

Summary
Cytochrome bo3 is the major respiratory oxidase located in the cytoplasmic membrane of E. coli when grown under high oxygen tension. The enzyme catalyzes the 2-electron oxidation of ubiquinol-8 and the 4-electron reduction of dioxygen to water. When solubilized and isolated using dodecylmaltoside, the enzyme contains one equivalent of ubiquinone-8, bound at a high affinity site (QH). The quinone bound at the QH site can form a stable semiquinone, and the amino acid residues which hydrogen bond to the semiquinone have been identified. In the current work, it is shown that the tightly bound ubiquinone-8 at the QH site is not displaced by ubiquinol-1 even during enzyme turnover. Furthermore, the presence of high affinity inhibitors, HQNO and aurachin C1-10, do not displace ubiquinone-8 from the QH site. The data clearly support the existence of a second binding site for ubiquinone, the QL site, which can rapidly exchange with the substrate pool. HQNO is shown to bind to a single site on the enzyme and to prevent formation of the stable ubisemiquinone, though without displacing the bound quinone. Inhibition of the steady state kinetics of the enzyme indicates that aurachin C1-10 may compete for binding with quinol at the QL site while, at the same time, preventing formation of the ubisemiquinone at the QH site. It is suggested that the two quinone binding sites may be adjacent to each other or partially overlap.
Keywords: ubiquinone, oxidase, E. coli, respiration, EPR
1. Introduction
Cytochrome bo3 is the predominant respiratory oxidase present in E. coli when grown under conditions of high aeration [1-3]. This enzyme is a member of the heme-copper superfamily [4, 5], which includes both oxygen reductases as well as NO reductases. The oxygen reductases are of particular interest because these enzymes couple the redox chemistry to pumping protons across the membrane bilayer and generate a proton motive force.
Cytochrome bo3 is in a sub-family of respiratory oxidases that directly catalyze the 2-electron oxidation of quinol within the membrane instead of the 1-electron oxidation of a soluble or membrane-bound cytochrome c. Cytochrome bo3 has one copy each of 4 subunits. Subunits I, II and III are homologous to the mitochondrial encoded subunits of the eukaryotic (mitochondrial) cytochrome c oxidase [6]. Whereas, in the mitochondrial oxidase subunit II contains the docking site of the cytochrome c substrate and the CuA redox center, neither of these features is present in the homologous subunit II of the quinol oxidases [7, 8]. The absence of the residues required for copper ligation to CuA is diagnostic of quinol oxidases and allows one to identify quinol oxidases in genome sequences. Phylogenetic analysis (manuscript in preparation) shows that cytochrome bo3 and related quinol oxidases are derived from the cytochrome c oxidases, so the CuA site can be considered to have been “lost”. In place of the CuA site, the quinol oxidases have acquired one or more sites for quinol oxidation. In the case of cytochrome bo3 from E. coli, the substrate is ubiquinol-8, which is confined to the cytoplasmic membrane. Closely related enzymes include the aa3-600 menaquinol oxidase from Bacillus subtilis [9, 10].
Purified cytochrome bo3 can be isolated with 0, 1 or 2 ubiquinones co-purifiying with the enzyme, depending upon the detergent used and the preparative protocol [11, 12]. This was the first indication that the enzyme may have more than one Q-binding site[11]. The effects of various inhibitors on the steady state kinetics of the enzyme have provided further evidence that there must be more than one Q-binding site [11, 13, 14], and the data have been interpreted in terms of two sites (QL or QH) with the inhibitors binding to one or the other Q-binding sites. The reactivity of the enzyme with synthetic quinone analogues has also been examined, showing the roles of the ring substituents and the hydrophobic side chain in determining the kinetic parameters [15, 16].
Cytochrome bo3 that is co-purified with at least one equivalent of ubiquinone exhibits a ubisemiquinone EPR signal when the solution potential is poised near 0 mV at pH 8, or when the enzyme is partially reduced [17, 18]. At pH 8.2, the 1-electron reduction of the oxidized ubiquinone has an Em2 = 15mV and the reduction of the semiquinone to the fully reduced dihydroubiquinol has an Em1 = 2.5mV [18]. At pH 9, the stability constant for the semiquinone species is ~10 [18]. The protein binding site which stabilizes the semiquinone is referred to as the high affinity Q-binding site, or QH. Stabilizing the 1-electron reduced form of the quinone is consistent with the quinone bound at this site acting as a “pair splitter”, which can be reduced by two electrons and then transfer the electrons one-at-a-time to a 1-electron acceptor, heme b [19, 20]. The rate of formation of the semiquinone upon mixing reduced ubiquinol-2 with the enzyme has been monitored by rapid freeze-quench EPR spectroscopy, and the results indicate that the semiquinone formed at the QH-site is an intermediate in the catalytic mechanism [21]. Transient formation of the semiquinone has also been examined by pulsed radiolysis, in which N-methylnicotinamide is used as a mediator for the one-electron reduction of the oxidized enzyme. The semiquinone at the QH-site is formed within 10 μs after pulsed radiolysis, and electron transfer from the semiquinone to heme b follows, with a first order rate constant of 1.5 × 103 s−1 [22]. Studies of the reaction of the fully reduced enzyme with O2 have shown that electron transfer from heme b to the heme o3/CuB binuclear center occurs with a rate of about 104 s−1 [19, 23]. A comparison of the intramolecular electron transfer events for enzyme containing the quinone bound at the QH-site with enzyme from which the quinone has been removed (prepared in Triton X-100) also supports the model that the tightly bound reduced quinone is capable of rapid electron transfer to heme b [12].
The intrinsic rate of “pure” electron transfer between the two hemes in cytochrome bo3, independent of coupled (slower) processes, is extremely fast (8 × 108 s−1) [24]. The slower rate (104 s−1) observed in the reaction of the reduced enzyme with O2 is due to coupled proton transfers and/or conformational changes.
The X-ray structure of cytochrome bo3 has been reported, but the detergent used (octylglucoside) resulted in stripping away the bound quinone, and substantial portions of the protein are not resolved [25]. Nevertheless, this structure was used to identify a reasonable candidate for a Q-binding site [25]. Subsequent EPR studies of site-directed mutants provided convincing evidence that this proposed site is the QH-site which stabilizes the observed semiquinone [26]. The residues implicated are R71, D75, H98 and Q101 [25, 26].
