

Abstract
We report a previously unknown recognition motif between the α-face of the steroid hydrocarbon backbone and π-electron-rich aromatic substrates. Our study is based on a systematic and comparative analysis of the solid-state complexation of four steroids with 24 aromatic molecules. By using the solid state as a medium for complexation, we circumvent solubility and solvent competition problems that are inherent to the liquid phase. Characterization is performed using powder and single crystal X-ray diffraction, infrared solid-state spectroscopy and is complemented by a comprehensive cocrystal structure prediction methodology that surpasses earlier computational approaches in terms of realism and complexity. Our combined experimental and theoretical approach reveals that the α⋯π stacking is of electrostatic origin and is highly dependent on the steroid backbone’s unsaturated and conjugated character. We demonstrate that the α⋯π stacking interaction can drive the assembly of molecules, in particular progesterone, into solid-state complexes without the need for additional strong interactions. It results in a marked difference in the solid-state complexation propensities of different steroids with aromatic molecules, suggesting a strong dependence of the steroid-binding affinity and even physicochemical properties on the steroid’s A-ring structure. Hence, the hydrocarbon part of the steroid is a potentially important variable in structure-activity relationships for establishing the binding and signaling properties of steroids, and in the manufacture of pharmaceutical cocrystals.
Keywords: steroids, molecular recognition, mechanosynthesis, crystal structure prediction
A wide variety of tools, including site-directed mutagenesis (1), binding and inhibition screening (2–4), computational modeling (5), and protein crystallography (6, 7) are commonly used in studying the interaction of biologically important molecules, such as steroids, with their respective receptor binding domains (8). Molecular recognition can also be effectively probed using the considerably simpler and inexpensive methodology of forming crystalline molecular complexes (9) (multicomponent crystals, also known as cocrystals). Similarly to binding on synthetic model receptors (10, 11), solid-state complexation with small molecules (cocrystallization) offers the possibility to separately screen and deconvolute a far larger space of molecular recognition motifs (12) that collectively account for the biological activity. However, formation of solid-state complexes from solution is not a reliable measure of molecular affinity due to the vexatious problems of solubility and solvent competition. Mechanosynthesis, in the form of liquid-assisted grinding (LAG) (13), avoids these adverse effects and offers the possibility to systematically explore molecular recognition in the solid state (14, 15).
The rationalization of statistical data from structure-activity binding (16) or cocrystallization studies (17) is often based on qualitative and intuitive arguments and an established inventory of molecular association patterns. We now demonstrate how mechanosynthesis, combined with modern methods of solid-state analysis and crystal structure prediction, can be used to identify novel intermolecular interactions of biologically important targets. We have selected steroids as model systems due to their prominent role in life sciences and their well-established use as pharmacophores. Whereas the formation of steroid hydrates and solvates is well-established (18) and cocrystallization of steroids has been proposed as a means to improve properties of pharmaceuticals (19–21), solid-state complexation of steroids with aromatic molecules has never been systematically studied. The exception is the work of Eger and Norton (22) who employed cocrystallization with 4-bromophenol to determine the steroid backbone stereochemistry using the heavy atom method (18, 22, 23). They also reported solid-state complexes of androsta-1,4-diene-3,17-dione and androst-4-ene-3,17-dione with naphthalene (22, 24). These complexes, although never structurally characterized, indicate that steroids can form cocrystals even in the absence of hydrogen bonds, suggesting a potentially complex recognition mechanism.
Our extensive investigation using mechanochemical and solution screening, single crystal and powder X-ray diffraction (PXRD) and Fourier-transform attenuated total reflectance (FTIR-ATR) spectroscopy, demonstrates a previously not described recognition mode of the steroid α-face by π-electron-rich systems. By using the four model steroids progesterone (pro, a progestagen), pregnenolone (pre, a prohormone), and two estrogens, β-estradiol (bes) and estrone (est), we show that α⋯π recognition, reflected in the diverse propensity to form solid-state complexes with 24 aromatic molecules (Scheme 1), is strongly dependent on the steroid backbone chemistry (18). The model steroids were selected with the intention of examining different types of A-ring: nonsaturated, saturated, and aromatic. The use of pharmaceutical excipients, such as xinafoic acid (10) and gentisic acid (15), as complexation partners illustrates how the structure of the A-ring could have wide implications in cocrystal-based drug discovery and manufacture (25).
Scheme 1.

(A) Model steroids pro, pre, bes, and est and (B) the library of cocrystallization partners.
Results
Mechanochemical Screening.
