

Abstract
The ability of cancer vaccines to induce tumor-specific CD8+ T cells in the circulation of cancer patients has been shown to poorly correlate with their clinical effectiveness. In this study, we report that although Ags presented by different types of mature dendritic cells (DCs) are similarly effective in inducing CD8+ T cell expansion, the acquisition of CTL function and peripheral-type chemokine receptors, CCR5 and CXCR3, requires Ag presentation by a select type of DCs. Both “standard” DCs (matured in the presence of PGE2) and type 1-polarized DCs (DC1s) (matured in the presence of IFNs and TLR ligands, which prevent DCs “exhaustion”) are similarly effective in inducing CD8+ T cell expansion and acquisition of CD45RO+IL-7R+IL-15R+ phenotype. However, granzyme B expression, acquisition of CTL activity, and peripheral tissue-type chemokine responsiveness are features exclusively exhibited by CD8+ T cells activated by DC1s. This advantage of DC1s was observed in polyclonally activated naive and memory CD8+ T cells and in blood-isolated melanoma-specific CTL precursors. Our data help to explain the dissociation between the ability of cancer vaccines to induce high numbers of tumor-specific CD8+ T cells in the blood of cancer patients and their ability to promote clinical responses, providing for new strategies of cancer immunotherapy.
Recent trials of cancer vaccines demonstrated that the induction of high numbers of circulating tumor-specific CD8+ T cells is not necessarily accompanied by acquisition of an effector function (1, 2), resulting in the limited ability of the current vaccines to induce tumor regression (3–6). This raises the question of whether the currently used vaccination strategies are optimal with regard to their ability to induce effector-type cytotoxic CD8+ T cells (CTLs) with tumor-relevant homing potential.
In the case of CD4+ T cells, extensive studies in human and mouse models have demonstrated that dendritic cells (DCs) maturing in different environments or preactivated for different periods of time can instruct naive CD4+ T cells to selectively acquire Th1 or Th2 effector functions (7–10), leading to the concept of “signal 3,” which selectively regulates the acquisition of T cell effector functions (7). Although the role of the functional status of DCs in the development of effector CD8+ T cells is less clear, in several in vivo mouse models of infections, it was demonstrated that inflammatory cytokines, such as IL-12, IFN-α, and IFN-γ, not only regulate the proliferation of CD8+ T cells but also their acquisition of CTL functions (11–13).
To directly test whether the induction of CTL functions and tumor-relevant chemokine responsiveness are differentially regulated by different DC types, we compared the phenotype and functions of human CD8+ T cells primed by different types of mature, highly stimulatory DCs, such as type 1-polarized DCs (DC1s) matured in the presence of IFNs and TLR ligands (including the clinicallyused TNF-α/IL-1β/polyinosinic:polycytidylic acid (poly-I:C)/IFN-γ/IFN-α–matured αDC1s; ClinicalTrials.gov: NCT00390338, NCT00099593, NCT00766753, NCT00558051, and NCT00970203) (14) and non-polarized DCs matured in the presence of PGE2 (including the clinically applied TNF-α/IL-1β/IL-6/PGE2–matured “standard” [s]DCs) (15) that were previously shown to induce different numbers of tumor-specific T cells, as determined by IFN-γ ELISPOT (14).
Our data indicate that although both type 1-polarized and non-polarized DCs induce similar CD8+ T cell expansion, the induction of functional CTLs with peripheral homing capacity requires “nonexhausted” DC1s. In contrast, nonpolarized DCs selectively induce CD8+ T cell expansion, without the accompanying development of CTL functions or peripheral homing potential.
Materials and Methods
Cell lines, media, and reagents
Serum-free AIM-V medium (Invitrogen, Carlsbad, CA) was to used to generate DCs and IMDM (Invitrogen) with 5% human serum (Atlanta Biologicals, Norcross, GA) was used for in vitro sensitization (IVS) experiments. The following factors were used to generate mature DCs: recombinant human (rhu) GM-CSF and IL-4 (gifts from Schering-Plough, Kenilworth, NJ), IFN-α (intron A), rhuTNF-α, rhuIFN-γ, rhuIL-1β (all from Strathmann Biotech, Hannover, Germany), rhuIL-6 (Genzyme, Cambridge, MA), LPS (Sigma-Aldrich, St. Louis, MO), PGE2 (Sigma-Aldrich), and poly-I:C (Sigma-Aldrich). IL-2 (Chiron, Emeryville, CA) and rhuIL-7 (Strathmann Biotech) were used to support the CD8+ T cell expansion.
Generation and maturation of DCs
PBMCs were obtained from the blood of healthy donors or melanoma patients using lymphocyte separation medium (Cellgro, Mediatech, Herndon, VA). Monocytes were isolated on density gradients, with Percoll (Sigma-Aldrich), followed by plastic adherence. Monocytes were cultured for 6 d in 24-well plates (BD Falcon, Franklin Lakes, NJ) at 5 ×105 cells/well in rhuGM-CSF and IL-4 (both 1000 U/ml). At day 6, maturation was induced by exposing the DCs to the following combinations of maturation stimuli: LPS (250 ng/ml) and IFN-γ (1000 U/ml), LPS and PGE2 (10−6 M), TNF-α (100 ng/ml) and IFN-γ, and TNF and PGE2 for 48 h (apart from Fig 1B, when 24–96-h maturation was used, as indicated). In addition, as representatives of clinically applicable polarized and nonpolarized DCs currently used as cancer vaccines, we used nonpolarized sDCs matured for 48 h in the presence of TNF-α (50 ng/ml), IL-1β (25 ng/ml), PGE2 (10−6 M), and IL-6 (1000 U/ml) (15), and αDC1 matured using the cytokine mixture composed of TNF-α (100 ng/ml), IL-1β (25 ng/ml), IFN-γ (1000 U/ml), poly-I:C (20 μg/ml), and IFN-α (3000 U/ml) (14).
