

Abstract
We investigated the ultrafast structural transitions of the heme induced by nitric oxide (NO) binding for several heme proteins by subpicosecond time-resolved resonance Raman and femtosecond transient absorption spectroscopy. We probed the heme iron motion by the evolution of the iron-histidine Raman band intensity after NO photolysis. Unexpectedly, we found that the heme response and iron motion do not follow the kinetics of NO rebinding. Whereas NO dissociation induces quasi-instantaneous iron motion and heme doming (< 0.6 ps), the reverse process results in a much slower picosecond movement of the iron toward the planar heme configuration after NO binding. The time constant for this primary domed-to-planar heme transition varies among proteins (∼30 ps for myoglobin and its H64V mutant, ∼15 ps for hemoglobin, ∼7 ps for dehaloperoxidase, and ∼6 ps for cytochrome c) and depends upon constraints exerted by the protein structure on the heme cofactor. This observed phenomenon constitutes the primary structural transition in heme proteins induced by NO binding.
Keywords: time-resolved Raman spectroscopy, allostery, structural dynamics
The role of diatomics endogenously generated in cells as messengers in various signaling pathways has been demonstrated for nitric oxide (NO) (1, 2) and carbon monoxide (CO) (3, 4) and various gas-sensors heme proteins have been discovered in the last two decades (5). In these signaling pathways, the binding of diatomics to their respective heme sensor domain triggers a response of the associated enzymatic domain by modulating the catalytic or the gene regulating activity (5). Recently, myoglobin (Mb) was proposed to be involved in the regulation of NO level (6), and neuroglobin was proposed as a sensor regulating the NO/O2 balance (7). Similarly to Hb (8–10), the signaling mechanisms are based at the molecular level on an allosteric structural change of the receptor/sensor protein coupled to a change of activity or affinity and are induced by diatomic ligation to the heme iron cofactor. The signal of diatomic binding/release can be transmitted to the overall protein structure either by in-plane/out-of-plane motion of the iron through the bound proximal histidine or by the cleavage of the iron-histidine (Fe-His) bond. In particular, for Hb and its monomeric counterpart Mb, the out-of-plane iron motion occurs in less than 1 ps after gaseous ligand release (11, 12) and represents the first event of the allosteric R → T transition (9). This primary heme iron motion is followed by overall tertiary changes, notably motion of α-helices (13). Upon gaseous ligand binding, the reverse T → R allosteric transition is coupled to the iron motion toward the heme plane, and this primary motion is also considered quasi-instantaneous since the stereochemical description of Hb allostery by Perutz (9, 10) although up to now it has never been observed.
The ultrafast events in the interaction of heme proteins with various gaseous ligands were studied extensively at molecular level in the last two decades by femtosecond transient absorption (TA) (14, 15) and time-resolved IR (TR-IR) (16, 17) spectroscopies. Time-resolved resonance Raman (18, 19) (TR3) and X-ray diffraction (TRXD) (20–23), two structure-sensitive techniques able to probe the heme iron position, focused so far on the heme-CO complex, studying the consequences of CO release. At the same time, the binding of a diatomic and subsequent T → R transition have not yet been studied by a time-resolved technique sensitive to heme structural changes. The first event following gaseous ligand binding, namely, the transition from domed-to-planar heme, is believed to occur in less than 0.6 ps, by analogy with the well-studied heme response to ligand release (11, 12, 24, 25), although the structural constraints and energy barriers for these two opposite processes are not necessarily the same.
We investigated the primary event related to the T → R transition by subpicosecond TR3 and report the observation of the in-plane movement of the ferrous heme iron induced by NO geminate rebinding, following a femtosecond photodissociating laser pulse. We studied various nitrosylated heme proteins: wild-type Mb and its distal mutant H64V, Hb, and dehaloperoxidase (DHP), all incorporating a b-type heme, and mitochondrial cytochrome c (Cyt c) having a c-type heme covalently linked to the protein. This allowed the evaluation of distinct structural parameters that could influence the iron motion. The conformational transition between domed and planar heme structure was monitored in TR3 spectra of the photoproduct through temporal evolution of the intensity of Fe-His stretching (26) (νFe-His), an important vibrational mode linking heme iron with the proximal histidine that transmits the binding information to the protein structure. Our approach is based on the findings that (i) the transition from planar to domed heme structure induces a change in Raman intensity of the low-frequency νFe-His stretching band (19, 26) located within the range 200–240 cm-1; (ii) the intensity of νFe-His resonantly excited within the Soret absorption band depends on the out-of-plane position of the iron with respect to the heme macrocycle plane (27) and is zero for the in-plane iron position. For this study, we have employed a specially designed home-built subpicosecond TR3 spectrometer having ∼0.7-ps temporal and ∼25-cm-1 spectral resolution (24, 28, 29), probing structural changes in the time range not yet accessible to TRXD.
Results
Difference Between Iron Motion and Dynamics of NO Binding.
