

Abstract
The identity of the histidine specific transfer RNA (tRNAHis) is largely determined by a unique guanosine residue at position −1. In eukaryotes and archaea, the tRNAHis guanylyltransferase (Thg1) catalyzes 3'-5' addition of G to the 5'-terminus of tRNAHis. Here, we show that Thg1 also occurs in bacteria. We demonstrate in vitro Thg1 activity for recombinant enzymes from the two bacteria Bacillus thuringiensis and Myxococcus xanthus and provide a closer investigation of several archaeal Thg1. The reaction mechanism of prokaryotic Thg1 differs from eukaryotic enzymes, as it does not require ATP. Complementation of a yeast thg1 knockout strain with bacterial Thg1 verified in vivo activity and suggests a relaxed recognition of the discriminator base in bacteria.
Keywords: tRNA-His guanylyltransferase, Thg1, tRNA processing, histidyl-tRNA synthetase, RNase P
1. Introduction
During tRNA maturation, the 5' leader sequence of precursor (pre)-tRNA is removed by the tRNA processing enzyme RNase P, which specifically cleaves at position 1 of almost all tRNAs [1,2]. In the case of tRNAHis, however, accurate translation of histidine codons relies on the presence of an additional guanosine nucleotide at the 5' end of nearly all mature tRNAHis species [3,4]. Other rare cases of −1 residues in mature tRNAs include a A−1 residue in tRNAPhe from Tetrahymena pyriformis as well as A−1 in tRNAiMet and a G−1 in tRNATyr from Nanoarchaeum equitans [5,6]. In Escherichia coli tRNAHis, the G−1 residue base pairs with the discriminator base at position C73, typically an unpaired base in most tRNAs, and is important for recognition by histidiny1-tRNA synthetase (HisRS) and efficient formation of His-tRNAHis [7,8]. G−1 is a critical identity element in tRNAHis as its absence was shown to result in a greater than 100-fold reduction in catalytic efficiency of HisRS in both yeast and E. coli systems [8–11].
While the G−1 residue is genome encoded in many organisms, the standard cleavage site selection of RNase P would remove this essential residue [12]. In many bacteria, RNase P displays an altered cleavage pattern maintaining the genome encoded G−1 [3,13]. In eukaryotes and some archaea, however, the G−1 residue is either not genome encoded or removed during cleavage by an RNase P without the ability to cleave at the −1 position. Therefore, G−1 has to be added posttranscriptionally [14,15]. To accomplish this, a tRNAHis guanylyltransferase (Thg1) catalyzes the addition of a guanylate to the 5' end of tRNAHis to guarantee functional mature tRNAs in these organisms [14,15]. Thg1 genes and proteins have been identified and studied in yeast [16–18] and more recently in archaea [19,20]. A deletion of thg1 in yeast was shown to be lethal, while a conditional depletion leads to the accumulation of tRNAHis lacking the G−1 residue [16]. Further investigation with Thg1 depleted yeast cells indicated that the absence of mature tRNAHis could be rescued by overexpression of both tRNAHisΔG−1 and HisRS, which then produces sufficient amounts of His-tRNAHis. This suggests that adding the G−1 residue to tRNAHis may be the only biological function of Thg1 [21].
Thg1 has been studied most intensively in yeast, where the unusual untemplated 3'-5' polymerization reaction is carried out in an ATP dependent manner [17,18]. Here, the anticodon of tRNAHis was shown to be the major identity element [18] by which Thg1 differentiates substrate from non-substrate tRNAs. A significant role has also been assigned to the discriminator base of tRNAHis for both eukaryotic and archaeal Thg1 [18,19]. Yeast Thg1 requires an adenosine as discriminator base in order to restrict catalytic activity to the addition of only a single guanidine residue. In contrast, archaeal Thg1s have been shown previously to only complement a yeast Thg1 depleted strain when a tRNAHis featuring a C as discriminator base is present [19]. Archaeal homologs do not complement the yeast knockout strain when only endogenous yeast tRNAHis with an A-discriminator base is present, thus demonstrating the requirement of a C -discriminator base for catalytic activity.
Here, we report that Thg1 activity is not restricted to eukaryotes and archaea, but is also found in bacteria. We provide the first characterization of the bacterial enzymes and demonstrate differences with the yeast enzyme involving cofactor dependence and substrate recognition.
2. Materials and Methods
2.1 General
Oligonucleotide synthesis and DNA sequencing was performed by Integrated DNA Technologies and the DNA sequencing facility on Science Hill at Yale. [α-32P]GTP (3000 Ci/mmol) and [α-32P]ATP (3000 Ci/mmol) were purchased from GE Healthcare.
