

Abstract
Drug resistance resulting from mutations to the target is an unfortunate common phenomenon that limits the lifetime of many of the most successful drugs. In contrast to the investigation of mutations after clinical exposure, it would be powerful to be able to incorporate strategies early in the development process to predict and overcome the effects of possible resistance mutations. Here we present a unique prospective application of an ensemble-based protein design algorithm, K∗, to predict potential resistance mutations in dihydrofolate reductase from Staphylococcus aureus using positive design to maintain catalytic function and negative design to interfere with binding of a lead inhibitor. Enzyme inhibition assays show that three of the four highly-ranked predicted mutants are active yet display lower affinity (18-, 9-, and 13-fold) for the inhibitor. A crystal structure of the top-ranked mutant enzyme validates the predicted conformations of the mutated residues and the structural basis of the loss of potency. The use of protein design algorithms to predict resistance mutations could be incorporated in a lead design strategy against any target that is susceptible to mutational resistance.
Keywords: negative design algorithm, DHFR, MRSA, K* design algorithm
Resistance has been observed for even the most reserved antibiotics, sometimes after only brief clinical exposure. One of the most common resistance mechanisms is the accumulation of mutations in an enzyme target, creating an active site that can no longer accommodate the inhibitor yet maintains function. When these resistance mutations are discovered in the clinic, the mutants must be identified and studied, forcing the drug design process to start anew. To address this problem in preclinical drug discovery, resistance mutants are generated and studied in vitro with labor-intensive experiments. In contrast, it would be useful to predict resistance mutations in silico during the very early stages of drug discovery, thus encouraging strategies to overcome these limitations during the design process. In response to this need, we have developed and experimentally tested a protocol to computationally predict resistance mutations in a protein target, using algorithms for positive and negative structure-based protein design.
Protein design algorithms have recently been developed and used to reengineer proteins and enzymes to bind unique ligands. In the case of enzymes, successful design requires catalytic activity as well as binding. A promising approach involves ensemble-based scoring and search algorithms for protein design, which have been applied to modify the substrate specificity of an antibiotic-producing enzyme in the nonribosomal peptide synthetase pathway (1). New algorithms for protein design, an example of which is called K∗, combine a statistical mechanics-derived ensemble-based approach to computing the binding constant with the speed and completeness of a branch-and-bound pruning algorithm (2–4). In addition, efficient deterministic algorithms include provable ϵ-approximation algorithms for estimating partition functions in order to model binding to arbitrary precision (4). Other examples of successful applications of computational protein design include design of unique enzymes (5, 6), enhancement of antibody affinity (7), and design of a unique protein fold (8).
There have been few previous attempts to predict potential resistance mutations; most of these have been applied retrospectively to predict known mutations that occur in response to clinically used antibiotics. Predictions have identified peptide sequences that would bind more tightly to a mutant HIV protease enzyme (9), profiled tyrosine kinase inhibitors (10), and estimated “vitality values” to account for the catalytic activity of resistant mutants of HIV protease (11). A recent review (12) summarizes structure- and sequence-based attempts to predict known mutations in HIV protease and reverse transcriptase.
Using structure-based design, we have been developing a unique class of propargyl-linked antifolate inhibitors that are active against several pathogenic species of dihydrofolate reductase (DHFR), specifically, methicillin-resistant Staphylococcus aureus (MRSA) DHFR (Sa DHFR) (13, 14). As MRSA has an extensive array of resistance mechanisms, it is critical to consider the likely development of resistance for any new inhibitors. Therefore, given that we have determined high resolution structures of wild-type Sa DHFR bound to these propargyl-linked antifolates (13, 14), we considered this to be an excellent case to apply structure-based protein design algorithms for resistance mutation prediction.
Here, we report a prospective study that uses the protein design algorithm, K∗, to predict resistance mutations in DHFR from MRSA toward a potent propargyl-linked antifolate. Structure-based negative design predicted mutations to destabilize the binding of the inhibitor; positive design predicted mutations that stabilize the native protein function. Intersecting the top-scoring positive and negative designs predicted candidate resistance mutations. Four of the top ten ranked mutant enzymes were created and evaluated. Three of the mutants indeed maintained activity and displayed lower (18-, 9- and 13-fold) affinity for the inhibitor. A crystal structure of the top-ranked mutant bound to the inhibitor shows a conformation of the ligand that clearly has significantly fewer interactions with the protein. This unique and expedient approach to resistance mutation prediction should be useful for the development of inhibitors toward other targets for which drug therapy is limited by mutational resistance.