The mechanochemical screen revealed significant differences in molecular association of the four model steroids (Table 1). Combined PXRD and FTIR-ATR solid-state characterization (SI Appendix, Sections S1, S2, and S3) showed that est formed a cocrystal with only the electron deficient octafluoronaphthalene (20), while, additionally, bes also formed solid-state complexes with phenanthrene (21) and pyrene (22). The most promiscuous steroid in terms of solid-state complexation was, by a large margin, pro that formed crystalline complexes with 19 arenes. The structurally similar pre formed four cocrystals. The overall shape of the model steroids is very similar* (26) and hence shape complementarity with the potential cocrystal formers is unlikely to be the discriminating factor. The intuitive differences in the A- and D-ring hydrogen-bonding functionalities are a more salient reason, but fail to fully explain the observed trend of solid-state complexation. The pro > pre > bes propensity for solid-state complexation suggests that keto groups may be more effective in facilitating hydrogen bonding than hydroxyl groups. However, this structure-recognition relationship does not explain the persistent pro cocrystallization with all fused-ring aromatic hydrocarbons; i.e., complexation in the absence of hydrogen bond donors. Moreover, the mechanochemical screen with est, that contains a keto instead of a hydroxyl group at position 17, did not increase the tendency for complexation compared to bes. Overall, the cocrystallization outcome does not obviously correlate with the purportedly different functionalities at positions 3 and 17.
Table 1.
Results of solid-state screening for complex formation*
| Steroid | ||||
| Complex former | pro | pre | bes | est |
| 1, bromobenzene | − | − | − | − |
| 2, 4-bromophenol | + | + | − | − |
| 3, phloroglucinol | + | − | − | − |
| 4, 1-naphthol | + | + | − | − |
| 5, 2-naphthol | + | + | − | − |
| 6, 1-naphthoic acid | + | − | − | − |
| 7, 2-naphthoic acid | + | − | − | − |
| 8, 2-hydroxy-1-naphthoic acid | + | − | − | − |
| 9, 3-hydroxy-2-naphthoic acid | + | − | − | − |
| 10, xinafoic acid | + | − | − | − |
| 11, decahydro-2-naphthol | − | − | − | − |
| 12, pyrenol | + | − | − | − |
| 13, 9-phenanthrol | + | − | − | − |
| 14, 2,7-dihydroxynaphthalene | + | + | − | − |
| 15, gentisic acid | + | − | − | − |
| 16, phthalimide | + | − | − | − |
| 17, theophylline | − | − | − | − |
| 18, theobromine | − | − | − | − |
| 19, naphthalene | + | − | − | − |
| 20, octafluoronaphthalene | − | − | + | + |
| 21, phenanthrene | + | − | + | − |
| 22, pyrene | + | − | + | − |
| 23, perylene | + | − | − | − |
| 24, benzocoronene | + | − | − | − |
*Formation of a solid-state complex is denoted by “+,” and was determined by comparison of PXRD patterns and FTIR-ATR spectra of the products and starting materials. The PXRD patterns of mechanosynthesis products were also compared to calculated patterns of known polymorphs and solvates of starting materials.
Crystal Structure Analysis.
Pro forms solid-state complexes with naphthalene (19), phenanthrene (21), pyrene (22), perylene (23), and benzocoronene (24), that all lack hydrogen bond donors. To investigate the interaction driving solid-state complexation we pursued single crystal X-ray diffraction structure analysis. Suitable single crystals were grown from solution for the cocrystals of pro with 4-bromophenol (2), pyrenol (12), 9-phenanthrol (13), 2,7-dihydroxynaphthalene (14), gentisic acid (15), phenanthrene (21), and pyrene (22). The cocrystals of pre with 2-naphthol (5) and of bes with pyrene (22) were also structurally characterized using single crystal X-ray diffraction, whereas the structure of (pre).(2) was determined from PXRD data (SI Appendix, Section S1). In all cases, except (pro)·(12), the PXRD pattern simulated for the determined crystal structure was identical to the one measured for the grinding product. Mechanosynthesis indicated the formation of two different solid-state pro∶12 complexes with 1∶1 and 2∶1 stoichiometric ratios, whereas only the 2∶1 stoichiometry complex was obtained from solution (27).
Key to understanding the interaction that drives the complexation of pro with aromatic molecules are the (pro)3·(21) and (pro)2·(22) structures. Both complexes are trimers with the arene “sandwiched” between the α-faces of two pro molecules (Fig. 1A). The same type of stacking persists in the presence of O-H⋯O hydrogen bonds, as manifested by the (pro)·(12) solid-state complex. Similarly, in the complex of pro with the diol 14, O-H⋯O hydrogen bonds are accompanied by the α⋯π stacking. The complex of pro with 13, a hydroxylated derivative of 21, also shows α⋯π stacking, but with only one side of the arene participating. Such an α⋯π dimer is repeated in the (pro)·(2) complex and the (pro)·(15) pharmaceutical cocrystal (20). Crystallographic data for all structures determined in this work have been deposited with the Cambridge Structural Database, deposition codes CCDC 753857–753869.
