

Abstract
Metabolic activation of estrogens to catechols and further oxidation to highly reactive o-quinones generates DNA damage including apurinic/apyrimidinic (AP) sites. 4-Hydroxyequilenin (4-OHEN) is the major catechol metabolite of equine estrogens present in estrogen replacement formulations, known to cause DNA strand breaks, oxidized bases, and stable and depurinating adducts. However, the direct formation of AP sites by 4-OHEN has not been characterized. In the present study, the induction of AP sites in vitro by 4-OHEN and the endogenous catechol estrogen metabolite, 4-hydroxyestrone (4-OHE) was examined by an aldehyde reactive probe assay. Both 4-OHEN and 4-OHE can significantly enhance the levels of AP sites in calf thymus DNA in the presence of the redox cycling agents, copper ion and NADPH. The B-ring unsaturated catechol 4-OHEN induced AP sites without added copper, whereas 4-OHE required copper. AP sites were also generated much more rapidly by 4-OHEN. For both catechol estrogens, the levels of AP sites correlated linearly with 8-oxo-dG levels, implying that depuriniation resulted from reactive oxygen species (ROS) rather than depurination of estrogen-DNA adducts. ROS modulators such as catalase which scavenges hydrogen peroxide and a Cu(I) chelator blocked the formation of AP sites. In MCF-7 breast cancer cells, 4-OHEN significantly enhanced the formation of AP sites with added NADH. In contrast, no significant induction of AP sites was detected in 4-OHE-treated cells. The greater redox activity of the equine catechol estrogen produces rapid oxidative DNA damage via ROS, which is enhanced by redox cycling agents and interestingly by NADPH-dependent quinone oxidoreductase (NQO1).
Keywords: apurinic/apyrimidinic sites; catechol estrogen; 4-hydroxyequilenin; 4-hydroxyestrone; 8-oxo-7, 8-dihydro-2′-deoxyguanosine; quinone; reactive oxygen species
Introduction
An increased risk of breast cancer in postmenopausal women is strongly linked to long-term exposure to estrogens including hormone/estrogen replacement therapy (HRT/ERT) (1). Observations from various clinical trials and epidemiological studies collectively support the hypothesis that estrogens enhance breast cancer risk (2–8). However, the mechanisms of estrogen carcinogenesis are not fully understood. It has been suggested that estrogens contribute to hormone-sensitive cancers through two mechanisms; the proliferative and anti-apoptotic hormonal effects of estrogens and the genotoxic effects of estrogen metabolites generated through P450 catalyzed metabolic activation (9–15).
The most widely prescribed estrogen replacement formulation in the United States is Premarin (Wyeth-Ayerst), which contains conjugated human endogenous estrogens and B-ring unsaturated equine estrogens (16). Human endogenous estrogens are hydroxylated by cytochrome P450s at the 2- and 4- positions to form the corresponding catechol estrogen metabolites. The 4-hydroxy catechol metabolites, not 2-hydroxy catechol metabolites, are proposed to be genotoxic as the result of oxidation to reactive o-quinones that alkylate DNA as well as generation of reactive oxygen species (ROS) through redox cycling (14, 17–19) (Scheme 1). The B-ring unsaturated equine estrogens [equilin (EQ)/equilenin (EN)] are selectively oxidized to the 4-hydroxy catechols (4-OHEQ/4-OHEN) which are readily autoxidized to the redox cycling 4-OHEN-o-quinone (4-OHEN-Q) (20–22). Data have shown that 4-OHEN can induce a variety of different types of DNA damage (14, 23), causing DNA mutations (24), induction of apoptosis (25), and cellular transformation (26).
Scheme 1.

Formation of AP sites by depurination of labile estrogen-DNA adducts and ROS via redox cycling between catechols and o-quinones.
Oxidation of catechol estrogens to semiquinones and quinones is generally catalyzed by oxidative enzymes (e.g., P450s) and/or metal ions (e.g., copper ion). Copper ion is present in many tissue at micromolar concentrations, and it has been found at higher concentration in breast tumors as compared with normal breast tissues (27). At the cellular level, approximately 20% of copper is in the nucleus associated with DNA bases (28). Previous studies have demonstrated that endogenous catechol estrogens can induce DNA strand breaks (29), oxidized bases (30), and apurinic/apyrimidinic (AP) sites (31) in the presence of copper ion, and this effect could be enhanced by reducing agents such as NAD(P)H (30, 31).
It has been proposed that formation of depurinating DNA adducts by endogenous estrogen o-quinones generates AP sites leading to cancer initiation (17, 19). For example, the 4-hydroxyestrone-o-quinone (4-OHE-Q) can react with DNA to form 4-OHE-N7-guanine and 4-OHE-N3-adenine depurinating adducts in vitro. However, data also suggested that AP sites might be derived from ROS via hydrogen abstraction from the deoxyribose moiety of nucleic acids (31, 32). Since 4-OHEN is considered to be more reactive and cytotoxic compared to endogenous catechol estrogens (14, 25, 26), it was of interest to investigate whether 4-OHEN forms more ROS-mediated AP sites compared to the endogenous catechol 4-OHE. Formation of AP sites was analyzed using the aldehydic reactive probe assay after incubation of the catechol estrogens with copper ion and NAD(P)H. The results demonstrated that 4-OHEN is more redox reactive than 4-OHE resulting in the induction of AP sites in calf thymus DNA and human breast cancer cells (MCF-7).
Experimental Procedures
Caution: The catechol estrogens are potentially hazardous materials. Protective clothing should be worn, and appropriate safety procedure should be followed.
Materials
4-OHEN was synthesized by treating equilin (EQ) with Fremy’s salt as described previously (33) with minor modifications (21). 4-OHE was purchased from Steraloids (Newport, RI) and the synthesis of 4-OHE-Q was accomplished following the published method (34). Aldehydic reactive probe (ARP), standard ARP-DNA conjugate, streptavidin-horseradish peroxidase (HRP) conjugate, and DNA binding solution were purchased from Dojindo Molecular Technologies (Gaithersburg, MA). Plasmid ΦX174 RFI DNA was purchased from New England Biolabs (Beverly, MA). Calf thymus DNA, β-nicotinamide adenine dinucleotide phosphate, reduced form (NADPH), cupric chloride (CuCl2), methoxyamine hydrochloride, 3,3′,5,5′-tetramethylbenzidine (TMB), sodium azide, bathocuproinedisulfonic acid (BCS), 2,3-dimethoxy-1,4-naphthoquinone (DMNQ) methional, catalase (23000 units/mg from bovine liver) and superoxide dismutase (SOD, 1933 units/mg from bovine liver) were purchased from Sigma (St. Louis, MO). All other chemicals were obtained from Sigma or Fisher Scientific (Itasca, IL) unless stated otherwise.
Preparation of Methoxyamine-Treated Calf Thymus DNA
Calf thymus DNA (2 mg/mL) in 10 mM Tris-HCl buffer (pH 7.4) was treated with 100 mM methoxyamine (pH 7.2) for 2 h at 37°C to block the endogenous AP sites. The methoxyamine treated-DNA was precipitated by adding sodium acetate (3 M, 0.1 volumes) and cold ethanol (2.5 volumes). The DNA pellets were washed with 70% ethanol four times, resuspended in 10 mM Tris-EDTA buffer (pH 7.4), and stored at −80 °C until use.