FIGURE 1.

Differential regulation of CD8+ T cell expansion versus the induction of CTL granules by DCs matured in different inflammatory conditions. Immature DCs were activated with different combination of cytokines (see Materials and Methods), resulting in different levels of IL-12p70 production (TNF-α/IFN-γ, 3610 ± 160 pg/ml; LPS/IFN-γ, 3960 ± 30 pg/ml; αDC1, 1900 ± 120 pg/ml; TNF-α/PGE2, 330 ± 80 pg/ml; LPS/PGE2 and sDC, both <20 pg/ml). For priming of naive CD8+ T cells, DCs were harvested after 48 h (additionally, 24-and 96-h matured DCs were tested in B), washed, pulsed with Ag (SEB), and coincubated with naive CD45RA+ CD8+ T cells (triplicates). On day 5, CD8+ T cells were counted to assess cell expansion (A, left) and stained for intracellular GrB (A, right). The fold increase in mean fluorescent intensity (MFI) of GrB was calculated as the ratio of GrB MFI to isotype control MFI (mean ± SE of three independent cultures). A, Selective induction of GrB-expressing CD8+ T cells by polarized DCs. B, Polarized DCs show persistent (although reduced) ability to induce GrB-expressing CD8+ T cells even after 96 h of DC maturation.
Isolation of peripheral blood CD8+ T cell populations
PBMCs were obtained from the blood of healthy donors or melanoma patients using lymphocyte separation medium (Mediatech). Naive CD8+ CD45RA+CD45RO− T cells were isolated from the lymphocyte fraction by negative selection with CD8 enrichment mixture with the addition of biotinylated anti-CD45RO Ab (StemCell Technologies, Vancouver, British Columbia, Canada) as a uniform population of CD8+CCR7+CD45RA+ CD45RO− cells (16, 17). CD8+CCR7+CD45RA− (CD45RO+) memory T cell population was flow-sorted using MoFlo high-speed cell sorter (DakoCytomation, Carpinteria, CA), after labeling with appropriate Abs.
Flow cytometry
Two- and three-color cell surface and intracellular immunostaining analysis was performed using Beckman Coulter Epics XL flow cytometer, after staining with the Abs against human granzyme B (GrB) (BD Pharmingen and CellSciences), CCR7 (R&D Systems, Minneapolis, MN), CCR5 (BD Pharmingen, San Diego, CA), or the corresponding isotypes IgG2a and IgG1. HLA-A2/MART-127–35 tetramer staining (Beckman Coulter, Immunomics, Fullerton, CA) was performed according to the manufacturer’s instructions.
IVS (polyclonal)
Naive CD8+CD45RA+CCR7high T cells (5 ×105 cells/well) were activated with staphylococcal enterotoxin B (SEB)-pulsed monocyte-derived DCs (5 ×104 cells/well), as described previously (16, 17). Autologous or allogeneic DCs were used with similar results. On days 5–6, expanded CD8+ T cells were counted and analyzed for the expression of chemokine receptors and chemokine responsiveness and for CTL phenotype and function (see Supplemental Fig. 1 for the kinetics of acquisition of CTL functions in the differentially primed CD8+ T cells). Alternatively, the cultures were fed with low-dose IL-2 and IL-7 (10 ng/ml) every 2 d and analyzed for cell surface and intracellular markers on days 16–20. When indicated, neutralizing IL-12 Ab (clone 24910; R&D Systems) was added at the beginning of the IVS culture. In preliminary experiments, we compared the outcome of naive CD8+ T cell priming by polarized and nonpolarized DCs in the additional presence of CD40L-expressing J558 cells. Because the presence of CD40L did not abolish the differences in the phenotype and function of the resulting T cells, all subsequent experiments were performed in the absence of CD40L.
IVS (melanoma specific)
Bulk CD8+ T cells (5 × 105 cells/well) were activated with the HLA-A2–restricted peptide MART-127–35–pulsed autologous DCs (5 ×104 cells/well). A total of 3000 rad-irradiated CD40L-J558 cells (5 ×104) were added as surrogates of CD40L-expressing CD4+ Th cells, as described previously (14). On day 4, rhuIL-2 (50 U/ml) and IL-7 (10 ng/ml) were added. CD8+ T cell cultures were expanded by an additional stimulation (day 14) with irradiated peptide-pulsed autologous PBMCs. At day 24, the differentially induced CD8+ T cell lines were stained for CCR5, GrB, and MART-1. CTL activity was determined by 51Cr release assays against HLA-A2+ melanoma (Fem X), with HLA-A2neg 397 melanoma cells serving as negative specificity control.
Chemotaxis assay
Chemotaxis assays were performed in 96-well Transwell plates with a 3-μm pore-size polycarbonate filter (Corning, Corning, NY). The lower chamber was filled with 200 μl of rhuCCL19 (100–1000 ng/ml) or rhuCCL5 (100–1000 ng/ml) in RPMI 1640 plus 0.5% FBS (chemotaxis media), and 50 μl (5 × 104 cells) of differentially activated CD8+ T cells was added in the upper chamber, and migration chambers were incubated for 3 h at 37°C. After 3 h, the cells from lower wells were harvested and counted. The number of cells that migrated in media alone was subtracted to normalize for background migration.
CTL assay
Cytolytic activity against HLA-A2+ melanoma cells (Fem X) was determined by standard 4-h 51Cr release assays, with HLA-A2neg 397 melanoma cell line serving as negative control of specificity. The results were calculated and recorded as percent target killing at individual E:T ratios, or percentage of cytolysis was converted to LUs (LU10/107) as described previously (18).
Statistical analysis
The data were analyzed using Student’s t test (with paired tests being used for comparisons including αDC1s- versus sDCs-induced responses from multiple donors). Values of p < 0.05 were considered significant.