The TR3 spectra of ferrous Mb-NO in the low-frequency range of Fe-His mode at various time delays after NO photodissociation are presented in Fig. 1A. The Raman spectrum of five-coordinate deoxy Mb possessing a domed heme is dominated by νFe-His at 219 cm-1. The spectrum of the ground-state nitrosylated complex recorded at negative time delay between the photolysis and Raman probe pulses (Δt = -5 ps) does not exhibit the νFe-His mode, as expected for a six-coordinate planar heme. After the femtosecond photolysis pulse, νFe-His appears already at Δt = 1 ps with high intensity due to NO dissociation and immediate out-of-plane motion of the iron toward proximal His, forming a domed heme. We emphasize that, because the difference TR3 spectra at positive delay in Fig. 1A originate solely from the photoproduct species (see Materials and Methods), the intensity decay kinetics of νFe-His (Fig. 1B) directly reflects the evolution of the photoproduct structure.
Fig. 1.

TR3 spectra of ferrous Mb in interaction with NO and kinetics of the νFe-His mode. (A) Ground-state Raman spectra of deoxy Mb and of six-coordinate NO-liganded Mb (-5 ps), and difference TR3 spectra after NO photodissociation at indicated time delays after photolysis. (B) Evolution of normalized area of the νFe-His mode contour (180–270 cm-1, between the vertical dotted lines in A for two Mb concentrations: 0.3 mM (circles) and 0.1 mM (squares). The red curve is the solution of rate equations for population decay of all nonplanar heme species, yielding the time of transition from domed-to-planar 6-c heme τD = 30 ± 10 ps. The blue and green dashed curves are, respectively, double-exponential decays after NO dissociation from transient visible (Table 1) and from IR absorption (τ1 = 5.3 ps [54%], τ2 = 133 ps [46%]; ref. 17). (C and D) The same for Mb-NO in 50% glycerol solution, yielding τD = 40 ± 15 ps.
The striking feature of this kinetics is that the intensity of the mode νFe-His (which is located at 225 cm-1 at Δt = 2 ps and gradually shifts to ∼222 cm-1 at Δt = 100 ps) remains nearly constant during the first 10 ps, contrary to absorption kinetics. Indeed, it is established from visible TA experiments on Mb-NO (Table 1 and Fig. S1; refs. 15 and 29) that about 40% of five-coordinate heme population converts into the six-coordinate form with time constant of ∼10 ps, followed by a second geminate phase in ∼150 ps. Moreover, TR-IR measurements (17) of the stretch of bound NO in Mb-NO have shown that ∼54% of NO rebinds to the iron with time constant of 5.3 ps. Remarkably, the decay of the νFe-His intensity compared with those of transient visible and IR absorption (Fig. 1B) is obviously different and much slower.
Table 1.
Kinetic parameters obtained from transient absorption measurements and fitting the νFe-His Raman intensity kinetics (τD)
| NO rebinding phases (from transient absorption) |
||||
| Protein | τ1 (amplitude) | τ2 (amplitude) | Constant | Heme iron motion τD (from νFe-His kinetics) |
| Myoglobin | 13 ps (0.40) | 148 ps (0.50) | 0.10 | 30 ± 10 ps |
| Myoglobin in 50% glycerol | 15.3 ps (0.42) | 71 ps (0.36) τ3 = 378 ps (0.21) | 0.01 | 40 ± 15 ps |
| Myoglobin H64V mutant | 10 ps (0.51) | 92 ps (0.43) | 0.06 | 30 ± 15 ps |
| Dehaloperoxidase | 14 ps (0.61) | 65 ps (0.38) | 0.01 | 7 ± 2 ps |
| Cytochrome c | 8.4 ps (0.99) | — | 0.01 | 6 ± 2 ps |
| Hemoglobin | 10.8 ps (0.74) | 61 ps (0.22) | 0.04 | 15 ± 6 ps |
Only for Mb in 50% glycerol, we found a third component.
Several phenomena could influence this unexpected νFe-His intensity kinetics. First, the accurate measurement of νFe-His intensity could be perturbed by reabsorption of scattered Raman light. However, sample illumination from the bottom of the cell near the inner surface has been specially designed in order to minimize this effect. Indeed, this possibility was ruled out by the observation that the νFe-His intensity kinetics for two different Mb concentrations (0.1 mM and 0.3 mM) exhibit exactly the same behavior (Fig. 1B). Second, the intensity of νFe-His depends not only on the population of the domed heme species but also on Raman resonance enhancement, so that time-dependent spectral shift of transient absorption may influence the result (30), along with the process of vibrational relaxation (28). However, these effects vanish in a few picoseconds, and therefore the kinetics of νFe-His was analyzed in the time range Δt = 2–200 ps, where TA spectra of Mb-NO show a clear isosbestic point (Fig. S1).