Reaction of Estrogen Catechol/O-Quinones with Calf Thymus DNA
The methoxyamine-treated calf thymus DNA was treated with estrogen catechol/o-quinones under various conditions. The DNA pellet (100 μg) was incubated with the indicated concentrations of catechols/o-quinones, CuCl2, NADPH, and various scavengers in 250 μL of 10 mM potassium phosphate buffer (PPB, pH 7.4) at 37 °C. Following incubation, the solutions were extracted twice with an equal volume of chloroform:isoamyl alcohol (24:1) to remove excess quinones. The DNA was then precipitated and washed three times with ethanol. The DNA pellets were dissolved in 10 mM Tris-EDTA buffer (pH 7.4). The concentrations were adjusted to 100 μg/mL DNA using a UV spectrometer assuming that one absorbance unit equals 50 μg/mL of DNA. ARP tagging was applied immediately to label the AP sites according to the manufacturer’s protocol (Dojindo Molecular Technologies, Rockville, MD). Briefly, a DNA sample (1 μg, 10 μL) was incubated with 10 μL of 10 mM ARP solution for 1 h at 37 °C. Tris-EDTA buffer (380 μL) was added to the reaction solution and the solution was transferred to a filtration tube (Dojindo). After centrifugation to isolate the ARP-tagged DNA, the resultant ARP-tagged DNA was dissolved in 400 μL Tris-EDTA buffer, and stored at 4 °C until analysis.
Incubation of Estrogen Catechol/O-Quinones in MCF-7 Cells
All cell culture reagents were purchased from Invitrogen (Carlsbad, CA) unless stated otherwise. The MCF-7 human breast cancer cells were maintained in RPMI 1640 containing 10% fetal bovine serum (Atlanta Laboratory, Atlanta, GA), 2 mM L-glutamine, 0.1 mM nonessential amino acids, and 6 μg/mL bovine insulin (Sigma). Estrogen-free media were prepared from phenol-red-free RPMI1640 media supplied with 10% charcoal-dextran-treated fetal bovine serum whereas other components remained the same. The cells were cultured in estrogen-free media for 3 days before the experiment. The cells (1.2 × 106) in estrogen-free media (4 mL) were plated into 6-well plates and cultured for 24 h before treatment. Compounds were freshly dissolved in DMSO with 0.1% final concentration in cell media. After treatment, the cells were washed with cold PBS and then trypsinized. DNA extraction was performed as previously published (31) with minor modification using a Gentra PureGene cell kit from Qiagen (Valencia, CA). Briefly, cell pellets were lysed in Cell Lysis Solution with RNases A (0.15 mg) at 37 °C for 5 min, and then Protein Precipitation Solution was added to precipitate protein. After centrifugation, the supernatant was extracted twice by an equal volume of chloroform/isoamyl alcohol (24:1, v/v), and then precipitated with isopropyl alcohol and washed with 70% ethanol. The DNA pellets were resuspended in Tris-EDTA buffer and the concentration was determined by UV spectrometer. The AP sites were immediately trapped using the ARP solution.
Quantification of AP Sites Using a Colorimetric Assay
Assays were performed according to the manufacturer’s protocol (Dojindo). Briefly, an aliquot of the diluted ARP-tagged DNA (60 μL) was added with DNA binding solution to each well of a microtiter plate, and then incubated overnight at room temperature. After discarding the solution, the plate was washed five times with 10 mM phosphate-buffered saline containing 0.05% Tween 20 (PBST, Sigma). An aliquot of diluted streptavidin-HRP solution (1:4000) was added and then the plate was incubated in the dark for 1 h at 37 °C. After discarding the excess streptavidin-HRP solution, the plate was washed five times with PBST. An aliquot of substrate TMB (100 μL) was added and incubated for 1 h at 37 °C for color development. The absorbance at 650 nm was read using PowerWave 200 Microplate Spectrophotometer (Bio-Tek, Vermont). The number of AP sites in samples was quantified by using the standard ARP-tagged DNA conjugates (Dojindo) with known amounts of AP sites from 0 per 40 per 105 base pairs.
Determination of NADPH Oxidation
The assay was conducted on the PowerWave 200 Microplate Spectrophotometer (Bio-Tek, Vermont). The mixture contained 200 μM NADPH, 10 μM catechol/o-quinones, with/without 10 μM CuCl2 in 10 mM PPB (pH 7.4, 200 μL) at 23 °C. The reaction was initiated by adding NADPH and monitored at 340 nm. The rate of NADPH oxidation was determined from the decrease in absorbance of NADPH (340 nm), and calculated by using a standard curve with known amounts of NADPH.
Detection of 8-oxo-dG by LC-MS/MS
The 8-oxo-dG analysis was carried out as described previously (35) with minor modifications. Briefly, DNA samples were extracted twice by chloroform/isoamyl alcohol, and then precipitated and washed with ethanol. After DNA hydrolysis, the stable isotopically labeled [15N5]-8-oxo-dG was added and the mixture was analyzed on a Thermo (San Jose, CA) TSQ Quantum triple quadrupole mass spectrometer coupled with Surveyor HPLC pump and PDA detector. The samples were separated using an ODS AQ C18 column (2.0 mm × 100 mm, YMC, Wilmington, NC) with a gradient methanol/water mobile phase; the native dG and 8-oxo-dG was determined as previously described (35).
DNA Strand Breaks Assay
DNA strand breaks were analyzed by the conversion of supercoiled plasmid ΦX174 RF I DNA to open circular and linear forms as described previously (36, 37). Briefly, ΦX174 RF I DNA (0.5 μg) was incubated with various concentrations of catechol/o-quinones in the presence of CuCl2 (10 μM) and NADPH (200 μM) in 10 mM PPB (pH 7.4), in a final volume of 50 μL. The solutions were incubated in the dark at 37 °C for the indicated times. Samples (20 μL) were immediately subjected to electrophoresis on a 1.0% agarose gel with 1.0 μg/mL ethidium bromide in 40 mM Tris-acetate buffer containing 1 mM EDTA. After electrophoresis, the DNA was visualized under UV irradiation at 312 nm and photographed with a Polaroid 3000 camera.
Statistical Analysis
All data of AP sites were obtained from at least two separate assays of triplicate determinations. The results represented the average ± standard deviation (S.D.). The statistical analysis of these results consisted of one-way ANOVA analysis with Dunnett’s or Tukey’s multiple comparison tests by using the GraphPad Prism version 5 for Windows.
Results
Formation of AP Sites in Calf Thymus DNA treated with Estrogen Metabolites
Previous studies have shown that 4-OHE can induce the formation of AP sites in calf thymus DNA in the presence of copper (31). To test whether 4-OHEN is also capable of inducing AP sites, comparisons were performed using calf thymus DNA in the presence of CuCl2 and/or NADPH (Figure 1). As shown in Figure 1A, 4-OHEN (10 μM) induced two fold higher AP sites over control in the presence of CuCl2 (1 μM), and increasing the CuCl2 concentration to 10 μM enhanced AP levels 7-fold. 4-OHE also significantly induced AP sites in the presence of CuCl2 and the levels depended on the concentration of CuCl2, consistent with previous data (31). At 100 μM CuCl2, the levels of AP sites produced by 4-OHE were significantly higher than those from 4-OHEN. In the absence of CuCl2, only 4-OHEN induced NADPH dependent AP site formation in a concentration dependent manner (Figure 1B). These results show that 4-OHE requires CuCl2 to stimulate AP site formation whereas 4-OHEN, which autoxidizes, does not require CuCl2 to generate AP sites, but at lower concentrations of catechol and copper both estrogens induce AP sites at similar levels.