The identity of the low affinity, or QL site, has been problematic to the extent that the existence of this site has been questioned [25]. Both the use of photo-reactive Q analogues designed to covalently attach to the protein at the QL site [27, 28], as well as the location of inhibitor-resistant mutations [29], have been used to identify the QL site. In each case, subunit II was implicated, which was satisfying since the CuA site and docking site for cytochrome c in the related cytochrome c oxidases are located within subunit II. Mutations in one residue in subunit II, WII136A, were shown to perturb the apparent Km of ubiquinol-1 [30]. However, closer examination of WII 136A and other specific residues suggested to be at or near the QL site [29, 30] show that they are not near the membrane interface in the structure [25] and, therefore, are not likely to be part of a quinone binding site. Furthermore, sequence alignments of the many quinol oxidases (>400) presently available shows that WII136 is not fully conserved. The residues identified by the photolabeling [28] and inhibitor-resistance [28, 29] are not near the site proposed to be the QH site nor to any reasonable location for binding ubiquinol-8 from within the membrane bilayer.
In the current work, new evidence is presented to confirm that cytochrome bo3 must contain two functionally distinct Q-sites. It is also shown that the inhibitor HQNO binds stoichiometrically to the enzyme and prevents formation of the ubisemiquinone at the QH-site, but does not displace the ubiquinone-8 bound at the QH-site. The inhibitor aurachin C1-10 also prevents formation of the ubisemiquinone at the QH-site, but appears to compete for quinol binding at the QL-site. The possibility is considered that perhaps the QH-, QL- and HQNO binding sites are adjacent or partially overlap, but that binding of ubiquinone-8 at the QH-site is not eliminated by simultaneous occupation of either the QL-site or the HQNO binding site.
2. Materials and Methods
2.1 Expression and purification of cytochrome bo3
Wild type and mutants of cytochrome bo3 were expressed and the proteins were purified using the detergent dodecylmaltoside, as previously described [31].
2.2 Quinone extraction and analysis
The amount of ubiquinone-8 in the purified cytochrome bo3 was determined by quinone extraction followed by reversed phase HPLC analysis. 10 nmole of purified enzyme, together with 10 nmole of ubiquinone-10 (Q10), were diluted in 50 mM K2HPO4, 0.1% dodecylmaltoside, pH 8.3 buffer, and extracted with 3 mL of 6:4 methanol:petroleum ether by vortexing vigorously for 1 min (3 times). The mixture was allowed to sit for 10 min before vortexing again for an additional min, followed by centrifuging at 1,000 rpm for 10 min. The upper, organic layer was removed to a fresh tube, and the remaining mixture was subjected to further extraction two more times. The organic phase was then evaporated under a nitrogen stream, and the dried, oily residue was re-dissolved in ethanol.
The extracted quinones were analyzed using a reversed phase Microsorb-MV 100-5 C18 HPLC column (Varian, Palo Alto, CA) on a Beckman System Gold HPLC system fitted with a diode array detector, and monitored at 278 nm. The mobile phase was 4:3:3 ethanol:methanol:acetonitrile. The extracted ubiquinone-8 was quantified from the ratio of the peak areas of ubiquinone-8 to an internal standard, ubiquinone-10.
2.3 Preparation of quinone-free cytochrome bo3
Purified cytochrome bo3 in dodecylmaltoside (0.1%) was diluted at least 50-fold in buffer containing 50 mM K2HPO4, 0.1% Triton X-100 (Anatrace), pH 8.3, and then incubated for several hours with Ni-NTA resin with gentle stirring at 4 °C. The resin (with bound enzyme) was then loaded onto a column and washed with several column volumes of 50 mM K2HPO4, 0.1% Triton X-100, pH 8.3, followed by 50 mM K2HPO4, 0.1% dodecylmaltoside, pH 8.3. The protein was then eluted with the same buffer plus 100 mM imidazole. The eluted protein was pooled, concentrated, and dialyzed against 50 mM K2HPO4, 10 mM EDTA, 5% glycerol, 0.1% dodecylmaltoside, pH 8.3, and then flash-frozen in liquid nitrogen and stored at −80 °C.
2.4 Steady state enzymatic activity assays
The steady state oxygen reductase activity of cytochrome bo3 was measured using two different methods.
Method I: Oxygen electrode
Oxygen concentration was monitored using a YSI model 53 oxygen electrode (Yellow Springs Instrument Co., Yellow Springs, OH) equipped with a temperature-controlled 1.8-mL electrode chamber at 37 °C. The reaction mixture for this assay consisted of 50 mM K2HPO4, 0.1% DM, 2 mM dithiothreitol and 200 μM ubiquinone-1, pH 7.0. The concentration of oxygen in the air-saturated buffer at this temperature was assumed to be 250 μM, and the reaction was initiated by injecting a few microliters of diluted enzyme.
Method II: Coupled enzyme assay
To determine the enzyme velocity as a function of the concentration of ubiquinol-1, it was advantageous to use a coupled enzyme assay that maintained the concentration of ubiquinol-1 constant. This was accomplished by using rat liver DT-diaphorase (kindly provided by Professor Gary Cecchini at the University of California, San Francisco), with NADH as the reductant. The reaction mixture for this assay consisted of 160 μM NADH and 20-80 μg/mL DT-diaphorase, and the reaction was followed by the decrease in absorption at 340 nm (oxidation of NADH) after the addition of cytochrome bo3 at 25°C.
2.5 Re-isolation of cytochrome bo3 after turnover with ubiquinol-1
In order to determine if ubiquinol-8 bound at the QH-site was displaced during enzyme turnover with ubiquinol-1 the following procedure was used. Purified cytochrome bo3 (5 mg), isolated in dodecylmaltoside, was diluted into 5 mL of 50 mM K2HPO4 buffer, pH 8.3, containing 0.1% dodecylmaltoside, 10.5 mM dithiothreitol and 1.05 mM ubiquinol-1 at 37 °C. The reaction mixture was incubated at 37 °C for 10 min, and then loaded onto a column containing the Ni-NTA resin (QIAGEN) pre-equilibrated with 50 mM K2HPO4, 0.1% dodecylmaltoside, pH 8.3. The Ni-NTA resin with bound cytochrome bo3 was washed with buffer containing 50 mM K2HPO4, 0.1% dodecylmaltoside at pH 8.3, until the excess ubiquinone-1 was removed (monitored by the absorbance at 275 nm). The bound cytochrome bo3 was eluted with the same buffer plus 100 mM imidazole, and then concentrated and dialyzed to remove the imidazole. The content of ubiquinone-8 was then determined as described above.
An alternative procedure was also used in which the enzyme was first bound to the Ni-NTA column and was washed by buffer containing dithiothreitol and ubiquinol-1. Any displaced ubiquinol-8 would be expected to be swept away by the buffer. Both procedures yielded the same results.
Variations of the same procedure were used to quantify the displacement of ubiquinone-8 after incubation in the presence of the inhibitors HQNO and AC1-10.
2.6 EPR spectroscopy
The CW EPR measurements were performed on an X-band Varian EPR-E122 spectrometer. Methods used for preparing samples and obtaining EPR spectra have been previously described [31, 32].