After having discarded these possible artifacts, because the intensity of νFe-His resonantly excited in the Soret absorption band depends on the out-of-plane position of the iron with respect to the heme plane (27; SI Text), we conclude that the kinetics of νFe-His intensity (Fig. 1B) directly represents the transition from domed-to-planar heme structure and consequently the movement of the iron triggered by NO binding. Thus, the νFe-His intensity decay would be identical to the kinetics measured by TA or TR-IR if the iron were moving instantaneously upon NO binding, which is not the case. Accordingly, because the parameters probed by TA/TR-IR and TR3 are not the same (NO coordination to the iron versus heme iron position), we interpret our observation as follows: The photolyzed NO molecules from the heme pocket bind to the iron of the domed five-coordinate heme and the resulting transient six-coordinate heme complex does not return immediately to a planar configuration, but remains domed for some time, providing enhancement for the νFe-His Raman intensity.
To analyze quantitatively this effect, the evolution of νFe-His intensity has been fitted by the convolution of Raman instrumental response function with the solution of rate equation (Eq. 1) for population changes of all nonplanar heme species (see Materials and Methods). We employed a kinetic model of NO recombination (Fig. 2A) based on TA measurements (15, 29) (Fig. S1) incorporating an additional transient six-coordinate species with nonplanar structure, not detected by TA as a separate component (Table 1). In Raman spectra (Δt = 1–200 ps) of Fig. 1A the intensity of the νFe-His band originates from two different domed species: the photodissociated five-coordinate domed heme (5c-His; species X, B, and Const. in Fig. 2A) and six-coordinate NO-liganded domed heme (6c-His-NO; species D), which can exist only transiently. The solid red curve through the data points in Fig. 1B is the best-fit kinetics yielding the time constant τD = 30 ± 10 ps for the domed-to-planar heme transition (D → A) using the TA time constants in Table 1. This picosecond heme iron motion represents the first structural event in wild-type Mb taking place after NO binding.
Fig. 2.

Scheme of NO recombination to Mb together with relevant transient species within the picosecond time range (A). The positions of His side chains are obtained from structures (2FRK and 2V1K) in the Protein Data Bank. After dissociation (thermal or photoinduced) or diffusing from solution, NO can bind to a domed heme whose iron is displaced toward the proximal His. The letters (X, B, D, A) refer to the kinetic model. This scheme is applicable to other proteins studied, with time constants given in Table 1. The bimolecular rebinding, occurring in microsecond time scale, is represented as a constant term. (B) Evolution of the population of the species A, D, B, and X, according to rate equations (see Materials and Methods) for Mb-NO. (C) Evolution of the population ratio of the six-coordinate (species D) to five-coordinate (B + X + const) nonplanar species, for Mb-NO.
Other Experimental Evidence for the Transient Population of Domed Six-Coordinate Heme.
It is essential to compare the behavior of νFe-His to that of the ν4 band, which is a π electron density marker also sensitive to spin and coordination state of the heme iron (31). Therefore, in another experiment we recorded simultaneously (with a less dispersive spectrograph grating) the temporal changes of the modes νFe-His and ν4 (Fig. 3 and Figs. S2 and S3). A straightforward separation of the mode ν4 corresponding specifically to species D is rather complicated by the facts that (i) the frequency difference between the limiting values for six-coordinate planar Mb-NO (1,374 cm-1) and five-coordinate domed deoxy-Mb (1,354 cm-1) is 20 cm-1 (Fig. 3), whereas our spectral resolution is 25 cm-1; (ii) because the ν4 band position is sensitive to polar heme environment, we may expect fine ν4-frequency difference between the species X (NO far from the heme) and B (NO in the vicinity of the heme), so that four different components (or a continuous distribution) are expected to contribute to the resulting Raman contour; (iii) in our experiments the total amount of the photoproduct species constitutes no more than 35% (determined by ground-state subtraction) of the probed molecules and from this amount, the maximal population of species D is about 24% (Fig. 2B).
Fig. 3.

TR3 spectra in the range 150–1,700 cm-1 of Mb-NO (a, probe only), deoxy-Mb (b, probe only), Mb-NO photoproducts (species D + B + X + const) at Δt = 2 ps (c) and Δt = 30 ps (d), and the weighted difference (e) of spectrum (d) minus spectrum (c). Note that trace (e) is magnified 3 times. The asterisk indicates Raman band of 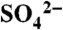 utilized for intensity normalization.
utilized for intensity normalization.
Therefore, we compared the photoproduct transient spectra at various time delays with that at 2 ps (minimal contribution of species D). The spectrum at 30 ps with maximal contribution of D (Fig. 2C) is highlighted in Fig. 3. Importantly, the intensity of the ν4 band (1,355 cm-1) clearly decreased at 30 ps, whereas that of νFe-His intensity is much less pronounced (Fig. 3). The difference (Fig. 3E) reveals a pronounced high-frequency shift of the 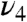 band (shift centered at ∼1,362 cm-1), whereas the bands ν7 (672–675 cm-1) and νFe-His (223–225 cm-1) experience only very small shifts. Fig. S3 shows transient spectra in the entire frequency range and difference traces with ν4 band shift for all time delays. Fig. S4 shows that the kinetics of band ν4 is well superimposed with the TA decay and not with that of νFe-His, within the accuracy of measurements. We thus conclude that the intensity of the band ν4 follows heme coordination, whereas that of the νFe-His band does not.
band (shift centered at ∼1,362 cm-1), whereas the bands ν7 (672–675 cm-1) and νFe-His (223–225 cm-1) experience only very small shifts. Fig. S3 shows transient spectra in the entire frequency range and difference traces with ν4 band shift for all time delays. Fig. S4 shows that the kinetics of band ν4 is well superimposed with the TA decay and not with that of νFe-His, within the accuracy of measurements. We thus conclude that the intensity of the band ν4 follows heme coordination, whereas that of the νFe-His band does not.