Results and Discussion
Compound 1 (Fig. 1) is a lead inhibitor of MRSA DHFR (Ki = 10 nM) and possesses antibacterial activity against isolates of MRSA in culture. In order to elucidate the basis of its potency, we determined a crystal structure of the enzyme bound to its cofactor, NADPH and compound 1. Crystals of the wild-type Sa DHFR showed diffraction amplitudes to 2.1 Å (Table 1); the structure was determined with difference Fourier methods using coordinates of Sa (F98Y)DHFR (15). The model reveals the overall characteristic “DHFR fold” that consists of an eight-stranded beta sheet and four alpha helices connected by flexible loop regions. The pyrimidine ring of compound 1 forms two conserved hydrogen bonds with Asp 27 and the 4-amino group forms two additional hydrogen bonds with the backbone carbonyl atoms of residues Leu 5 and Phe 92 (Fig. 1). The propargyl linker and propargylic methyl group form van der Waals contacts with Phe 92 and Leu 20. The methyl group at the C6 position forms van der Waals interactions with Leu 20 and Leu 28 and the meta-biphenyl ring extends into a hydrophobic pocket formed primarily by residues Ile 50, Leu 28, and Leu 54.
Fig. 1.

Stereo view of the superposition of the ternary crystal structures of Sa (WT) DHFR (green) and Sa(V31Y/F92I) DHFR (magenta) bound to NADPH and compound 1 (A) as B) stick models and C) surface rendering.
Table 1.
Statistics of data collection and refinement
| Complex | Sa (WT)/ cmpd 1/NADPH | Sa (V31Y,F92I)/ cmpd 1/NADPH |
| PDB ID | 3F0Q | 3LG4 |
| Space group | P6122 | P61 |
| Unit cell (a,b,c in Å) | a = b = 79.16, c = 108.80 | a = b = 88.752, c = 103.167 |
| Resolution, (last shell, Å) | 25.9 - 2.10 (2.29 - 2.08) | 42.8 - 3.15 (3.23 - 3.15) |
| Completeness, % (last shell,%) | 87.9 (95.3) | 91.2 (99.7) |
| Redundancy (last shell) | 10.40 (12.2) | 5.0 (5.4) |
| Rsym (last shell) | 0.050 (0.143) | 0.061 (0.379) |
| 〈I/σ〉 (last shell) | 8.6 (2.7) | 14.5 (3) |
| Refinement statistics | ||
| R-factor/Rfree | 0.196; 0.231 | 0.26; 0.292 |
| Rms deviation bond lengths (Å), angles (°) | 0.010; 1.386 | 0.007; 1.219 |
| Average B factors (Å2): overall; NADPH; compound 1 | 20.6; 16.7; 21.8 | 88.7; 94.7; 68.5 |
| Residues in most favored regions, allowed regions (%) | 90.4, 9.6 | 87.5; 12.5 |
In order to identify residues that when mutated may incur a binding penalty for compound 1 but that would maintain binding to the substrate, dihydrofolate, K∗ searches using the minDEE/A ∗ /K∗ algorithms (1, 4) were performed. For these searches, the algorithm used as input the crystal structure of Sa DHFR:compound 1∶NADPH as well as a model of Sa DHFR bound to NADPH and dihydrofolate, which was adapted from coordinates of a single mutant Sa DHFR bound to the same ligands (15). Ten active site residues (Leu 5, Val 6, Leu 20, Asp 27, Leu 28, Val 31, Thr 46, Ile 50, Leu 54, and Phe 92) were modeled as flexible using rotamers (16) and allowed to keep their wild-type identity or to mutate within a restricted group of amino acids (see Materials and Methods) that conserve the amino acid property or correlate with a different DHFR species. In order to comprehensively test the algorithm, the group included mutations that represent both single- and double-nucleotide polymorphisms. While single-nucleotide polymorphisms are the most prevalent mechanism of mutational resistance, more complex mutations do evolve such as those in HIV-1 reverse transcriptase (17–19), hepatitis C protease (20), hepatitis B reverse transcriptase (21), and Helicobacter pylori rpoB (22).
Based on the input model, K∗ searches designed to identify up to 2-point mutations were performed separately for dihydrofolate and compound 1. K∗ scores are computed as a ratio of Boltzmann-weighted partition functions over rotamer-based conformational ensembles for the bound protein-ligand complex, the free protein, and free ligand (see Materials and Methods). Since higher K∗ scores imply better affinity, we focused our attention on pairs of mutations that scored highly for dihydrofolate and poorly for compound 1, particularly on those that returned a score of zero for compound 1 (Table 2) and a resultant score ratio of infinity. A mutant K∗ score of 0 for compound 1 implies one of two scenarios: (1) K∗ pruned all rotameric conformations for these mutants; or (2) the computed partition function for the bound protein-ligand complex for a given mutant was significantly less (in the current redesigns: more than eight orders of magnitude) than the product of the partition functions for the free protein and the free ligand. Typically, scenario (1) occurs when significant steric interference exists either within the protein or between the protein and the ligand for all possible rotameric conformations, even after minimization. An initial steric overlap (before minimization) between two rotamers of more than 1.5 Å was considered significant and the corresponding pair of rotamers was pruned from further consideration (Materials and Methods). Scenario (2) is typically caused by significantly unfavorable interactions between the protein and the ligand, as opposed to the protein in its unbound form (Table 2). The different mutants with a score of 0 for compound 1 are thus not equivalent with respect to the predicted destabilized interactions with compound 1. There were 105 mutants with a score ratio of infinity; the top ten exhibited a dihydrofolate score considerably better than the others and were further investigated. These ten mutants fell into two groups: the first group contained mutations at residues 31 and 92, the second group contained mutations at positions 50 and 92 (Table 2). The computed partition functions for the bound protein-ligand complex (with compound 1) and the free protein (Table 2 D, E), along with the lowest energy for the respective ensemble of conformations (Table S1) suggest that compound 1 has significantly destabilized interactions with these mutants.