2.2 Preparation and purification of RNA transcripts
The E. coli, Methanothermobacter thermautotrophicus and Pyrobaculum aerophilum tRNAHis genes and M. thermautotrophicus tRNAphe genes were cloned into a pUC19 vector that allowed for in vitro T7 RNA polymerase run-off transcription after plasmid cleavage with BstNI. Anticodon exchanges were introduced by QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) according to the manufacturer's instructions. The tRNAs were purified via 12% polyacrylamide gel electrophoresis in the presence of 8 M urea as described [15].
2.3 Preparation and purification of Thg1 enzyme
For the archaeal enzymes, the genes Mbar_A1746 (Methanosardna barkeri), MTH972 (M. thermautotrophicus) and PAE0886 (P. aerophilum) were amplified from genomic DNA and cloned into the NdeI/XhoI sites of pet20b(+) vector to facilitate expression of the proteins in the E. coli BL21-codon plus (DE3)-RIL strain (Stratagene, La Jolla, CA). Genes encoding Bacillus thuringiensis Thg1 (RBTH_06728) and Myxococcus xanthus Thg1 (MXAN_5968) were codon optimized for E. coli codon usage and synthesized by GenScript Corporation (Piscataway, NJ). Genes without a stop codon were cloned between EcoRI and SalI restriction sites into pET20b vector (Novagen, Madison, WI) with a C-terminal His6 tag. Cultures were grown at 37°C in Luria–Bertani medium supplemented with 100 μg/mL ampicillin and 34 μg/mL chloramphenicol and the recombinant proteins were produced by autoinduction as described [16]. Cells were harvested by centrifugation and resuspended in 1 × Thg1 buffer (50 mM Tris–HCl (pH 7.0), 200 mM NaCl, 20 mM MgCl2, 5 % glycerol and 3 mM DTT), and broken by sonication. The M. thermautotrophicus and P. aerophilum enzymes were flocculated at 80°C for 30 min, and then centrifuged for 30 min at 20,000 g. Cell lysates were applied to Ni-NTA metal affinity resin and purified according to the manufacturer's instructions. The eluted enzymes were dialyzed into 1 × Thg1 buffer. SDS–PAGE electrophoresis followed by staining with Coomassie blue revealed greater than 90% purity.
2.4 Thg1 activity assays
Activity of various Thg1 proteins was assayed monitoring the incorporation of [α-32P]GTP into tRNAHisΔG−1 of as described previously [16,22]. Reactions were incubated at 37°C for the M. barkeri and bacterial enzymes, at 45°C for M. thermautotrophicus Thg1 and at 65°C for P. aerophilum Thg1. Bacterial and archaeal enzymes were assayed with P. aerophilum, M. thermautotrophicus or E. coli tRNAHisΔG−1, and E. coli tRNAHis.
2.5 Construction of yeast expression plasmids
To express bacterial or archaeal Thg1 in yeast, plasmids were constructed by cloning the respective ORFs into the yeast centromeric plasmid (CEN) pRS416 expression vector [23], which carries the URA3 marker. This plasmid allows the expression of the ORF under the control of the alcohol dehydrogenase (ADH1) promoter. The ORFs of M. barkeri and P. aerophilum and the codon optimized sequence for B. thuringiensis were amplified by PCR and the fragments were digested and cloned between EcoRI and SacII restriction sites in the pRS416 plasmid.
2.6 Saccharomyces cerevisiae complementation assays
A THG1/thg1Δ∷KanMX4 heterozygous diploid (ATCC 4026977) was transformed using the lithium acetate method [24] with empty plasmid or pRS416 containing THG1, which expresses the THG1 ORF under control of the constitutive yeast ADH promoter. Transformants were selected on synthetic medium lacking uracil, and colonies arising were sporulated and dissected according to standard yeast genetic manipulations [25]. Tetrads were incubated at 30°C for three days before imaging. The genotypes of the germinating spores were determined by replica plating onto plates containing 300 μg/mL G418 sulfate (Calbiochem).
2.7 Phylogenetic analysis
Sequences were downloaded from the National Center for Biotechnology Information (NCBI) and from the Integrated Microbial Genomes database [26] from the Joint Genome Institute. Sequence alignment and alignment editing was carried out using Muscle [27] and the Multiseq alignment editor from VMD 1.8.7 [28]. A maximum likelihood phylogeny for Thg1 sequences was determined using Phyml [29]. The starting tree was generated with BioNJ, and the tree space was searched with the SPR followed by the NNI algorithm to find the best tree. The JTT+Γ model with 4 rate categories was applied. Likelihood parameters were initially estimated from the alignment, Shimodaira–Hasegawa boot strap values were computed as implemented in PHYML.
3. Results
3.1 Thg1 is present in all three domains of life
Thg1 activity has been described in eukaryotes [16] and archaea [19,20]. Bacteria were generally thought not to rely on the posttranscriptional addition of a G residue to tRNAHis, as most bacteria retain a genome encoded G−1 residue of tRNAHis due to an altered cleavage pattern of bacterial RNase P, making Thg1 activity redundant. Only few bacteria belonging to a small group of α -proteobacteria do not encode G−1 in their tDNA, but this is compensated by an alteration in HisRS, resulting in an enzyme that does not require a G−1 [30,31].