Figure 1. Formation of AP sites in calf thymus DNA treated with catechol estrogens.

Calf thymus DNA (100 μg) was treated with 4-OHEN (10 μM) or 4-OHE (10 μM) with various concentrations of A) CuCl2 or B) NADPH. Control experiment was performed with catechol estrogens only. Assays were performed in 10 mM PPB (pH 7.4) at 37 °C for 4 h. DNA was isolated and labeled immediately with ARP reagent as described in Experimental Procedures. Data represents the mean ± S.D. (n = 3). Asterisk: p < 0.05, significant difference as compared to control.
The dependence of AP site formation on catechol estrogen concentration was also studied (Figure 2). The maximum induction of AP sites was observed at a concentration of 5 μM 4-OHEN within 4 h. The number of AP sites induced by 4-OHE or 4-OHE-Q was also dose-dependent, while the levels were lower than observed with 4-OHEN under the same conditions. As expected, in the absence of CuCl2 and NADPH, none of the catechol/o-quinones caused significant increases in AP sites relative to control (Figure 2A). Similar experiments were conducted with the para-quinone 2,3-dimethoxy-1,4-naphthoquinone (DMNQ), which is considered to be a pure redox-cycling ROS generator (38). In contrast to the estrogen o-quinone, DMNQ did not enhance the levels of AP sites in the presence of CuCl2 and NADPH (data not shown). Kinetic studies on NADPH oxidation showed that NADPH (200 μM) did not reduce DMNQ (10 μM). Similar results have also been reported for 1,4-naphthoquinone which also did not significantly increase the levels of AP sites in calf thymus DNA (39).
Figure 2. Dose- and time-dependent formation of AP sites by catechol estrogens in calf thymus DNA through redox cycling.

Calf thymus DNA (100 μg) was treated with catechol/o-quinones with/without CuCl2 (10 μM) and NADPH (200 μM). Solid lines, with CuCl2 and NADPH; dashed lines, without CuCl2 and NADPH. Opened squares, 4-OHEN treatment; closed circles, 4-OHE-Q treatment; opened circles, 4-OHE treatment. Assays were performed in 10 mM PPB (pH 7.4) at 37 °C for 4 h. Control experiment was performed with CuCl2 and NADPH only. Data represents the mean ± S.D. (n = 3). A) Formation of AP sites with micromolar catechol/o-quinones. B) Linear increase of AP sites with nonamolar catechol/o-quinones. C) Time-dependent increase of AP sites with 1 μM of 4-OHEN or 4-OHE.
Using estrogens at more relevant submicromolar concentrations, 4-OHEN was found to be considerably more reactive in generation of AP sites in calf thymus DNA compared to 4-OHE (Figure 2B, C). 4-OHEN induced ~5-fold higher levels of AP sites compared to 4-OHE or 4-OHE-Q under the same conditions. Moreover the rate of AP site formation by 4-OHEN was ~5-fold higher than that of the human estrogen homologs. These data clearly suggest that 4-OHEN is much more reactive in generation of AP sites through redox cycling at concentrations approaching physiological levels.
Comparison of Nonenzymatic NADPH Oxidation
To determine whether NADPH consumption is correlated with formation of AP sites, NADPH oxidation by catechols/o-quinones was monitored in the presence/absence of CuCl2 (Figure 3). NADPH was spontaneously oxidized by 4-OHEN (10 μM) in the absence of CuCl2. In contrast, CuCl2 was required for NADPH oxidation in incubations with 4-OHE (10 μM) because of the requirement for catalysis in oxidation of 4-OHE to 4-OHE-Q. The lower rate of NADPH oxidation for 4-OHE-Q compared to 4-OHE might be due to the loss of 4-OHE-Q (t1/2 ~ 12 min) in aqueous solution from disproportionation, dimerization, and other reactions (14). These findings confirmed that 4-OHEN was more efficient at oxidizing NADPH as compared to 4-OHE/4-OHE-Q, and did not require CuCl2.
Figure 3. Nonenzymatic NADPH oxidation by catechol estrogens.

Solutions contained NADPH (200 μM), catechol/o-quinones (10 μM) with/without CuCl2 (10 μM) in 10 mM PPB (pH 7.4) at 23 °C. The total volume was 200 μL. The reaction was initiated by adding NADPH and the decrease in absorbance was monitored at 340 nm.
Effect of ROS Modulators on the Formation of AP Sites
To further explore the role of ROS in AP site formation by 4-OHEN and 4-OHE, a series of ROS modulators was assessed. As shown in Figure 4A, the presence of catalase abolished the induction of AP sites for both catechols indicating requirement role for hydrogen peroxide. SOD also inhibited AP site formation compatible with the importance of superoxide. Similarly, sodium azide, used in such studies as a hydroxyl radical and singlet oxygen scavenger, significantly inhibited formation of AP sites from both 4-OHEN and 4-OHE (Figure 4B). In contrast, mannitol that scavenges hydroxyl radicals, only partially reduced 4-OHEN-induced AP site formation (Figure 4B). Methional, used in such studies as a copper-hydroperoxo complex [Cu(I)OOH] chelator (40), was more effective at blocking 4-OHEN-mediated AP site formation as compared to 4-OHE. The selective Cu(I) chelator BCS abolished AP site formation for both catechols (Figure 4B). These results demonstrate that Cu(I) is involved in the formation of AP sites, and formation of Cu(I)OOH complex is important for 4-OHEN-mediated AP site formation.
Figure 4. The effects of ROS scavengers on the formation of AP sites by catechol estrogens.

Calf thymus DNA (100 μg) was treated with catechols in the presence of CuCl2 and NADPH, and ROS scavengers were added as indicated. Control experiment was performed with catechols [4-OHEN (1 μM), 4-OHE (10 μM)] in the presence of CuCl2 (10 μM) and NADPH (200 μM). A) Concentrations of enzymes were catalase (800 U/mL), SOD (200 U/mL), and NQO1 (10 μg/mL). B) Concentrations of ROS scavengers were sodium azide (0.1 M), mannitol (0.1 M), methional (0.1 M), and BCS (0.1 mM). Data represent the mean ± S.D. (n = 3). Asterisk: p < 0.05, compared to control.
Finally, we examined whether NADPH:quinone oxidoreductase 1 (NQO1), a detoxification enzyme that catalyzes the two election reduction of quinones, could prevent catechol estrogen-induced AP site formation. The data showed that AP sites were actually enhanced by NQO1 under redox cycling conditions with both 4-OHE and 4-OHEN (Figure 4A). As a result, NQO1 may not represent a detoxification pathway but instead could catalyze redox cycling of catechol estrogens generating AP sites.
