

Abstract
Here we explore the potential power of denaturation mapping as a single-molecule technique. By partially denaturing YOYO®-1-labeled DNA in nanofluidic channels with a combination of formamide and local heating, we obtain a sequence-dependent “barcode” corresponding to a series of local dips and peaks in the intensity trace along the extended molecule. We demonstrate that this structure arises from the physics of local denaturation: statistical mechanical calculations of sequence-dependent melting probability can predict the barcode to be observed experimentally for a given sequence. Consequently, the technique is sensitive to sequence variation without requiring enzymatic labeling or a restriction step. This technique may serve as the basis for a new mapping technology ideally suited for investigating the long-range structure of entire genomes extracted from single cells.
Keywords: DNA barcoding, DNA denaturation, DNA optical mapping, nanochannel
Nanofabrication technology, developed originally for the integrated circuit industry, has over the past decade been increasingly applied to build small-scale fluidic devices, giving rise to a science of “nanofluidics.” Nanofluidics aims to exploit the ability of nanoscale devices to manipulate and analyze single molecules (1). Single-molecule analysis devices obviate the need for cloning and molecular amplification steps and will enable researchers to detect phenomena that in the past would have been obscured by ensemble averaging. In particular, nanofluidics aims to develop on-chip methods for rapidly and cheaply reading out sequence information from single large intact DNA molecules (hundreds of kbp long), with the ultimate goal of directly analyzing single genomes extracted from single cells. For example, such a single-molecule analysis capability could potentially be used to characterize the extent and dynamics of genetic heterogeneity, i.e., local variation of genotype among a population of cells from the same bacterial strain/tissue. In particular, substantial genetic heterogeneity can be present in bacterial colonies (biofilms) (2) and cancer (3), reflecting local adaptations of the cells to varying conditions within the evolving biofilm/tumor (e.g., oxygen and nutrient level). Genetic heterogeneity in these systems is believed to be a crucial factor in determining resistance to therapy.
At the heart of our device are nanochannel structures in which DNA molecules spontaneously stretch out (4–7), creating an extension of the molecule along the channel linear with molecule contour length (6). Nanochannel arrays interfaced with microfluidic loading channels serve as a highly parallel platform for performing physical mapping of DNA (5). Nano-confinement-based stretching does not require complicated setups or additional microfluidic plumbing to create an external stretching force, enabling easy automation, rapid acquisition of high statistics, and, eventually, savings for commercial realizations of the technique.
Nanochannel stretching of DNA can be combined with denaturation mapping and fluorescence microscopy to create a single-molecule optical barcoding technique. DNA can be melted, i.e., denatured from a double-stranded to a single-stranded form, by increasing temperature or adjusting solution chemistry (8, 9). Regions of the molecule rich in A’s and T’s will melt at lower temperature than regions rich in G’s and C’s. Consequently, at certain temperatures the molecule will have ds regions interspersed with ss regions. This pattern of partial melting reveals the underlying sequence in a coarse-grained manner, limited fundamentally by the average size of the “cooperatively melting regions” (typically ∼100 's bp)
If the DNA is uniformly stained with a dye that unbinds when the DNA melts, local fluorescence of the melted region will decrease. Thus, the partial melting will create a grayscale barcode consisting of brighter and darker regions along the nanochannel-extended molecule. This barcode is unique to the sequence of the specific DNA molecule analyzed. The strength of this concept lies in the simplicity and robustness with which it can detect sequence variation along long (∼100 kbp) DNA molecules to a resolution limited by optical diffraction to around 1 kbp. In contrast to earlier barcoding methods, which are based on restriction mapping (5), protein-nucleic acid labeling of the chain (10), or labeling through single-stranded nicks filled in with fluorescent nucleotides (4), our method requires no enzymatic pretreatments of the DNA, only an easily applied generic stain.