Fig. 1.

Dominant intermolecular interactions in cocrystals of (A) pro, (B) pre, and (C) bes. For clarity, the framework of bes molecules in (C) is shown in wireframe and molecules of 22 in space-filling representation.
Electrostatic Nature of the α⋯π Interaction.
The strength of the α⋯π interaction should differ for the model steroids, given their contrasting arene complexation propensities. The distances between the carbon atoms of the pro α-face and the aromatic carbon atoms in α⋯π dimers and trimers vary between 3.8 and 4.2 Å (Fig. 1 A and B). Such separations are at the upper limit of C-H⋯π hydrogen bond lengths that are typically shorter than 3.8 Å (28). Consequently, the stabilization gained by α⋯π stacking originates from the overall complementarity of positive charge over the steroid α-face and the negative charge of the arene. The contribution of α⋯π interaction to the overall stability of the cocrystal must depend on the degree of unsaturation of the steroid backbone. We expect that reducing the π-electron density of the arene through electron-withdrawing substituents should weaken the α⋯π interaction. This argument is substantiated by the outcome of complexation experiments of pro with naphthalene (19) and octafluoronaphthalene (20). Whereas the complex with 19 forms readily, there was no evidence of complexation between pro and 20.
The nature of α⋯π interaction is elucidated by the electrostatic surface potentials (ESP, Fig. 2A) of pro, pre, and bes α-faces, modeled from the distributed multipole expansion (29) up to hexadecapole of the B3LYP/6-31G(d,p) charge density on the molecular surface defined by twice the van der Waals radii. The ESPs drawn using ORIENT (30) reveal qualitative differences in the electron distribution of the model steroids. The area of positive charge is most pronounced over the pro backbone, consistent with the electrostatic stabilization of pro cocrystals with π-electron-rich molecules.
Fig. 2.

(A) Calculated B3LYP/6-31G(d,p) ESPs for representative, low-energy gas-phase conformations (< 1 kJ mol-1 less stable than global conformational minimum, for further information see SI Appendix, Section S4) of pro, pre, bes, and est on 2 × vdW surface; (B) crystal structure of the (pro).(13) complex displaying an α⋯π dimer and a hydrogen-bonded neighboring pro molecule (blue); (C) overlap of B3LYP/6-31G(d,p) ESPs (in V) on the 2 × vdW surface of the pro α-face (Upper) and 13 (ball-and-stick) for the conformations, relative position and orientation of the two molecules in the experimental crystal structure; ESP on both sides of 13 is shown for comparison (Lower), with the side facing pro labeled; (D) comparison of PXRD patterns measured for the grinding product (Upper), simulated for the predicted global minimum structure (Center) and simulated for the experimental structure (pro).(13) (Lower); (E) and (F) lattice energy vs. density landscapes for (pro).(13) and 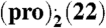 . The open and filled squares correspond to rigid-body and flexible-molecule lattice energy minimizations, respectively. The horizontal black and red lines denote the sum of the lattice energies of the least and most stable polymorphs of the components, respectively, obtained with the same computational model that was used to minimize the experimental crystal structures (solid red circle). For
. The open and filled squares correspond to rigid-body and flexible-molecule lattice energy minimizations, respectively. The horizontal black and red lines denote the sum of the lattice energies of the least and most stable polymorphs of the components, respectively, obtained with the same computational model that was used to minimize the experimental crystal structures (solid red circle). For 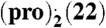 the search structure (shown with a solid blue circle) that resembled most closely the minimized experimental solid-state complex differs by 0.49 Å in the 20 molecule coordination sphere and is 2 kJ mol-1 less stable.
the search structure (shown with a solid blue circle) that resembled most closely the minimized experimental solid-state complex differs by 0.49 Å in the 20 molecule coordination sphere and is 2 kJ mol-1 less stable.
Relocation of the C = C bond from the A- to the B-ring going from pro to pre introduces a region of negative charge above the central part of the molecule, and shrinks the area of positive α-face potential. Additionally, the lack of conjugation with the electron-withdrawing keto group makes the C = C bond π-electron density of pre more visible. Consequently, solid-state complexation with arenes is thermodynamically less favorable. The structure of the solid-state complex between pre and 2-naphthol (5) is consistent with this rationalization (Fig. 1B). In this complex, α⋯π stacking is absent and stabilization is attained by multiple C-H⋯π hydrogen bonds between pre and neighboring molecules of 5. Aromatization of the A-ring in bes and est further reduces the area and the intensity of positive potential, which suppresses α⋯π stacking. The cocrystals (bes)·(22) and (bes)·(21) stand out as the only solid-state complexes of an estrogen with an electron-rich aromatic hydrocarbon. Structure determination reveals that (bes)·(22) is a lattice inclusion compound, resulting from a serendipitous fit of molecular shapes, with tapes of 22 filling square-grid channels formed by hydrogen-bonded bes molecules (Fig. 1C). The complementarity of ESPs also explains the switching of the pro-arene-pro trimer to the simpler arene-pro dimer motif on changing the arene from 21 to 13. In (pro)·(13), hydrogen bonding to a pro 3-keto group, shown in Fig. 2B, twists the OH group of 13 out of the aromatic plane and differentiates the two faces of 13 in terms of electrostatic potential. Fig. 2C illustrates that the side of 13 facing pro exhibits more negative electrostatic potential and overlaps almost perfectly with the most positive region of pro α-face, so as to maximize the α⋯π stacking stabilization.