Candidate Thg1 genes were previously identified in bacterial genomes [19,20]. In order to investigate the relationship between the bacterial, archaeal and eukaryotic Thg1 variants, we performed a detailed phylogenetic analysis of Thg1 from all three domains of life (Figure 1). Almost all eukaryotic Thg1 variants are represented in a major phylogenetic group, whereas a second clade contains archaeal, bacterial and few eukaryotic representatives. In eukaryotes and archaea, the distribution of Thg1 is mainly characterized by vertical inheritance, i.e. organismal groupings are essentially in accordance with accepted taxonomy.
Fig. 1.

Phylogenetic tree of Thg1. Organism names are color-coded: Archaea (blue), Eukarya (green), Bacteria (red) and viruses (purple). Bootstrap values for major branches are shown. Sequence data were downloaded from the Integrated Microbial Genomes database [26]. The tree was calculated with PHYML [29] using a BioNJ starting tree and SPR tree search followed by NNI branch swapping to optimize the tree. Likelihood parameters were estimated from the alignment, and the JTT+Γ model with four rate categories was applied. Bootstrap values were computed according to the Shimodaira–Hasegawa re-estimation of log-likelihood test implemented in PHYML.
In contrast, grouping of bacterial ORFs does not support Thg1 being present in an early bacterial ancestor. The bacterial Thg1s do not form a single phylogenetic group but rather cluster in a larger group containing mostly archaeal variants. The identified bacterial variants are distributed randomly into two major subgroups (Figure 1, group 1 and group 2). Within each bacterial subgroup, members of the different phyla are found, but representatives of a single phylum are not restricted to one subgroup. For example, Thg1 variants from phylogenetically close organisms Eubacterium rectale (group 1) and Clostridiales bacterium (group 2) are found to have highly diverged Thg1s (Figure 1). The sparce distribution of Thg1 among bacteria and the unusual phylogenetic pattern observed suggest that Thg1 occurrence in bacteria might be the result of at least two independent horizontal gene transfer events from archaea to bacteria, and further horizontal gene transfers within bacteria.
Thg1 candidates from Bacillus thuringiensis (group 1) and Myxococcus xanthus (group 2) were cloned and produced to investigate whether Thg1s from either group display guanylyltransferase activity. A closer look at the genomes of these bacterial species that potentially contain Thg1s confirmed that they encode a G−1 in the respective tRNAHis gene. Therefore, Thg1 might not be required for posttranscriptional GMP addition in these organisms. This is also the case in Methanosarcina species, which have been shown to contain functional Thg1 [19,20]. In parallel, representatives from the respective archaeal groups, i.e. Pyrobaculum aerophilum, a crenarchaeotal variant and Methanothermobacter thermautotrophicus and Methanosarcina barkeri, both euryarchaeal representatives, were analyzed.
We first tested the ability of the purified bacterial enzymes to add a G residue to tRNAHisΔG−1 from E. coli in the presence of ATP. Radiolabeled [α-32P]-GTP, substrate tRNA and purified enzymes were incubated for 1 h and then separated by polyacrylamide gel electrophoresis. Only reacted product yields a band of radiolabeled tRNA. Both the B. thuringiensis and the M. xanthus Thg1 efficiently add a GMP residue to this substrate tRNA (Figure 2, panels labeled 60).
Fig. 2.
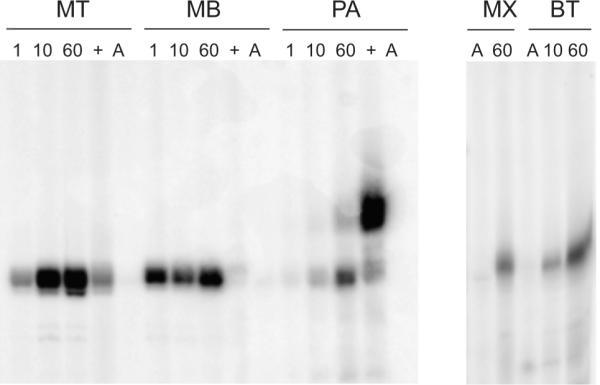
Thg1 assay with [α-32P]GTP and tRNAHisΔG−1. Autoradiography of Thg1 reaction products separated on a 12% polyacrylamide gel with 8M Urea. Thg1 from various archaeal and bacterial origins add guanine nucleotides to the 5' end of p-tRNAHisΔG−1. Escherichia coli tRNAHis transcripts lacking a −1 base were incubated with [α-32P]GTP and Thg1 from Methanosarcina barkeri (MB), Methanothermobacter thermophilus (MT), Pyrobaculum aerophilum (PA), Myxococcus xanthus (MX) and Bacillus thuringiensis (BT) for 1 min (1), 10 min (2) or 60 min (3,+,A). A tRNAHis that contains already a G−1 (+) is either a very weak substrate (for MT and MB Thg1) or promotes an artificial elongation reaction (for PA Thg1). [α-32P]ATP (A) is not a Thg1 substrate.