Results
Independent regulation of CD8+ T cell expansion and acquisition of CTL functions by polarized and nonpolarized DCs
In order to delineate the requirements for the effective expansion of CD8+ T cells and their acquisition of effector functions, we compared the outcome of CD8+ T cell priming by DCs induced to mature by mediators of acute inflammation (combination of IFNs and TLR ligands) or by mediators of chronic inflammation (presence of PGE2 [19–21]). Although the DC maturation in the presence of PGE2 is associated with an irreversible process of DC “exhaustion” manifested by reduced ability to produce IL-12, the key mediator of inflammatory-type responses (22), and reduced ability to induce Th1 responses of CD4+ Th cells (10, 23, 24), DC1s induced in the conditions of early inflammation avoid the maturation-associated DC “exhaustion”, retaining their ability to produce IL-12 and to induce Th1 responses of CD4+ Th cells (7, 14, 23, 24).
As shown in Fig. 1A, left, both polarized and nonpolarized DCs induced similar rates of expansion of naive CD8+ T cells. However, only naive CD8+ T cells primed by the polarized DC1s in our previously established model of priming of naive CD8+ T cells (16, 17) demonstrated an effective induction of GrB (Fig. 1A, right), a marker of effector T cell differentiation (25). In sharp contrast, the low IL-12–producing nonpolarized DCs (14, 23, 24) did not prime naive CD8+ T cells to express GrB (Fig. 1A, right), despite inducing a similar or higher T cell expansion (Fig. 1A, left).
Importantly for their use as therapeutic agents in vivo, DC1s retained a significant (although reduced) ability to induce GrB expression in expanding CD8+ T cells, even at later times (96 h) after the induction of their maturation (Fig. 1B). These latter observations indicate that the maturation of DCs in the conditions mimicking early inflammation allows them to at least partially avoid or delay the acquisition of an “exhausted” status (10, 24), previously shown to be associated with abrogated ability to induce functional Th1 responses in the population of CD4+ T cells (10).
Using the clinically-relevant TNF-α/IL-1β/poly-I:C/IFN-γ/IFN-α–matured αDC1s (14) and TNF-α/IL-1β/IL-6/PGE2–matured sDCs (15) as representatives of type 1-polarized versus non-polarized DCs, we observed that the induction of GrB correlated with the superior cytolytic function of CD8+ T cells primed by the polarized DCs (Fig. 2A). In contrast, priming of CD8+ T cells by the PGE2-matured sDCs led to low levels of GrB and poor ability to kill Ag-pulsed target cells (Fig. 2A–C), despite effective proliferation of T cells in these cultures and induction of CD45RO (Figs. 1, 2B). In accordance with the central role of IL-12 in the development of CTL activity in CD8+ T cells, neutralization of that factor abrogated GrB induction by DC1s (Fig. 2D).
FIGURE 2.
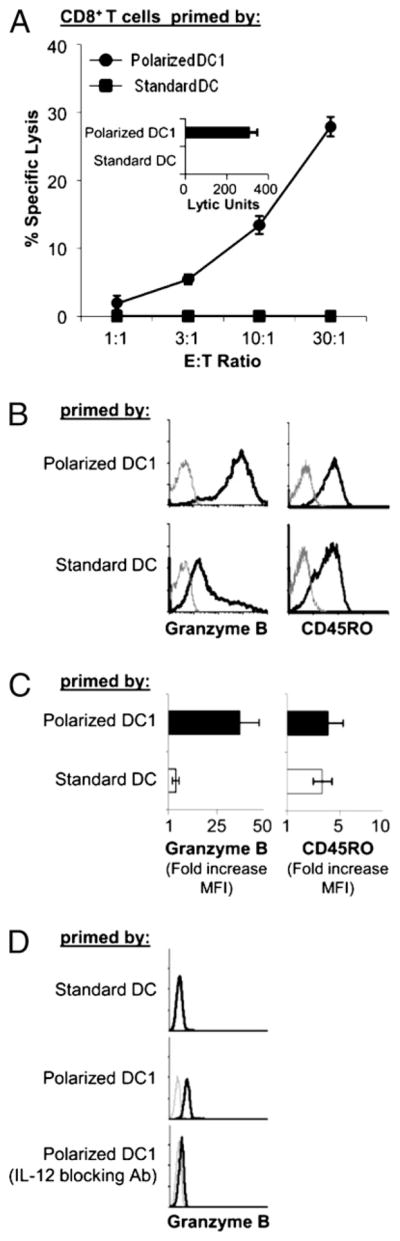
Induction of cytolytic and noncytolytic pathway of CD8+ T cell differentiation by polarized DC1s and sDCs. Naive CD8+ T cells primed with either polarized or non-polarized DCs, using αDC1 (matured for 48 h in TNF-α, IL-1β, IFN-γ, poly-I:C, and IFN-α) and sDCs (matured for 48 h in TNF-α, IL-1β, IL-6, and PGE2 [15]), as the respective representatives. A, Cytolytic function of day 5-primed CD8+ T cells was assessed by standard 51 Cr release assay using SEB-pulsed JY cells as targets (17). Inset, Data calculated as LUs. B and C, Intracellular expression of GrB and surface expression of CD45RO were determined by flow cytometry on day 5. B, Data from a representative donor. Gray lines indicate isotype controls. C, Summary of data from three different donors. Fold increase in MFI of GrB and CD45RO was calculated as in Fig. 1. Data are shown as mean and SEM of three independent experiments that all showed advantage of polarized αDC1s in the induction of GrB (p < 0.02). D, Neutralization of IL-12 abrogates the induction of GrB-positive CD8+ T cells by polarized αDC1s. Representative data from three experiments that all yielded similar results.