Table 2.
The top ten amutants as ranked by the score ratio criterion are shown with the respective K∗ scores with bdihydrofolate (DHF)
| Compound 1 | ||||
| Partition functionc | ||||
| Rank | Mutationsa | DHF K∗ Scoreb | FPd | PLe |
| 1 | V31Y/F92I | 4.30 × 1040 | 6.6 × 10292 | 2.1 × 10132 |
| 2 | V31Y/F92V | 3.81 × 1040 | 2.9 × 10301 | 4.3 × 10143 |
| 3 | V31Y/F92S | 3.13 × 1040 | 1.2 × 10331 | 2.4 × 10173 |
| 4 | V31Y/F92A | 2.94 × 1040 | 1.3 × 10330 | 4.4 × 10171 |
| 5 | V31Y/F92M | 6.77 × 1038 | 3.4 × 10332 | 1.0 × 10169 |
| 6 | V31Y/F92L | 6.38 × 1038 | 2.3 × 10320 | 2.7 × 10150 |
| 7 | V31F/F92L | 6.01 × 1033 | 2.3 × 10327 | 2.3 × 10310 |
| 8 | I50W/F92M | 7.70 × 1032 | 8.8 × 10340 | 5.0 × 10327 |
| 9 | I50W/F92S | 2.74 × 1032 | 1.9 × 10339 | 3.2 × 10325 |
| 10 | I50W/F92A | 2.10 × 1032 | 1.9 × 10338 | 3.9 × 10323 |
For each of the mutants, the computed cpartition function for the dfree protein (FP) and eprotein-ligand (PL) complex (with compound 1) are also shown; all of the top 10 mutants had a K∗ score of 0 for compound 1. Mutants shown in bold were selected for experimental validation.
Interestingly, the wild-type sequence was ranked #306 out of 1,173 with dihydrofolate and #369 out of 1,173 with compound 1, suggesting that a subset of the higher-ranked sequences may have improved binding to dihydrofolate or compound 1.
Experimental Validation of the Predicted Mutants.
To test the validity of the results of the algorithm, we created four of the mutants that show diversity at each of the positions (ranked by the algorithm #1, 3, 7, and 9) using site-directed mutagenesis and purification procedures similar to the wild-type enzyme (13, 14). Enzyme activity assays show that there is only a modest 3-fold reduction in KM values for dihydrofolate (Table 3) for the Val31/Phe92 mutants. While the kcat and kcat/KM values are lower than those of the wild-type enzyme (Table 3), the losses are within the range of other clinically observed DHFR mutants. For example, the F57L mutation in Plasmodium vivax DHFR (pyrimethamine, cycloguanil and WR99210 resistance) (23), the L22R mutation in human DHFR (methotrexate resistance) (24) and the A16V mutation in Plasmodium falciparum DHFR (cycloguanil resistance) (25) suffer 220-, 250-, and 680-fold losses, respectively, in kcat/KM. The ninth-ranked mutant, Sa (I50W, F92S) DHFR, was not active, suggesting that the Ile50Trp mutation prevents binding to the substrate. The results for the mutants ranked #1, 3, and 7 by the algorithm experimentally validate the positive design component of the K∗ algorithm and its success in predicting mutants that retain catalytic activity.
Table 3.
Kinetic parameters for the wild-type and mutant enzymes
| Enzyme | KM, units in μM | kcat, units in 1/s | kcat/KM (fold decrease) |
| Sa (WT) | 14.5 ± 3.5 | 31 | 2.14 (1.00) |
| Sa (V31Y, F92I) | 43 ± 2.6 | 2.8 | 0.06 (36) |
| Sa (V31Y, F92S) | 58 ± 3.0 | 1.4 | 0.02 (107) |
| Sa (V31F, F92L) | 45 ± 4.3 | 0.31 | 0.007 (306) |
In order to assess the results of the negative design component of the algorithm, Ki values were measured for the wild-type and Sa (V31Y, F92I) DHFR enzymes with compound 1. Dixon plots show that the inhibitor binds competitively (Figs. S1 and S2); analysis of the plots yields Ki values of 7.5 nM and 128 nM for the wild type and mutant, respectively. Ki values were also calculated from IC50 values and KM values (26) for all active mutants (Table 4). The top-ranked resistance mutant, Sa (V31Y, F92I) DHFR, shows the greatest (18-fold) loss in affinity for compound 1. Mutants Sa (V31Y, F92S) and Sa (V31F, F92L) DHFR also show significant (9-fold and 13-fold, respectively) losses in potency, suggesting that the algorithm is also successful in its negative design component. The success of the algorithm prompted the investigation of a structure of the mutant to determine why the resistance mutations at positions 31 and 92 retain activity but lose affinity for compound 1.