Our method is also distinct from currently existing DNA melting analysis techniques. Melting analysis has historically been performed by measuring the UV absorption of a DNA sample as temperature is increased (8, 9). For DNA in the 1- to 100-kbp range, the differential melting curve contains structure relating to the cooperative melting of sequence domains of varying GC content. In contrast, our technique can directly map the spatial position of these domains along an extended single DNA molecule. A second technique, denaturating gradient gel electrophoresis (DGGE), involves electrophoresis of DNA on a gel containing a gradient of a chemical denaturant. The DNA mobility will decrease during partial melting enabling separation of fragments identical in size but varying in sequence composition (11). In contrast to our technique, DGGE compares the melting temperature of different DNA fragments present in a sample: DGGE does not map spatial variation of melting along single molecules.
Results and Discussion
The denaturation mapping experiments were performed with fused silica nanochannel devices fabricated in a clean-room facility via a standard process combining electron beam and photo lithography (see SI Text). The devices were mounted in a chuck allowing for pressure-actuated DNA transport, heating of the device, and simultaneous optical imaging in an inverted optical microscope setup (see Fig. 1 A–F for experimental setup and device design). Formamide—a well-known DNA denaturant (12, 13)—was added to the running buffer to reduce the DNA melting temperature to a range accessible via optical microscopy. Formamide reduces DNA melting temperature via a simple empirical relationship: ΔTm = -0.62F °C, where F is the percentage of formamide by volume (12). In order to minimize the heating required, we have used solutions of 50% formamide to reduce the melting temperature to just a few degrees above room temperature.
Fig. 1.

Experimental setup and chip design. (A) Schematic of how the chip, fluidic chuck, and heater are assembled and integrated in an experiment. A retaining ring held the chip clamped against a fluidic chuck via eight o-ring seals. The chuck was designed so that pneumatic pressure could be applied to bring fresh DNA into the nanoslit from the loading channels. In addition, the chuck was designed with a built-in “hot plate” so that the chip could be heated during experiments. A cartridge heater (Omega) was inserted into a cylindrical aluminum insert that was then gently pressed against the chip’s back surface. In order to monitor the temperature in situ, a thermocouple was threaded through the insert and placed in a hole drilled halfway through the silica sample. (This hole was located 2 mm away from the nanogroove region.) The hot-plate surface was painted with thermally conductive grease (Omegatherm) to enhance heat transfer. Thermocouple measurements of the chip surface temperature show that the measured thermal difference across the chip is comparable to the expected absolute accuracy of our thermocouple readings (∼1–2 °C). The fluctuations in thermocouple readings are ∼0.1 °C. (B) Three-dimensional diagram of the nanogroove and nanochannel arrays interfaced to microscale loading channels (1 μm deep, 50 μm wide). The nanochannels are etched first (120 nm), followed by the nanoslit (30 nm), to create nanogrooves within the open nanoslit region. Cartoon DNA molecules are shown in red. (C) Photograph of the chuck-chip-heater assembly on the microscope stage with external tubing for applying pressure. (D) Photograph of a chip clamped to the chuck via the aluminum retaining ring. (E) Low-magnification SEM micrograph of the nanochannels. (F) High-magnification SEM micrograph of a nanochannel (150 nm wide, 120 nm deep). (G) Raw image of λ-DNA molecules in nanochannels at 28 °C in 50% formamide (with background removed). (H) Example of single λ-DNA molecule from G with accompanying “time trace” of intensity integrated transverse to nanochannel axis plotted for all frames. (I Bottom) Time trace for molecule shown in H with thermal fluctuations “smoothed out” via rescaling procedure. (I Top) Intensity profile obtained by averaging over rescaled frames. The barcode shown below the plot is graphed data displayed as a grayscale plot. The scale bars are equal to 2 μm.