2.7 HQNO binding monitored by fluorescence anisotropy
HQNO in water fluoresces with an emission maximum at 480 nm, but when bound to proteins the fluorescence is often completely quenched. Examples are the cytochrome bc1 complex [33], fumarate reductase [34-36], nitrate reductase [37, 38] and dimethylsulfoxide reductase [39]. However, attempts to monitor HQNO by changes in the fluorescence intensity revealed that the fluorescence of HQNO is not quenched upon binding to cytochrome bo3. For this reason, the binding of HQNO to cytochrome bo3 was monitored by the change in the fluorescence anisotropy of the emission from HQNO, using a Varian Cary Eclipse™ fluorescence spectrophotometer (Varian, Palo Alto, CA) fitted with automated polarizers. The anisotropy is defined in terms of the ratio of the intensity of light emitted parallel and perpendicular to the plane of polarization of the incident beam.
The titration was performed by increasing the enzyme concentration with a fixed concentration of HQNO to avoid problems of HQNO aggregation and to limit the amount of ethanol (used to prepare the stock solution of HQNO) which caused the protein to precipitate. Cytochrome bo3 was titrated in 1-4 μL aliquots from a stock solution into 50 mM K2HPO4, 0.1% dodecylmaltoside, pH 8.3 buffer containing 1 μM HQNO. The excitation wavelength was 341 nm and the emission wavelength was 480 nm. As a control, enzyme was titrated into buffer in the absence of HQNO. The data were fit with a model assuming independent binding sites.
2.8 Synthesis of aurachin C1-10
The inhibitor aurachin C1-10 was synthesized based on a protocol described previously [40]. The most important modification was that the order of the first two steps of the 7-step synthesis was reversed. The methylation of crotonate was followed by the condensation of aniline and 3-methylcrotonate instead of carrying out the condensation of aniline and crotonate followed by methylation of 3-anilinocrotonate.
3. Results
3.1 One or two Q-binding sites?
The main argument supporting a two-site model is that the quinone at the QH-site does not exchange with exogenous ubiquinone-8 and, once removed, is difficult to replace the ubiquinone-8 [11, 12]. This is consistent with the idea that the QH site cannot function as a catalytic site, which demands rapid exchange during catalytic turnover. It is noted that the bc1 complex is isolated with tightly bound quinones at the Qo and Qi sites but, under turnover conditions, both sites are involved in catalysis with rapid binding of substrate and release of products [41].
To provide a more convincing test for the existence of the QL-site, the ability to exchange the bound ubiquinone-8 for exogenous quinone was examined under turnover conditions with ubiquinol-1. Figure 1 illustrates the two models to be distinguished. The one-site model assumes that the quinone bound at the high affinity site can readily exchange under conditions of catalytic turnover and, therefore, is the only site required. The two-site model assumes that oxidation of the substrate occurs at the QL-site and that electrons are transferred, presumably in a two-electron transfer to the quinone at the QH-site. In order to distinguish between these two alternatives, the enzyme with one equivalent of bound ubiquinone-8 was allowed to incubate under aerobic conditions with reduced ubiquinol-1, a soluble analogue of the natural substrate, ubiquinol-8 (see Figure 2). The incubation time was sufficient to allow multiple turnovers of ubiquinol-1. If the one-site model is correct, then multiple turnovers with ubiquinol-1 must displace the initially bound ubiquinol-8, which would become diluted into the detergent micelles. To determine whether the bound ubiquinone-8 had been displaced, the His-tagged enzyme was bound to the Ni-NTA column and extensively washed with buffer to remove free or weakly bound quinone. After elution of the enzyme, the quinone was extracted and quantified by HPLC analysis. The results (Figure 3B) show that the ubiquinone-8 remained bound to the enzyme with the same stoichiometry as measured prior to the turnover with ubiquinol-1. The same experiment was done by allowing the enzyme to turnover with ubiquinol-1 while bound to the Ni-NTA column, during which buffer was continuously flowing. The results were identical. As a control, the enzyme was washed with Triton X-100 detergent to remove the quinone, and the quinone content determined by HPLC analysis (Figure 3A). The data support the two Q-site model.
Figure 1.
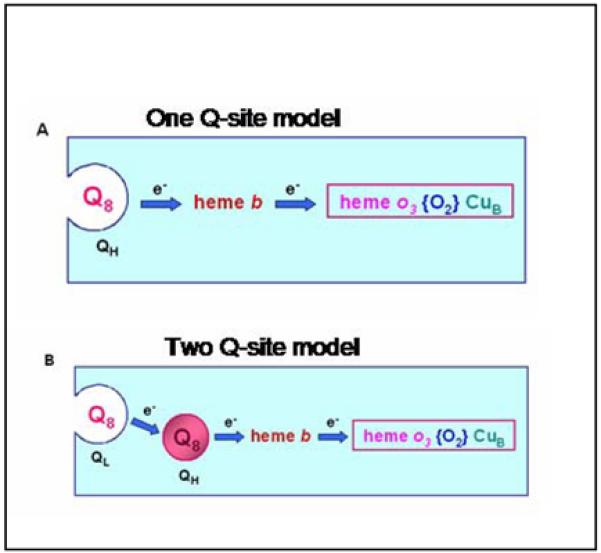
Schematic illustrating the 1-site and 2-site models for quinone binding to cytochrome bo3.
Figure 2.
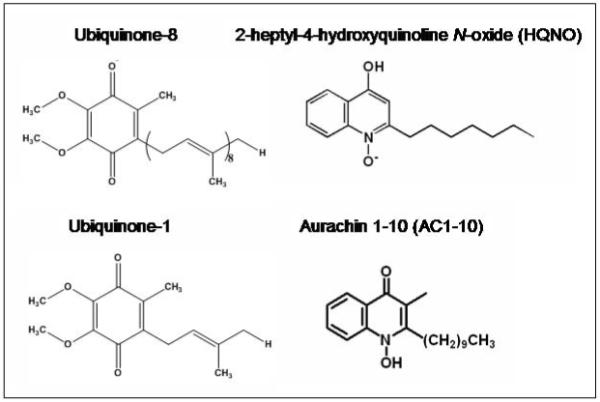
Structures of quinone substrates and quinone-like inhibitors referred to in the text.
Figure 3.