Table 4.
Inhibition assays for enzymes and compound 1
| Enzyme | Ki, nM | Fold loss* |
| Sa wild-type (WT) | 10 | 1.0 |
| Sa (V31Y,F92I) | 180 | 18 |
| Sa (V31Y, F92S) | 87 | 8.7 |
| Sa (V31F, F92L) | 130 | 13 |
*Fold loss = Ki value for enzyme/Ki value for WT.
Determination of a Crystal Structure of Sa (V31Y, F92I) DHFR, NADPH and Compound 1.
Crystals of Sa (V31Y, F92I) DHFR showed diffraction amplitudes to 3.15 Å (Table 1); the structure of the mutant was determined using difference Fourier methods based on the wild-type structure bound to NADPH and compound 3 (Table S2) as a model (PDB ID: 3FQC) (13). There is a high degree of similarity between Sa (wild-type) and Sa (V31Y, F92I) DHFR, reflected in a root mean square deviation for 157 Cα atoms of 0.355 Å. The similarity of the enzymes is also reflected in their melting temperatures, as determined by circular dichroism (Tm wild-type = 42.5 °C, Tm Sa(V31Y,F92I) = 36.3 °C, graphs shown in Fig. S3). The Sa (V31Y, F92I) DHFR mutant structure exhibits the standard, extended conformation of NADPH, in contrast to the alternate conformation observed in several structures of the Sa (F98Y) DHFR mutant (13). In contrast to the wild-type structure in which the ligand fully occupies the site, compound 1 binds the mutant active site with 50% occupancy, suggesting that the V31Y and F92I mutations affect ligand binding.
Despite the moderate resolution of the data for the mutant enzyme, the electron density maps revealed significant structural details including side chain and ligand orientations in the active site that disclose the basis of the lower affinity of compound 1 (Fig. 1). Strong hydrophobic interactions made with the native Phe92 residue and propargyl linker of compound 1 are reduced with the mutation to Ile92. The Val31Tyr mutation introduces steric bulk in the active site that interferes with the 2-methyl substitution on the distal phenyl ring, causing the substituted biphenyl of the ligand to contort around the propargyl position and reorient by approximately 60°. Reorientation positions the two phenyl rings outside the main hydrophobic pocket, causing the loss or reduction of strong hydrophobic interactions with residues Leu 28, Val 31, Leu 54, and Phe 92. In the new position, the distal phenyl ring maintains interactions only with Leu 20. While it appears that the mutant enzyme may have bound the opposite enantiomer compared to that bound in the wild-type structure, the resolution of the electron density does not permit exact evaluation.
KM values suggest that active sites mutated at the Val31 and Phe92 positions retain affinity for the substrate, dihydrofolate. In order to understand why these mutations allow substrate binding, we compared the Sa (V31Y, F92I) DHFR crystal structure bound to compound 1 and NADPH with the lowest energy predicted structure for Sa (V31Y, F92I) DHFR bound to dihydrofolate (Fig. 2A). In contrast to compound 1, which relies on the interaction between the propargyl linker and Phe 92, dihydrofolate lacks a propargyl linker and is minimally affected by the Ile 92 mutation. In addition, the para-aminobenzoic acid moiety of dihydrofolate can be accommodated in the limited space near the Tyr 31 mutation. This accommodation differs from compound 1, where the biphenyl changes orientation to avoid steric repulsion with Tyr 31. Although a crystal structure for the Sa (V31F, F92L) DHFR mutant was not obtained, it appears that the dihydrofolate molecule could also be accommodated in the space near the Phe 31 mutation.
Fig. 2.

Stereo views of the superposition of the Sa(V31Y/F92I) DHFR crystal structure (magenta) with A) model of dihydrofolate in the active site (cyan) and B) the lowest energy model from the predicted ensemble (yellow).
To assess whether the K∗ algorithm predicted similar rotamer conformations as were observed in the crystal structure, the crystal structure of Sa (V31Y, F92I) DHFR was compared with the lowest energy predicted structure (Fig. 2B). The Tyr 31 mutation observed in the crystal structure has the same conformation as each of the top ten members of the ensemble of predicted structures. Although the rotamer for Ile 92 in the crystal structure differs from the predicted structure, the predicted residue is located in the same location and space as both the mutant Sa (V31Y, F92I) and Sa (WT) DHFR structures. Similarity of rotamer conformations suggests that the algorithm is accurate in predicting not only the identity of residues targeted for mutation, but also the proper orientation of mutated residues.