The chip is wetted in formamide-containing buffer and applied pressure is used to bring fluorescently labeled DNA molecules from the microchannel into the nanochannel/nanogrooves (see SI Text for description of DNA labeling protocol and buffer chemistry). In order to increase the number of molecules available for analysis in a single field of view, we have developed a strategy for concentrating DNA in the nanogroove arrays after initial loading (see SI Text, Fig. S1, and Movie S1). Equally distributed pressure is applied to the four reservoirs adjoining the nanochannels to symmetrically bring DNA from the nanochannels into the nanogrooves. As the nanogrooves are deeper than the surrounding slit, at sufficiently low pressures the molecules will be trapped and accumulate in the nanogrooves. After concentration, the molecules are imaged in the nanogrooves and then ejected. This protocol (load, concentrate, image, and eject) is an efficient way to consistently maximize the number of molecules imaged at a fixed device location, lending itself to easy iteration and automation. Movies of nanochannel-extended molecules are obtained during the image acquisition phase of device operation (Fig. 1G). When the device temperature is elevated, the molecules exhibit characteristic banding patterns, a “barcode,” clearly visible when the intensity data are plotted as time traces (Fig. 1H).
Thermal fluctuations in contour density and center-of-mass diffusion create visible distortions of the raw barcodes. In order to remove these distortions, we use an analysis procedure that aligns the center of mass in time and “smoothes out” internal density fluctuations (Fig. 1I, SI Text, and Fig. S2 A and B). The corrected traces are then averaged to create a single-molecule intensity profile (Fig. 1I). In order to compare and average the intensity profiles for an ensemble of molecules, we use an algorithm to systematically align the individual profiles (SI Text and Fig. S2 C–E). The aligned profiles are then averaged to create “consensus barcode profiles.”
As a proof of principle demonstration of our technique we have applied our methodology to four DNA sequences, λ-phage, T7, T4GT7, and a BAC construct from chromosome 12 (RP11-125C7) with an 11.6-kbp cloning vector. The λ-phage and T7 sequences (Fig. 2) give rise to distinct barcode profiles with characteristic temperature dependence. The λ-phage DNA develops a single central dip with a satellite dip that grows as the temperature is increased. The T7-phage sequence develops a three-dip structure. The profiles are distinct and can easily be distinguished, even for single molecules side by side in the nanochannels (Fig. 2F). The T4GT7 profiles can be aligned, but it is clear that they must be shifted relative to each other in order to achieve alignment: The barcode appears to be circularly permutated between molecules (Fig. 3F). This agrees with the classic result of phage genetics that T4 has a linear, but circularly permutated, genome (14). The circularly permutated T4GT7 sequence is an excellent test of our alignment procedure: We can align the permutated T4GT7 barcodes to construct a circular consensus melting map (Fig. 3 A and B). In a second test of our alignment procedure, the RP11-125C7 construct was run in the devices prior to linearization of its plasmid structure. While circular DNA cannot be easily mapped, a certain fraction of the circular BACs will break during chip loading. These linearized BACs were then aligned and converted into a circular map (Fig. 3 C and D). In addition, the ability to align the barcodes of different molecules allows for relaxation of the requirement of DNA monodispersity. DNA fragments can be aligned to positions on overlapping molecules via the barcodes without a priori knowledge of their size and sequence position (Fig. S2 C–E).
Fig. 2.

λ-phage and T7 consensus barcodes in 50% formamide. (A) λ-phage barcode at 27 °C with three example time traces (n = 13 molecules total in consensus). (B) λ-phage at 28 °C (n = 56 molecules in consensus). (C) λ-phage at 30 °C (n = 47 molecules in consensus). (D) λ-phage at 31 °C (n = 38 molecules in consensus). (E) λ-phage at 32 °C (n = 19 molecules in consensus). (F) A raw fluorescence micrograph showing λ-phage and T7 molecules side by side: It is possible to easily distinguish the two from the melting barcode. (G) T7 at 30 °C (n = 20 molecules in consensus). (H) T7 at 30.6 °C (n = 10 molecules in consensus). (I) T7 at 31.2 °C (n = 17 molecules in consensus). The example time traces have 10-s duration. The scale bar in all images is 2 μm. The error bars in the plots are standard errors on the ensemble mean (SEM), calculated as the SD of ensemble values divided by 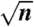 .
.
Fig. 3.

