotides with the sequence “CAGAATTGAG.” pPUR (Clontech, Palo Alto, CA), a plasmid that contains the puromycin-resistance gene under the control of the SV40 early promoter, was used as the backbone to create a series of replication reporter plasmids. Two oligonucleotides, AGO19 and AGO20, were used to amplify all of oriP except FR. This PCR fragment was introduced into pPUR digested with BamHI and BsrGI, to create AGP83. Both sets of sFRs were introduced into AGP83 in lieu of FR to create the following set of replication reporter plasmids described in Fig. 1. These plasmids were sequenced to confirm there were no errors in DS, sequences between sFR and DS, and that they contained the requisite number of EBNA1-binding sites with the desired spacer between adjacent binding sites.
Transcription reporters were constructed using AGP47 as the plasmid backbone. AGP47 contains the firefly luciferase gene under the control of the thymidine kinase (TK) promoter from herpes simplex virus type 1 and was constructed by replacing the wild-type FR in plasmid 985 (Middleton and Sugden, 1992) with a multiple cloning site. Derivatives of AGP47 were constructed that contained either set of sFRs introduced into a multiple cloning site immediately 5′ to the TK-luciferase cassette, to create the transcription reporter plasmid described in Fig. 1. All constructed transcription reporter plasmids were sequenced to ensure there were no errors in the TK promoter and that the sFRs contained the requisite number of EBNA1-binding sites with the desired spacer between adjacent binding sites. Sequences for all the plasmids used in this study can be found at the URL http://ebv.mimnet.northwestern.edu/plasmids.
Cell culture and transfections
The human EBNA1 expressing cell lines C33A/1553 and 293/EBNA1 were used in this study. 293/EBNA1 cells are a derivative of 293 cells (Graham et al., 1977) that stably express wild-type EBNA1. They were propagated in DMEM supplemented with 10% fetal bovine serum, G418 (200 mg/L), and antibiotics. C33A/1553 cells are a derivative of C33A cells (Yee et al., 1985) that stably express wild-type EBNA1 by carrying integrated copies of the EBNA1 expression plasmid 1553 (Aiyar and Sugden, 1998). C33A/1553 were grown in DMEM/F12 (1:1) supplemented with 5% calf serum, hygromycin (100 mg/L), and antibiotics. All cells were grown at 37°C in a humidified 5% CO2 atmosphere. Plasmids were introduced into cells by the calcium phosphate method as described previously (Sambrook et al., 1989). Transfections were normalized by the inclusion of 1 μg of a CMV-EGFP expression plasmid, 2145, in each transfection. Upon harvest, a fraction of the cells were profiled using a Becton–Dickinson FAC-Scan or FACScalibur (Franklin Lakes, NJ). Transfection efficiency was measured as the fraction of GFP-expressing, live cells quantified using CellQuest software from Becton–Dickinson.
Transcription reporter assays
One microgram of AGP47, or an equivalent number of moles of AGP48, AGP50, AGP51, AGP52, AGP53, AGP92, AGP93, AGP94, or AGP95, was cotransfected with 1 μg 2145 into C33A/1553 cells. Cells were split 8 hours posttransfection so that they would not have reached confluence when harvested 48 or 96 h posttransfection. Harvested cells were then counted twice using a Coulter counter, pelleted, and lysed in reporter lysis buffer (provided along with a luciferase assay kit from Promega, Madison, WI) at a concentration of 1 × 105 cells/μl. Lysates were spun for 5 min at 1000 g to remove nuclei and then frozen at −80°C until assay. Luminescence assays were performed as per manufacturer’s instructions, using a Zylux FB 15 luminometer.
Colony formation assays to assess plasmid maintenance and partitioning
Fifteen micrograms of AGP83 or an equivalent number of moles of plasmids AGP72, AGP73, AGP74, AGP100, AGP127, AGP128, AGP88, AGP89, AGP90, and AGP91 were cotransfected with 1 μg of 2145 into 1 × 107 293/EBNA1 cells on a 10-cm dish. Cells were split 8 hours posttransfection so that they would not have reached confluence at 48 h posttransfection. At this time, cells were harvested, FACS profiled to measure GFP expression, and replated in duplicate at 2 × 105, 2 × 104, and 2 × 103 GFP-positive, live cells per culture dish. Cells were placed under selection with 0.5 μg/ml puromycin 4 days posttransfection. After 2 weeks of selection, the resulting puromycin-resistant colonies were fixed with formamide and subsequently stained with methylene blue. Colonies that were at least 2 mm in size were scored as positive.