Crystal Structure Prediction (CSP).
The inference of the importance of the α⋯π interaction drawn from a limited number of single crystal X-ray structures may be flawed due to undetected polymorphism in our screen. To alleviate this potential drawback, we conducted structure prediction calculations of solid-state complexes to examine whether there are alternative but thermodynamically competitive intermolecular motifs besides the α⋯π stacking. We selected the systems (pro).(13) and 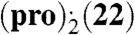 , that exhibit α⋯π dimer and trimer motifs in the solid state, respectively. We improved our methodology, that previously led to the successful blind prediction of the racemic pro crystal structure (31), to ensure an extensive structure search suitable for highly complex asymmetric units comprising large, flexible molecules (SI Appendix, Section S5).
, that exhibit α⋯π dimer and trimer motifs in the solid state, respectively. We improved our methodology, that previously led to the successful blind prediction of the racemic pro crystal structure (31), to ensure an extensive structure search suitable for highly complex asymmetric units comprising large, flexible molecules (SI Appendix, Section S5).
The (pro).(13) lattice energy landscape showed a variety of packing motifs, with the hydroxyl donor bonded to either pro carbonyl group with equal frequency. The three most stable predicted structures are additionally stabilized by α⋯π stacking. The experimentally observed solid-state complex corresponds to the densest and most stable predicted structure (Fig. 2 D and E). This structure is also the only one with a small thermodynamic advantage over the most stable predicted polymorphs of pro and 13 crystallizing independently. The most stable predicted cocrystal structure that lacks α⋯π stacking is ca. 5 kJ mol-1 less stable. Hence, the predicted (pro)·(13) lattice energy landscape clearly shows that α⋯π stacking not only does not disrupt close packing and hydrogen bonding, but also provides the extra stabilization necessary for solid-state complexation. To the best of our knowledge, the generated lattice energy landscape for 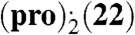 is the most demanding crystal structure prediction reported to date in terms of molecular size and asymmetric unit complexity. The lattice energy landscape shows limited packing diversity with most low-energy structures exhibiting clear α⋯π stacking on both sides of 22 (SI Appendix). The global minimum structure is marginally less favorable to the pure component crystals (Fig. 2F). However, the lattice energy differences involved are small and likely to change sign depending on the model for the intermolecular forces and entropy effects (32). Many of the predicted structures differ mainly in the in-plane rotation of 22 within the (pro)2·(22) sandwich. Such rotations are likely to be labile and to correspond to low-frequency librations contributing to entropic stabilization. This view is supported by the thermal ellipsoids of 22 in the experimental solid-state complex, which become laterally elongated toward the periphery of the molecule (SI Appendix). Hence, our static lattice energy results are informative in providing all thermodynamically plausible molecular arrangements in the solid-state complex and establishing the dominance of α⋯π stacking in crystalline complexes of pro with both 13 and 22.
is the most demanding crystal structure prediction reported to date in terms of molecular size and asymmetric unit complexity. The lattice energy landscape shows limited packing diversity with most low-energy structures exhibiting clear α⋯π stacking on both sides of 22 (SI Appendix). The global minimum structure is marginally less favorable to the pure component crystals (Fig. 2F). However, the lattice energy differences involved are small and likely to change sign depending on the model for the intermolecular forces and entropy effects (32). Many of the predicted structures differ mainly in the in-plane rotation of 22 within the (pro)2·(22) sandwich. Such rotations are likely to be labile and to correspond to low-frequency librations contributing to entropic stabilization. This view is supported by the thermal ellipsoids of 22 in the experimental solid-state complex, which become laterally elongated toward the periphery of the molecule (SI Appendix). Hence, our static lattice energy results are informative in providing all thermodynamically plausible molecular arrangements in the solid-state complex and establishing the dominance of α⋯π stacking in crystalline complexes of pro with both 13 and 22.