Increasing the viscosity of the solvent enhances the yield and rates of geminate rebinding and can slow down the structural protein relaxation (15, 32). We thus also performed TR3 measurements for Mb-NO in 50% glycerol solution (Fig. 1C) in order to investigate a change of τD for Mb as a function of viscosity. We found that the corresponding TA kinetics were best fitted with a triple-exponential (Table 1) and best-fit analysis of TR3 data using modified rate equations yielded τD = 40 ± 15 ps (Fig. 1D) revealing slower kinetics of νFe-His band intensity in glycerol in spite of faster geminate recombination. This is consistent with the hypothesis of a slower iron motion as increased viscosity slows down the protein fluctuations.
The difference in kinetics of TA and TR-IR (Fig. 1B) may be explained, along the same line of reasoning, by the fact that TR-IR directly probes NO coordination, whereas visible TA kinetics reflect the change in the electronic configuration of the heme, not only the Fe coordination. However, because TA in the range 400–650 nm is quite insensitive to small nonplanar porphyrin distortions (33), the effect is rather weak. We have performed TA measurements of the charge transfer Band III (755–760 nm) whose intensity was hypothesized to depend upon the Fe position and thus may reflect heme doming (34). The main observation (Fig. S4) is that the Band III kinetics comprises a phase slower than those of the background (tail of the Q bands). This latter result, as well as the simultaneous measurement of νFe-His and ν4 modes, supports our conclusion that the observed difference in the kinetics of iron motion and NO rebinding is not due to instrumental differences between TR3 and TA experiments, but is intrinsic to the protein. To further study the retarded domed-to-planar heme transition after NO binding, we investigated various nitrosylated proteins.
The configuration of the NO binding to the domed heme in species D could in principle be studied by detection of the kinetics of the νFe-NO (∼554 cm-1) and νN-O (∼1,613 cm-1) stretching modes. However, such studies are as yet compromised by the combination of (i) extremely small amplitudes, (ii) spectral congestion, in particular for νN-O, (iii) the presence of similar bands in the planar six-coordinate species, which is the most populated species in the experiments at any time and whose contribution must be subtracted, and (iv) the lack of any reference spectra and a priori knowledge of these bands in domed six-coordinate heme. By contrast the νFe-His and ν4 Raman bands (Fig. S3) on which we focus in this study provide strong and background free markers of the heme configuration that are well suited for quantitative analysis.
Effect of Hydrogen Bonding Between Nitric Oxide and Distal Histidine.
One of the most important distal side chains is His64, which stabilizes NO (35) and which experiences a motion within 10 ps after diatomic ligand dissociation (21). To evaluate a possible role of electrostatic interaction between NO and His64 in the observed effect, we have studied the H64V Mb mutant, whose apolar Valine side chain does not interact with NO. The general behavior of νFe-His intensity in the mutant protein (Fig. 4 A and B and Table 1) was found similar to that of wild-type Mb (Fig. 1) with the same time constant τD = 30 ± 15 ps, despite faster NO geminate rebinding in H64V Mb. We conclude that the domed-to-planar heme transition does not depend on the stabilizing interaction between NO and His64, which is involved only in diatomic stabilization after binding (35) and not in heme relaxation.
Fig. 4.

TR3 spectra of various heme proteins studied and corresponding kinetics of the νFe-His mode during NO rebinding: Mb-H64V mutant (A), DHP (C), Cyt c (E), Hb (G), and the corresponding kinetics of the mode νFe-His (B, D, F, and H) calculated according to rate equations using time constants and yielding τD as in Table 1. The blue dashed curve in the lower panels represents the absorption kinetics to be compared to that of the mode νFe-His.
Effect of Sequence with the Same Globin Fold.
In order to investigate the influence of the sequence in the constraints on heme iron relaxation, we similarly studied the ferrous nitrosylated DHP, an invertebrate heme protein involved in catalytic oxidation of various halophenols (36) whose fold is similar to that of Mb. This allowed us to vary the sequence, but keeping a similar tertiary globin fold. Femtosecond TA measurement of DHP revealed that NO geminate rebinding is double-exponential (Table 1 and Fig. S1), the first component being the same as in Mb, whereas the second is about two times faster. TR3 spectra and kinetics of the νFe-His intensity after NO dissociation from DHP (Fig. 4 C and D) exhibit both similarity and differences as compared to Mb. Before photodissociation (Δt = -5 ps), the spectrum of the six-coordinate DHP-NO complex (Fig. 4C) does not show the νFe-His stretching, which appears at 230 cm-1 faster than 1 ps after NO photolysis, as in Mb. The kinetics of νFe-His intensity also deviates from expectation based on TA measurement (Fig. 4D) indicating noninstantaneous domed-to-planar heme transition. However, the fit using the model of Fig. 2 yields τD = 7 ± 2 ps for the transition toward a planar heme, which is much faster for DHP than for Mb.