In order to investigate nucleotide specificity of bacterial Thg1, we further tested the ability of the enzymes to utilize [α-32P]-ATP as substrate. This reaction, however, did not yield any radiolabeled product, indicating that bacterial Thg1 incorporates GTP but not ATP.
Activity assays with archaeal Thg1s were carried out utilizing substrate tRNAHisΔG−1 from M. thermautotrophicus, P. aerophilum and mature E. coli tRNAHis species containing the G−1 residue. Archaeal Thg1s displayed activity in vitro on archaeal tRNAHisΔG−1 substrates (Figure 2 panels labeled 1, 10, 60). The mature tRNAHis from E. coli (already containing the G−1 residue) was also assayed to test if these Thg1 variants were capable of addition of nucleotides beyond the −1 position. The mature tRNAHis from E. coli already is either a very weak substrate (for M. thermautotrophicus Thg1 and M. barkeri Thg1) or promotes an artificial elongation reaction (for P. aerophilum Thg1), suggesting a specific addition of a single GMP as the primary function of Thg1 (Figure 2, panels labeled +). Similar to the bacterial variants, the archaeal homologs did not show any reaction product when incubated with [α-32P]-ATP. (Figure 2, panels labeled A).
3.2 Prokaryotic Thg1 catalyze ATP-independent guanylation
S. cerevisiae Thg1 transfers G onto the 5'end of mature tRNAHisΔG−1 in an ATP dependent manner [17]. Here, prior to GMP addition, binding of ATP activates tRNAHisΔG−1, which is then replaced by the guanylate. ATP activation is required only when the substrate tRNA contains a 5'monophosphate, but not when it contains a 5'triphosphate [17]. We probed archaeal and bacterial Thg1 homologs in the absence of ATP, in order to investigate whether this ATP dependence is a shared feature with the yeast enzyme. Interestingly, the prokaryotic Thg1 reaction mechanism differs from the one described for the yeast enzyme. We demonstrated that G−1 addition to tRNAHisΔG−1 is ATP independent. No difference between [α-32P]-GTP labeling of tRNA was seen with or without ATP even when the tRNA transcription mix included an excess of GMP to ensure a 5'-monophosphate start of the transcription product (Figure 3). Furthermore, in vitro transcribed substrate tRNAs were treated with tobacco acid pyrophosphatase (Sigma Aldrich, St Louis, MO), which specifically removes triphosphates to yield a monophosphate at the 5'end of substrate tRNA.
Fig. 3.
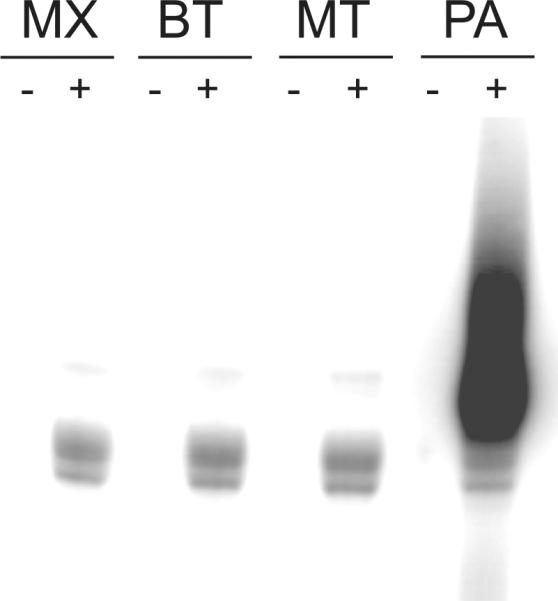
ATP-dependence analysis of Thg1. ATP-independence of the Thg1 reaction is shown for bacterial variants Myxococcus xanthus (MX) and Bacillus thuringiensis (BT) and archaeal variants Methanothermobacter thermautotrophicum (MT) and Pyrobaculum aerophilum (PA). Escherichia coli tRNAHisΔG−1 was employed as substrate for bacterial enzymes and M. thermautotrophicum or P. aerophilum tRNAHisΔG−1 as substrate for archaeal enzymes. The reaction was carried out in the presence (+) and absence (−) of a Apyrase. G−1 transfer for the archaeal enyzmes was restored when the reaction mix was heated to 80°C (to denature Apyrase) before tRNA and [α-32P]GTP were added. Following treatment with Apyrase, bacterial enzymes were subjected to an additional purification step, which restored G−1 addition. A double band is observed due to partial tRNA degradation resulting from necessary extended incubation time.