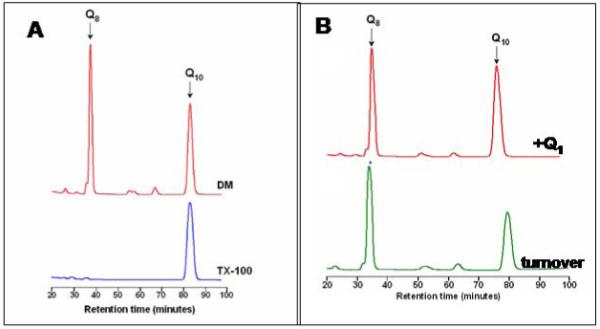
HPLC elution profiles following extraction of ubiquinone-8 from purified cytochrome bo3. A known amount of ubiquinone-10 is included in each sample as an internal standard. Panel A: Enzyme isolated in dodecylmaltoside (DM) contains 1 equivalent of ubiquinone-8, whereas enzyme washed with Triton X-100 has no bound ubiquinone-8. Panel B: Enzyme incubated with oxidized ubiquinone-1 (500 μM) retains bound ubiquinone-8 (top), as does enzyme which has been incubated under conditions of catalytic turnover with ubiquinol-1 (see text).
3.2 Binding of HQNO to cytochrome bo3
It has been shown previously that HQNO (Figure 2) is an effective inhibitor of cytochrome bo3 [13, 20]. Figure 4A shows the inhibition of cytochrome bo3 (with ubiquinone-8 bound to the QH site) by HQNO using the coupled assay to maintain the ubiquinol-1 substrate reduced. The Ki is 0.74 μM. Figure 4B shows a double-reciprocal plot, indicating a pattern of inhibition that is noncompetitive, consistent with previous reports [13, 20]. The noncompetitive inhibition pattern suggests that HQNO does not bind to the substrate (QL) binding site.
Figure 4.
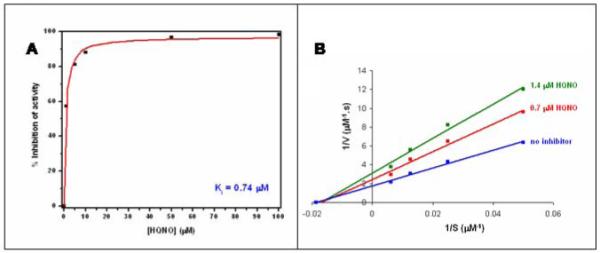
Inhibition of steady state ubiquinol-1 oxidase activity by HQNO. Panel (A) is a direct plot of the data, and Panel (B) is a Lineweaver-Burke plot showing noncompetitive inhibition.
To extend these observations, the stoichiometry and binding affinity of HQNO to the enzyme were measured directly by fluorescence spectroscopy. HQNO is itself fluorescent, and the fluorescence is often quenched upon binding to proteins. However, upon binding to cytochrome bo3, the fluorescence intensity is not substantially perturbed. Therefore, binding was monitored by measuring the fluorescence anisotropy, which increases substantially when HQNO binds to the enzyme. In order to avoid artifacts due to the aggregation of HQNO at high concentrations, the titration was performed by maintaining the total concentration of HQNO constant while increasing the amount of the protein. A representative binding isotherm is shown in Figure 5. Fitting the data to a model, assuming independent sites, yields a best fit of 1.1 sites and a Kd of 4.0 μM. The reason why the kinetic inhibition constant and the dissociation constant are different by about a factor of 5 is not clear, but may be due to differences in assay conditions or enzyme preparation. The main point is that the data show that HQNO binds to a single site on cytochrome bo3. The experiment was repeated in the presence of a large excess of oxidized ubiquinol-1 (Table 1) and the same result was obtained, suggesting that at least the oxidized form of ubiquinol-1 does not compete for binding at the same site.
Figure 5.
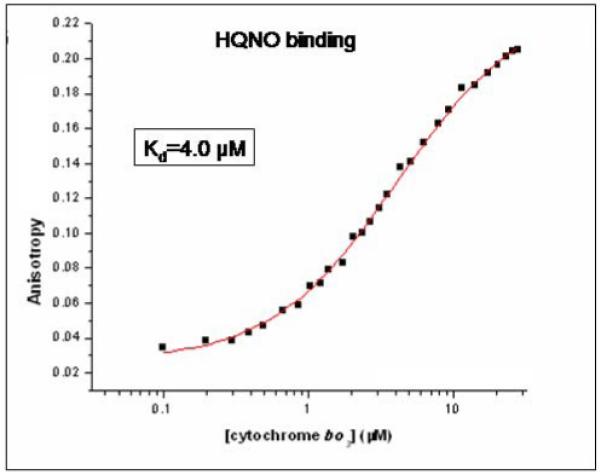
HQNO binding to cytochrome bo3 which has been isolated with one equivalent of ubiquinone-8 bound to the QH-site. Binding is monitored by fluorescence anisotropy, titrating the enzyme into a solution containing 1 μM HQNO. The points are experimental data and the line is a fit to 1 binding site with a Kd of 4 μM.
Table 1.
Characterization of wild type and mutants of cytochrome bo3. Shown are the steady state kinetic parameters for ubiquinol-1 oxidase activity (Vmax and KM); the dissociation constant for HQNO determined by fluorescence anisotropy titration; and the concentration of AC1-10 required for 50% inhibition of ubiquinol-1 oxidase activity (Ki)
| Cytochrome bo3 | Relative | |||
|---|---|---|---|---|
| Vmax (%) |
KM UQ1H2 (μM) |
Kd HQNO (μM) |
Ki AC1-10 (nM) |
|
| wild type | 100 | 53 | 4.0 | 15 |
| wild type + 300 μM Q1 |
- | - | 4.1 | - |
| wild type + 1500 μM Q1 |
- | - | 6.8 | - |
| wild type (no bound Q8) |
- | - | 5.8 | - |
| D188A | 69 | 56 | 5.0 | 25 |
| D188N | 71 | 52 | 4.0 | ND |
| R257Q | 47 | 60 | 4.0 | 12 |
| WII136A | 75 | 63 | 4.1 | 23 |
| H98S | - | - | 3.6 | - |
It has been previously reported that upon the addition of an excess of HQNO, the EPR signal of the pre-formed semiquinone at the QH-site is inhibited [18]. Figure 6A shows that the presence of an excess of HQNO prevents formation of the semiquinone. If HQNO were binding at the QH site, it would displace the tightly bound ubiquinone-8. However, this does not occur. The enzyme was bound to the Ni-NTA column in the presence of HQNO and then washed with several column volumes of buffer containing HQNO. Following this, the enzyme was eluted and the quinone was extracted and quantified. Cytochrome bo3 retains one equivalent of ubiquinone-8 in the presence of HQNO (Figure 6B).
Figure 6.