Consensus barcodes for T4GT7 and BAC RP11-125C7. (A) Consensus barcode for T4GT7 DNA (blue) with theoretical fit (bold curve). Data acquired with 50% formamide at 27 °C. Consensus obtained from 26 molecules. (B) T4GT7 barcode displayed as a circular wheel (color corresponds to grayscale value, white maximum values, dark-red minimum). The inner wheel is the theoretical prediction; the outer wheel is the experimental consensus barcode. (C) Consensus barcode for BAC RP11-125C7 (blue) with theoretical fit (bold curve). Data acquired with 48% formamide at 27 °C. Consensus obtained from 32 molecules. The shaded region is the location of the cloning vector. (D) BAC RP11-125C7 barcode displayed as a circular wheel. The inner wheel is the theoretical prediction; the outer wheel is the experimental consensus barcode. The error bars in the plots are standard deviations of the mean arising from profile variation across the ensemble. (E) Raw time traces for RP11-125C7 arranged to emphasize barcode overlap. The scale bar is 2 μm. (F) Raw time traces for T4GT7 arranged to emphasize barcode overlap. The scale bar is 2 μm.
The observed barcode structure in the DNA fluorescence profile is consistent with the presence of local denaturation. Computer algorithms exist, based on the Poland–Scheraga model (8, 9), which can predict the sequence-dependent melting probability of dsDNA pds(s) for a particular temperature and salt concentration (15). These codes use as inputs the measured enthalpies and entropies of the 10 independent base-pair duplexes (16), resulting from a combination of hydrogen bonding between bases and stacking interactions between nearest-neighbor base pairs. In addition, the codes contain a description of polymer looping energetics to correctly distinguish between internally melted regions (bubbles) and melted end segments (17).
These calculated melting probabilities can be transformed into an expected fluorescence profile (Fig. 4A). We argue that an approximate average sequence-dependent intensity I(s) and extension r(s) along the channel can be obtained from
| [1] |
| [2] |
The quantity Ids is the intensity of the double-stranded regions, Iss the intensity of single-stranded regions, rds is the average extension per base pair of dsDNA along the channel, and rss is the average extension per base pair of ssDNA. The intensity profile I(r) obtained from this calculation is our theory for the emitted intensity. The observed intensity is the emitted intensity convoluted with the point-spread function of the microscope objective. We approximate the latter with a Gaussian with standard deviation σ ∼ 200 nm. The results of this calculation for the example case of λ-DNA, using the algorithm developed by Hovig and coworkers (15), are shown in Fig. 4A. The melting-probability calculation uses as inputs the 10 independent enthalpies and entropies of the stacked and paired base-pair duplexes measured by Blake and Delcourt (16) with the loop entropy weight suggested by Blossey and Carlon (17). The computed profiles clearly reproduce qualitatively the structure and temperature dependence evident in the λ-DNA consensus profiles. The theoretical profiles can be directly fitted to the experimental data upon appropriate specification of Ids, Iss, rds, rss, and σ. The results of the fitting procedure for λ-DNA and T7 DNA are shown in Fig. 2.
Fig. 4.

Theoretical prediction of barcode structure from dsDNA melting probability (A). Theoretical melting traces can be obtained directly from melting-probability calculations by assuming values for ds/ss DNA stretching, fluorescence, and convolving the profile with a Gaussian to simulate optical broadening. (B) Evolution of λ-phage melting traces with temperature across the melting transition: by scanning temperature, both high AT and high GC regions are imaged. Details in high AT regions are visible at low temperature, wheras at higher temperature details in high GC regions can be imaged. (C) Experimental barcode profile (red) for single BAC RP11-125C7 molecule aligned to theoretical estimate of RP11-125C7 barcode (blue). (D) Experimental barcode profile (red) for single T4GT7 molecule (red) aligned to theoretical estimate of T4GT7 barcode (blue). The theoretical estimates for T4GT7 and BAC RP11-125C7 are created from two identical and adjoining sequences to capture trace permutation. (E) Fitted λ-phage helicities vs. temperature; circles, open squares, and triangles indicate measurements with different chips/setups. The variation between experimental runs arises primarily from variations in thermocouple calibration (∼1 °C error). The bold curve is a theoretical estimate of the helicity at 5 mM NaCl.