The conformation of compound 1 from the top K∗-predicted structure lacks the reorientation of the biphenyl observed in the Sa (V31Y, F92I) DHFR crystal structure. This reoriented conformation is significantly out of the range of ligand conformations input to the software to be modeled by K∗. For example, in the K∗ input ligand rotamer (see Materials and Methods section) closest to the ligand conformation in the Sa (V31Y, F92I) DHFR crystal structure, the biphenyl of compound 1 is rotated by approximately 60°.
The goal of the K∗ algorithm was to block binding to compound 1 by introducing mutations to DHFR, based on the selected set of input ligand conformations. Using this input model, the algorithm successfully identified mutations that have significantly diminished binding to compound 1. The ligand flexibility model used by K∗ significantly improves on models where ligands are treated as rigid or in which rigid ligand rotamers are used. However, due to the expense of the computation, the number of rotamers and the span of ligand conformations were still significantly limited. While, ideally, the entire feasible conformation space around a ligand should be evaluated by the computational procedure, this would come at the cost of a combinatorial increase in compute times. Optimal balance should thus be sought between the computational requirements and the comprehensiveness of the flexibility models.
Nevertheless, it is interesting to determine how a ligand conformation mimicking the one observed in the crystal structure of the Sa (V31Y, F92I) mutant complex would be scored by our algorithm. To this end, we set up and performed a K∗ score computation for the Sa (V31Y, F92I) mutant, with the ligand dihedrals constrained to a conformation close to the one observed in the mutant crystal structure. Interestingly, the computed lowest energy (approx. -179.4 kcal/mol) in the K∗ conformation ensemble for the bound protein-ligand complex was virtually identical to the lowest energy of the original K∗ ensemble (approx. -180.0 kcal/mol, see Table S1). Thus, even with the ligand conformation mimicking the one observed in the crystal structure complex, K∗ predicts the Sa (V31Y, F92I) mutant to have significantly destabilized interactions with compound 1 as compared to the free protein (see Table S1).
Elucidating Structure-Activity Relationships of Additional Antifolate Compounds.
The structural evidence suggests that the Val31Tyr and Phe92Ile mutations decrease affinity by reducing hydrophobic interactions with Phe 92 and introducing steric bulk at Val 31. In order to validate these hypotheses, we tested the activity of four other propargyl-based antifolates for the mutant enzymes and probed the structure-activity relationships. All four additional antifolates (compounds 2, 3, 4, and 5 shown in Table S2 with inhibition values) are potent inhibitors for the wild-type enzyme, but lose potency for all of the mutant enzymes. Compound 2, with an unsubstituted distal phenyl ring relative to the dimethyl substitution on compound 1, loses only 7-fold affinity for the Sa (V31Y, F92I) DHFR enzyme, validating that the unsubstituted phenyl is less affected by mutations at the Val 31 position. In contrast, compound 4, with an ethyl group at the C6 position of the diaminopyrimidine ring, is affected by the Val 31 mutation (exhibiting 10.5-, 16-, and 28-fold losses with each of the enzymes mutated at Val 31) because the ethyl group has extensive van der Waals contacts with Val 31.
Mutations at the Phe 92 position affect the interactions with the propargyl substitutions on the compounds. As such, these mutations reduce the affinity of compound 3, with a gem-dimethyl at the propargylic position, by 7–18-fold. Compound 5, with a para-biphenyl ring instead of the meta-biphenyl of compound 1, has lower affinity for the wild-type enzyme (Ki = 24 nM); analysis of cocrystal structures shows that the para-biphenyl ring juts out of the active site (13). Interestingly, the orientation of the para-biphenyl ring in these structures is similar to the new orientation adopted by the meta-biphenyl ring of compound 1 in the Sa (V31Y, F92I) DHFR structure.
Conclusions
Predicting potential resistance mutations in an enzyme target in silico would enable the design of compounds early in the drug design process that overcome these limitations. Towards that goal, we used the protein design algorithm, K∗, to predict mutations in DHFR from S. aureus. Positive design selected mutations that maintain binding to the substrate, dihydrofolate; negative design selected mutations that lower the affinity for a lead inhibitor and the intersection of these sets resulted in a ranked series of double mutants. Four of the ten top-ranked mutants were chosen for experimental validation. Excitingly, as predicted, three of the mutants maintain catalytic activity and show lower affinity for the inhibitor. A crystal structure of the top-ranked mutant further validates the predicted conformations of the mutated residues and reveals a conformation of the ligand with many fewer interactions than are apparent in the wild-type structure.
Materials and Methods
K∗.