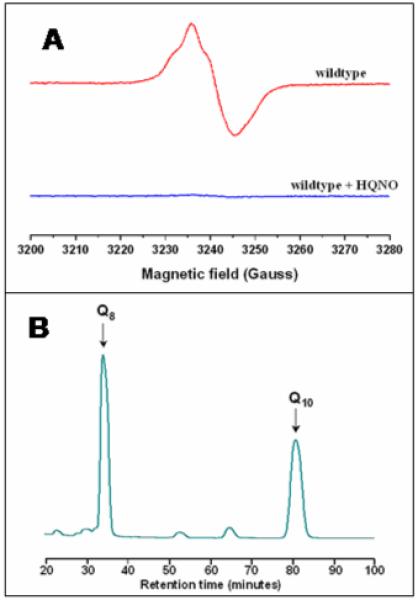
(A) EPR spectrum of the semiquinone formed by ubiquinone-8 bound at the QH-site of wild type cytochrome bo3. The presence of a 5-fold excess of HQNO prevents formation of the semiquinone. (B) HPLC elution profile of the ubiquinone-8 extracted from enzyme incubated under the same conditions as used for the EPR experiment in the top panel. The data show approximately 1 equivalent of ubiquinone-8 remains bound to the enzyme, unchanged from prior to the addition of HQNO. (B) EPR spectrum of the semiquinone formed by ubiquinone-8 bound at the QH-site of wild type cytochrome bo3. The presence of excess HQNO prevents formation of the semiquinone.
The binding of HQNO to enzyme from which the tightly bound ubiquinone-8 had been removed was also examined. The results (Table 1) show one HQNO binds with a Kd = 5.8 μM, similar to the data obtained with enzyme containing ubiquinone-8 bound at the QH site.
3.3 The binding of AC1-10 to cytochrome bo3
Previous work has shown that aurachin C and analogues are potent inhibitors of cytochrome bo3 [40, 42]. The inhibition of ubiquinol-1 oxidase activity by AC1-10 [40] is shown in Figure 7. The Ki was determined to be about 15 nM, and the pattern of inhibition (Figure 7A) indicates that AC1-10 is a competitive inhibitor. Previous work concluded that aurachin C binds to one site on cytochrome bo3 [42]. Incubation of a 50 μM solution of cytochrome bo3 with a 5-fold excess of AC1-10 prevents formation of the semiquinone EPR signal (Figure 7B) but, as was the case with HQNO, the tightly bound ubiquinone-8 is not displaced from the QH site (Figure 7C).
Figure 7.
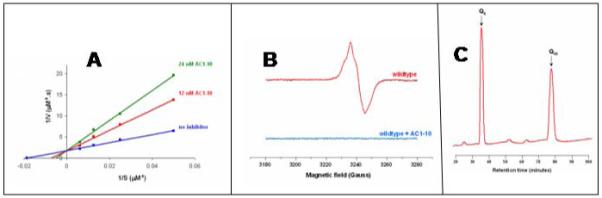
Inhibition of cytochrome bo3 by AC1-10. (A) A double reciprocal plot of (enzyme velocity)−1 vs (ubiquinol-1concentration)−1 showing competitive inhibition. (B) EPR spectrum of the semiquinone formed by ubiquinone-8 bound at the QH-site of wild type cytochrome bo3. The presence of a 5-fold excess of AC1-10 prevents formation of the semiquinone. (C) HPLC profile showing the ubiquinone-8 remains bound to cytochrome bo3 in the presence of AC1-10. The ubiquinone-10 is an internal standard used to quantify the amount of ubiquinone-8 extracted from the enzyme.
Site-directed mutagenesis of D188, R257 and WII136
The location of the QL-site is not known. It is a reasonable assumption that residues that directly interact with ubiquinol at the QL-site will be conserved among the many ubiquinol oxidases. Residues that are conserved include R71, D75, M78 and H98, which are proposed to constitute the QH-site. Two additional residues in subunit I that are conserved in all the quinol oxidases are D188 and R257. These residues are located near each other on the surface of subunit I at a level expected to be within the membrane bilayer and reasonably close to the QH-site. Hence, mutants were made to test whether these residues might be part of the QL site: D188A, D188N and R257Q. The results are summarized in Table 1, along with data from WII136A. In each case, the enzymes were purified and examined using 1) ubiquinol-1 oxidase activity (Vmax), including the KM of ubiquinol-1; 2) HQNO binding, measured by fluorescence anisotropy; and 3) inhibition of ubiquinol-1 oxidase activity by AC1-10. Although there is a modest loss of activity due to each mutation, there is no indication that either the QL quinol binding site, or the HQNO binding site, or the AC1-10 binding site involves these residues. It is noted that both D188 and R257 are also present in many cytochrome c oxidases, indicating a function other than quinol binding. One unusual observation is that neither the D188A nor R257Q mutant oxidases were able to support aerobic growth when expressed in a strain of E. coli without a genomically encoded respiratory oxidase. However, the isolated proteins were active (Table 1).
3.4 Additional mutations at the QH-site
A number of mutations at the QH-site (R71, D75, H98 and Q101) have been described [25, 26]. In those cases where measurements were made, it was shown that these mutations do not reduce the amount of bound ubiquinone [25]. Table 2 shows result obtained with a number of additional mutations at the QH-site. The EPR spectroscopy of the D75E and D75H mutants has been previously reported [32]. Besides the conservative replacement D75E, all of the other mutants have very low or no quinol oxidase activity. Apart from D75E, D75H and D75N, the semiquinone is also not formed. Table 2 shows that none of these mutations result in loss of the tightly bound ubiquinone-8 from the QH-site. This is also true for the double mutation, R71D/D75R, in which the aspartate and arginine have been switched.
Table 2.
Characterization of mutants at the QH-site of cytochrome bo3. Shown are the relative ubiquinol-1 oxidase activity, the content of ubiquinol-8 and whether the semiquinone is formed upon partial reduction by ascorbate
| Cytochrome bo3 | Relative activity (%) |
Quinone content (Q8/enzyme) |
Semiquinone formation |
|---|---|---|---|
| wild type | 100 | 1.1 | Yes |
| D75E | 135 | 1.2 | Yes |
| D75H | 4 | 0.9 | Yes |
| D75N | ~0 | 1.3 | Yes |
| D75R | ~0 | 1.2 | No |
| R71D | ~0 | 1.4 | No |
| R71K | ~0 | 1.3 | No |
| R71Q | ~0 | 1.3 | No |
| H98N | 1 | 1.4 | No |
| H98S | 2 | 1.4 | No |
| H98T | 1 | 1.5 | No |
| Q101N | 5 | 1.3 | No |
| R71D/D75R | ~0 | 1.3 | No |
For one mutation at the QH-site, H98S, the binding of HQNO was also examined. The results (Table 1) show that the dissociation constant is essentially the same as for the wild type enzyme, indicating that this residue is not important for the affinity of HQNO to the enzyme.