Effect of Covalent Links Between Protein and Heme.
In order to evaluate the influence of covalent links between protein and heme, we performed measurements on ferrous mitochondrial Cyt c whose heme is covalently linked to the protein. The heme iron in Cyt c is six-coordinated, so that the binding of NO necessitates the thermal displacement of the distal Met (28). The induced absorption corresponds to that of five-coordinate heme and the transient difference spectrum (Fig. S1) corresponds to geminate rebinding of NO. The νFe-His stretching is absent in the spectrum of nitrosylated Cyt c at Δt = -5 ps (Fig. 4E) due to planarity of the six-coordinate heme and appears at 233 cm-1 immediately after NO photodissociation due to heme doming. The population of domed species decreases rapidly as probed by difference TR3 spectra from 2 to 25 ps (Fig. 4E) and the kinetics of the νFe-His band intensity deviates again from that measured by TA (Fig. 4F). Contrary to Mb, NO geminate rebinding in Cyt c is monoexponential and fast (τ1 = 8.4 ps); solving the rate equations for a monoexponential behavior yields a relaxation time constant τD = 6 ± 2 ps, revealing less constraints on the iron through the proximal histidine than in Mb. Thus, neither the Cyt c rigidity due to heme-protein covalent links nor the monoexponential character of NO rebinding leads to a quasi-instantaneous (femtosecond) domed-to-planar heme transition that again occurs with a picosecond delay.
Heme Iron Relaxation in the Allosteric Protein Hemoglobin.
Because the T → R allosteric transition in hemoglobin is triggered by the iron motion toward the heme plane, we aimed at verifying whether the same behavior is observed in the tetrameric Hb. As a result of TR3 and TA measurements on Hb-NO (Fig. 4 G and H, Fig. S1E, and Table 1), we have also found the retarded νFe-His kinetics in Hb, but with a time constant τD = 15 ± 6 ps, faster than in Mb, showing that the interaction between monomers does not increase the constraints exerted on the heme iron in the T state of Hb with respect to its homologous monomeric counterpart. The implications are discussed below.
Discussion
We report that the heme iron does not move instantaneously into the macrocycle plane after binding NO as a sixth coordination, but experiences a delayed picosecond transition from domed-to-planar heme for all proteins studied here, with the time constant depending upon the protein. This iron motion represents the very first structural event in heme proteins taking place after NO binding. This finding has several implications.
First, NO can bind to a domed five-coordinate high-spin ferrous heme. Such a possibility has been hypothesized (15) from the absence of energy barrier for NO rebinding and agrees with theoretical calculation indicating that NO can bind to an out-of-plane iron (37). The ability to bind to a domed heme contributes to the difference in reactivity between NO and CO (SI Text), and we carried out preliminary TR3 experiments that revealed the same behavior for Mb-O2 interaction as Mb-NO.
Second, the delayed iron motion reveals structural constraints exerted by the protein on the heme. Such constraints imply that structural changes of the heme pocket and/or its close vicinity have taken place after the dissociation of diatomics (faster than τD). A time-resolved circular dichroism (TRCD) study on Mb-CO (38) reported a motion of the proximal histidine with 7-ps time constant after CO dissociation, and a TR3 study (39), probing the aromatic Mb side chains in the heme vicinity, observed localized structural motions with 8-ps time constant. Furthermore, another TRCD study (40) has shown that the primary protein structural change after gaseous ligand release is the same for Hb-CO and Mb-CO and remarkably, for both Mb and Hb a similar “scissoring” concerted motion of the α-helices positioned on each side of the heme pocket has been observed (13, 41) during the R → T transition. Perutz described the unliganded deoxy-Hb as a “tensed” structure, having a strain between the proximal histidine and the heme iron (9, 10), which relaxes in the liganded oxy-Hb state. The constraints in Hb were demonstrated in conditions where the Fe-His bond is broken upon diatomic binding (42). The removal of the underlying constraints in the heme vicinity during the T → R transition, first proposed by Perutz for Hb (9), necessitates overcoming an energy barrier and thus cannot be immediate, but requires some time, causing the retardation of iron movement exactly as we observed in the present study for Hb-NO. The observed delayed motion from domed-to-planar heme (τD = 15 ps) may constitute the very first step triggering the T → R allosteric transition in hemoglobin.
An evident effect of the protein structure on the heme iron motion is revealed by the DHP versus Mb experiment. Sharing the same globin fold but different sequence, DHP experiences faster domed-to-planar heme transition than Mb (7 ps versus 30 ps). This contrasting behavior suggests less constraint in DHP and a difference in energy barrier to be overcome before iron motion. Remarkably, a photoacoustic calorimetric study found that DHP is more flexible than Mb (36). Taken together, these findings reveal the importance of side-chain interactions for the flexibility of the protein passed onto the ultrafast iron motion. It is important to note that both Mb and Hb experience concerted motions of entire helices (13, 41), whereas the peroxidase function of DHP does not require such a motion. It is also noteworthy that DHP and Cyt c have similar time constant τD despite their different structure and heme linking, revealing that several factors, not only the protein flexibility, can influence the heme response kinetics.