Induction of DNA Strand Breaks by Estrogen Catechol/O-Quinones
The conversion of supercoiled ΦX174 RFI double stranded DNA by catechol estrogens/o-quinones to open circular and linear forms was examined in an in vitro DNA strand break model (Figure 5) (41). 4-OHEN induced both open circular and linear forms in a dose- and time-dependent manner, which was consistent with the AP site formation data (Figure 2). The supercoiled DNA was converted to the open circular form after 4-OHEN (1 μM) treatment for 1 h. By increasing the concentration of 4-OHEN, the linear form was increased indicating the formation of double strand breaks. 4-OHE or 4-OHE-Q were much less reactive compared to 4-OHEN in generation of DNA strand breaks under the same conditions. 4-OHE required higher concentrations (10 μM) to completely convert supercoiled form to open circular form within 1 h (Figure 5A). In the time-course experiment, a complete conversion of supercoiled to open circular form by 4-OHEN (10 μM) required 30 min incubation, while 4-OHE or 4-OHE-Q (10 μM) required more than 1 h for full conversion (Figure 5B). These data confirmed that 4-OHEN is more reactive than 4-OHE or 4-OHE-Q in induction of DNA strand breaks under redox cycling conditions, entirely compatible with observations on AP site formation. In the absence of CuCl2 and NADPH, none of these estrogen metabolites alone were capable of inducing DNA strand breaks (data not shown).
Figure 5. Electrophoresis analysis of DNA strand breaks in RFI plasmid treated with catechol/o-quinones.
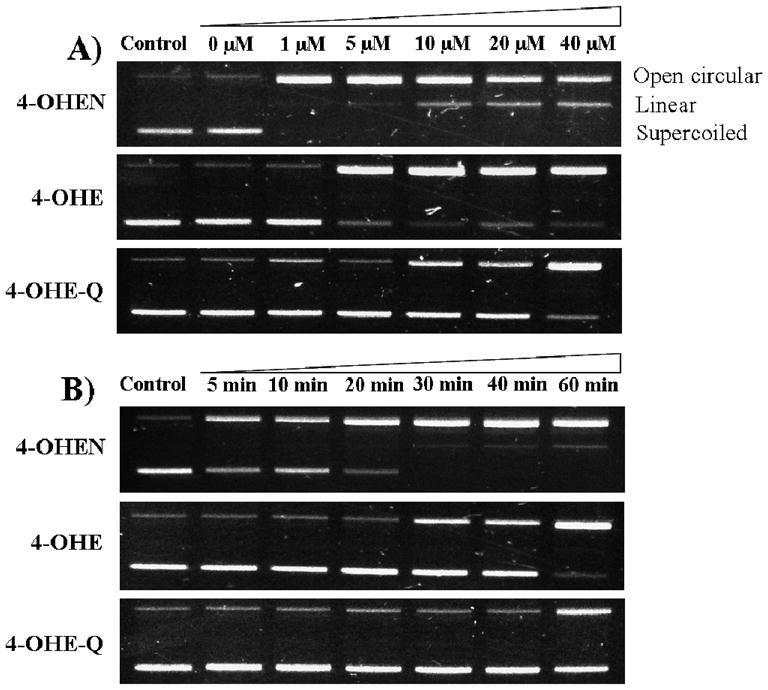
A) Concentration-dependent formation. RFI plasmid DNA (0.5 μg) was incubated with various concentrations of catechol/o-quinones in presence of CuCl2 (10 μM) and NADPH (200 μM) at 37 °C for 60 min. B) Time-dependent formation. Plasmid DNA was incubated with catechol/o-quinones (10 μM) in presence of CuCl2 and NADPH at 37 °C for various incubation times. The images shown are representative of at least three independent experiments.
Parallel Induction of 8-oxo-dG by Estrogen Catechol/O-Quinones
As 8-oxo-dG is a classical biomarker for oxidative stress, it was of interest to compare the levels of this biomarker with AP sites in calf thymus DNA. The basal level of 8-oxo-dG in untreated DNA was found to be 1.5 per 105 dG. In the presence of CuCl2 and NADPH, 4-OHEN increased the levels of 8-oxo-dG in a concentration-dependent manner, and the level of 8-oxo-dG was almost 5-fold higher than 4-OHE or 4-OHE-Q treatment under the same conditions (Figure 6A). Of substantial interest was the correlation of AP site formation with 8-oxo-dG formation. The absolute amount of 8-oxo-dG formation was 10-fold higher than AP sites, however, over interpretation is ill-advised given the well known operator and lab variability in 8-oxo-dG measurements (42). More importantly, regression analysis revealed that the levels of 8-oxo-dG correlated linearly with the number of AP sites (Figure 6B). These data may suggest that 8-oxo-dG and AP sites are formed from a common ROS.
Figure 6. Formation of 8-oxo-dG in calf thymus DNA by catechol estrogens; correlation with AP sites.

A) Concentration-dependent formation of 8-oxo-dG. Calf thymus DNA (100 μg) was treated with catechol/o-quinones in the presence of CuCl2 (10 μM) and NADPH (200 μM). Assays were performed in 10 mM PPB (pH 7.4) at 37 °C for 4 h. DNA was isolated and enzymatically hydrolyzed to deoxynucleosides for LC-MS/MS analysis. B) The regression analysis of 8-oxo-dG versus the number of AP sites. Each point represents the average ± S.D. of three determinations.
Formation of AP Sites in Breast Cancer MCF-7 Cells
To investigate whether 4-OHEN induces AP sites in human breast cancer cells, MCF-7 cells were incubated with 4-OHEN (10 μM) for 1 h (Figure 7A). 4-OHEN slightly increased the number of AP sites in MCF-7 cells; however, this number was not statistically significant as compared to control. The presence of added CuCl2 (50 μM) did not significantly enhance the levels of AP sites in 4-OHEN treated cells. However, the levels of AP sites induced by 4-OHEN were significantly increased 2-fold over control by addition of NADH (200 μM), and the presence of CuCl2 and NADH resulted in a 3-fold induction of AP sites. Trends in increasing levels of AP site with added redox cycling agents were apparent with both 4-OHEN and 4-OHE, however, the effects of 4-OHE (10 μM) in MCF-7 cells did not reach significance (Figure 7B). These data showed that 4-OHEN induced ROS-mediated AP sites in MCF-7 cells.
Figure 7. Formation of AP sites in human breast cancer MCF-7 cells treated with catechol estrogens.

MCF-7 cells (3 × 105 cells/mL) were treated with A) 4-OHEN (10 μM) or B) 4-OHE (10 μM) with/without CuCl2 (50 μM), NADH (200 μM) as indicated at 37 °C for 60 min. Control experiment was performed with DMSO (0.2% final concentration) treatment only. Data represents the mean ± S.D. (n = 3). Asterisk: p < 0.05 (*) or p < 0.01 (**), compared to control.
Discussion
Metabolic activation of estrogens to form o-quinones has been correlated with the carcinogenicity of estrogens. The major estrogen components in Premarin are endogenous estrogens and B-ring unsaturated equine estrogens that are converted to genotoxic o-quinones by P450 catalyzed 4-hydroxylation and subsequent two electron oxidation (14). Aromatization of the B-ring affects the reactivity of the equine estrogen catechol metabolite, 4-OHEN, stabilizing the electron deficient quinone form and hence leading to increased redox activity over endogenous catechol estrogens, and a much longer lived quinone form, 4-OHEN-Q (t1/2 ~ 2 h) compared to 4-OHE-Q (t1/2 ~ 12 min) (43). We confirmed that NADPH was spontaneously oxidized by 4-OHEN, and the presence of copper ion accelerated NADPH consumption (Figure 3). NADPH was rapidly oxidized by 4-OHE-Q; however, oxidation of NADPH terminated without addition of copper to catalyze redox cycling (Figure 3). The enhanced redox activity of 4-OHEN suggests that the extent and type of DNA damage might differ from endogenous catechol estrogens.