Southern hybridization analysis to assess plasmid replication
Fifteen micrograms of AGP83 or an equivalent number of moles of AGP72, AGP73, AGP74, AGP100, AGP127, AGP128, AGP88, AGP89, AGP90, AGP91 were cotransfected with 1 μg 2145 into 1 × 107 293/EBNA1 cells on a 10-cm dish. Cells were split 8 h posttransfection so that they would not be confluent at 96 h posttransfection. At that time, episomal DNAs were harvested from 2 × 107 transfected cells, while 1 × 106 GFP-positive, live cells, as measured by FACS profiling, were plated per 10-cm dish for puromycin selection as described above. After 3 weeks of selection, episomal DNAs were extracted from cells in puromycin-resistant colonies that were pooled. Episomal DNAs were extracted from 2 × 108–108 puromycin-resistant cells as described previously (Aiyar et al., 1998). Extracted DNAs were digested with 200 units of DpnI, 20 units of BamHI, and 20 units of XbaI in a final volume of 100 μl overnight at 37°C. Restriction endonucleases were purchased from New England Biolabs (Beverly, MA) and used as per the manufacturer’s instructions. Digestions were extracted with phenol:chloroform (1:1), precipitated, and electrophoresed on a 0.8% agarose gel. DNAs were transferred from the gel to Hybond membrane (Amersham, Buckinghamshire, UK) using an Appligene vacuum transfer apparatus (Boekel Scientific, Feasterville, PA). Radioactive probes were prepared by the incorporation of [α-32P]dCTP (6000 Ci/mmol) (Amersham) during Klenow synthesis using random primers and PstI-digested AGP83 as template. Probe-specific activities ranged from 1 × 109 to 3 × 109 cpm/μg. Southern hybridization was performed as described by Hubert and Laimins (Hubert et al., 1999). Southern blots were visualized and quantified by phosphorimage analysis using a Molecular Dynamics Storm phosphorimager (Molecular Dynamics, Sunnyvale, CA).
Acknowledgments
Some constructions used in this study were made by N. Pinchott. We thank Richard Longnecker, Patricia Spear, Lou Laimins, Neil Clipstone, and members of the Aiyar, Laimins, Longnecker, and Spear laboratories for discussions and assistance while this work was conducted. We thank Hank Seifert and Kathy Rundell for critically reviewing this manuscript. FACS analyses were performed at the Immunobiology Center of Northwestern University. C.H. is a student in the Integrated Graduate Program at Northwestern University and was supported by a fellowship from the Northwestern University Graduate School. A.A. was supported by awards from the Leukemia Research Foundation, the Leukemia and Lymphoma Society, and the National Cancer Institute.
References
- Adams A. Replication of latent Epstein–Barr virus genomes in Raji cells. J. Virol. 1987;61(5):1743–1746. doi: 10.1128/jvi.61.5.1743-1746.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiyar A, Sugden B. Fusions between Epstein–Barr viral nuclear antigen-1 of Epstein–Barr virus and the large T-antigen of simian virus 40 replicate their cognate origins. J. Biol. Chem. 1998;273(49):33073–33081. doi: 10.1074/jbc.273.49.33073. [DOI] [PubMed] [Google Scholar]
- Aiyar A, Tyree C, Sugden B. The plasmid replicon of EBV consists of multiple cis-acting elements that facilitate DNA synthesis by the cell and a viral maintenance element. EMBO J. 1998;17(21):6394–6403. doi: 10.1093/emboj/17.21.6394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambinder RF, Shah WA, Rawlins DR, Hayward GS, Hayward SD. Definition of the sequence requirements for binding of the EBNA-1 protein to its palindromic target sites in Epstein-Barr virus DNA. J. Virol. 1990;64(5):2369–2379. doi: 10.1128/jvi.64.5.2369-2379.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baer R, Bankier AT, Biggin MD, Deininger PL, Farrell PJ, Gibson TJ, Hatfull G, Hudson GS, Satchwell SC, Seguin C, et al. DNA sequence and expression of the B95-8 Epstein-Barr virus genome. Nature. 1984;310(5974):207–211. doi: 10.1038/310207a0. [DOI] [PubMed] [Google Scholar]
- Bouallaga I, Massicard S, Yaniv M, Thierry F. An enhanceosome containing the Jun B/Fra-2 heterodimer and the HMG-I(Y) architectural protein controls HPV 18 transcription. EMBO Rep. 2000;1(5):422–427. doi: 10.1093/embo-reports/kvd091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccarelli DF, Frappier L. Functional analyses of the EBNA1 origin DNA binding protein of Epstein-Barr virus. J. Virol. 2000;74(11):4939–4948. doi: 10.1128/jvi.74.11.4939-4948.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri B, Xu H, Todorov I, Dutta A, Yates JL. Human DNA replication initiation factors, ORC and MCM, associate with oriP of Epstein–Barr virus. Proc. Natl. Acad. Sci. USA. 2001;98(18):10085–10089. doi: 10.1073/pnas.181347998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gahn TA, Schildkraut CL. The Epstein–Barr virus origin of plasmid replication, oriP, contains both the initiation and termination sites of DNA replication. Cell. 1989;58(3):527–535. doi: 10.1016/0092-8674(89)90433-9. [DOI] [PubMed] [Google Scholar]