The following mutations (one-letter amino acid codes) were allowed for the ten active site residues: Leu5, Val6, Leu20, Leu28, Val31, and Ile50—AVLIMFWY; Asp27—DE; Phe92—AVLIMFWYS. Residues Thr46 and Leu54 were modeled using rotamers but were not allowed to mutate. K∗ performed 2-point mutation searches, in which any two of the ten active site residues were allowed to simultaneously mutate, while the other eight residues were allowed to change rotamers. These searches yielded a total of 1,173 candidate mutants (that included the wild-type, 1-, and 2-point mutations), corresponding to 4.37 × 1010 (DHFR∶NADPH∶dihydrofolate) and 2.49 × 1012 (DHFR∶NADPH∶compound 1) conformations, for which K∗ scores were computed (see below). The use of a factor of ∼100 more conformations to predict mutants with reduced binding to compound 1 reflects the need for greater conformational flexibility for negative design.
NADPH and a steric shell of residues within 5 Å from the active site or 8 Å from dihydrofolate were included as part of the input structure. The substrate was modeled as flexible using rotamers and was allowed to rotate/translate. Modal values from the Penultimate rotamer library (16) were used for the amino acid side chains. Ligand rotamers were defined for dihydrofolate and compound 1 by sampling sets of rotatable bonds (see SI Text). For all rotamers, each dihedral was allowed to minimize within ± 9 ° from its initial value.
For each of the candidate mutants, positive design (dihydrofolate as substrate) and negative design (compound 1 as substrate) computations were performed and scored. The ratio of the K∗ scores for a given mutant with dihydrofolate and compound 1 was used to rank the candidate mutants; mutants with high ratios were predicted to be good dihydrofolate and poor compound 1 binders. The energy function consisted of the Amber vdW, electrostatic, and dihedral energies (27), and the EEF1 implicit pairwise solvation model (28) (see SI Text).
A K∗ score for a given mutant was computed as the ratio of the partition functions for the bound protein-ligand complex and the free protein and free ligand. Partition functions were computed over Boltzmann-weighted ensembles of conformations: a partition function q for a given ensemble S of conformations was computed as: 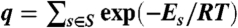 , where Es is the energy of conformation s, T is the temperature in Kelvin, and R is the gas constant. For each partition function computation, initial rotamer pruning based on the MinDEE algorithm (4) was first applied as a preprocessing step to reduce the number of candidate rotamers and rotamer-based conformations. MinDEE is provably accurate with continuously flexible rotamers defined over a bounded (but continuous) region of side-chain conformation space. Following, A∗ enumerated the remaining unpruned conformations in order of increasing lower bounds on their energies. For computational efficiency, a provably accurate ϵ-approximation algorithm was applied to guarantee the accuracy of the computed partition function based only on a small fraction of the remaining unpruned conformations (4). Using this ϵ-approximation algorithm, the A∗ enumeration could be provably halted once the computed partition function was guaranteed to be within ϵ from the full partition function (when all rotameric conformations are taken into account). An ϵ value of 0.03 guaranteed that the computed partition functions were at least 97% of the corresponding full partition functions (1, 4). The MinDEE/A∗ and K∗ algorithms return a gap-free list of predictions in the order of the predicted score (either empirical molecular mechanics energy [MinDEE], or free energy based on molecular ensembles [K∗]).
, where Es is the energy of conformation s, T is the temperature in Kelvin, and R is the gas constant. For each partition function computation, initial rotamer pruning based on the MinDEE algorithm (4) was first applied as a preprocessing step to reduce the number of candidate rotamers and rotamer-based conformations. MinDEE is provably accurate with continuously flexible rotamers defined over a bounded (but continuous) region of side-chain conformation space. Following, A∗ enumerated the remaining unpruned conformations in order of increasing lower bounds on their energies. For computational efficiency, a provably accurate ϵ-approximation algorithm was applied to guarantee the accuracy of the computed partition function based only on a small fraction of the remaining unpruned conformations (4). Using this ϵ-approximation algorithm, the A∗ enumeration could be provably halted once the computed partition function was guaranteed to be within ϵ from the full partition function (when all rotameric conformations are taken into account). An ϵ value of 0.03 guaranteed that the computed partition functions were at least 97% of the corresponding full partition functions (1, 4). The MinDEE/A∗ and K∗ algorithms return a gap-free list of predictions in the order of the predicted score (either empirical molecular mechanics energy [MinDEE], or free energy based on molecular ensembles [K∗]).
Mutant Enzyme Preparation.
Sa DHFR mutants ranked #1, 3, 7, and 9 by the K∗ algorithm were prepared with site-directed mutagenesis using the DNA encoding Sa (WT) DHFR as a template. All clones were verified by DNA sequencing. Mutant enzymes were expressed in E. coli BL21(DE3) cells and purified using procedures previously adapted for the wild-type protein (13).
Enzyme Assays.