So far, it has been a commonly accepted idea that diatomic ligand binding produces an instantaneous planar heme. As our data show, this is not the case. The difference in time constant of heme structural response depending on ligand release or binding must be put in context of allostery. The quasi-instantaneous motion of heme iron upon gaseous ligand dissociation can be considered as the very primary triggering event for further conformational changes toward the equilibrium unliganded deoxy state (11). By contrast, for the reverse process of NO binding, the primary structural event of domed-to-planar heme transition takes ∼30 ps in Mb and ∼15 ps in Hb. Therefore, the view that in-plane motion of the heme iron following gaseous ligand binding is an instantaneous mere mechanistic event, like pulling a switch (9) whose motion is subsequently propagated through the protein structure bond after bond, should be revised. Our results support the view that ligand binding changes the protein potential energy curve (43) so that thermally driven (44, 45) fluctuations facilitate overcoming an energy barrier toward a new energy minimum, leading to the motion of the heme iron. We suggest that these constraints, revealed by the delayed domed-to-planar heme transition, play a crucial role in switching from one allosteric state into another in Hb and possibly also in heme-based sensors.
Materials and Methods
Time-Resolved Raman Spectroscopy.
The TR3 apparatus has 0.7-ps temporal resolution and 25-cm-1 spectral resolution (28, 46, 47). The broad band photolysis pulse (560–570 nm; 2 μJ; 100 fs) is delayed and collinearly superimposed to the probe beam (435 nm; 25 cm-1; 25 nJ). The pure transient TR3 spectrum of photoproduct at a given time delay is obtained as described in SI Text. Time-resolved absorption spectroscopy and sample preparation are described in SI Text.
Calculation of Time Constant τD from the Kinetics of νFe-His Intensity.
To obtain the time constant τD for the transition from domed 6-c to planar 6-c heme (D → A, Fig. 2), we resolved the rate equations for population decay of all nonplanar heme species (see SI Text). The kinetic model employed was similar to that of ref. 15 but with the additional species domed 6c-heme (D). The set of following rate equations was solved:
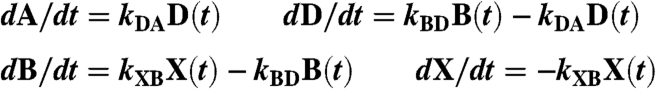 |
[1] |
with the total population A(t) + D(t) + B(t) + X(t) + const = 1. The only planar heme species is A(t) so that the total nonplanar population is 1 - A(t) = D(t) + B(t) + X(t) + const. The initial conditions are A(0) = 0, D(0) = 0. X(0) and B(0) are normalized amplitudes of rebinding phases after NO photodissociation (Table 1). The rate constants kXB and kBD were measured by transient absorption. kDA = 1/τD is to be determined and was varied until the kinetics of the nonplanar population D(t) + B(t) + X(t) + const fits the experimental points obtained from the normalized area of the νFe-His Raman band.
Supplementary Material
Acknowledgments.
We thank A. Jasaitis and S. Cianetti for their help in preparing the samples and J.M. Sintes and X. Solinas for technical assistance. S.G.K. was supported by Ecole Polytechnique and Institut National de la Santé et de la Recherche Médicale during his work at Laboratoire d’Optique et Biosciences.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.0912938107/-/DCSupplemental.
References
- 1.Ignarro LJ, Buga GM, Wood KS, Byrns RE, Chaudhuri G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc Natl Acad Sci USA. 1987;84:9265–9269. doi: 10.1073/pnas.84.24.9265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Knowles RG, Palacios M, Palmer RMJ, Moncada S. Formation of nitric oxide from L-arginine in the central nervous system—A tranduction mechanism for stimulation of the soluble guanylate cyclase. Proc Natl Acad Sci USA. 1989;86:5159–5162. doi: 10.1073/pnas.86.13.5159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Verma A, Hirsch DJ, Glatt CE, Ronnet GV, Snyder SH. Carbon monoxide—a putative neuronal messenger. Science. 1993;259:381–384. doi: 10.1126/science.7678352. [DOI] [PubMed] [Google Scholar]
- 4.Kim HP, Ryter SW, Choi AMK. CO as a cellular signaling molecule. Annu Rev Pharmacol Toxicol. 2006;46:411–449. doi: 10.1146/annurev.pharmtox.46.120604.141053. [DOI] [PubMed] [Google Scholar]
- 5.Ghosh A, editor. The Smallest Biomolecules: Diatomics and Their Interactions with Heme Proteins. Amsterdam: Elsevier; 2008. [Google Scholar]