- Gahn TA, Sugden B. An EBNA-1-dependent enhancer acts from a distance of 10 kilobase pairs to increase expression of the Epstein-Barr virus LMP gene. J. Virol. 1995;69(4):2633–2636. doi: 10.1128/jvi.69.4.2633-2636.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham FL, Smiley J, Russell WC, Nairn R. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 1977;36(1):59–74. doi: 10.1099/0022-1317-36-1-59. [DOI] [PubMed] [Google Scholar]
- Heinzel SS, Krysan PJ, Tran CT, Calos MP. Autonomous DNA replication in human cells is affected by the size and the source of the DNA. Mol. Cell Biol. 1991;11(4):2263–2272. doi: 10.1128/mcb.11.4.2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollander M, Wolfe DA. In: Nonparametric statistical methods. Bradley RA, Hunter JS, Kendall DG, Watsonn GS, editors. John Wiley & Sons; New York: 1973. pp. 67–82. (series in probability and mathematical statistics). [Google Scholar]
- Hubert WG, Kanaya T, Laimins LA. DNA replication of human papillomavirus type 31 is modulated by elements of the upstream regulatory region that lie 5′ of the minimal origin. J. Virol. 1999;73(3):1835–1845. doi: 10.1128/jvi.73.3.1835-1845.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung SC, Kang MS, Kieff E. Maintenance of Epstein-Barr virus (EBV) oriP-based episomes requires EBV-encoded nuclear antigen-1 chromosome-binding domains, which can be replaced by high-mobility group-I or histone H1. Proc. Natl. Acad. Sci. USA. 2001;98(4):1865–1870. doi: 10.1073/pnas.031584698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TK, Maniatis T. The mechanism of transcriptional synergy of an in vitro assembled interferon-beta enhanceosome. Mol. Cell. 1997;1(1):119–129. doi: 10.1016/s1097-2765(00)80013-1. [DOI] [PubMed] [Google Scholar]
- Kirchmaier AL, Sugden B. Plasmid maintenance of derivatives of oriP of Epstein–Barr virus. J. Virol. 1995;69(2):1280–1283. doi: 10.1128/jvi.69.2.1280-1283.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krysan PJ, Haase SB, Calos MP. Isolation of human sequences that replicate autonomously in human cells. Mol. Cell. Biol. 1989;9(3):1026–1033. doi: 10.1128/mcb.9.3.1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krysan PJ, Smith JG, Calos MP. Autonomous replication in human cells of multimers of specific human and bacterial DNA sequences. Mol. Cell Biol. 1993;13(5):2688–2696. doi: 10.1128/mcb.13.5.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langle-Rouault F, Patzel V, Benavente A, Taillez M, Silvestre N, Bompard A, Sczakiel G, Jacobs E, Rittner K. Up to 100-fold increase of apparent gene expression in the presence of Epstein-Barr virus oriP sequences and EBNA1: implications of the nuclear import of plasmids. J. Virol. 1998;72(7):6181–6185. doi: 10.1128/jvi.72.7.6181-6185.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leight ER, Sugden B. Establishment of an oriP replicon is dependent upon an infrequent, epigenetic event. Mol. Cell Biol. 2001;21(13):4149–4161. doi: 10.1128/MCB.21.13.4149-4161.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb DD, Sung NS, Pesano RL, Sexton CJ, Hutchison C, 3rd, Pagano JS. Plasmid origin of replication of herpesvirus papio: DNA sequence and enhancer function. J. Virol. 1990;64(6):2876–2883. doi: 10.1128/jvi.64.6.2876-2883.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupton S, Levine AJ. Mapping genetic elements of Epstein-Barr virus that facilitate extrachromosomal persistence of Epstein-Barr virus-derived plasmids in human cells. Mol. Cell Biol. 1985;5(10):2533–2542. doi: 10.1128/mcb.5.10.2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackey D, Sugden B. The linking regions of EBNA1 are essential for its support of replication and transcription. Mol. Cell Biol. 1999;19(5):3349–3359. doi: 10.1128/mcb.19.5.3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin K, Huo L, Schleif RF. The DNA loop model for ara repression: AraC protein occupies the proposed loop sites in vivo and repression-negative mutations lie in these same sites. Proc. Natl. Acad. Sci. USA. 1986;83(11):3654–3658. doi: 10.1073/pnas.83.11.3654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merika M, Williams AJ, Chen G, Collins T, Thanos D. Recruitment of CBP/p300 by the IFN beta enhanceosome is required for synergistic activation of transcription. Mol. Cell. 1998;1(2):277–287. doi: 10.1016/s1097-2765(00)80028-3. [DOI] [PubMed] [Google Scholar]