3.5 Generating the semiquinone radical at the QH site using ascorbate
The semiquinone form of ubiquinone-8 bound at the QH site can be generated by using ascorbate as a reducing agent and, after an empirically determined time, freezing the partially reduced (mixed valence) enzyme [31, 32, 43-45]. The continuous wave X-band EPR spectra of the oxidized and the partially reduced forms of wild type enzyme are shown in Figure 8A. The spectra show features previously assigned [46, 47] to high spin heme o3 (g = 5.81) and low spin heme b (g = 2.98, 2.26 and 1.45) as well as the signal from the semiquinone radical (g = 2). As previously discussed, the signal from the high spin heme o3 (g = 5.81) is due to a small fraction of the heme (≈ 5% to10%) which is no longer coupled to CuB [46, 47]. The spectra of the partially reduced forms of two mutants in the QH site, D75E and D75H, are also shown in Figure 8B. The D75E mutant is catalytically active and the EPR spectrum is similar to that of the wild type oxidase. The D75H mutant is catalytically inactive, and the EPR spectrum of the partially reduced enzyme lacks the signals from the low spin heme b, indicating that heme b in this mutant is completely reduced by the ascorbate.
Figure 8.

(A) Continuous-wave X-band EPR spectrum of oxidized (red) and reduced (black) cytochrome bo3 (see text for details). (B) EPR spectra of the reduced D75E and D75H mutants. Temperature 15 K, microwave power ~ 10 mWatt, modulation amplitude 0.5 mT, reduction time 6 hours.
The peak intensities of the EPR features for the wild type oxidase as a function of time after the addition of ascorbate are shown in Figure 9. The intensity of the low spin heme b signal decreases with time. In contrast, the amplitude of the high spin heme o3 signal as well as the amplitude of the semiquinone signal increase in parallel, reaching a maximum at about 6 hr. The simplest explanation is that the amount of reduced CuB (revealing the EPR signal from the oxidized heme o3) varies in proportion to the amount of semiquinone present. In the D75H mutant, the reduction of heme b occurs substantially more rapidly than in either the wild type or D75E mutant oxidase. As a result, after 6 hours, the EPR signal from heme b is not observed.
Figure 9.
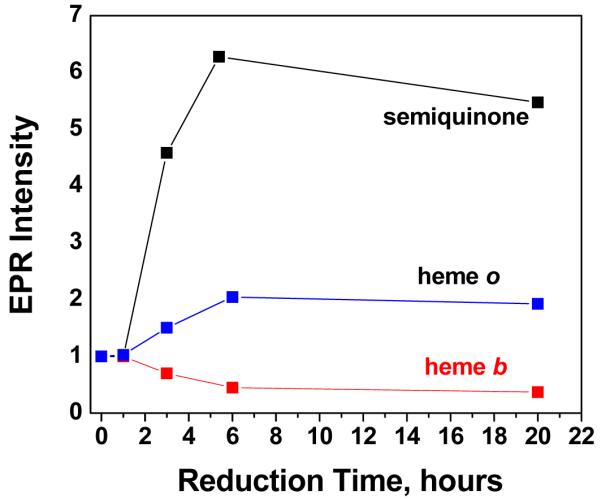
Behavior of the EPR intensities of the semiquinone, heme o3 and heme b as a function of reduction time. The peak height of each sample in the spectrum recorded at 1 hour is arbitrarily set to a value of 1, and relative intensities are plotted as a function of time. The amplitudes of the signals due to ferric heme o3 (g=5.81) and ferric heme b (g=2.26) were measured from the spectra recorded at 15 K. The amplitude of the semiquinone signal was measured at 90 K with low microwave power to avoid saturation conditions and significant overlap with other signals around g~2. The relative intensities of the heme b, heme o3 and semiquinone signals are not necessarily proportional to the relative concentrations of the three species.
4. Discussion
The motivation of the current work was primarily to test the validity of the two-site model of ubiquinone binding to cytochrome bo3. It was previously shown that a large excess of ubiquinone-1 does not displace the bound ubiquinone-8 under conditions when all the species remain oxidized [11], and this has been repeated (Figure 3). The critical test, however, is to examine displacement under conditions when the enzyme is turning over ubiquinol-1. The fact that ubiquinol-1 does not displace ubiquinol-8 at the QH site under turnover conditions clearly shows that ubiquinone-8 at this site does not readily exchange. These new data definitively demonstrate that there must be more than one quinone binding site, thus supporting the two-site model (Figure 1) [11].
Arguments for the two-site model based on the effect of inhibitors on steady state kinetics [13, 14] have assumed that some of the inhibitors, including HQNO, can bind to the QH-site. However, data presented in this paper as well as previously published data [11] show that HQNO does not displace the ubiquinone-8 at the QH-site. New data in the current work show, in addition, that only one equivalent of HQNO binds to the enzyme, and that the affinity is not significantly altered by removing the tightly bound ubiquinone-8 at the QH-site.
Most of the information about the interaction of ubiquinone at the QH-site is from EPR studies of the stabilized ubisemiquinone that forms at this site [26, 31, 32, 45]. Data in Figures 8 and 9 show the full EPR spectrum of the enzyme and the changes that occur during the partial reduction by ascorbate. Under the conditions utilized, the reduction of the wild type enzyme by ascorbate is slow (hours) and, in addition to formation of the ubisemiquinone, heme b and CuB are partially reduced the final product.
HQNO also prevents the formation of the ubisemiquinone at the QH-site when the enzyme is partially reduced. This is shown in the present work, and was reported previously [18]. The addition of HQNO to the fully reduced enzyme also prevents the oxidation of ubiquinol-8 at the QH-site by O2 [48]. Hence, HQNO functionally disrupts the reactions of the ubiquinone at the QH-site, though this quinone is not displaced from the enzyme. One possibility, previously discussed [13], is that the ubiquinone-8 bound at the QH-site is held in place primarily by interactions with the hydrophobic isoprene side chain. Therefore, inhibitors such as HQNO can displace the quinone ring without displacing the molecule as a whole. This would be consistent with the observation that the mutations at the QH-site which eliminate catalytic activity as well as prevent formation of the semiquinone species, retain the bound ubiquinone-8, shown in Table 2 as well as in previous work [26, 48]. Arguing against this interpretation is the fact that the dissociation constant of HQNO to the enzyme is only slightly increased (4.0 μM to 5.8 μM) by removal of the ubiquinone-8 from the QH-site (Table 1). If HQNO were competing for the same site with a locally tethered head group of ubiquinone-8, one would expect a larger effect on the Kd of HQNO by the presence of the bound ubiquinone-8. Nevertheless, the competition between HQNO and ubiquinone-8 for binding to the QH-site needs to be examined under conditions where the enzyme is partially or fully reduced to further test this model. What can definitely be concluded at this point is that H98, although part of the QH-site, does not interact with HQNO, as shown by the fact that the H98S mutant has the same affinity for HQNO as does the wild type (Table 2).