As this assay still cannot exclude ATP contamination resulting from protein purification, we treated purified Thg1 homologs with Apyrase that catalyses the hydrolysis of ATP and GTP. No activity in the presence of Apyrase was observed (Figure 3, panels labeled “−”). For the archaeal enzymes, we took advantage of the thermostability of M. thermautotrophicus Thg1 and P. aerophilum Thg1 to heat-inactivate Apyrase at 80°C for 20 min just before the addition of [α-32P]-GTP. This heat inactivation restored Thg1 activity (Figure 3, panels labeled “+”). Since the bacterial enzymes do not feature this kind of heat stability, an additional His-tag purification step was employed to remove Apyrase. The purified Thg1 was then incubated with [α-32P]-GTP and substrate tRNAHisG−1. As shown in Figure 3, all assayed enzymes displayed activity solely dependent on GTP and not ATP. Thus, the bacterial and archaeal Thg1 variants differ from the eukaryotic enzymes in their reaction mechanism and do not require ATP for catalytic activity.
3.3 Anticodon recognition by archaeal and bacterial Thg1
The G−1 residue of tRNAHis is a unique identity element recognized by most HisRSs [3,4]. In order to make sure that only tRNHis exhibits this specific identity element, the guanylation reaction must be restricted to the substrate tRNAHisΔG−1. For the yeast enzyme, the recognition of tRNAHisΔG−1 by Thg1 has been investigated in some detail, with the anticodon being identified as the major identity element for tRNAHis recognition [18]. In order to determine whether archaeal and bacterial Thg1 variants recognize the anticodon in a manner similar to yeast Thg1, guanylyation activity of Thg1 on tRNAPhe was assayed. tRNAPhe was either a very weak substrate (M. xanthus Thg1) or did not promote GMP addition to tRNAPhe by Thg1 (M. thermautotrophicus Thg1) (Figure 4, panels 4). Weak guanylyation of tRNAPhe by M. xanthus Thg1 was observed in vitro; this may be due to the heterologous substrate tRNAPhe from M. thermautotrophicus. Transplantation of the tRNAPhe anticodon GAA to tRNAHis resulted in the loss of guanylyation activity (Figure 4, lanes 2), whereas the mutation of the tRNAPhe anticodon to the His-anticodon GUG converted tRNAPhe into a valid substrate for Thg1 (Figure 4, panels 3). From these results we conclude that the tested bacterial and archaeal Thg1 homologs recognize the anticodon of tRNAHis and are thus able to restrict their activity to their substrate tRNA and prevent mis-guanylation of other tRNAs.
Fig. 4.
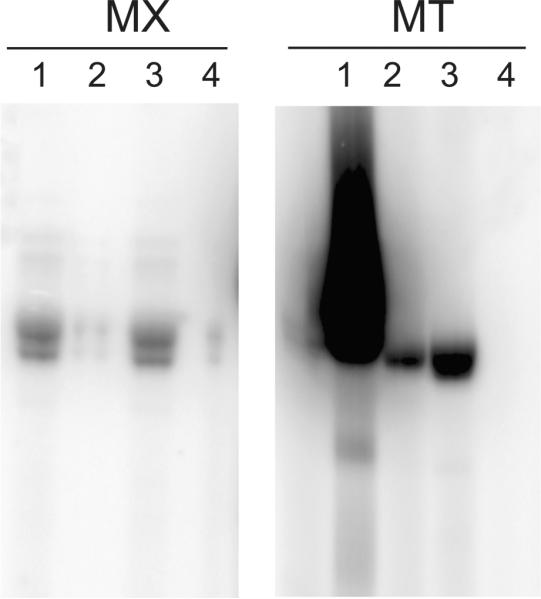
Recognition of the tRNAHisΔG−1 anticodon by Thg1. Myxococcus xanthus (MX) and Methanothermobacter thermautotrophicus (MT) Thg1 were incubated with [α-32P]GTP and different p-tRNA substrates from M. thermautotrophicus. Both enzymes efficiently add G−1 to tRNAHisΔG−1 (1). Transplanting the Phe GAA anticodon to tRNAHisΔG−1 results in little or no guanylation activity (2), whereas a tRNAPhe with a His-anticodon GUG provides a valid substrate for Thg1 (3). Unaltered tRNAPhe is not a substrate for Thg1 (4). A double band is observed due to partial tRNA degradation resulting from necessary extended incubation time for M. xanthus.