Enzyme activity and inhibition assays were performed in triplicate for each DHFR mutant by monitoring the rate of NADPH oxidation by the DHFR enzyme at an absorbance of 340 nm (13). Kinetic parameters were measured by performing triplicate enzyme activity assays at varying substrate concentrations of dihydrofolate and analyzed with Lineweaver-Burk plots. KM values, in addition to the obtained IC50 values, were used to calculate Ki values for each enzyme and inhibitor (26). Ki values were also determined experimentally for compound 1 and the wild-type and Sa (V31Y, F92I) enzymes using four concentrations of substrate (50, 100, 150, and 200 μM) and four concentrations of inhibitor near the IC50 value (50, 75, 100, and 150 nM for wild-type and 150, 300, 450, and 600 nM for Sa(V31Y,F92I)DHFR). The inhibition data were analyzed with Dixon plots.
Enzyme Stability by Circular Dichroism.
Temperature-induced unfolding experiments were performed for both Sa (WT) and Sa (V31Y, F92I) DHFR by increasing the temperature from 5 °C to 70 °C and monitoring circular dichroism at 222 nm. CD measurements were taken at 1 °C temperature increments with an equilibration time of 2 min. Protein concentrations used in these experiments were 12 μM for Sa (WT) DHFR and 17 μM for Sa (V31Y, F92I) DHFR. To account for the difference in enzyme concentration, the data points were converted from millidegrees to molar ellipticity and then plotted to determine Tm values.
Crystallization.
Both Sa DHFR and Sa (V31Y, F92I) DHFR were crystallized using hanging-drop vapor diffusion. The purified enzymes (12 mg/mL) were incubated with compound 1 (1 mM) and NADPH (2 mM) for 2 h on ice. Crystals of the protein:ligand:NADPH complex were optimized in a crystallization solution containing 15% PEG MW10,000, 150 mM sodium acetate, and 100 mM MES pH 6.5.
Data Collection and Refinement.
Diffraction data with amplitudes extending to 2.1 Å or 3.15 Å were collected at National Synchrotron Light Source (beamline X29 or X25) for complexes of Sa DHFR and Sa (V31Y, F92I) DHFR, respectively. Data were indexed and scaled using HKL 2000 (29). Programs Coot (30) and Refmac (31) were used to build and refine the structure until an acceptable Rcryst and Rfree were achieved. The geometry of the structure was validated using Procheck (32) and Ramachandran plots. Data collection and refinement statistics are reported in Table 1.
Software.
All software is freely available open-source upon publication.
Supplementary Material
Acknowledgments.
The authors thank Pablo Gainza-Cirauqui, Nanda Karri, and Michael Lombardo as well as all members of the Donald and Anderson labs for thoughtful suggestions. We thank Dr. Dennis Wright's laboratory for providing compounds 1–5, Drs. Carol Teschke and Juliana Cortines for assistance with the circular dichroism experiments and the National Institutes of Health (NIH) for funding (GM78031 to B.R.D. and GM067542 to A.C.A.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 3F0Q and 3LG4).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1002162107/-/DCSupplemental.
References
- 1.Chen C, Georgiev I, Anderson A, Donald B. Computational structure-based redesign of enzyme activity. Proc Natl Acad Sci USA. 2009;106:3764–3769. doi: 10.1073/pnas.0900266106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Georgiev I, Donald B. Dead-end elimination with backbone flexibility. Bioinformatics. 2007;23:i185–194. doi: 10.1093/bioinformatics/btm197. [DOI] [PubMed] [Google Scholar]
- 3.Georgiev I, Keedy D, Richardson J, Richardson D, Donald B. Algorithm for backrub motions in protein design. Bioinformatics. 2008;24:i196–204. doi: 10.1093/bioinformatics/btn169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Georgiev I, Lilien R, Donald B. The minimized dead-end elimination criterion and its application to protein redesign in a hybrid scoring and search algorithm for computing partition functions over molecular ensembles. J Comput Chem. 2008;29:1527–1542. doi: 10.1002/jcc.20909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jiang L, et al. De Novo computational design of retro-aldol enzymes. Science. 2008;319:1387–1391. doi: 10.1126/science.1152692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rothlisberger D, et al. Kemp elimination catalysts by computational enzyme design. Nature. 2008;453:190–195. doi: 10.1038/nature06879. [DOI] [PubMed] [Google Scholar]
- 7.Lippow S, Wittrup K, Tidor B. Computational design of antibody affinity improvement beyond in vivo maturation. Nat Biotechnol. 2007;25:1171–1176. doi: 10.1038/nbt1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuhlman B, et al. Design of a novel globular protein fold with atomic-level accuracy. Science. 2003;302:1364–1368. doi: 10.1126/science.1089427. [DOI] [PubMed] [Google Scholar]