Because certain conditions of effector T cell induction can be associated with their irreversible differentiation into short-lived, terminally differentiated effector cells (26), we tested the ability of the DC1-induced effector cells to respond to secondary activation and undergo secondary CTL differentiation. As shown in Fig. 3, after completing the effector phase of activation (>2 wk after priming), the αDC1-primed CD8+ T cells downregulated the levels of GrB expression and their cytolytic activity. Consistent with the ability of polarized αDC1s to induce long-lived CD8+ T cells (14), such resting αDC1-primed CD8+ T cells expressed high levels of IL-15Rα and IL-7Rα (CD127) (Fig 3A; see Supplemental Fig. 2 for the levels of both receptors in naive CD8+ T cells), the memory cell-associated receptors for the homeostatic cytokines mediating long-term survival of CD8+ T cells (27, 28) and were fully capable of rapidly reacquiring high levels of CTL activity upon restimulation with polarized αDC1s (Fig. 3B).
FIGURE 3.

CD8+ T cells primed by polarized DC1s revert to memory status and can be reactivated to undergo secondary CTL differentiation. A, Induction of memory CD8+ T cells at later stages of activation with αDC1s. Naive CD8+ T cells were primed with αDC1s. After 3 wk, the cells were analyzed for the expression of GrB, CD45RO, IL-7Rα, and IL-15Rα. B, Effective induction of secondary CTL function in αDC1-primed resting CD8+ T cells. Three weeks after priming with αDC1s, resting CD8+ T cells were (re)stimulated for 24 h with αDC1s and reassessed for CTL function. SEB-pulsed JY cells were used as target population for chromium release assay (17). Similar data were obtained in three independent experiments, with the observed range of killing between 0 and 8.9% (at the maximal 30:1 ratio) for the resting CD8+ T cells and between 34.8 and 72% for the restimulated CD8+ T cells.
Polarized DC1s induce a switch in chemokine receptor expression and peripheral tissue-associated chemokine responsiveness in expanding CD8+ T cells: key role of IL-12
Because polarized αDC1s and sDCs both promoted the expansion of naive T cells but had a differential impact on the induction of their CTL function, we tested their influence on the CD8+ T cell expression of CCR7 and CCR5, the respective lymphoid versus peripheral effector-type chemokine receptors, and the migratory responsiveness to their respective ligands, lymph node-associated CCL19/MIP3β (29–31) and CCL5/RANTES, a ubiquitous peripheral tissue-produced chemokine (29, 30) known to be over-expressed in cancer tissues (30, 32).
As shown in Fig. 4A and 4B, αDC1s effectively induced the expression of CCR5, the chemokine receptor typical for effector (and effector-memory) CD8+ T cells (33–35), with a concomitant loss of CCR7 on 50–70% of CD8+ T cells. In contrast, CD8+ T cells stimulated by sDCs retained high levels of CCR7 expression and did not acquire CCR5.
FIGURE 4.

Polarized DC1s induce a switch in chemokine receptor expression and chemokine responsiveness. Naive CD8+ T cells were primed by αDC1s or sDCs. Differentially primed CD8+ T cells were harvested on day 5 and analyzed for the expression of chemokine receptors. A, Data from a representative donor: Levels of expression of CCR7 and CCR5 (black lines), compared with isotope controls (gray lines). B, Cumulative data from three donors. Fold increase in MFI of CCR7 and CCR5 were calculated as in Fig. 1. Data are shown as mean ± SEM of three independent experiments that all showed advantage of polarized αDC1s in promoting the loss of CCR7 expression (p < 0.0005) and induction of CCR5 (p < 0.005). C, Differentially primed CD8+ T cells were analyzed for their responsiveness to chemokine receptor ligands CCL19 and CCL5 by chemotaxis assay (mean ± SEM of three independent experiments). In the three donors tested, at the maximal concentrations of the two chemokines, the migration of αDC1s to CCL19 was 3.4- to 5.2-fold lower than the migration of sDCs to CCL19, whereas the migration of αDC1s in response to CCL5 was 3.6- to 11.8-fold higher that the migration of sDCs. *, Undetectable. D, IL-12–blocking Ab was added during the priming of naive CD8+ T cells by polarized αDC1s. CCR7 and CCR5 expression (black lines) was assessed by flow cytometry on day 5. Gray line indicates isotype control in all histograms. Similar data were observed in two additional experiments.
In accordance with their differential expression of CCR7 and CCR5, the differentially activated CD8+ T cells showed reciprocal patterns of migratory responsiveness to the lymph node-associated versus peripheral tissue-associated chemokines (CCL19 and CCL5, respectively [29, 30, 32, 35]) with αDC1-primed CTLs preferentially migrating toward the peripheral tissue chemokine CCL5 (RANTES), whereas the sDC-primed T cells preferentially responded to the lymphoid chemokine CCL19 (Fig. 4C).
Because in the CD4+ T cell system, the levels of DC-produced IL-12 were shown to be the key to the differential ability of DCs to induce a Th1 or Th2 pattern of differentiation in naive CD4+ T cells (7, 8, 10) and rIL-12 was shown to directly affect the expression of Th1- and Th2-associated chemokine receptors (36, 37), we tested the role of IL-12 in the DC-induced switch in chemokine receptor expression of CD8+ T cells. As shown in Fig. 4D, the neutralization of IL-12 during T cell priming abrogated the above differences, preventing the downregulation of CCR7 and elevation of CCR5 on CD8+ T cells activated by the polarized DCs. These data indicate that IL-12, originally identified as a factor supporting killer activities of CD8+ T cells and NK cells (reviewed in Ref. 22), is also a key DC-produced factor responsible for the switch from central to peripheral chemokine receptor pattern in the differentiating naive CD8+ T cells.