3.4 Untemplated and templated 3'-5' polymerase activity of archaeal and bacterial Thg1
In order to investigate the role of the discriminator base for bacterial Thg1, we employed in vitro and in vivo activity assays. We investigated activity of bacterial Thg1 in vitro with the E. coli tRNAHisΔG−1 substrate containing a C discriminator base and could show that bacterial Thg1 does not require a specific discriminator base as B. thuringiensis Thg1 displays activity with the E. coli tRNAHisΔG−1 substrate (Figure 2, panel BT, 60). Furthermore, in vivo complementation of a Thg1-deficient yeast strain was performed, by expressing B. thuringiensis Thg1 in a yeast Thg1 knockout strain (Figure 5). Yeast endogenous tRNAHis features an A discriminator base, which was not a substrate for archaeal Thg1 variants [19]. Expression of bacterial Thg1 in the knockout strain restored growth to near wild type levels. The data reveal not only that B. thuringiensis Thg1 is active in vivo, but that it also accepts an adenosine at the discriminator base position, even though bacterial tRNAHis genes usually contain a C in this position. This indicates a less stringent recognition of the discriminator base compared to both archaeal and yeast enzymes, which both have strict preferences at this position [19,20].
Fig. 5.

Complementation of yeast thg1Δ by Bacillus thuringiensis Thg1. A THG1/thg1Δ∷KanMX4 heterozygous diploid was transformed using the lithium acetate method [24] with pRS416 containing B. thuringiensis Thg1 under control of the constitutive yeast ADH promoter. Transformants were selected, sporulated and dissected according to standard yeast genetic manipulations. Three representative tetrads are shown. White boxes denote thg1Δ segregants.
4. Discussion
4.1 Thg1 activity is present in all three domains of life
Accurate translation is dependent on the stringent recognition of tRNAs by their cognate aminoacyl-tRNA synthetases. In the case of tRNAHis, nearly all organisms require a unique G−1 residue to accomplish this. This residue is added post-transcriptionally by Thg1, an essential tRNA processing enzyme in eukaryotes, which encode at least one, if not several copies in their genomes. For example, the plants Arabidopsis thaliana and Orzya sativa contain several copies of tandem Thg1 genes and the slime mold Dictyostelium discoideum contains four Thg1 homologs, some of which are phylogenetically more closely related to the prokaryotic Thg1 than the eukaryotic Thg1 variants (Figure 1).
In contrast, most bacteria utilize an alternate way to ensure the presence of the G−1 residue. These organisms contain a genome encoded G−1 residue and display an alternate RNase P cleavage pattern for tRNAHis, to spare the G−1 from removal. A few α-proteobacteria contain a HisRS with unique peptide insertion and do not require the G−1 residue. Since the so far identified Thg1 encoding bacteria also encode the G−1 residue for tRNAHis, a biological role for Thg1 in these organisms is not obvious. Nevertheless, Thg1 homologs have also been found and proven to be enzymatically active in G−1 encoding archaea, without the apparent necessity for the function of the eukaryotic variant [19,20].
In this study, we showed that tRNAHis guanylation activity is present in all three kingdoms of life, including enzymatically active examples from bacteria. Open reading frames encoding Thg1 are found in a variety of bacteria and are not restricted to particular phyla, but only very few of the currently sequenced bacterial genomes contain a potential tRNAHis guanylyltransferase.
Interestingly, a few bacterial Thg1 genes appear to be split, opening the possibility of the presence of split Thg1 variants that function in trans in vivo. Thg1 has been shown to function as two halves in vitro as described for M. acetivorans Thg1 [20]. Three examples of potentially split Thg1 genes can be found in bacteria. In Cyanthece sp. and Coprococcus eutactus, Thg1 split genes have been annotated to code for two proteins due to a frameshift in the ORF, resulting in two almost equal halves, whereas in Verrumicrobium spinosum the ORF for Thg1 is disrupted by an ORF for a transposase (ZP_02926794). All three examples could result in the expression of two proteins corresponding to N- and C- terminal halves of Thg1, which could lead to potentially functional Thg1s in vivo. Whether these organisms require or have Thg1 activity in vivo remains to be elucidated.
2. Prokaryotic Thg1 differs from eukaryotic Thg1
Bacterial and archaeal Thg1 proteins share certain features with eukaryotic Thg1, such as the recognition of the anticodon and the addition of GMP but not AMP to their substrate tRNA. Nevertheless, the prokaryotic Thg1s differ from their eukaryotic counterparts by not requiring an ATP cofactor for the activation of substrate tRNAHisΔG−1 prior to GMP addition. Yeast Thg1 has been shown to rely on ATP for tRNA activation and does not catalyze GMP addition without activation of the tRNA by ATP. However, this feature is not shared by the prokaryotic variants, which display Thg1 activity in the absence of ATP. Possibly, prokaryotic Thg1 can utilize GTP to activate monophosphorylated tRNAHisΔG−1. This observation suggests that both ATP and GTP could be effective for tRNA activation in vivo.