The current work strongly supports the model that cytochrome bo3 contains two quinone binding sites, QH and QL. The interactions critical for the high affinity of the tightly bound quinone at the QH-site remain unknown, since mutations of residues at the QH-site which are known to interact with the ubisemiquinone ring [31, 32, 45] do not physically eliminate the bound quinone (Table 2).
The location of the QL site is also not known. It is likely that the QL-site is adjacent to the QH-site. This would be consistent with the fact that AC1-10, which is a competitive inhibitor for the QL-site, also perturbs the QH-site, evidenced by blocking the formation of the ubisemiquinone at the QH-site upon reduction by ascorbate. It cannot be excluded that some of the residues involved at the QH-site are also involved in the QL-site.
Further examination of HQNO binding to mutants should be helpful to identify the HQNO binding site, which appears to be separate from both the QH-site and the QL-site. The possibility that the ubiquinone-8 bound at the QH-site is tethered by the hydrophobic side chain and has several modes of interaction between the quinone ring and the protein, depending on the redox state, should be seriously considered. In this model, HQNO could displace the ubiquinone head group at the QH-site without actually displacing the bound quinone. This would be consistent with the fact that no mutants have been found yet which increase the KM for ubiquinol-1, since such mutations would also alter the QH-site and eliminate catalytic function. Clearly, further work is required.
5. Conclusions
It is definitively demonstrated that cytochrome bo3 must have two ubiquinone binding sites. Ubiquinone-8 bound at the QH-site acts as a cofactor and is not displaced during enzyme turnover, nor in the presence of the inhibitors HQNO or AC1-10. AC1-10 appears to compete for ubiquinol-1 binding at the QL-site, which is the substrate binding site. HQNO binds stoichiometrically to one site, but is a noncompetitive inhibitor at the QL site. The QH-site, QL-site and the HQNO binding site are likely to be adjacent and may be partially overlapping.
Acknowledgements
We would like to that Dr. James Hemp for many helpful discussions and for pointing out the extent of conservation of residues in the quinol oxidases. This investigation was supported by Grants from Chemical Sciences, Geosciences and Biosciences Division, Office of Basic Energy Sciences, Office of Sciences, US DOE: DE-FG02-87ER13716 (to R.B.G.) and DE-FG02-08ER15960 (to S.A.D.), and the National Institutes of Health GM062954 Grant (to S.A.D.) and NIH/Fogarty Grant RP3 TW01495 (to R.I.S.).
References
- [1].Mogi T, Tsubaki M, Hori H, Miyoshi H, Nakamura H, Anraku Y. J. Biochem. Mol. Biol. & Biophys. 1998;2:79–110. [Google Scholar]
- [2].Cotter PA, Gunsalus RP. FEMS Lett. 1992;91:31–36. doi: 10.1016/0378-1097(92)90558-6. [DOI] [PubMed] [Google Scholar]
- [3].Tseng C-P, Albrecht J, Gunsalus RP. J. Bact. 1996;178:1094–1098. doi: 10.1128/jb.178.4.1094-1098.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Pereira MM, Santana M, Teixeira M. Biochim. Biophys. Acta. 2001;1505:185–208. doi: 10.1016/s0005-2728(01)00169-4. [DOI] [PubMed] [Google Scholar]
- [5].Hemp J, Gennis RB. Results Probl Cell Differ. 2008;45:1–31. doi: 10.1007/400_2007_046. [DOI] [PubMed] [Google Scholar]
- [6].Chepuri V, Lemieux LJ, Au DC-T, Gennis RB. J. Biol. Chem. 1990;265:11185–11192. [PubMed] [Google Scholar]
- [7].Saraste M, Raito M, Jalli T, Chepuri V, Lemieux L, Gennis RB. Annals of the New York Academy of Sciences. 1989;550:314–324. doi: 10.1111/j.1749-6632.1988.tb35346.x. [DOI] [PubMed] [Google Scholar]
- [8].van der Oost J, Pappalainen P, Musacchio A, Warne A, Lemieux L, Rumbley J, Gennis RB, Aasa R, Pascher T, Malmström BG, Saraste M. EMBO J. 1992;11:3209–3217. doi: 10.1002/j.1460-2075.1992.tb05398.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Santana M, Kunst F, Hullo MF, Rapoport G, Danchin A, Glaser P. J. Biol. Chem. 1992;267:10225–10231. [PubMed] [Google Scholar]
- [10].Mattatall NR, Cameron LM, Hill BC. Biochemistry. 2001;40:13331–13341. doi: 10.1021/bi011116q. [DOI] [PubMed] [Google Scholar]
- [11].Sato-Watanabe M, Mogi T, Ogura T, Kitagawa T, Miyoshi H, Iwamura H, Anraku Y. J. Biol. Chem. 1994;269:28908–28912. [PubMed] [Google Scholar]
- [12].Puustinen A, Verkhovsky MI, Morgan JE, Belevich NP, Wikström M. Proc. Natl. Acad. Sci. USA. 1996;93 doi: 10.1073/pnas.93.4.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Musser SM, Stowell MHB, Lee HK, Rumbley JN, Chan SI. Biochemistry. 1997;36:894–902. doi: 10.1021/bi961723r. [DOI] [PubMed] [Google Scholar]
- [14].Sato-Watanabe M, Mogi T, Miyoshi H, Iwamura H, Matsushita K, Adachi O, Anraku Y. J. Biol. Chem. 1994;269:28899–28907. [PubMed] [Google Scholar]
- [15].Sakamoto K, Miyoshi H, Takegami K, Mogi T, Anraku Y, Iwamura H. J. Biol. Chem. 1996;271:29897–29902. doi: 10.1074/jbc.271.47.29897. [DOI] [PubMed] [Google Scholar]