Discussion
Increasing the level of unsaturation and aromaticity of the steroid A-ring changes the character of the recognition with arenes from α⋯π stacking to C-H⋯π hydrogen bonding in (pre)·(5) and, presumably, to π⋯π stacking with electron deficient aromatic molecules like octafluoronaphthalene in (bes)·(20) and (est)·(20). The Cambridge Structural Database (CSD, version 5.30, November 2008, update 1) contains a limited number of steroid cocrystals with π-electron-rich molecules (33,34) to confirm or disprove our hypothesis. Of the cocrystals for which crystallographic atomic positions are reported, the two solid-state complexes of androsta-1,4-diene-3,17-dione, an androgen with a strongly positive α-face region (SI Appendix), with naphthols 4 and 5 exhibit clear α⋯π stacking (Fig. 3 A and B) (34). As an additional test of α⋯π generality, we have prepared and structurally characterized the solid-state complex of 5 with androsterone, another member of the androgen family. The ESP of androsterone is dependent on the rotation of the 3-OH group (SI Appendix) that determines the position of the oxygen lone pairs. In the complex with 5 (Fig. 3C) the rotation of the hydroxyl group deviates from its value in the B3LPYP/6-31G(d,p) optimized molecule by 35°, in that way maximizing the positive region of the α-face that stacks with the π-electron density of 5.
Fig. 3.

The α⋯π stacking in solid-state complexes of (A) androsta-1,4-diene-3,17-dione with 4 (33), (B) androsta-1,4-diene-3,17-dione with 5 (34) and (C) androsterone with 5.
The crystal structures of the steroid-binding domains of pro (35) and estrogen receptors (7) do not contradict our analysis, within the limitations of resolution and dynamic character of protein crystal structures. The structure of the ligand-binding domain of the pro receptor shows that only the A-ring keto group of pro forms a clear hydrogen bond with adjacent glutamine and arginine residues and a water molecule (35). The D-ring acetyl group lies in the vicinity of a threonine hydroxyl but not at sufficiently short distance for hydrogen bonding (36, 37). Such counterintuitive nonutilization of a keto acceptor in hydrogen bonding suggests that the steroid backbone should be, at least partially, responsible for recognition and may even contribute to binding specificity. Indeed, binding of pro to a1-acid glycoprotein occurs at the hydrophobic part of the protein and involves one π-electron-rich tryptophane residue (38). Androstenedione is similar to pro in exhibiting a strongly positive, although more localized, region of positive α-face potential (SI Appendix). When bound to the human aromatase receptor (6), the α-face of androstenedione is snugly enclosed by two π-electron-rich phenylalanine and one tryptophane residue. In the estrogen receptor (7), the A-ring of bes is once more the backbone element in close contact with the cavity. The aromatic residue in the proximity of the α-face does not exhibit α⋯π stacking, but instead forms a C-H⋯π interaction with the bes A-ring (SI Appendix).
In summary, efficient solid-state methodologies to construct molecular adducts, complemented by modern solid-state characterization and modeling techniques, provide a simple and viable means to decipher the molecular recognition of biomolecules. This is achieved with the added benefit of producing solids of potentially pharmaceutical significance. The pervasive α⋯π stacking of pro with aromatic molecules is present in the majority of experimental and also theoretically predicted pro solid-state complexes. On the other hand, from the solid-state complexation propensity and molecular modeling we infer that the energetic stabilization due to α⋯π stacking diminishes and eventually vanishes with increasing unsaturation and aromaticity of the steroid backbone. This is observed by contrasting pro to pre and finally bes and est. This previously undocumented dependence of steroid recognition on the backbone structure suggests the degree of saturation of the A-ring as a potentially important variable that should not be overlooked in structure-based and structure-activity modeling.
Materials and Methods
Mechanochemical Screening.
Mechanochemical screening was conducted (13) by liquid-assisted grinding of a 1∶1 stoichiometric mixture (200 mg) of the model steroid and the potential complexation partner in the presence of a small amount of liquid (50 μL nitromethane), corresponding to the η factor (39) of 0.25 μL mg-1. Grinding was performed for 20 min in stainless steel cylinders of 10 mL volume, using two stainless steel grinding balls of 7 mm diameter. The experiments were performed using a Retsch MM200 grinder mill operating at 30 Hz. The samples after LAG were left to dry in air and were subsequently analyzed using PXRD and FTIR-ATR spectroscopy. Further details of experimental procedures and instrumentation are provided in the SI Appendix.
Computational Methodology.