Polarized and nonpolarized DCs differentially regulate CTL activity and chemokine receptor expression on tumor Ag-specific CD8+ T cells
Prompted by the results of the experiments with polyclonally activated naive CD8+ T cells (Fig 2A) and similar data obtained using memory cells (Supplemental Fig. 3), we have compared the outcome of IVS of HLA-A2–restricted melanoma-specific CD8+ T cells using MART-127–35–loaded autologous αDC1s or sDCs, currently applied as cancer vaccines.
In contrast to the short-term experiments performed in the polyclonal system, the generation of high numbers of MART-1–specific T cells required prolonged cultures of the differentially sensitized CD8+ T cells. Although in these long-term cultures we could not detect the differences in CCR7 expression between the differentially-sensitized CD8+ T cells (CCR7 was low on both populations; data not shown), exclusively the MART-1–specific (tetramer positive) CD8+ T cells sensitized with polarized αDC1s showed high GrB expression and high CTL activity against MART-1–expressing HLA-A2+ melanoma cells (but not against HLA-A2− melanoma cells; Fig. 5A–C). Although in contrast to their inability to induce CTL activity in naive CD8+ T cell population (see Figs. 1 and 2), nonpolarized DCs showed significant ability to induce CTL function in tumor-specific T cells from melanoma patients, though DC1s were clearly more efficient (Fig. 5C), with the level of advantage comparable to that observed in the polyclonal model of (re)activation of “bulk” (memory and naive) CD8+ T cells (Supplemental Fig. 4). In accordance with the data obtained in the polyclonal models (Fig 4), MART-1–specific CD8+ T cells sensitized by polarized αDC1s also showed elevated levels of CCR5 (Fig. 5D).
FIGURE 5.

Polarized αDC1s and non-polarized sDCs induce differential expression of GrB and melanoma-relevant chemokine receptors on MART-1-specific CD8+ T cells. αDC1s and sDCs from HLA-A2+ melanoma patients were pulsed with the HLA-A2–restricted MART-1 peptide and used to stimulate autologous CD8+ T cells in an IVS system (see Materials and Methods). A, Intracellular expression of GrB in MART-1–tetramer+CD8+ T cells. Similar data were obtained in case of two donors. B and C, High CTL activity of αDC1-sensitized CD8+ T cells against melanoma cells. Cytotoxic activity of the differentially primed CD8+ T cells was measured against MART-1–expressing HLA-A2+ melanoma cell line (Fem-X). Inset, HLA-A2− melanoma cell line (melanoma 397) was used as a negative control of antigenic specificity. B, Representative data from one patient. C, Combined data from four different patients expressed as LUs (p < 0.005). D, Surface expression of CCR5 and CXCR3 were measured in MART-1–tetramer+CD8+ T cells. Left, Representative data from a single melanoma patient. Right, Cumulative data from three melanoma patients expressed as the mean ± SEM. αDC1-sensitized CD8+ T cells show enhanced expression levels of both CCR5 and CXCR3 (p < 0.05 in both cases).
In addition to CCR5, which shows high effectiveness in attracting mouse effector cells to melanoma lesions (38) and was recently implicated in the responsiveness of melanoma patients to immunotherapy (39), another CTL-associated chemokine receptor, CXCR3, has been recently implicated in melanoma regression (40) and prolonged survival of patients with advanced disease (41). Therefore, we compared the expression of CXCR3 on MART-1–specific CD8+ T cells presensitized with polarized DC1s and sDCs. As shown in Fig. 5D, polarized αDC1s induced strongly elevated levels of CXCR3 in MART-1–specific CD8+ T cells from melanoma patients.
Discussion
Our data demonstrate that the ability of DCs to activate T cells and to efficiently induce their expansion does not predict their ability to induce CTL activity and the ability to respond to peripheral-type chemokines. In contrast, we observed that although the expansion of CD8+ T cells can be driven efficiently by the DCs matured in a wide spectrum of inflammatory conditions, the induction of the CD8+ T cell effector functions in naive CD8+ T cells and a switch in their chemokine responsiveness was a sole property of the “nonexhausted” IL-12–producing DCs matured in the conditions that mimic acute inflammation (presence of IFNs and TLR ligands). This “inflammatory” pathway of activation of CD8+ T cells, associated with the IL-12–dependent induction of GrBhigh CTLs, eventually results in a resting population of memory-type (CD8+CD45RO+GrBlow) cells. In accordance with the previously reported long-lived character of cells activated by the high IL-12–producing DC1s (14) and with the ability of rIL-12 to promote CTL survival (42, 43), the CD8+ T cells undergoing such “inflammatory” pathway of differentiation expressed high levels of IL-7R and IL-15R (Fig. 3), known to be essential for the homeostatic proliferation and long-term survival of CD8+ T cells in vivo (27, 28), and could effectively reacquire CTL function following restimulation with polarized DCs.
The effectiveness of polarized DC1s in inducing functional CCR5 (and CXCR3)-expressing CTLs suggest that these cells can be useful tools to direct the vaccination-induced T cells to tumors in therapeutic conditions. Because melanomas are known to over-express CCL5/RANTES (44, 45), on which they rely as an autocrine growth factor (45, 46), CCL5-responsive, αDC1-induced T cells are likely to show improved therapeutic activity, not only because of their higher per-cell killer activity, but also because of their ability to preferentially home to tumor tissues. In support of the opposite roles of tumor-expressed CCR5 versus T cell expressed CCR5 in melanoma progression (respectively, tumor promoting versus tumoricidal), it was recently shown that although overall populations of melanoma patients lacking functional CCR5 (CCR5Delta32+ individuals) and CCR5-competent melanoma patients have similar course of disease, functional CCR5 is needed for positive response to immunotherapy (39). Similarly, in accordance with high expression of CXCR3 ligands (CXCL9/MIG and CXCL10/IP10) in melanoma tissues (47) and the presence of CXCR3 on tumor-infiltrating lymphocytes in regressing melanoma lesions (40), high levels of CXCR3 on circulating CD8+ T cells has been recently implicated in effective control of advanced melanoma (41).