- Middleton T, Sugden B. EBNA1 can link the enhancer element to the initiator element of the Epstein–Barr virus plasmid origin of DNA replication. J. Virol. 1992;66(1):489–495. doi: 10.1128/jvi.66.1.489-495.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puglielli MT, Woisetschlaeger M, Speck SH. OriP is essential for EBNA gene promoter activity in Epstein–Barr virus-immortalized lymphoblastoid cell lines. J. Virol. 1996;70(9):5758–5768. doi: 10.1128/jvi.70.9.5758-5768.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawlins DR, Milman G, Hayward SD, Hayward GS. Sequence-specific DNA binding of the Epstein-Barr virus nuclear antigen (EBNA-1) to clustered sites in the plasmid maintenance region. Cell. 1985;42(3):859–868. doi: 10.1016/0092-8674(85)90282-x. [DOI] [PubMed] [Google Scholar]
- Reisman D, Sugden B. Trans activation of an Epstein–Barr viral transcriptional enhancer by the Epstein–Barr viral nuclear antigen 1. Mol. Cell Biol. 1986;6(11):3838–3846. doi: 10.1128/mcb.6.11.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reisman D, Yates J, Sugden B. A putative origin of replication of plasmids derived from Epstein–Barr virus is composed of two cis-acting components. Mol. Cell Biol. 1985;5(8):1822–1832. doi: 10.1128/mcb.5.8.1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 2 ed Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1989. [Google Scholar]
- Schepers A, Ritzi M, Bousset K, Kremmer E, Yates JL, Harwood J, Diffley JF, Hammerschmidt W. Human origin recognition complex binds to the region of the latent origin of DNA replication of Epstein-Barr virus. EMBO J. 2001;20(16):4588–4602. doi: 10.1093/emboj/20.16.4588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleif R. Two positively regulated systems, ara and mal. In: Neidhardt FC, Curtis R III, Ingraham JL, Lin ECC, Low KB, Magasanik B, Reznikoff WS, Riley M, Schaechter M, Umbarger HE, editors. Escherichia coli and Salmonella: Cellular and Molecular Biology. Second ed ASM Press; Washington, DC: 1996. pp. 1300–1309. [Google Scholar]
- Su W, Middleton T, Sugden B, Echols H. DNA looping between the origin of replication of Epstein-Barr virus and its enhancer site: stabilization of an origin complex with Epstein–Barr nuclear antigen 1. Proc. Natl. Acad. Sci. USA. 1991;88(23):10870–10874. doi: 10.1073/pnas.88.23.10870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugden B, Warren N. Plasmid origin of replication of Epstein, Barr virus, oriP, does not limit replication in cis. Mol. Biol. Med. 1988;5(2):85–94. [PubMed] [Google Scholar]
- Watson JD, Crick FHC. A structure for DNA. Nature. 1953;171:737–738. doi: 10.1038/171737a0. [DOI] [PubMed] [Google Scholar]
- Wysokenski DA, Yates JL. Multiple EBNA1-binding sites are required to form an EBNA1-dependent enhancer and to activate a minimal replicative origin within oriP of Epstein-Barr virus. J. Virol. 1989;63(6):2657–2666. doi: 10.1128/jvi.63.6.2657-2666.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates J, Warren N, Reisman D, Sugden B. A cis-acting element from the Epstein–Barr viral genome that permits stable replication of recombinant plasmids in latently infected cells. Proc. Natl. Acad. Sci. USA. 1984;81(12):3806–3810. doi: 10.1073/pnas.81.12.3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates JL, Camiolo SM, Ali S, Ying A. Comparison of the EBNA1 proteins of Epstein–Barr virus and herpesvirus papio in sequence and function. Virology. 1996;222(1):1–13. doi: 10.1006/viro.1996.0392. [DOI] [PubMed] [Google Scholar]
- Yates JL, Camiolo SM, Bashaw JM. The minimal replicator of Epstein-Barr virus oriP. J. Virol. 2000;74(10):4512–4522. doi: 10.1128/jvi.74.10.4512-4522.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates JL, Guan N. Epstein-Barr virus-derived plasmids replicate only once per cell cycle and are not amplified after entry into cells. J. Virol. 1991;65(1):483–488. doi: 10.1128/jvi.65.1.483-488.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee C, Krishnan-Hewlett I, Baker CC, Schlegel R, Howley PM. Presence and expression of human papillomavirus sequences in human cervical carcinoma cell lines. Am. J. Pathol. 1985;119(3):361–366. [PMC free article] [PubMed] [Google Scholar]