While archaeal and eukaryotic Thg1 display a strong preference for either a C or A - discriminator base, and show altered or abolished activity with a different base in this position, bacterial Thg1 activity does not rely on a specific base in this position and accepts both A and C, indicating that the discriminator base is not a significant recognition element. Archaeal Thg1 catalytic activity strictly relies on the base-paring of the G−1 residue with a C discriminator base [19]. Bacterial Thg1, however, accepts the yeast endogenous substrate tRNAHis containing an A in this position, suggesting that a templated reaction mechanism including Watson/Crick base pairing as described for archaeal Thg1 might not be essential for bacterial Thg1 catalytic activity. This is surprising, as bacterial Thg1 variants are phylogenetically closely related to the archaeal homologs and share other features such as ATP independence of reaction with archaeal Thg1.
Yeast Thg1 does not require a certain discriminator base in order to display catalytic activity, since it is capable of adding GMP residues to both A73 and C73 containing tRNAs. The presence of an A73 is absolutely essential in order to restrict the enzymes activity to the addition of a single GMP residue and prevent it from adding multiple GMPs [17]. Bacterial Thg1 shows no capability for multiple GMP addition events in vitro, suggesting that it must employ a different mechanism to restrict its catalytic activity to the addition of a single GMP residue. In this regard, bacterial Thg1 mechanisms remains to be elucidated and will be the subject of future investigations. It will be interesting to see if the presented tRNAHis maturation activity of bacterial Thg1 is the enzyme's sole function in vivo, or if the enzyme is used in other RNA processing reactions. The precise cellular role of Thg1 is yet unknown, and it is unclear if the tRNA processing activity of Thg1 is required for compensating RNase P cleavage at base 1 or if the evolutionary advantage of its presence lies in the ability to “heal” 5'termini after somewhat ambiguous RNase P cleavage.
Acknowledgments
This work was supported by grants (to D.S.) from the National Institute of General Medical Sciences and the National Science Foundation. I.U.H. is a Postdoctoral Fellow of the Deutsche Forschungsgemeinschaft (HE5802/1-1). We thank Jing Yuan, Patrick O'Donoghue, Markus Englert and Michael Hohn for help and encouragement, and Imke Schroeder for a gift of P. aerophilum cells.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Guerrier-Takada C, Altman S. Catalytic activity of an RNA molecule prepared by transcription in vitro. Science. 1984;223:285–286. doi: 10.1126/science.6199841. [DOI] [PubMed] [Google Scholar]
- [2].Altman S, Bowman EJ, Garber RL, Kole R, Koski RA, Stark BC. In: Transfer RNA: Biological Aspects. Söll D, Abelson J, Schimmel P, editors. Vol. 09B. Cold Spring Harbor Press; 1980. pp. 71–82. [Google Scholar]
- [3].Burkard U, Willis I, Söll D. Processing of histidine transfer RNA precursors. Abnormal cleavage site for RNase P. J. Biol. Chem. 1988;263:2447–2451. [PubMed] [Google Scholar]
- [4].Connolly SA, Rosen AE, Musier-Forsyth K, Francklyn CS. G-1:C73 recognition by an arginine cluster in the active site of Escherichia coli histidyl-tRNA synthetase. Biochemistry. 2004;43:962–969. doi: 10.1021/bi035708f. [DOI] [PubMed] [Google Scholar]
- [5].Randau L, Schröder I, Söll D. Life without RNase P. Nature. 2008;453:120–123. doi: 10.1038/nature06833. [DOI] [PubMed] [Google Scholar]
- [6].Schnare MN, Heinonen TY, Young PG, Gray MW. Phenylalanine and tyrosine transfer RNAs encoded by Tetrahymena pyriformis mitochondrial DNA: primary sequence, post-transcriptional modifications, and gene localization. Curr. Genet. 1985;9:389–393. doi: 10.1007/BF00421610. [DOI] [PubMed] [Google Scholar]
- [7].Francklyn C, Schimmel P. Enzymatic aminoacylation of an eight-base-pair microhelix with histidine. Proc. Natl. Acad. Sci. USA. 1990;87:8655–8659. doi: 10.1073/pnas.87.21.8655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Himeno H, Hasegawa T, Ueda T, Watanabe K, Miura K, Shimizu M. Role of the extra G-C pair at the end of the acceptor stem of tRNAHis in aminoacylation. Nucleic Acids Res. 1989;17:7855–7863. doi: 10.1093/nar/17.19.7855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Gu W, Hurto RL, Hopper AK, Grayhack EJ, Phizicky EM. Depletion of Saccharomyces cerevisiae tRNAHis guanylyltransferase Thg1p leads to uncharged tRNAHis with additional m5C. Mol. Cell Biol. 2005;25:8191–8201. doi: 10.1128/MCB.25.18.8191-8201.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Rudinger J, Florentz C, Giegé R. Histidylation by yeast HisRS of tRNA or tRNA-like structure relies on residues −1 and 73 but is dependent on the RNA context. Nucleic Acids Res. 1994;22:5031–5037. doi: 10.1093/nar/22.23.5031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Yan W, Augustine J, Francklyn C. A tRNA identity switch mediated by the binding interaction between a tRNA anticodon and the accessory domain of a class II aminoacyl-tRNA synthetase. Biochemistry. 1996;35:6559–6568. doi: 10.1021/bi952889f. [DOI] [PubMed] [Google Scholar]