In contrast to such “proinflammatory/effector” pathway of differentiation driven by polarized DCs, naive CD8+ T cells activated by standard nonpolarized DCs did not acquire CTL functions and remained responsive to lymph node-associated chemokines, even though they vigorously expanded. Although our preliminary data indicate that such cells can be effectively reactivated by polarized DC1s (data not shown) to undergo secondary CTL differentiation, the identity and functional role of such “non-effector” CD8+ T cells induced by standard “exhausted” DCs remains a subject of our follow up studies. Interestingly, although nonpolarized DCs were unable to induce the de novo effector function in naive CD8+ T cells, they showed a significant (although lesser than polarized DCs) ability to induce CTL function in (expectedly previously primed) tumor-specific T cells from melanoma patients, type (Fig. 5A–C), and in the polyclonal model of (re)activation of “bulk” (memory and naive) CD8+ T cells (Supplemental Fig. 4).
The current demonstration that the ability of DCs to induce proliferation and expansion of tumor-specific CD8+ T cells is independent from their ability to induce their tumor-relevant homing properties and tumoricidal effector functions helps to interpret the limited effectiveness of cancer vaccines observed in recent clinical trials (3–6) and aids in designing corrective measures to enhance the efficacy of cancer immunotherapies. Several recently tested cancer vaccines involving antigenic peptides or tumor Ag-expressing viral vectors were shown to promote massive increase of blood-circulating tumor-specific CD8+ T cells but not clinical responses (1, 2, 4–6). Interestingly, at least one study indicated that such split effectiveness of cancer vaccines can be corrected by a follow up treatment of the vaccinated patients with IFN-α (1). Although our current data (Fig. 2D) demonstrate the key role of IL-12 in the induction of functional CTLs by DC1s, it remains to be tested whether other factors may supplement or replace the function of IL-12 in differentially matured DCs.
Our current data suggest that the limitations of current cancer vaccines, including “standard” DC-based vaccines (48), may result from their selective deficit in inducing the effector functions in tumor-specific T cells and may be corrected by the modification of the current therapeutic vaccines, or their combination with proinflammatory factors, capable of inducing tumoricidal function and tumor-homing ability in tumor-specific T cells.
Acknowledgments
This work was supported by the National Institutes of Health Grants CA095128, CA114931, CA101944, CA121973, and CA132714.
We thank Drs. Robert Hendricks, Michael Lotze, and Kyle McKenna for stimulating discussions and critical comments.
Abbreviations in this paper
- DC
dendritic cell
- DC1
type 1-polarized dendritic cell
- GrB
granzyme B
- IVS
in vitro sensitization
- MFI
mean fluorescent intensity
- rhu
recombinant human
- poly-I
C, polyinosinic:polycytidylic acid
- sDC
standard dendritic cell
- SEB
staphylococcal enterotoxin B
Footnotes
The online version of this article contains supplemental material.
Disclosures
αDC1s, used as one of the models in this paper, are a topic of a current patent application. None of the authors involved in this study receives any form of support or remuneration related to that platform. The clinical trials and other studies involving αDC1s are supported by the National Cancer Institute and the Department of Defense.
References
- 1.Astsaturov I, Petrella T, Bagriacik EU, de Benedette M, Uger R, Lumber G, Berinstein N, Elias I, Iscoe N, Hammond C, et al. Amplification of virus-induced antimelanoma T-cell reactivity by high-dose interferon-α2b: implications for cancer vaccines. Clin Cancer Res. 2003;9:4347–4355. [PubMed] [Google Scholar]
- 2.Rosenberg SA, Sherry RM, Morton KE, Scharfman WJ, Yang JC, Topalian SL, Royal RE, Kammula U, Restifo NP, Hughes MS, et al. Tumor progression can occur despite the induction of very high levels of self/tumor antigen-specific CD8+ T cells in patients with melanoma. J Immunol. 2005;175:6169–6176. doi: 10.4049/jimmunol.175.9.6169. [DOI] [PubMed] [Google Scholar]
- 3.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10:909–915. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Srivastava PK. Therapeutic cancer vaccines. Curr Opin Immunol. 2006;18:201–205. doi: 10.1016/j.coi.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 5.Nestle FO, Farkas A, Conrad C. Dendritic-cell-based therapeutic vaccination against cancer. Curr Opin Immunol. 2005;17:163–169. doi: 10.1016/j.coi.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 6.Banchereau J, Palucka AK. Dendritic cells as therapeutic vaccines against cancer. Nat Rev Immunol. 2005;5:296–306. doi: 10.1038/nri1592. [DOI] [PubMed] [Google Scholar]
- 7.Kaliński P, Hilkens CM, Wierenga EA, Kapsenberg ML. T-cell priming by type-1 and type-2 polarized dendritic cells: the concept of a third signal. Immunol Today. 1999;20:561–567. doi: 10.1016/s0167-5699(99)01547-9. [DOI] [PubMed] [Google Scholar]