Given that naturally occurring steroids are generally chiral, the CSP search was limited to the most frequently observed enantiomorphous space groups P1, P21, C2, P212121, and P21212. The search was performed using Crystal Predictor (40) with the optimized B3LYP/6-31G(d,p) conformations held rigid. For 13 we only used the low-energy conformation that was 9 kJ mol-1 less stable than the alternative configuration of the hydroxyl proton. The intermolecular forces were modeled with CHELPG (41) atomic charges fitted to the B3LYP/6-31G(d,p) electrostatic potential and an empirical exp-6 repulsion-dispersion model with C, N, O, H(-C) and H(-O) parameters obtained from Coombes et al. (42). The search for solid-state complexes of pro and 22 included both 1∶1 (results in the SI Appendix) and 2∶1 stoichiometries. The five thousand most stable, distinct crystal structures from each solid-state complex search were minimized using DMACRYS (43) with the same computational model, apart from the intermolecular electrostatic interactions that were modeled using atomic multipoles up to hexadecapole. For consistency with the ESP calculations, the multipole moments were derived from a distributed multipole analysis (29) of the B3LYP/6-31G(d,p) isolated-molecule charge density. The minimised crystal structures were clustered by comparing their simulated X-ray powder diffraction patterns and molecular coordination spheres (44) and the 20 most stable, distinct crystal structures reminimized using Crystal Optimizer (45), a substantially revised version of DMAflex (46) to account for the effect of molecular flexibility on the relative lattice energy of the predicted crystal structures. This procedure simultaneously minimized the cell angles, cell lengths, the position and orientation of the molecules in the asymmetric unit and all torsion angles with the exception of torsions defining methylene and aromatic hydrogen atoms. For pro and 13 we also included the C-C-C bond angle of the exocyclic carbonyl chain and H-O-C hydroxyl hydrogen respectively. The conformational energy in the course of lattice energy minimization was modeled using a series of Taylor expansions of second order, with the intramolecular energy and its first and second gradients with respect to the intramolecular degrees of freedom computed at the B3LYP/6-31G(d,p) level of theory. Each Taylor expansion was considered valid for up to 8° and 5° change in torsion and bond angles respectively, and repeated for larger conformational variations. The atomic multipole moments were rotated with the local environment of the atoms for up to 5° and 3° change in torsion and bond angles respectively, beyond which the B3LYP/6-31G(d,p) isolated-molecule charge density and distributed multipole analysis calculations were repeated. The same crystal structure prediction methodology was also applied to pure solids pro, 13 and 22, discussed in the SI Appendix, Section S5.
Calculations were performed on the High Performance Computing Cluster of Imperial College (www.imperial.ac.uk/ict/services/teachingandresearchservices/highperformancecomputing). The computed low-energy crystal structures are stored on the STFC e-Science Centre data portal and are available from P.G.K. (p.karamertzanis@imperial.ac.uk) on request.
Supplementary Material
Acknowledgments.
We gratefully acknowledge Professor William Jones, Professor Sally Price, Professor Douglas Covey, Dr. Petra Bombicz, and Mr. Nizar Issa for useful discussions. We acknowledge Dr. John Davies for collecting single crystal X-ray diffraction data. We acknowledge the Herchel Smith fund for a research fellowship (T.F.). Funding to the Molecular Systems Engineering group from the Engineering and Physical Sciences Research Council (EPSRC) of the United Kingdom (EP/E016340) is gratefully acknowledged (P.G.K.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. J.L.A. is a guest editor invited by the Editorial Board.
Data deposition: All crystallographic data have been deposited within the Cambridge Structural Database under deposition codes CCDC 753857–753869.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.0915142107/-/DCSupplemental.
*The comparison of molecular shapes for pro, pre, and bes using the recently introduced molecular shape descriptors (26) indicated that the propensity the model steroids towards cocrystallization should be very similar.