- 6.Brunori M. Nitric oxide moves myoglobin centre stage. Trends Biochem Sci. 2001;26:209–210. doi: 10.1016/s0968-0004(01)01824-2. [DOI] [PubMed] [Google Scholar]
- 7.Brunori M, Vallone B. A globin for the brain. FASEB J. 2006;20:2192–2197. doi: 10.1096/fj.06-6643rev. [DOI] [PubMed] [Google Scholar]
- 8.Monod J, Wyman J, Changeux JP. On nature of allosteric transitions—a plausible model. J Mol Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 9.Perutz MF. Stereochemistry of cooperative effects in haemoglobin. Nature. 1970;228:726–793. doi: 10.1038/228726a0. [DOI] [PubMed] [Google Scholar]
- 10.Perutz MF, Wilkinson AJ, Paoli M, Dodson GG. The stereochemical mechanism of the cooperative effects in hemoglobin revisited. Annu Rev Biophys Biomol Struct. 1998;27:1–34. doi: 10.1146/annurev.biophys.27.1.1. [DOI] [PubMed] [Google Scholar]
- 11.Franzen S, Lambry JC, Bohn B, Poyart C, Martin JL. Direct evidence for the role of heme doming as the primary event in the cooperative transition of hemoglobin. Nat Struct Biol. 1994;1:230–233. doi: 10.1038/nsb0494-230. [DOI] [PubMed] [Google Scholar]
- 12.Zhu L, Sage JT, Champion PM. Observation of coherent reaction dynamics in heme-proteins. Science. 1994;266:629–632. doi: 10.1126/science.7939716. [DOI] [PubMed] [Google Scholar]
- 13.Kachalova GS, Popov AN, Bartunik HD. A steric mechanism for inhibition of CO binding to heme proteins. Science. 1999;284:473–476. doi: 10.1126/science.284.5413.473. [DOI] [PubMed] [Google Scholar]
- 14.Vos MH. Ultrafast dynamics of ligands within heme proteins. Biochim Biophys Acta Bioenergetics. 2008;1777:15–31. doi: 10.1016/j.bbabio.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 15.Ionascu D, et al. Temperature-dependent studies of NO recombination to heme and heme proteins. J Am Chem Soc. 2005;127:16921–16934. doi: 10.1021/ja054249y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lim MH, Jackson TA, Anfinrud PA. Ultrafast rotation and trapping of carbon monoxide dissociated from myoglobin. Nat Struct Biol. 1997;4:209–214. doi: 10.1038/nsb0397-209. [DOI] [PubMed] [Google Scholar]
- 17.Kim S, Jin G, Lim M. Dynamics of geminate recombination of NO with myoglobin in aqueous solution probed by femtosecond mid-IR spectroscopy. J Phys Chem B. 2004;108:20366–20375. [Google Scholar]
- 18.Mizutani Y, Kitagawa T. Direct observation of cooling of heme upon photodissociation of carbonmonoxy myoglobin. Science. 1997;278:443–446. doi: 10.1126/science.278.5337.443. [DOI] [PubMed] [Google Scholar]
- 19.Findsen EW, Friedman JM, Ondrias MR, Simon SR. Picosecond time resolved resonance Raman studies of hemoglobin—implications for reactivity. Science. 1985;229:661–665. doi: 10.1126/science.4023704. [DOI] [PubMed] [Google Scholar]
- 20.Schlichting I, Berendzen J, Phillips GN, Sweet RM. Crystal structure of photolyzed caronmonoxy-myoglobin. Nature. 1994;371:808–812. doi: 10.1038/371808a0. [DOI] [PubMed] [Google Scholar]
- 21.Schotte F, et al. Watching a protein as it functions with 150-ps time-resolved X-ray crystallography. Science. 2003;300:1944–1947. doi: 10.1126/science.1078797. [DOI] [PubMed] [Google Scholar]
- 22.Bourgeois D, et al. Extended subnanosecond structural dynamics of myoglobin revealed by Laue crystallography. Proc Natl Acad Sci USA. 2006;103:4924–4929. doi: 10.1073/pnas.0508880103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schotte F, Soman J, Olson JS, Wulff M, Anfinrud PA. Picosecond time-resolved X-ray crystallography: probing protein function in real time. J Struct Biol. 2004;147:235–246. doi: 10.1016/j.jsb.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 24.Kruglik SG, et al. Subpicosecond oxygen trapping in the heme pocket of the oxygen sensor FixL observed by time-resolved resonance Raman spectroscopy. Proc Natl Acad Sci USA. 2007;104:7408–7413. doi: 10.1073/pnas.0700445104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mizutani Y, Kitagawa T. Ultrafast structural relaxation of myoglobin following photodissociation of carbon monoxide probed by time-resolved resonance Raman spectroscopy. J Phys Chem B. 2001;105:10992–10999. [Google Scholar]
- 26.Kitagawa T, Li X-Y. Heme protein structure and the iron-histidine stretching mode. In: Spiro TG, editor. Biological Applications of Raman Spectroscopy. Vol. 3. New York: Wiley; 1988. pp. 97–131. [Google Scholar]