Using the barcode alignment technique, circularly permutated sequences can also be aligned to an estimated theoretical barcode—see Fig. 4 C and D—enabling us to compare the T4GT7 and BAC construct data to theory and calibrate the x axis in units of kbp referenced to a known position on the true sequence (see SI Text). The theoretical profiles convincingly capture the structure of the experimental barcodes, suggesting that the alignment procedure is effective. For example, we are able to map the position of the 11.6-kbp vector sequence, which results in the strong central peak, due to its relatively higher GC content (47.4%, compared to 37% on RP11-125C7).
The key fitting parameters are the ratio of the ssDNA to dsDNA extension, rss/rds, and the ratio of the intensities, Iss/Ids. We expect that rss/rds < 1 because ssDNA is more flexible than dsDNA. At the ionic strength of 5 mM used in the experiment, the persistence length of dsDNA Pds lies in the range of 60–80 nm (18, 19) and the persistence length of ssDNA Pss = 6 ± 2 nm (20). The extension of DNA in a nanochannel is r ∼ P1/3, so a crude estimate is that rss/rds ∼ (Pss/Pds)1/3 ∼ 0.4–0.5. In fact, we have found in solutions of 50% formamide that the best fits are obtained with rss/rds ∼ 0.7–0.9. This result is plausible as formamide is known to extend ssDNA, a fact well established from early electron microscopy studies (21). For definiteness, in the theoretical calculations we have fixed rss/rds = 0.85, the average value obtained from the λ-phage fits.
Least-squares fitting yields values of Iss consistent with zero. Consequently, we have fixed Iss = 0 for all the plots shown. Physically, this implies that the stain completely leaves the melted regions of the molecule. While it is known that the ratio of the ssDNA and dsDNA quantum yields in bulk is 0.66 (22), it is likely that electrostatic dye-silica surface interactions might promote the loss of stain from single-stranded regions of the molecule (we have, in fact, observed stain binding to the nanochannel surface during the melting cycle). From an application point of view, this implies that the contrast between the melted and nonmelted regions is significantly higher than expected from the bulk fluorescence ratios.
It is convenient to define the probability plots as a function of the helicity (total percentage of paired bases, i.e., DNA not melted). Best-fit helicities from the profiles can then be compared to theoretical estimations of the expected helicity at the experimental temperature, ionic strength, and formamide concentration (Fig. 4E). The deviations from theory observed (< 3 °C) likely arise from complexities of the buffer composition not taken into account in the model, such as beta-mercaptoethanol, the O2 scavenging system components, and possible effects of the fluorescent stain used (YOYO®-1) on the duplex enthalpies and melting temperatures. In addition, the melting-probability calculations do not take into account how confinement might modify the polymer looping statistics, an effect that would be worthwhile clarifying in future theoretical studies of DNA melting.
Conclusion
The denaturation mapping technique could have a number of important implications for the field of single-molecule genomic DNA analysis. As a final demonstration of the technique, we show that it is feasible to align experimental melting barcodes within a denaturation map calculated from the full sequence of human chromosome 12 (23) (Fig. 5). The true location of RP11-125C7, determined from comparison to the human reference sequence for chromosome 12 (130 Mbp), is between 81,654,422 and 81,806,391. Using an automatic least-squares alignment procedure (see SI Text), the BAC was positioned from site 81,655,000 to 81,807,000 with a 1-sigma error ∼1 kbp. Note that the alignment is not sensitive to exact knowledge of the temperature as the experimental profile is compared to denaturation maps calculated for a range of temperatures (and the best fitting temperature and sequence position found in a single search). This procedure suggests that it may be possible to identify the global genomic position of a DNA fragment based on measuring the fragment’s denaturation profile. Moreover, this example—taken together with our ability to align circularly permutated DNA sequences—suggests that it may be possible to construct genome-wide physical maps based on identifying and aligning the barcodes of overlapping DNA fragments.
Fig. 5.