Swenberg and co-workers have compared the relative abilities of the endogenous catechol estrogens, 2-hydroxyestradiol (2-OHE2) and 4-hydroxyestradiol (4-OHE2), to induce AP sites in calf thymus DNA (31). Both catechols at submicromolar doses (0.1 μM) significantly induced AP sites in the presence of NADPH and Cu(II). In good agreement with their findings, we also found 4-OHE induced AP sites; however, 4-OHEN was found to be a much more effective AP site generator due to its enhanced redox cycling ability.
It has been proposed that depurination of labile estrogen o-quinone adducts is the major pathway resulting in AP sites, and these AP sites by error-prone base excision repair can lead to the critical mutations that initiate breast and other human cancers (17, 19, 44). The depurinating adducts have been detected in vitro and in vivo (15, 17, 45), although the levels of these adducts in vivo were very low. In addition, lack of direct comparisons between the levels of depurinating adducts and AP sites weakens the hypothesis that depurinating adducts are the predominant source for AP site formation. It is known that AP sites could arise from ROS by nonspecific hydrogen atom abstraction from the C-1′, C-2′, and C-4′ positions of deoxyribose (46). Additionally, 8-oxo-dG, which is formed under these conditions, is more reactive than dG and can undergo secondary oxidation leading to destabilization of the N-glycosidic bond, and production of the aldehydic sites. Each of the resultant AP sites can be detected by ARP probe because they all generate an aldehydic group at the ROS-targeted carbon. As a result, the ARP assay is unable to distinguish between these lesions.
It has been shown that AP sites can be derived from ROS via redox cycling between catechol estrogens and o-quinones (31). Similar experiments with naphthalene, polycyclic aromatic hydrocarbon, and pentachlorophenol o-quinones also support ROS derived AP site formation (39, 47, 48). In these studies, the levels of AP sites generated by the thermal depurination of labile o-quinone adducts in calf thymus DNA were determined and compared to the levels of AP sites generated by ROS. The data showed that formation of AP sites from depurinating adducts appears to be a minor pathway. Our findings also support AP site formation by a ROS-mediated pathway although our data do not directly inform on AP sites from depurinating DNA adducts. A linear correlation of 8-oxo-dG with AP site formation was observed for both catechol estrogens, the simplest explanation being that a common ROS intermediate causes both types of DNA damage.
In order to investigate the possible mechanisms involved in generation of AP sites by ROS, the effect of a series of ROS scavengers were examined on AP site formation (Figure 4). Both superoxide and hydrogen peroxide are common intermediates in formation of DNA damaging ROS (Scheme 2), therefore unsurprisingly catalase abolished and SOD attenuated AP site formation. Mannitol, azide ion, and methional differentially affected the levels of AP sites induced by 4-OHEN versus 4-OHE. However, both catechol estrogens were equally sensitive to chelation of cuprous ion by bathocuproine, demonstrating the requirement for copper in AP site formation. A function of copper ion is to oxidize the catechol to o-quinone (29, 40, 48). In the absence of copper ion, 4-OHEN autoxidizes, forming AP sites in the presence of reducing agent (NADPH) amplified by redox cycling. Copper ion is a requirement for oxidation of 4-OHE to yield AP sites. Although 4-OHEN autoxidizes to 4-OHEN-Q, the presence of copper ion increases the levels of AP sites. In addition to a role in catechol oxidation and redox cycling, copper ion reacts with ROS to form a reactive intermediate, Cu(I)-hydroperoxy [Cu(I)OOH] complex, which has been proposed as the proximal DNA-damaging agent (41, 49). The localized generation of a copper-peroxide complex on DNA represents a hydroxyl radical equivalent bound in close proximity to DNA rather than a free hydroxyl radical. This complex has been proposed to have similar reactivity to singlet oxygen. The significant inhibitory effect of methional suggests that the [Cu(I)OOH] complex is involved in AP site formation by catechol estrogens.
Scheme 2.

Diagram of a proposed model for copper ion-mediated AP site formation by catechol estrogens
It has been proposed that NQO1 represents a detoxification mechanism for catechol estrogen o-quinones through reduction of the DNA damaging quinone to the catechol estrogen, leading to a decrease in apurinic DNA adducts (44, 50, 51). 4-OHE-Q has been shown to be a substrate for NQO1, although the rate of reduction of catechol estrogen o-quinones is only modestly enhanced by NQO1 over the non-enzymatic reduction by the reducing cofactors NADPH/NADH (34). In the current study, the addition of NQO1 would be predicted to increase redox cycling by enhancing the rate of quinone reduction by NAD(P)H; this prediction was borne out by the observed significant increases in AP site formation for both 4-OHE and 4-OHEN (Figure 4). These results indicate that NQO1 may not catalyze detoxification of catechol estrogen o-quinones but instead may represent a bioactivation mechanism leading to enhanced AP site formation.
The addition of NADH promotes redox cycling of 4-OHEN and the generation of ROS (43), thereby increasing 4-OHEN-induced DNA cleavage and apoptosis in breast cancer cells (25). When we investigated the induction of AP sites in MCF-7 cells exposed to 4-OHEN and 4-OHE, only 4-OHEN induced a modest increase in AP site formation (Figure 7). The addition of NADH and Cu(II)/NADH significantly increased the levels of AP sites induced by 4-OHEN. Finally, it should be noted that the repair of oxidized bases (e.g. 8-oxo-dG) by the base excision repair system forms AP sites by removing damaged bases, and thus it is possible that the levels of AP sites detected in MCF-7 cells were partially contributed from the repair of oxidized bases derived from oxidative stress.
The formation of 8-oxo-dG has been detected in breast cells from both 4-OHE2 (52) and 4-OHEN (53). Studies in MCF-7 cells required depletion of GSH levels prior to detection of significant increases in 8-oxo-dG after treating cells with 2-OHE2 or 4-OHE2 (10 μM) for 30 min (54). Similarly, pretreatment of MCF-7 cells with the P450 inducer dioxin as well as the COMT inhibitor Ro 41-0960 was shown to allow detection of increased 8-oxo-dG levels after treatment with 0.1 μM estradiol (55). Micromolar concentrations of 4-OHE2 in MCF-10A cells or E2 in MCF-7 cells were also required to generate time- and concentration-dependent increases in DCF-fluorescence staining which is indicative of induced intracellular accumulation of ROS (56, 57). Given the limited number of reports of oxidative damage to DNA induced by catechol estrogens and the high concentrations necessary to achieve significant increase in these oxidized lesions, it is unclear if oxidative damage to DNA plays a significant role in the carcinogenesis mechanisms of endogenous estrogens. The data for oxidative DNA damage induced by 4-OHEN in vitro and in vivo is much more compelling (25, 53, 58, 59). Injection of 4-OHEN into the mammary fat pads of Sprague-Dawley rats resulted in a dose-dependent increase in single strand breaks and oxidized bases as analyzed by the comet assay (58). In addition, extraction of mammary tissue DNA, hydrolysis to deoxynucleosides, and analysis by LC-MS-MS showed the formation of 8-oxo-dG as well as 8-oxo-dA. In mice treated with equilenin, the levels of 8-oxo-dG were increased 1.5-fold in the uterus (59). Taken together, these results suggest that 4-OHEN is more efficient in induction of ROS-mediated AP sites and 8-oxo-dG compared to 4-OHE in breast cancer tissues and cells.