- 9.Altman M, Nalivaika E, Prabu-Jeyabalan M, Schiffer C, Tidor B. Computational design and experimental study of tighter binding peptides to an inactivated mutant of HIV-1 protease. Proteins. 2008;70:678–694. doi: 10.1002/prot.21514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Verkihiver G. Computational proteomics of biomolecular interactions in the sequence and structure space of the tyrosine kinome: Deciphering the molecular basis of the kinase inhibitors selectivity. Proteins. 2007;66:912–929. doi: 10.1002/prot.21287. [DOI] [PubMed] [Google Scholar]
- 11.Ishikita H, Warshel A. Predicting drug-resistant mutations of HIV protease. Angewandte Chemie International Edition. 2008;47:697–700. doi: 10.1002/anie.200704178. [DOI] [PubMed] [Google Scholar]
- 12.Cao Z, et al. Computer prediction of drug resistance mutations in proteins. Drug Discov Today. 2005;10:521–529. doi: 10.1016/S1359-6446(05)03377-5. [DOI] [PubMed] [Google Scholar]
- 13.Frey K, et al. Crystal structures of wild-type and mutant methicillin-resistant Staphylococcus aureus dihydrofolate reductase reveal an alternative conformation of NADPH that may be linked to trimethoprim resistance. J Mol Biol. 2009;387:1298–1308. doi: 10.1016/j.jmb.2009.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frey K, Lombardo M, Wright D, Anderson A. Towards the understanding of resistance mechanisms in clinically isolated, trimethoprim-resistant, methicillin-resistant Staphylococcus aureus dihydrofolate reductase. J Struc Biol. 2010;170:93–97. doi: 10.1016/j.jsb.2009.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dale G, et al. A single amino acid substitution in Staphylococcus aureus dihydrofolate reductase determines trimethoprim resistance. J Mol Biol. 1997;266:23–30. doi: 10.1006/jmbi.1996.0770. [DOI] [PubMed] [Google Scholar]
- 16.Lovell S, Word J, Richardson J, Richardson D. The penultimate rotamer library. Proteins. 2000;40:389–408. [PubMed] [Google Scholar]
- 17.Hu Z, et al. Fitness comparison of thymidine analog resistance pathways in human immunodeficiency virus type 1. J Virol. 2006;80:7020–7027. doi: 10.1128/JVI.02747-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Larder B, Coates K, Kemp S. Zidovudine-resistant human immunodeficiency virus selected by passage in cell culture. J Virol. 1991;65:5232–5236. doi: 10.1128/jvi.65.10.5232-5236.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Larder B, Kemp S. Multiple mutations in HIV-1 reverse transcriptase confer high-level resistance to zidovudine (AZT) Science. 1989;246:1155–1158. doi: 10.1126/science.2479983. [DOI] [PubMed] [Google Scholar]
- 20.Rong L, Dahari H, Ribeiro R, Perelson A. Rapid emergence of protease inhibitor resistance in hepatitis C virus. Science Translational Medicine. 2010;2 doi: 10.1126/scitranslmed.3000544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yatsuji H, et al. Emergence of a novel lamivudine-resistant hepatitis B virus variant with a substitution outside the YMDD motif. Antimicrob Agents Chemother. 2006;50:3867–3874. doi: 10.1128/AAC.00239-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wueppenhorst N, Stueger H, Kist M, Glocker E. Identification and characterization of triple- and quadruple-resistant Helicobacter pylori clinical isolates in Germany. J Antimicrob Chemother. 2009;63:648–653. doi: 10.1093/jac/dkp003. [DOI] [PubMed] [Google Scholar]
- 23.Leartsakulpanich U, et al. Molecular characterization of dihydrofolate reductase in relation to antifolate resistance in Plasmodium vivax. Mol Biochem Parasitol. 2002;119:63–73. doi: 10.1016/s0166-6851(01)00402-9. [DOI] [PubMed] [Google Scholar]
- 24.Ercikan-Abali E, et al. Active site-directed double mutants of dihydrofolate reductase. Cancer Res. 1996;56:4142–4145. [PubMed] [Google Scholar]
- 25.Sirawaraporn W, Sathitkul T, Sirawaraporn R, Yuthavong Y, Santi D. Antifolate-resistant mutants of Plasmodium falciparum dihydrofolate reductase. Proc Natl Acad Sci USA. 1997;94:1124–1129. doi: 10.1073/pnas.94.4.1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng Y, Prusoff W. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 percent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 27.Cornell W, et al. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J Am Chem Soc. 1995;117:5179–5197. [Google Scholar]
- 28.Lazaridis T, Karplus M. Effective energy function for proteins in solution. Proteins. 1999;35:133–152. doi: 10.1002/(sici)1097-0134(19990501)35:2<133::aid-prot1>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 29.Otwinowski Z, Minor W. Macromolecular Crystallography. In: Carter CW, Sweet RM, editors. Method Enzymol. Vol. 276. New York: Academic Press; 1997. pp. 307–326. [Google Scholar]
- 30.Emsley P, CoWTan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 31.Murshudov G, Vagin A, Dodson E. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 32.Laskowski R, MacArthur M, Moss D, Thornton J. PROCHECK: A program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.