Alignment of RP11-125C7 to chromosome 12. (A) The melting profile of human chromosome 12. (B) The melting profile of chromosome 12 between position 79,500,000 and 85,500,000 with aligned RP11-125C7 (blue). Profile calculated at 59 °C for 5 mM NaCl (corresponding to an estimated melting temperature of 29 °C in 48% formamide). (C) Selected 500 kbp of melting profile around the position of RP11-125C7.
In terms of future potential applications, the key advantage of the denaturation mapping technique is the ease with which it can detect long-range sequence variation along single DNA molecules. Because the technique does not necessitate DNA fragmentation, large-scale organization of the genome is left intact and observed intact. For example, the melting profiles of a nonsequenced genome may provide a scaffold for ordering sequence contigs assembled via the shotgun approach (24). Typically, a first round of shotgun sequencing of a genome results in hundreds to thousands of contigs, which could be turned into hypothesized melting profiles via the bioinformatics methodology and compared to the experimental fluorescence trace. In addition, the melting barcode could be used to identify the global binding position in the genome of additional sequence-specific markers for genome-wide haplotyping. Finally, we suggest that no fundamental obstacle exists to measuring barcodes along extremely long DNA molecules stretched in the channels, e.g., mega-base pairs in size. Undulating channels could be used to display such large molecules across a single field of view. While fragmentation of long molecules is a challenge (6), techniques designed to solve this difficulty are being developed, such as direct gradual funneling of nanochannels (25) and on-chip lysis of cells (26) with direct extraction of chromosomes into nanochannels. In the future, coupling such techniques with the denaturation mapping methodology might enable direct single cell genome mapping, whereby the genomes of individual cells in large populations could be rapidly screened to identify disease-related structural variants and as a precursor to targeted sequencing.
Supplementary Material
Acknowledgments.
We gratefully acknowledge Peixiong Shi for assistance with e-beam lithography at the Danchip clean-room facility. We also thank Eivind Tøstesen and Eivind Hovig for generously providing the melting-probability map for human chromosome 12. In addition, we thank Mattias Höglund, Samuel Gebre-Medhin, and Eivind Hovig for fruitful discussions. This work was supported by the Danish Research Council for Technology and Production Sciences through Grant 274-05-0375, the Human Frontier Science Program, Young Investigator, Grant RGY78/2007, the Swedish Research Council (Grants 2002-5972 and 2007-584), and the Wilhelm Johannsen Centre for Functional Genome Research (established by the Danish National Research Foundation). In addition, the research leading to these results has received funding from the European Community’s Seventh Framework Programme (FP7/2007-2013) under grant agreement no [HEALTH-F4-2008-201418] entitled READNA.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1007081107/-/DCSupplemental.
References
- 1.Craighead H. Nature. 2006;442:387–393. doi: 10.1038/nature05061. [DOI] [PubMed] [Google Scholar]
- 2.Stewart P, Franklin MJ. Physiological heterogeneity in biofilms. Nat Rev Microbiol. 2008;6:199–210. doi: 10.1038/nrmicro1838. [DOI] [PubMed] [Google Scholar]
- 3.Salk JJ. Mutational heterogeneity in human cancers: Origin and consequences. Annu Rev Pathol Mech. 2010;5:121808–102113. doi: 10.1146/annurev-pathol-121808-102113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jo K, et al. A single-molecule barcoding system using nanoslits for DNA analysis. Proc Natl Acad Sci USA. 2007;104:2673–2678. doi: 10.1073/pnas.0611151104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Riehn R, et al. Restriction mapping in nanofluidic devices. Proc Natl Acad Sci USA. 2005;102:10012–10016. doi: 10.1073/pnas.0503809102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tegenfeldt JO, et al. The dynamics of genomic-length DNA molecules in 100 nm channels. Proc Natl Acad Sci USA. 2004;101:10979–10983. doi: 10.1073/pnas.0403849101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reisner W, et al. Statics and dynamics of single DNA molecules confined in nanochannels. Phys Rev Lett. 2005;94:196101. doi: 10.1103/PhysRevLett.94.196101. [DOI] [PubMed] [Google Scholar]