- 8.Kaliński P, Hilkens CM, Snijders A, Snijdewint FG, Kapsenberg ML. IL-12-deficient dendritic cells, generated in the presence of prostaglandin E2, promote type 2 cytokine production in maturing human naive T helper cells. J Immunol. 1997;159:28–35. [PubMed] [Google Scholar]
- 9.Spörri R, Reis e Sousa C. Inflammatory mediators are insufficient for full dendritic cell activation and promote expansion of CD4+ T cell populations lacking helper function. Nat Immunol. 2005;6:163–170. doi: 10.1038/ni1162. [DOI] [PubMed] [Google Scholar]
- 10.Langenkamp A, Messi M, Lanzavecchia A, Sallusto F. Kinetics of dendritic cell activation: impact on priming of TH1, TH2 and nonpolarized T cells. Nat Immunol. 2000;1:311–316. doi: 10.1038/79758. [DOI] [PubMed] [Google Scholar]
- 11.Curtsinger JM, Valenzuela JO, Agarwal P, Lins D, Mescher MF. Type I IFNs provide a third signal to CD8 T cells to stimulate clonal expansion and differentiation. J Immunol. 2005;174:4465–4469. doi: 10.4049/jimmunol.174.8.4465. [DOI] [PubMed] [Google Scholar]
- 12.Whitmire JK, Tan JT, Whitton JL. Interferon-γ acts directly on CD8+ T cells to increase their abundance during virus infection. J Exp Med. 2005;201:1053–1059. doi: 10.1084/jem.20041463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pearce EL, Shen H. Generation of CD8 T cell memory is regulated by IL-12. J Immunol. 2007;179:2074–2081. doi: 10.4049/jimmunol.179.4.2074. [DOI] [PubMed] [Google Scholar]
- 14.Mailliard RB, Wankowicz-Kalinska A, Cai Q, Wesa A, Hilkens CM, Kapsenberg ML, Kirkwood JM, Storkus WJ, Kalinski P. α-Type-1 polarized dendritic cells: a novel immunization tool with optimized CTL-inducing activity. Cancer Res. 2004;64:5934–5937. doi: 10.1158/0008-5472.CAN-04-1261. [DOI] [PubMed] [Google Scholar]
- 15.Jonuleit H, Kühn U, Müller G, Steinbrink K, Paragnik L, Schmitt E, Knop J, Enk AH. Pro-inflammatory cytokines and prostaglandins induce maturation of potent immunostimulatory dendritic cells under fetal calf serum-free conditions. Eur J Immunol. 1997;27:3135–3142. doi: 10.1002/eji.1830271209. [DOI] [PubMed] [Google Scholar]
- 16.Mailliard RB, Egawa S, Cai Q, Kalinska A, Bykovskaya SN, Lotze MT, Kapsenberg ML, Storkus WJ, Kalinski P. Complementary dendritic cell-activating function of CD8+ and CD4+ T cells: helper role of CD8+ T cells in the development of T helper type 1 responses. J Exp Med. 2002;195:473–483. doi: 10.1084/jem.20011662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Watchmaker PB, Urban JA, Berk E, Nakamura Y, Mailliard RB, Watkins SC, van Ham SM, Kalinski P. Memory CD8+ T cells protect dendritic cells from CTL killing. J Immunol. 2008;180:3857–3865. doi: 10.4049/jimmunol.180.6.3857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Friberg DD, Bryant JL, Whiteside TL. Measurements of natural killer (NK) activity and NK-cell quantification. Methods. 1996;9:316–326. doi: 10.1006/meth.1996.0037. [DOI] [PubMed] [Google Scholar]
- 19.Balkwill F, Charles KA, Mantovani A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell. 2005;7:211–217. doi: 10.1016/j.ccr.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 20.Clevers H. At the crossroads of inflammation and cancer. Cell. 2004;118:671–674. doi: 10.1016/j.cell.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 21.Phipps RP, Stein SH, Roper RL. A new view of prostaglandin E regulation of the immune response. Immunol Today. 1991;12:349–352. doi: 10.1016/0167-5699(91)90064-Z. [DOI] [PubMed] [Google Scholar]
- 22.Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. 2003;3:133–146. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- 23.Vieira PL, de Jong EC, Wierenga EA, Kapsenberg ML, Kaliński P. Development of Th1-inducing capacity in myeloid dendritic cells requires environmental instruction. J Immunol. 2000;164:4507–4512. doi: 10.4049/jimmunol.164.9.4507. [DOI] [PubMed] [Google Scholar]
- 24.Kaliński P, Schuitemaker JHN, Hilkens CMU, Wierenga EA, Kapsenberg ML. Final maturation of dendritic cells is associated with impaired responsiveness to IFN-γ and to bacterial IL-12 inducers: decreased ability of mature dendritic cells to produce IL-12 during the interaction with Th cells. J Immunol. 1999;162:3231–3236. [PubMed] [Google Scholar]
- 25.Berke G. The CTL’s kiss of death. Cell. 1995;81:9–12. doi: 10.1016/0092-8674(95)90365-8. [DOI] [PubMed] [Google Scholar]
- 26.Tsukishiro T, Donnenberg AD, Whiteside TL. Rapid turnover of the CD8+CD28− T-cell subset of effector cells in the circulation of patients with head and neck cancer. Cancer Immunol Immunother. 2003;52:599–607. doi: 10.1007/s00262-003-0395-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schluns KS, Kieper WC, Jameson SC, Lefrançois L. Interleukin-7 mediates the homeostasis of naive and memory CD8 T cells in vivo. Nat Immunol. 2000;1:426–432. doi: 10.1038/80868. [DOI] [PubMed] [Google Scholar]
- 28.Tan JT, Ernst B, Kieper WC, LeRoy E, Sprent J, Surh CD. Interleukin (IL)-15 and IL-7 jointly regulate homeostatic proliferation of memory phenotype CD8+ cells but are not required for memory phenotype CD4+ cells. J Exp Med. 2002;195:1523–1532. doi: 10.1084/jem.20020066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Campbell DJ, Kim CH, Butcher EC. Chemokines in the systemic organization of immunity. Immunol Rev. 2003;195:58–71. doi: 10.1034/j.1600-065x.2003.00067.x. [DOI] [PubMed] [Google Scholar]