References
- 1.Zhuo D, Pompon D, Chen S. Structure-function studies of human aromatase by site-directed mutagenesis: Kinetic properties of mutants Pro-308 → Phe, Tyr-361 → Phe, Tyr-361 → Leu and Phe-406 → Arg. Proc Natl Acad Sci USA. 1991;88:410–414. doi: 10.1073/pnas.88.2.410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pakhomova S, Buck J, Newcomer ME. The structure of the unique sulfotranferase retinol dehydratase with product and inhibitors provide insight into enzyme mechanism and inhibition. Protein Sci. 2005;14:176–182. doi: 10.1110/ps.041061105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Katzenellenbogen JA, Muthyala R, Katzenellenbogen BS. Nature of the ligand-binding pocket of estrogen receptor α and β: The search for subtype-selective ligands and implications for the prediction of estrogenic activity. Pure Appl Chem. 2003;75:2397–2403. [Google Scholar]
- 4.Covey DF. ent-Steroids: Novel tools for studies of signaling pathways. Steroids. 2009;74:577–585. doi: 10.1016/j.steroids.2008.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morrill GA, Kostellow AB, Askari A. Progesterone binding to the α1-subunit of the Na/K-ATPase on the cell surface: Insights from computational modelling. Steroids. 2008;73:27–40. doi: 10.1016/j.steroids.2007.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ghosh D, Griswold J, Erman M, Pangborn W. Structural basis for androgen specificity and oestrogen synthesis in human aromatase. Nature. 2009;457:219–224. doi: 10.1038/nature07614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brzozowski AM, et al. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature. 1997;389:753–758. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- 8.Anstead GM, Carlson KE, Katzenellenbogen JA. The estradiol pharmacophore: Ligand structure-estrogen receptor binding affinity relationships and a model for the receptor binding site. Steroids. 1997;62:268–303. doi: 10.1016/s0039-128x(96)00242-5. [DOI] [PubMed] [Google Scholar]
- 9.Nguyen KL, Friščić T, Day GM, Gladden LF, Jones W. Terahertz time-domain spectroscopy and the quantitative monitoring of mechanochemical cocrystal formation. Nat Mater. 2007;6:206–209. doi: 10.1038/nmat1848. [DOI] [PubMed] [Google Scholar]
- 10.Cacciarini M, Azov VA, Seiler P, Diederich F. Selective steroid recognition by a partially bridged resorcin[4]arene cavitand. Chem Commun. 2005:5269–5271. doi: 10.1039/b509990k. [DOI] [PubMed] [Google Scholar]
- 11.Kelly TR, Zhao C, Bridger GJ. A bisubstrate reaction template. J Am Chem Soc. 1989;111:3744–3745. [Google Scholar]
- 12.Auffinger P, Hays FA, Westhof E, Ho PS. Halogen bonds in biological molecules. Proc Nat Acad Sci USA. 2004;101:16789–16794. doi: 10.1073/pnas.0407607101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Friščić T, Trask AV, Jones W, Motherwell WDS. Screening for inclusion compounds and systematic construction of three-component solids via liquid-assisted grinding. Angew Chem Int Edit. 2006;45:7546–7550. doi: 10.1002/anie.200603235. [DOI] [PubMed] [Google Scholar]
- 14.Etter MC, Reutzel SM, Choo CG. Self-organization of adenine and thymine in the solid state. J Am Chem Soc. 1993;115:4411–4412. [Google Scholar]
- 15.Friščić T, Trask AV, Jones W, Motherwell WDS. Guest-directed assembly of caffeine and succinic acid into topologically different heteromolecular host networks upon grinding. Cryst Growth Des. 2008;8:1605–1609. [Google Scholar]
- 16.Goldstein RA, Katzenellenbogen JA, Luthey-Schulten ZA, Seielstad DA, Wolynes PG. Three-dimensional model for the hormone binding domains of steroid receptors. Proc Natl Acad Sci USA. 1993;90:9949–9953. doi: 10.1073/pnas.90.21.9949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aakeröy CB, Beatty AM, Hilfrich BA. “Total synthesis” supramolecular style: Design and hydrogen-bond-directed assembly of ternary supermolecules. Angew Chem Int Edit. 2001;40:3240–3242. doi: 10.1002/1521-3773(20010903)40:17<3240::AID-ANIE3240>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 18.Duax WL, Weeks CM, Rohrer DC. Crystal structures of steroids. Top Stereochem. 1976;9:271–383. [Google Scholar]
- 19.Takata N, Shiraki K, Takano R, Hayashi Y, Terada K. Cocrystal screening of stanolone and mestanolone using slurry crystallization. Cryst Growth Des. 2008;8:3032–3037. [Google Scholar]
- 20.Vishweshwar P, McMahon JA, Bis JA, Zaworotko MJ. Pharmaceutical co-crystals. J Pharm Sci. 2006;95:499–516. doi: 10.1002/jps.20578. [DOI] [PubMed] [Google Scholar]
- 21.Karki S, et al. Improving mechanical properties of crystalline solids by cocrystal formation: New compressible forms of paracetamol. Adv Mater. 2009;21:3905–3909. [Google Scholar]