In conclusion, we have shown that catechol estrogens can induce ROS-mediated AP sites through redox cycling in calf thymus DNA, which is correlated with corresponding increases in 8-oxo-dG levels. In addition, the data give strong evidence that 4-OHEN is more redox-reactive than 4-OHE generating AP sites in vitro. AP site formation from ROS must be considered as an alternative to AP formation from depurination of labile DNA adducts. These oxidative AP sites might be important in the induction of DNA mutations, which supports the hypothesis that oxidative DNA damage induced by estrogen o-quinones could contribute to estrogen carcinogenesis and increased breast cancer risk associated with long-term high-dose HRT.
Acknowledgments
This work was supported by NIH Grant (CA130037 J. L. B.). We thank Dr. Zhihui Qin for the synthesis of 4-OHEN. In addition, we thank to Dr. V.C. Jordan (Fox Chase Cancer Center, Philadelphia, PA) for the MCF-7 WS8 human breast cancer cells.
Abbreviations
- AP sites
apurinic/apyrimidinic sites
- ARP
aldehydic reactive probe
- BCS
bathocuproinedisulfonic acid
- DMNQ
2,3-dimethoxy-1,4-naphthoquinone
- DMSO
dimethyl sulfoxide
- E2
17β-estradiol
- EN
equilenin
- EQ
equilin
- ER
estrogen receptor
- ERE
estrogen response element
- GST
glutathione S-transferase
- HRP
horseradish peroxidase
- HRT/ERT
hormone/estrogen replacement therapy
- NADPH
β-nicotinamide adenine dinucleotide phosphate, reduced form
- NQO1
NAD(P)H: quinone oxidoreductase 1
- 2-OHE2
2-hydroxyestradiol
- 4-OHE2
4-hydroxyestradiol
- 4-OHE
4-hydroxyestrone
- 4-OHEN
4-hydroxyequilenin
- 4-OHEN-Q
4-hydroxyequilenin-o-quinone
- 4-OHEQ
4-hydroxyequilin
- 4-OHE-Q
4-hydroxyestrone-o-quinone
- 8-oxo-dG
8-oxo-7,8-dihydro-2′-deoxyguanosine
- PBS
phosphate buffered saline
- PBST
phosphate-buffered saline containing 0.05% Tween 20
- PPB
potassium phosphate buffer
- ROS
reactive oxygen species
- SOD
superoxide dismutase
- TMB
3,3′,5,5′-tetramethylbenzidine
References
- 1.Chen WY. Exogenous and endogenous hormones and breast cancer. Best Pract Res Clin Endocrinol Metab. 2008;22:573–585. doi: 10.1016/j.beem.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Colditz GA, Hankinson SE, Hunter DJ, Willett WC, Manson JE, Stampfer MJ, Hennekens C, Rosner B, Speizer FE. The use of estrogens and progestins and the risk of breast cancer in postmenopausal women. N Engl J Med. 1995;332:1589–1593. doi: 10.1056/NEJM199506153322401. [DOI] [PubMed] [Google Scholar]
- 3.Key T, Appleby P, Barnes I, Reeves G. Endogenous sex hormones and breast cancer in postmenopausal women: reanalysis of nine prospective studies. J Natl Cancer Inst. 2002;94:606–616. doi: 10.1093/jnci/94.8.606. [DOI] [PubMed] [Google Scholar]
- 4.Grodstein F, Stampfer MJ, Colditz GA, Willett WC, Manson JE, Joffe M, Rosner B, Fuchs C, Hankinson SE, Hunter DJ, Hennekens CH, Speizer FE. Postmenopausal hormone therapy and mortality. N Engl J Med. 1997;336:1769–1775. doi: 10.1056/NEJM199706193362501. [DOI] [PubMed] [Google Scholar]
- 5.Lupulescu A. Estrogen use and cancer incidence: a review. Cancer Invest. 1995;13:287–295. doi: 10.3109/07357909509094464. [DOI] [PubMed] [Google Scholar]
- 6.Service RF. Cancer: new role for estrogen in cancer? Science. 1998;279:1631–1633. doi: 10.1126/science.279.5357.1631. [DOI] [PubMed] [Google Scholar]
- 7.Vogel VG, Yeomans A, Higginbotham E. Clinical management of women at increased risk for breast cancer. Breast Cancer Res. 1993;28:195–210. doi: 10.1007/BF00666431. [DOI] [PubMed] [Google Scholar]
- 8.Zumoff B. Does postmenopausal estrogen administration increase the risk of breast cancer? Contributions of animal, biochemical, and clinical investigative studies to a resolution of the controversy. Proc Soc Exp Biol Med. 1998;217:30–37. doi: 10.3181/00379727-217-44202. [DOI] [PubMed] [Google Scholar]
- 9.Feigelson HS, Henderson BE. Estrogens and breast cancer. Carcinogenesis. 1996;17:2279–2284. doi: 10.1093/carcin/17.11.2279. [DOI] [PubMed] [Google Scholar]
- 10.Henderson BE, Feigelson HS. Hormonal carcinogenesis. Carcinogenesis. 2000;21:427–433. doi: 10.1093/carcin/21.3.427. [DOI] [PubMed] [Google Scholar]
- 11.Nandi S, Guzman R, Yang J. Hormones and mammary carcinogenesis in mice, rats, and humans: a unifying hypothesis. Proc Natl Acad Sci U S A. 1995;92:3650–3657. doi: 10.1073/pnas.92.9.3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Flototto T, Djahansouzi S, Glaser M, Hanstein B, Niederacher D, Brumm C, Beckmann MW. Hormones and hormone antagonists: mechanisms of action in carcinogenesis of endometrial and breast cancer. Horm Metab Res. 2001;33:451–457. doi: 10.1055/s-2001-16936. [DOI] [PubMed] [Google Scholar]
- 13.Yager JD, Davidson NE. Estrogen carcinogenesis in breast cancer. N Engl J Med. 2006;354:270–282. doi: 10.1056/NEJMra050776. [DOI] [PubMed] [Google Scholar]
- 14.Bolton JL, Thatcher GRJ. Potential mechanisms of estrogen quinone carcinogenesis. Chem Res Toxicol. 2008;21:93–101. doi: 10.1021/tx700191p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gaikwad NW, Yang L, Muti P, Meza JL, Pruthi S, Ingle JN, Rogan EG, Cavalieri EL. The molecular etiology of breast cancer: evidence from biomarkers of risk. Int J Cancer. 2008;122:1949–1957. doi: 10.1002/ijc.23329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li JJ, Li SA, Oberley TD, Parsons JA. Carcinogenic activities of various steroidal and nonsteroidal estrogens in the hamster kidney: relation to hormonal activity and cell proliferation. Cancer Res. 1995;55:4347–4351. [PubMed] [Google Scholar]