- 30.Moser B, Loetscher P. Lymphocyte traffic control by chemokines. Nat Immunol. 2001;2:123–128. doi: 10.1038/84219. [DOI] [PubMed] [Google Scholar]
- 31.Sallusto F, Mackay CR, Lanzavecchia A. The role of chemokine receptors in primary, effector, and memory immune responses. Annu Rev Immunol. 2000;18:593–620. doi: 10.1146/annurev.immunol.18.1.593. [DOI] [PubMed] [Google Scholar]
- 32.Mantovani A, Allavena P, Sozzani S, Vecchi A, Locati M, Sica A. Chemokines in the recruitment and shaping of the leukocyte infiltrate of tumors. Semin Cancer Biol. 2004;14:155–160. doi: 10.1016/j.semcancer.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 33.Qin S, Rottman JB, Myers P, Kassam N, Weinblatt M, Loetscher M, Koch AE, Moser B, Mackay CR. The chemokine receptors CXCR3 and CCR5 mark subsets of T cells associated with certain inflammatory reactions. J Clin Invest. 1998;101:746–754. doi: 10.1172/JCI1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sallusto F, Lenig D, Förster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–712. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- 35.Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu Rev Immunol. 2004;22:745–763. doi: 10.1146/annurev.immunol.22.012703.104702. [DOI] [PubMed] [Google Scholar]
- 36.Kim CH, Nagata K, Butcher EC. Dendritic cells support sequential reprogramming of chemoattractant receptor profiles during naive to effector T cell differentiation. J Immunol. 2003;171:152–158. doi: 10.4049/jimmunol.171.1.152. [DOI] [PubMed] [Google Scholar]
- 37.Sallusto F, Lenig D, Mackay CR, Lanzavecchia A. Flexible programs of chemokine receptor expression on human polarized T helper 1 and 2 lymphocytes. J Exp Med. 1998;187:875–883. doi: 10.1084/jem.187.6.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gough M, Crittenden M, Thanarajasingam U, Sanchez-Perez L, Thompson J, Jevremovic D, Vile R. Gene therapy to manipulate effector T cell trafficking to tumors for immunotherapy. J Immunol. 2005;174:5766–5773. doi: 10.4049/jimmunol.174.9.5766. [DOI] [PubMed] [Google Scholar]
- 39.Ugurel S, Schrama D, Keller G, Schadendorf D, Bröcker EB, Houben R, Zapatka M, Fink W, Kaufman HL, Becker JC. Impact of the CCR5 gene polymorphism on the survival of metastatic melanoma patients receiving immunotherapy. Cancer Immunol Immunother. 2008;57:685–691. doi: 10.1007/s00262-007-0407-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wenzel J, Bekisch B, Uerlich M, Haller O, Bieber T, Tüting T. Type I interferon-associated recruitment of cytotoxic lymphocytes: a common mechanism in regressive melanocytic lesions. Am J Clin Pathol. 2005;124:37–48. doi: 10.1309/4EJ9KL7CGDENVVLE. [DOI] [PubMed] [Google Scholar]
- 41.Mullins IM, Slingluff CL, Lee JK, Garbee CF, Shu J, Anderson SG, Mayer ME, Knaus WA, Mullins DW. CXC chemokine receptor 3 expression by activated CD8+ T cells is associated with survival in melanoma patients with stage III disease. Cancer Res. 2004;64:7697–7701. doi: 10.1158/0008-5472.CAN-04-2059. [DOI] [PubMed] [Google Scholar]
- 42.Lee SW, Park Y, Yoo JK, Choi SY, Sung YC. Inhibition of TCR-induced CD8 T cell death by IL-12: regulation of Fas ligand and cellular FLIP expression and caspase activation by IL-12. J Immunol. 2003;170:2456–2460. doi: 10.4049/jimmunol.170.5.2456. [DOI] [PubMed] [Google Scholar]
- 43.Valenzuela JO, Hammerbeck CD, Mescher MF. Cutting edge: Bcl-3 up-regulation by signal 3 cytokine (IL-12) prolongs survival of antigen-activated CD8 T cells. J Immunol. 2005;174:600–604. doi: 10.4049/jimmunol.174.2.600. [DOI] [PubMed] [Google Scholar]
- 44.Mattei S, Colombo MP, Melani C, Silvani A, Parmiani G, Herlyn M. Expression of cytokine/growth factors and their receptors in human melanoma and melanocytes. Int J Cancer. 1994;56:853–857. doi: 10.1002/ijc.2910560617. [DOI] [PubMed] [Google Scholar]
- 45.Mrowietz U, Schwenk U, Maune S, Bartels J, Küpper M, Fichtner I, Schröder JM, Schadendorf D. The chemokine RANTES is secreted by human melanoma cells and is associated with enhanced tumour formation in nude mice. Br J Cancer. 1999;79:1025–1031. doi: 10.1038/sj.bjc.6690164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Payne AS, Cornelius LA. The role of chemokines in melanoma tumor growth and metastasis. J Invest Dermatol. 2002;118:915–922. doi: 10.1046/j.1523-1747.2002.01725.x. [DOI] [PubMed] [Google Scholar]
- 47.Kunz M, Toksoy A, Goebeler M, Engelhardt E, Bröcker E, Gillitzer R. Strong expression of the lymphoattractant C-X-C chemokine Mig is associated with heavy infiltration of T cells in human malignant melanoma. J Pathol. 1999;189:552–558. doi: 10.1002/(SICI)1096-9896(199912)189:4<552::AID-PATH469>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 48.Schadendorf D, Ugurel S, Schuler-Thurner B, Nestle FO, Enk A, Bröcker EB, Grabbe S, Rittgen W, Edler L, Sucker A, et al. DC study group of the DeCOG. Dacarbazine (DTIC) versus vaccination with autologous peptide-pulsed dendritic cells (DC) in first-line treatment of patients with metastatic melanoma: a randomized phase III trial of the DC study group of the DeCOG. Ann Oncol. 2006;17:563–570. doi: 10.1093/annonc/mdj138. [DOI] [PubMed] [Google Scholar]