- 22.Eger C, Norton DA. Androgenic steroid complexes with p-Bromophenol. Nature. 1965;208:997–999. doi: 10.1038/208997a0. [DOI] [PubMed] [Google Scholar]
- 23.Bhatt PM, Desiraju GR. Co-crystal formation and the determination of absolute configuration. CrystEngComm. 2008;10:1747–1749. [Google Scholar]
- 24.Kádárné PJ. Néhány szterin-molekulavegyület vizsgálata. Magy Kem Foly. 1964;70:325–327. [Google Scholar]
- 25.Trask AV. An overview of pharmaceutical cocrystals as intellectual property. Mol Pharm. 2007;4:301–309. doi: 10.1021/mp070001z. [DOI] [PubMed] [Google Scholar]
- 26.Fábián L. Cambridge Structural Database analysis of molecular complementarity in cocrystals. Cryst Growth Des. 2009;9:1436–1443. [Google Scholar]
- 27.Trask AV, van de Streek J, Motherwell WDS, Jones W. Achieving polymorphic and stoichiometric diversity in cocrystal formation: Importance of solid-state grinding, powder X-ray structure determination, and seeding. Cryst Growth Des. 2005;5:2233–2241. [Google Scholar]
- 28.Nishio M. CH/π hydrogen bonds in crystals. CrystEngComm. 2004;6:130–158. [Google Scholar]
- 29.Stone AJ. Distributed multipole analysis: Stability for large basis sets. J Chem Theory Comput. 2005;1:1128–1132. doi: 10.1021/ct050190+. [DOI] [PubMed] [Google Scholar]
- 30.Stone AJ, et al. Orient: A program for studying interactions between molecules, version 4.5 University of Cambridge; version 4.6 obtainable from: http://www-stone.ch.cam.ac.uk/programs.html
- 31.Lancaster RW, et al. Racemic progesterone: Predicted in silico and produced in the solid state. Chem Commun. 2006:4921–4923. doi: 10.1039/b611599c. [DOI] [PubMed] [Google Scholar]
- 32.Price SL. Computed crystal energy landscapes for understanding and predicting organic crystal structures and polymorphism. Acc Chem Res. 2009;42:117–126. doi: 10.1021/ar800147t. [DOI] [PubMed] [Google Scholar]
- 33.Duax WL, Griffin JF, Rohrer DC. Conformation of progesterone side chain: Conflict between X-ray data and force-field calculations. J Am Chem Soc. 1981;103:6705–6712. [Google Scholar]
- 34.Böcskei Z, Simon K, Ambrus G, Ilköy É. On the isostructural molecular compound formation of a steroid with α- and β-naphthols. Acta Crystallogr. 1995;C51:1319–1322. [Google Scholar]
- 35.Williams SP, Sigler PB. Atomic structure of progesterone complexed with its receptor. Nature. 1998;393:392–396. doi: 10.1038/30775. [DOI] [PubMed] [Google Scholar]
- 36.Ellmann S, et al. Estrogen and progesterone receptors: From molecular structures to clinical targets. Cell Mol Life Sci. 2009;66:2405–2426. doi: 10.1007/s00018-009-0017-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matias PM, et al. Structural evidence for ligand specificity in the binding domain of the human androgen receptor. J Biol Chem. 2000;275:26164–26171. doi: 10.1074/jbc.M004571200. [DOI] [PubMed] [Google Scholar]
- 38.Albani JR. Progesterone binding to the tryptophan residues of human α1-acid glycoprotein. Carbohyd Res. 2006;341:2557–2564. doi: 10.1016/j.carres.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 39.Friščić T, Childs SL, Rizvi SAA, Jones W. The role of solvent in mechanochemical and sonochemical cocrystal formation: A solubility-based approach for predicting cocrystallisation outcome. CrystEngComm. 2009;11:418–426. [Google Scholar]
- 40.Karamertzanis PG, Pantelides CC. Ab initio crystal structure prediction—I Rigid molecules. J Comput Chem. 2005;26:304–324. doi: 10.1002/jcc.20165. [DOI] [PubMed] [Google Scholar]
- 41.Breneman CM, Wiberg KB. Determining atom-centered monopoles from molecular electrostatic potentials. The need for high sampling density in formamide conformation analysis. J Comp Chem. 1990;11:361–373. [Google Scholar]
- 42.Coombes DS, Price SL, Willock DJ, Leslie M. Role of electrostatic interactions in determining crystal structures of polar organic molecules: A distributed multipole study. J Phys Chem. 1996;100:7352–7360. [Google Scholar]
- 43.Welch GWA, Karamertzanis PG, Price SL, Leslie M. DMACRYS v1.05, is a substantial revision of DMAREL http://www.chem.ucl.ac.uk/cposs/dmacrys/2008.
- 44.Chisholm JA, Motherwell S. COMPACK: A program for identifying crystal structure similarity using distances. J Appl Crystallogr. 2005;38:228–231. [Google Scholar]
- 45.Kazantsev AV, Karamertzanis PG, Pantelides CC, Adjiman CS. CrystalOptimizer: An Efficient Algorithm for Lattice Energy Minimisation of Organic Crystals Using Isolated-Molecule Quantum Mechanical Calculations. In: Adjiman CS, Galindo A, editors. Process Systems Engineering Volume 6: Molecular Systems Engineering. Weinheim: Wiley VCH; 2010. [Google Scholar]
- 46.Karamertzanis PG, Price SL. Energy minimization of crystal structures containing flexible molecules. J Comp Theor Comp. 2006;2:1184–1199. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.