- 27.Stavrov SS. The effect of iron displacement out of the porphyrin plane on the resonance Raman spectra of heme-proteins and iron porphyrins. Biophys J. 1993;6:1942–1950. doi: 10.1016/S0006-3495(93)81265-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Negrerie M, Cianetti S, Vos MH, Martin JL, Kruglik SG. Photoinduced coordination dynamics of cytochrome c: Ferrous versus ferric species studied by time-resolved resonance Raman and transient absorption spectroscopies. J Phys Chem B. 2006;110:12766–12781. doi: 10.1021/jp0559377. [DOI] [PubMed] [Google Scholar]
- 29.Negrerie M, et al. Role of heme iron coordination and protein structure in the dynamics and geminate rebinding of nitric oxide to H93G myoglobin: Implications for NO-sensors. J Biol Chem. 2006;281:10389–10398. doi: 10.1074/jbc.M513375200. [DOI] [PubMed] [Google Scholar]
- 30.Ye X, et al. Investigations of heme protein absorption line shapes, vibrational relaxation, and resonance Raman scattering on ultrafast time scales. J Phys Chem A. 2003;107:8156–8165. [Google Scholar]
- 31.Spiro TG. Resonance Raman spectroscopy of metalloporphyrins. In: Spiro TG, editor. Biological Applications of Raman Spectroscopy. Vol. 3. New York: Wiley; 1988. pp. 1–37. [Google Scholar]
- 32.Shreve AP, Franzen S, Simpson MC, Dyer RB. Dependence of NO recombination dynamics in horse myoglobin on solution glycerol content. J Phys Chem B. 1999;103:7969–7975. [Google Scholar]
- 33.Haddad RE, et al. Origin of the red shifts in the optical absorption bands of nonplanar tetraalkylporphyrins. J Am Chem Soc. 2003;125:1253–1268. doi: 10.1021/ja0280933. [DOI] [PubMed] [Google Scholar]
- 34.Stavrov SS. Optical absorption band III of deoxyheme proteins as a probe of their structure and dynamics. Chem Phys. 2001;271:145–154. [Google Scholar]
- 35.Olson JS, Phillips GN. Myoglobin discriminates between O2, NO, and CO by electrostatic interactions with the bound ligand. J Biol Inorg Chem. 1997;2:544–552. [Google Scholar]
- 36.Miksovska J, Horsa S, Davis MF, Franzen S. Conformational dynamics associated with photodissociation of CO from dehaloperoxidase studied using photoacoustic calorimetry. Biochemistry. 2008;47:11510–11517. doi: 10.1021/bi8012033. [DOI] [PubMed] [Google Scholar]
- 37.Franzen S. Spin-dependent mechanism for diatomic ligand binding to heme. Proc Natl Acad Sci USA. 2002;99:16754–16759. doi: 10.1073/pnas.252590999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dartigalongue T, Hache F. Observation of sub-100 ps conformational changes in photolyzed carbonmonoxy-myoglobin probed by time-resolved circular dichroism. Chem Phys Lett. 2005;415:313–316. [Google Scholar]
- 39.Sato A, Gao Y, Kitagawa T, Mizutani Y. Primary protein response after ligand photodissociation in carbonmonoxy myoglobin. Proc Natl Acad Sci USA. 2007;104:9627–9632. doi: 10.1073/pnas.0611560104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dartigalongue T. Palaiseau, France: Ecole Polytechnique; 2005. Conformational dynamics of myoglobin studied by time-resolved circular dichroism. PhD thesis. [Google Scholar]
- 41.Rodgers KR, Spiro TG. Nanosecond dynamics of the R → T transition in hemoglobin: Ultraviolet Raman studies. Science. 1994;265:1697–1699. doi: 10.1126/science.8085153. [DOI] [PubMed] [Google Scholar]
- 42.Paoli M, Dodson G, Liddington RC, Wilkinson AJ. Tension in haemoglobin revealed by Fe-His(F8) bond rupture in the fully liganded T-state. J Mol Biol. 1997;271:161–167. doi: 10.1006/jmbi.1997.1180. [DOI] [PubMed] [Google Scholar]
- 43.Goodey NM, Benkovic SJ. Allosteric regulation and catalysis emerge via a common route. Nat Chem Biol. 2008;4:474–482. doi: 10.1038/nchembio.98. [DOI] [PubMed] [Google Scholar]
- 44.Armstrong MR, Ogilvie JP, Cowan ML, Miller RJD. Observation of the cascaded atomic-to-global length scales driving protein motion. Proc Natl Acad Sci USA. 2003;100:4990–4994. doi: 10.1073/pnas.0936507100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Popovych N, Sun S, Ebright RH, Kalodimos CG. Dynamically driven protein allostery. Nat Struct Mol Biol. 2006;13:831–838. doi: 10.1038/nsmb1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kruglik SG, et al. Molecular basis for nitric oxide dynamics and affinity with Alcaligenes xylosoxidans cytochrome c′. J Biol Chem. 2007;282:5053–5062. doi: 10.1074/jbc.M604327200. [DOI] [PubMed] [Google Scholar]
- 47.Kruglik SG, Lambry J-C, Martin J-L, Vos MH, Negrerie M. Sub-picosecond Raman spectrometer for time-resolved studies of structural dynamics in heme proteins. J Raman Spectrosc. 2010 doi: 10.1002/jrs.2685. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.