- [16].Sakamoto K, Miyoshi H, Ohshima M, Kuwabara K, Kano B, Akagi T, Mogi T, Iwamura H. Biochemistry. 1998;37:15106–15113. doi: 10.1021/bi981193u. [DOI] [PubMed] [Google Scholar]
- [17].Sato-Watanabe M, Itoh S, Mogi T, Matsuura K, Miyoshi H, Anraku Y. FEBS Lett. 1995;374:265–269. doi: 10.1016/0014-5793(95)01125-x. [DOI] [PubMed] [Google Scholar]
- [18].Ingledew WJ, Ohnishi T, Salerno JC. Eur. J. Biochem. 1995;227:903–908. doi: 10.1111/j.1432-1033.1995.tb20217.x. [DOI] [PubMed] [Google Scholar]
- [19].Mogi T, Sato-Watanabe M, Miyoshi H, Orii Y. FEBS Letters. 1999;457:61–64. doi: 10.1016/s0014-5793(99)01007-8. [DOI] [PubMed] [Google Scholar]
- [20].Sato-Watanabe M, Mogi T, Miyoshi H, Anraku Y. Biochemistry. 1998;37:5355–5361. doi: 10.1021/bi9727592. [DOI] [PubMed] [Google Scholar]
- [21].Schultz BE, Edmondson DE, Chan SI. Biochemistry. 1998;37:4160–4168. doi: 10.1021/bi971714y. [DOI] [PubMed] [Google Scholar]
- [22].Kobayashi K, Tagawea S, Mogi T. Biochemistry. 2000;39:15620–15625. doi: 10.1021/bi0014094. [DOI] [PubMed] [Google Scholar]
- [23].Svensson-Ek M, Thomas JW, Gennis RB, Nilsson T, Brzezinski P. Biochemistry. 1996;35:13673–13680. doi: 10.1021/bi961466q. [DOI] [PubMed] [Google Scholar]
- [24].Jasaitis A, Johansson MP, Wikstrom M, Vos MH, Verkhovsky MI. Proc Natl Acad Sci U S A. 2007;104:20811–4. doi: 10.1073/pnas.0709876105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Abramson J, Riistama S, Larsson G, Jasaitis A, Svensson-Ek M, Laakkonen L, Puustinen A, Iwata S, Wikström M. Nature Struct. Biol. 2000;7:910–917. doi: 10.1038/82824. [DOI] [PubMed] [Google Scholar]
- [26].Hellwig P, Yano T, Ohnishi T, Gennis RB. Biochemistry. 2002;41:10675–10679. doi: 10.1021/bi012146w. [DOI] [PubMed] [Google Scholar]
- [27].Welter R, Gu L-Q, Yu L, Yu C-A, Rumbley J, Gennis RB. J. Biol. Chem. 1994;269:28834–28838. [PubMed] [Google Scholar]
- [28].Tsatsos PH, Reynolds K, Nickels EF, He D-Y, Yu C-A, Gennis RB. Biochemistry. 1998;37:9884–9888. doi: 10.1021/bi9809270. [DOI] [PubMed] [Google Scholar]
- [29].Sato-Watanabe M, Mogi T, Sakamoto K, Miyoshi H, Anraku Y. Biochemistry. 1998;37:12744–12752. doi: 10.1021/bi981184l. [DOI] [PubMed] [Google Scholar]
- [30].Ma J, Puustinen A, Wikström M, Gennis RB. Biochemistry. 1998;37:11806–11811. doi: 10.1021/bi9809977. [DOI] [PubMed] [Google Scholar]
- [31].Yap LL, Samoilova RI, Gennis RB, Dikanov SA. J Biol Chem. 2006;281:16879–87. doi: 10.1074/jbc.M602544200. [DOI] [PubMed] [Google Scholar]
- [32].Yap LL, Samoilova RI, Gennis RB, Dikanov SA. J Biol Chem. 2007;282:8777–85. doi: 10.1074/jbc.M611595200. [DOI] [PubMed] [Google Scholar]
- [33].Brandt U, von Jagow G. Eur J Biochem. 1991;195:163–70. doi: 10.1111/j.1432-1033.1991.tb15690.x. [DOI] [PubMed] [Google Scholar]
- [34].Rothery RA, Seime AM, Spiers AM, Maklashina E, Schroder I, Gunsalus RP, Cecchini G, Weiner JH. Febs J. 2005;272:313–26. doi: 10.1111/j.1742-4658.2004.04469.x. [DOI] [PubMed] [Google Scholar]
- [35].Zhao Z, Rothery RA, Weiner JH. Eur J Biochem. 1999;260:50–6. doi: 10.1046/j.1432-1327.1999.00116.x. [DOI] [PubMed] [Google Scholar]
- [36].Rothery RA, Weiner JH. Eur J Biochem. 1998;254:588–95. doi: 10.1046/j.1432-1327.1998.2540588.x. [DOI] [PubMed] [Google Scholar]
- [37].Rothery RA, Blasco F, Magalon A, Asso M, Weiner JH. Biochemistry. 1999;38:12747–12757. doi: 10.1021/bi990533o. [DOI] [PubMed] [Google Scholar]
- [38].Rothery RA, Blasco F, Weiner JH. Biochemistry. 2001;40:5260–8. doi: 10.1021/bi002393k. [DOI] [PubMed] [Google Scholar]
- [39].Zhao Z, Weiner JH. J Biol Chem. 1998;273:20758–63. doi: 10.1074/jbc.273.33.20758. [DOI] [PubMed] [Google Scholar]
- [40].Miyoshi H, Takegami K, Sakamoto K, Mogi T, Iwamura H. J. Biochem. 1999;125:138–142. doi: 10.1093/oxfordjournals.jbchem.a022250. [DOI] [PubMed] [Google Scholar]
- [41].Crofts AR. Annu Rev Physiol. 2004;66:689–733. doi: 10.1146/annurev.physiol.66.032102.150251. [DOI] [PubMed] [Google Scholar]
- [42].Meunier B, Madgwick SA, Reil E, Oettmeier W, Rich PR. Biochemistry. 1995;34:1076–1083. doi: 10.1021/bi00003a044. [DOI] [PubMed] [Google Scholar]
- [43].Grimaldi S, MacMillan F, Ostermann T, Ludwig B, Michel H, Prisner T. Biochemistry. 2001;40:1037–1043. doi: 10.1021/bi001641+. [DOI] [PubMed] [Google Scholar]
- [44].Grimaldi S, Ostermann T, Weiden N, Mogi T, Miyoshi H, Ludwig B, Michel H, Prisner T, MacMillan F. Biochemistry. 2003;42:5632–5639. doi: 10.1021/bi034010z. [DOI] [PubMed] [Google Scholar]
- [45].Lin MT, Samoilova RI, Gennis RB, Dikanov SA. J Am Chem Soc. 2008;130:15768–9. doi: 10.1021/ja805906a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Salerno JC, Bolgiano B, Poole RK, Gennis RB, Ingledew WJ. J. Biol. Chem. 1990;265:4364–4368. [PubMed] [Google Scholar]
- [47].Cheesman MR, Watmough NJ, Pires CA, Turner R, Brittain T, Gennis RB, Greenwood C, Thomson AJ. Biochem. J. 1993;289:709–718. doi: 10.1042/bj2890709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Ek MS, Brzezinski P. Biochemistry. 1997;36:5425–5431. doi: 10.1021/bi962478e. [DOI] [PubMed] [Google Scholar]