- [12].Altman S, Baer MF, Bartkiewicz M, Gold H, Guerrier-Takada C, Kirsebom LA, Lumelsky N, Peck K. Catalysis by the RNA subunit of RNase P–a minireview. Gene. 1989;82:63–64. doi: 10.1016/0378-1119(89)90030-9. [DOI] [PubMed] [Google Scholar]
- [13].Orellana O, Cooley L, Söll D. The additional guanylate at the 5' terminus of Escherichia coli tRNAHis is the result of unusual processing by RNase P. Mol. Cell Biol. 1986;6:525–529. doi: 10.1128/mcb.6.2.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Jahn D, Pande S. Histidine tRNA guanylyltransferase from Saccharomyces cerevisiae. II. Catalytic mechanism. J. Biol. Chem. 1991;266:22832–22836. [PubMed] [Google Scholar]
- [15].Pande S, Jahn D, Söll D. Histidine tRNA guanylyltransferase from Saccharomyces cerevisiae. I. Purification and physical properties. J. Biol. Chem. 1991;266:22826–22831. [PubMed] [Google Scholar]
- [16].Gu W, Jackman JE, Lohan AJ, Gray MW, Phizicky EM. tRNAHis maturation: an essential yeast protein catalyzes addition of a guanine nucleotide to the 5' end of tRNAHis. Genes Dev. 2003;17:2889–2901. doi: 10.1101/gad.1148603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Jackman JE, Phizicky EM. tRNAHis guanylyltransferase catalyzes a 3'-5' polymerization reaction that is distinct from G-l addition. Proc. Natl. Acad. Sci. USA. 2006;103:8640–8645. doi: 10.1073/pnas.0603068103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Jackman JE, Phizicky EM. tRNAHis guanylyltransferase adds G-l to the 5' end of tRNAHis by recognition of the anticodon, one of several features unexpectedly shared with tRNA synthetases. RNA. 2006;12:1007–1014. doi: 10.1261/rna.54706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Abad MG, Rao BS, Jackman JE. Template-dependent 3'-5' nucleotide addition is a shared feature of tRNAHis guanylyltransferase enzymes from multiple domains of life. Proc. Natl. Acad. Sci. USA. 2010;107:674–679. doi: 10.1073/pnas.0910961107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Heinemann IU, O'Donoghue P, Madinger C, Benner J, Randau L, Noren C, Söll D. The appearance of pyrrolysine in tRNAHis guanylyltransferase by neutral evolution. Proc. Natl. Acad. Sci. USA. 2009;10:21103–21108. doi: 10.1073/pnas.0912072106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Preston MA, Phizicky EM. The requirement for the highly conserved G-l residue of Saccharomyces cerevisiae tRNAHis can be circumvented by overexpression of tRNAHis and its synthetase. RNA. 2010;16:1068–1077. doi: 10.1261/rna.2087510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Cooley L, Appel B, Söll D. Post-transcriptional nucleotide addition is responsible for the formation of the 5' terminus of histidine tRNA. Proc. Natl. Acad. Sci. USA. 1982;79:6475–6479. doi: 10.1073/pnas.79.21.6475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ito H, Fukuda Y, Murata K, Kimura A. Transformation of intact yeast cells treated with alkali cations. J. Bacteriol. 1983;153:163–168. doi: 10.1128/jb.153.1.163-168.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Guthrie C, Fink GR. In: Guide to Yeast Genetics and Molecular Cell Biology (Methods in Enzymology) Guthrie C, Fink GR, editors. Vol. 350. Academic Press; San Diego: 2002. p. 623. [Google Scholar]
- [26].Markowitz VM, et al. The integrated microbial genomes (IMG) system. Nucleic Acids Res. 2006;34:D344–348. doi: 10.1093/nar/gkj024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Roberts E, Eargle J, Wright D, Luthey-Schulten Z. MultiSeq: unifying sequence and structure data for evolutionary analysis. BMC Bioinformatics. 2006;7:382. doi: 10.1186/1471-2105-7-382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- [30].Ardell DH, Andersson SG. TFAM detects co-evolution of tRNA identity rules with lateral transfer of histidyl-tRNA synthetase. Nucleic Acids Res. 2006;34:893–904. doi: 10.1093/nar/gkj449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wang C, Sobral BW, Williams KP. Loss of a universal tRNA feature. J. Bacterial. 2007;189:1954–1962. doi: 10.1128/JB.01203-06. [DOI] [PMC free article] [PubMed] [Google Scholar]