- 17.Cavalieri E, Chakravarti D, Guttenplan J, Hart E, Ingle J, Jankowiak R, Muti P, Rogan E, Russo J, Santen R, Sutter T. Catechol estrogen quinones as initiators of breast and other human cancers: implications for biomarkers of susceptibility and cancer prevention. Biochim Biophys Acta. 2006;1766:63–78. doi: 10.1016/j.bbcan.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 18.Bolton JL. Quinoids, quinoid radicals, and phenoxyl radicals formed from estrogens and antiestrogens. Toxicology. 2002;177:55–65. doi: 10.1016/s0300-483x(02)00195-6. [DOI] [PubMed] [Google Scholar]
- 19.Cavalieri EL, Stack DE, Devanesan PD, Todorovic R, Dwivedy I, Higginbotham S, Johansson SL, Patil KD, Gross ML, Gooden JK, Ramanathan R, Cerny RL, Rogan EG. Molecular origin of cancer: catechol estrogen-3,4-quinones as endogenous tumor initiators. Proc Natl Acad Sci U S A. 1997;94:10937–10942. doi: 10.1073/pnas.94.20.10937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sarabia SF, Zhu BT, Kurosawa T, Tohma M, Liehr JG. Mechanism of cytochrome P450-catalyzed aromatic hydroxylation of estrogens. Chem Res Toxicol. 1997;10:767–771. doi: 10.1021/tx970021f. [DOI] [PubMed] [Google Scholar]
- 21.Zhang F, Chen Y, Pisha E, Shen L, Xiong Y, van Breemen RB, Bolton JL. The major metabolite of equilin, 4-hydroxyequilin, autoxidizes to an o-quinone which isomerizes to the potent cytotoxin 4-hydroxyequilenin-o-quinone. Chem Res Toxicol. 1999;12:204–213. doi: 10.1021/tx980217v. [DOI] [PubMed] [Google Scholar]
- 22.Spink DC, Zhang F, Hussain MM, Katz BH, Liu X, Hilker DR, Bolton JL. Metabolism of equilenin in MCF-7 and MDA-MB-231 human breast cancer cells. Chem Res Toxicol. 2001;14:572–581. doi: 10.1021/tx000219r. [DOI] [PubMed] [Google Scholar]
- 23.Wang Z, Edirisinghe P, Sohn J, Qin Z, Geacintov NE, Thatcher GR, Bolton JL. Development of a liquid chromatography electrospray ionization tandem mass spectrometry method for analysis of stable 4-hydroxyequilenin-DNA adducts in human breast cancer cells. Chem Res Toxicol. 2009;22:1129–1136. doi: 10.1021/tx900063g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yasui M, Matsui S, Laxmi YR, Suzuki N, Kim SY, Shibutani S, Matsuda T. Mutagenic events induced by 4-hydroxyequilin in supF shuttle vector plasmid propagated in human cells. Carcinogenesis. 2003;24:911–917. doi: 10.1093/carcin/bgg029. [DOI] [PubMed] [Google Scholar]
- 25.Chen Y, Liu X, Pisha E, Constantinou AI, Hua Y, Shen L, van Breemen RB, Elguindi EC, Blond SY, Zhang F, Bolton JL. A metabolite of equine estrogens, 4-hydroxyequilenin, induces DNA damage and apoptosis in breast cancer cell lines. Chem Res Toxicol. 2000;13:342–350. doi: 10.1021/tx990186j. [DOI] [PubMed] [Google Scholar]
- 26.Pisha E, Lui X, Constantinou AI, Bolton JL. Evidence that a metabolite of equine estrogens, 4-hydroxyequilenin, induces cellular transformation in vitro. Chem Res Toxicol. 2001;14:82–90. doi: 10.1021/tx000168y. [DOI] [PubMed] [Google Scholar]
- 27.Margalioth EJ, Schenker JG, Chevion M. Copper and zinc levels in normal and malignant tissues. Cancer. 1983;52:868–872. doi: 10.1002/1097-0142(19830901)52:5<868::aid-cncr2820520521>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 28.Prutz WA, Butler J, Land EJ. Interaction of copper(I) with nucleic acids. Int J Radiat Biol. 1990;58:215–234. doi: 10.1080/09553009014551581. [DOI] [PubMed] [Google Scholar]
- 29.Li Y, Trush MA, Yager JD. DNA damage caused by reactive oxygen species originating from a copper-dependent oxidation of the 2-hydroxy catechol of estradiol. Carcinogenesis. 1994;15:1421–1427. doi: 10.1093/carcin/15.7.1421. [DOI] [PubMed] [Google Scholar]
- 30.Mobley JA, Bhat AS, Brueggemeier RW. Measurement of oxidative DNA damage by catechol estrogens and analogues in vitro. Chem Res Toxicol. 1999;12:270–277. doi: 10.1021/tx980128i. [DOI] [PubMed] [Google Scholar]
- 31.Lin PH, Nakamura J, Yamaguchi S, Asakura S, Swenberg JA. Aldehydic DNA lesions induced by catechol estrogens in calf thymus DNA. Carcinogenesis. 2003;24:1133–1141. doi: 10.1093/carcin/bgg049. [DOI] [PubMed] [Google Scholar]
- 32.Breen AP, Murphy JA. Reactions of oxyl radicals with DNA. Free Radic Biol Med. 1995;18:1033–1077. doi: 10.1016/0891-5849(94)00209-3. [DOI] [PubMed] [Google Scholar]
- 33.Han X, Liehr JG. Microsome-mediated 8-hydroxylation of guanine bases of DNA by steroid estrogens: correlation of DNA damage by free radicals with metabolic activation to quinones. Carcinogenesis. 1995;16:2571–2574. doi: 10.1093/carcin/16.10.2571. [DOI] [PubMed] [Google Scholar]
- 34.Chandrasena RE, Edirisinghe PD, Bolton JL, Thatcher GR. Problematic detoxification of estrogen quinones by NAD(P)H-dependent quinone oxidoreductase and glutathione-S-transferase. Chem Res Toxicol. 2008;21:1324–1329. doi: 10.1021/tx8000797. [DOI] [PubMed] [Google Scholar]
- 35.Wang Z, Wijewickrama GT, Peng KW, Dietz BM, Yuan L, van Breemen RB, Bolton JL, Thatcher GR. Estrogen receptor alpha enhances the rate of oxidative DNA damage by targeting an equine estrogen catechol metabolite to the nucleus. J Biol Chem. 2009;284:8633–8644. doi: 10.1074/jbc.M807860200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ubezio P, Civoli F. Flow cytometric detection of hydrogen peroxide production induced by doxorubicin in cancer cells. Free Radic Biol Med. 1994;16:509–516. doi: 10.1016/0891-5849(94)90129-5. [DOI] [PubMed] [Google Scholar]
- 37.Thibodeau PA, Paquette B. DNA damage induced by catecholestrogens in the presence of copper (II): generation of reactive oxygen species and enhancement by NADH. Free Radic Biol Med. 1999;27:1367–1377. doi: 10.1016/s0891-5849(99)00183-5. [DOI] [PubMed] [Google Scholar]
- 38.Tan AS, Berridge MV. Differential effects of redox-cycling and arylating quinones on trans-plasma membrane electron transport. BioFactors. 2009;34:183–190. doi: 10.3233/BIO-2009-1071. [DOI] [PubMed] [Google Scholar]