- 8.Wartell M, Benight A. Thermal denaturation of DNA molecules: A comparison of theory with experiment. Phys Rep. 1985;126:67–107. [Google Scholar]
- 9.Gotoh O. Prediction of melting profiles and local helix stability for sequenced DNA. Adv Biophys. 1983;16:1–52. doi: 10.1016/0065-227x(83)90007-2. [DOI] [PubMed] [Google Scholar]
- 10.Chan YC, et al. DNA mapping using microfluidic stretching and single-molecule detection of fluorescent site-specific tags. Proc Natl Acad Sci USA. 2004;14:1137–1146. doi: 10.1101/gr.1635204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fischer SG, Lerman LS. DNA fragments differing by single base-pair substitutions are separated in denaturing gradient gels: Correspondence with melting theory. Proc Natl Acad Sci USA. 1983;80:1579–1583. doi: 10.1073/pnas.80.6.1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sadhu C, Dutta S, Gopinathan KP. Influence of formamide on the thermal stability of DNA. J Biosci. 1984;6:817–821. [Google Scholar]
- 13.Blake RD, Delcourt SG. Thermodynamic effects of formamide on DNA stability. Nucl Acid Res. 1996;24:2095–2103. doi: 10.1093/nar/24.11.2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hershey AD. Idiosyncrasies of DNA structure. Nobel lecture. 1969. [DOI] [PubMed]
- 15.Tøstesen E, Jerstad GI, Hovig E. Stitchprofiles.uio.no: Analysis of partly melted DNA conformations using stitch profiles. Nucl Acid Res. 2005;33:W573–W576. doi: 10.1093/nar/gki424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blake RD, Delcourt SG. Thermal stability of DNA. Nucl Acid Res. 1998;26:3323–3332. doi: 10.1093/nar/26.14.3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blossey R, Carlon E. Reparametrizing the loop entropy weights: Effect on DNA melting curves. Phys Rev E. 2003;68:061911. doi: 10.1103/PhysRevE.68.061911. [DOI] [PubMed] [Google Scholar]
- 18.Baumann CG, Smith SB, Bloomfield VA, Bustamante C. Ionic effects on the elasticity of single DNA molecules. Proc Natl Acad Sci USA. 1997;94:6185–6190. doi: 10.1073/pnas.94.12.6185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wenner JR, Williams MC, Rouzina I, Bloomfield VA. Salt dependence of the elasticity and overstretching transition of single DNA molecules. Biophys J. 2002;82:3160–3169. doi: 10.1016/S0006-3495(02)75658-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tinland B, Pluen A, Sturm J, Gilbert W. Persistence length of single-stranded DNA. Macromolecules. 1997;30:5763–5765. [Google Scholar]
- 21.Banfield HY, Inman RB. The electronmicroscopy of DNA. Annu Rev Biochem. 1974;43:605–619. doi: 10.1146/annurev.bi.43.070174.003133. [DOI] [PubMed] [Google Scholar]
- 22.Cosa G, Focsaneanu KS, McLean JRN, McNamee JP, Scalano JC. Photophysical properties of fluorescent DNA-dyes bound to single- and double-stranded DNA in aqueous buffered solution. Photochem Photobiol. 2001;73:585–599. doi: 10.1562/0031-8655(2001)073<0585:PPOFDD>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 23.Fang L, et al. Human genome melting map. PLOS Comput Biol. 2007;3:0874–0886. [Google Scholar]
- 24.Lin J, et al. Whole-genome shotgun optical mapping of Deinococcus radiodurans. Science. 1999;285:1558–1562. doi: 10.1126/science.285.5433.1558. [DOI] [PubMed] [Google Scholar]
- 25.Cao H, Tegenfeldt JO, Austin RH, Chou SY. Gradient nanostructures for interfacing microfluidics and nanofluidics. Appl Phys Lett. 2002;81:3058–3060. [Google Scholar]
- 26.Morton KJ, Loutherback K, Inglis DW, Tsui OK, Sturm JC. Crossing microfluidic streamlines to lyse, label and wash cells. Lab Chip. 2008;8:1448–1453. doi: 10.1039/b805614e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.