- 39.Lin PH, Pan WC, Kang YW, Chen YL, Lin CH, Lee MC, Chou YH, Nakamura J. Effects of naphthalene quinonoids on the induction of oxidative DNA damage and cytotoxicity in calf thymus DNA and in human cultured cells. Chem Res Toxicol. 2005;18:1262–1270. doi: 10.1021/tx050018t. [DOI] [PubMed] [Google Scholar]
- 40.Park JH, Gopishetty S, Szewczuk LM, Troxel AB, Harvey RG, Penning TM. Formation of 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxo-dGuo) by PAH o-quinones: involvement of reactive oxygen species and copper(II)/copper(I) redox cycling. Chem Res Toxicol. 2005;18:1026–1037. doi: 10.1021/tx050001a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li Y, Trush MA. DNA damage resulting from the oxidation of hydroquinone by copper: role for a Cu(II)/Cu(I) redox cycle and reactive oxygen generation. Carcinogenesis. 1993;14:1303–1311. doi: 10.1093/carcin/14.7.1303. [DOI] [PubMed] [Google Scholar]
- 42.Son J, Pang B, McFaline JL, Taghizadeh K, Dedon PC. Surveying the damage: the challenges of developing nucleic acid biomarkers of inflammation. Mol BioSyst. 2008;4:902–908. doi: 10.1039/b719411k. [DOI] [PubMed] [Google Scholar]
- 43.Shen L, Pisha E, Huang Z, Pezzuto J, Krol E, Alam Z, van Breemen R, Bolton J. Bioreductive activation of catechol estrogen-ortho-quinones: aromatization of the B ring in 4-hydroxyequilenin markedly alters quinoid formation and reactivity. Carcinogenesis. 1997;18:1093–1101. doi: 10.1093/carcin/18.5.1093. [DOI] [PubMed] [Google Scholar]
- 44.Cavalieri EL, Rogan EG. Depurinating estrogen-DNA adducts in the etiology and prevention of breast and other human cancers. Future Oncol. 2010;6:75–91. doi: 10.2217/fon.09.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang Q, Aft RL, Gross ML. Estrogen carcinogenesis: specific identification of estrogen-modified nucleobase in breast tissue from women. Chem Res Toxicol. 2008;21:1509–1513. doi: 10.1021/tx8001737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Povirk LF, Steighner RJ. Oxidized apurinic/apyrimidinic sites formed in DNA by oxidative mutagens. Mutat Res. 1989;214:13–22. doi: 10.1016/0027-5107(89)90193-0. [DOI] [PubMed] [Google Scholar]
- 47.Lin CH, Leow HT, Huang SC, Nakamura J, Swenberg JA, Lin PH. Induction of cytotoxicity, aldehydic DNA lesions, and poly(ADP-ribose) polymerase-1 activation by catechol derivatives of pentachlorophenol in calf thymus DNA and in human breast cancer cells. Chem Res Toxicol. 2005;18:257–264. doi: 10.1021/tx0498511. [DOI] [PubMed] [Google Scholar]
- 48.Spencer WA, Lehmler HJ, Robertson LW, Gupta RC. Oxidative DNA adducts after Cu2+-mediated activation of dihydroxy PCBs: role of reactive oxygen species. Free Radic Biol Med. 2009;46:1346–1352. doi: 10.1016/j.freeradbiomed.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yamamoto K, Kawanishi S. Hydroxyl free radical is not the main active species in site-specific DNA damage induced by copper (II) ion and hydrogen peroxide. J Biol Chem. 1989;264:15435–15440. [PubMed] [Google Scholar]
- 50.Gaikwad NW, Rogan EG, Cavalieri EL. Evidence from ESI-MS for NQO1-catalyzed reduction of estrogen ortho-quinones. Free Radic Biol Med. 2007;43:1289–1298. doi: 10.1016/j.freeradbiomed.2007.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Singh S, Zahid M, Saeed M, Gaikwad NW, Meza JL, Cavalieri EL, Rogan EG, Chakravarti D. NAD(P)H:quinone oxidoreductase 1 Arg139Trp and Pro187Ser polymorphisms imbalance estrogen metabolism towards DNA adduct formation in human mammary epithelial cells. J Steroid Biochem Mol Biol. 2009;117:56–66. doi: 10.1016/j.jsbmb.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen ZH, Na HK, Hurh YJ, Surh YJ. 4-Hydroxyestradiol induces oxidative stress and apoptosis in human mammary epithelial cells: possible protection by NF-[kappa]B and ERK/MAPK. Toxicol Appl Pharmacol. 2005;208:46–56. doi: 10.1016/j.taap.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 53.Liu X, Yao J, Pisha E, Yang Y, Hua Y, van Breemen RB, Bolton JL. Oxidative DNA damage induced by equine estrogen metabolites: role of estrogen receptor. Chem Res Toxicol. 2002;15:512–519. doi: 10.1021/tx0101649. [DOI] [PubMed] [Google Scholar]
- 54.Mobley JA, Brueggemeier RW. Increasing the DNA Damage Threshold in Breast Cancer Cells. Toxicol Appl Pharmacol. 2002;180:219–226. doi: 10.1006/taap.2002.9391. [DOI] [PubMed] [Google Scholar]
- 55.Lavigne JA, Goodman JE, Fonong T, Odwin S, He P, Roberts DW, Yager JD. The effects of catechol-O-methyltransferase inhibition on estrogen metabolite and oxidative DNA damage levels in estradiol-treated MCF-7 cells. Cancer Res. 2001;61:7488–7494. [PubMed] [Google Scholar]
- 56.Felty Q, Xiong WC, Sun D, Sarkar S, Singh KP, Parkash J, Roy D. Estrogen-induced mitochondrial reactive oxygen species as signal-transducing messengers. Biochemistry. 2005;44:6900–6909. doi: 10.1021/bi047629p. [DOI] [PubMed] [Google Scholar]
- 57.Park SA, Na HK, Kim EH, Cha YN, Surh YJ. 4-Hydroxyestradiol induces anchorage-independent growth of human mammary epithelial cells via activation of I{kappa}B kinase: potential role of reactive oxygen species. Cancer Res. 2009;69:2416–2424. doi: 10.1158/0008-5472.CAN-08-2177. [DOI] [PubMed] [Google Scholar]
- 58.Zhang F, Swanson SM, van Breemen RB, Liu X, Yang Y, Gu C, Bolton JL. Equine estrogen metabolite 4-hydroxyequilenin induces DNA damage in the rat mammary tissues: formation of single-strand breaks, apurinic sites, stable adducts, and oxidized bases. Chem Res Toxicol. 2001;14:1654–1659. doi: 10.1021/tx010158c. [DOI] [PubMed] [Google Scholar]
- 59.Okamoto Y, Chou PH, Kim SY, Suzuki N, Laxmi YR, Okamoto K, Liu X, Matsuda T, Shibutani S. Oxidative DNA damage in XPC-knockout and its wild mice treated with equine estrogen. Chem Res Toxicol. 2008;21:1120–1124. doi: 10.1021/tx700428m. [DOI] [PubMed] [Google Scholar]