

Abstract
Parkinson’s disease [PD], a progressive neurodegenerative disease, results in abnormal accumulation of insoluble alpha-synuclein [α-Syn] in dopaminergic neurons. Here we examined tauopathic changes and the α-Syn/p-GSK-3β/proteasome pathway in postmortem striata and inferior frontal gyri [IFG] from patients with PD and PD with dementia [PDD]. In both PD and PDD, α-Syn levels were high, especially the insoluble form of this protein; in PDD, insoluble α-Syn levels were persistently higher than PD across both brain regions. Levels of p-GSK-3β phosphorylated at Tyr 216, which hyperphosphorylates Tau to produce toxic pathological forms of p-Tau, were higher in striata of both PD and PDD compared to controls, but were unaltered in IFG. While proteasomal activity was unchanged in striatum of PD and PDD, such activity was diminished in the IFG of both PD and PDD. A decrease in 19S subunit of the proteasomes was seen in IFG of PDD, while lower levels of 20S subunits were seen in striatum and IFG of both PD and PDD patients. Parkin levels were similar in PD and PDD, suggesting lack of involvement of this protein. Most interestingly, tauopathic changes were noted only in striatum of PD and PDD, with increased hyperphosphorylation seen at Ser262 and Ser396/404; increases in Ser202 levels were seen only in PD but not in PDD striatum. We were unable to detect any tauopathy in IFG in either PD or PDD despite increased levels of α-Syn, and decreased proteasomal activity, and is probably due to lack of increase in p-GSK-3β in IFG. Unlike Alzheimer’s disease where tauopathy is more globally observed in diverse brain regions, our data demonstrates restricted expression of tauopathy in brains of PD and PDD, probably limited to dopaminergic neurons of the nigrostriatal region.
Keywords: synucleinopathies, tauopathies, neurodegeneration, Alzheimer’s disease, dementia, hyperphosphorylated Tau, p-GSK-3β, parkin, neurofibrillary tangles, Lewy bodies
INTRODUCTION
α-Synuclein (α-Syn) is ubiquitously expressed in brain and is highly enriched in presynaptic nerve terminals, where its chief physiological function is the regulation of synaptic levels of monoamine neurotransmitters through modulation of vesicular release (Murphy et al., 2000) and their cognate transporters (Wersinger et al., 2005; Wersinger et al., 2006a; Wersinger et al., 2006b). Overexpression of α-Syn, through its gene duplication and triplication, is linked to idiopathic Parkinson’s disease (PD), while its A30P and A53T mutants cause the autosomal dominant forms of familial PD (Hofer et al., 2004; Kruger et al., 1998; Polymeropoulos et al., 1997). In pathological states, α-Syn becomes misfolded, aggregates and accumulates in neuronal inclusion bodies, Lewy bodies (LBs), seen in PD and other synucleinopathies (Goedert et al., 1998; Hofer et al., 2004). Tau, a microtubule binding protein, is most commonly linked to Alzheimer’s disease [AD] and other tauopathies, where, after hyperphosphorylation, it accumulates in neurons as neurofibrillary tangles [NFTs] (Joachim et al., 1987).
Despite differences in clinical features, pathological and experimental evidence increasingly indicates that there is considerable overlap between tauopathies and synucleinopathies, reinforcing the notion that these diseases may be mechanistically linked. Thus, in AD, 50–60% of patients with NFTs have α-Syn-containing LBs (Lippa et al., 1998; Iseki et al., 1999; Arai et al., 2001; Szpak et al., 2001; Burns et al., 2005; Griffin et al., 2006) and in AD patients with clinically detected extrapyramidal signs, 50% of the patients showed extensive α-Syn pathology co-localized in the Substantia nigra with p-Tau (Mori et al., 2002). In PD and in dementia with LBs, co-staining of p-Tau has been observed (Duda et al., 2002; Yamaguchi et al., 2005) in 30–40% of the LBs in the nucleus basalis of Meynert and locus coerulus, and in 10–30% of LBs in the medulla (Yamaguchi et al., 2005). Extensive overlap in α-Syn and p-Tau pathology has been noted in patients with the A53T mutation (Yamazaki et al., 2000; Kotzbauer et al., 2004), in the Parkinsonism-Dementia complex of Guam (Forman et al., 2002), in dementia with LBs (Yancopoulou et al., 2005) and in familial frontotemporal dementia and progressive aphasia (Hishikawa et al., 2003).
Despite this wealth of pathological information, the molecular and cellular interplay between α-Syn and p-Tau leading to their pathological co-deposition is not understood. Under normal physiological conditions, α-Syn is highly soluble. Under pathological conditions, which include oxidative stress and overexpression, the protein becomes insoluble, self-aggregates and accumulates into LBs (Nemes et al., 2004; Lippa et al., 1998; Iseki et al., 1999; Mori et al., 2002). Similar to α-Syn, Tau is a highly soluble protein that becomes insoluble by pathological hyperphosphorylation at specific sites with ensuing conformational changes and accumulation of the protein into NFTs. Molecular evidence suggests a direct interaction between these proteins, and when incubated together in vitro, α-Syn serves as a seed to accelerate the aggregation of Tau (Kotzbauer et al., 2004).
Using the MPTP mouse neurotoxin model of PD, as well as MPP+ cellular models of PD, we recently demonstrated that increases in α-Syn can initiate and sustain Tau hyperphosphorylation both in vivo and in vitro, with co-precipitation of these proteins (Duka et al., 2006; Duka & Sidhu 2006; Kozikowski et al., 2006; Duka et al., 2009). The hyperphosphorylation of Tau was absolutely dependent on the presence of α-Syn, as indicated by lack of any p-Tau formation in MPTP-treated α-Syn−/− mice or in neuronal cells lacking α-Syn (Duka et al., 2006; Duka et al., 2009). Moreover, we found that upon oxidative stress by MPTP or MPP+, α-Syn induced p-Tau formation through specific activation and recruitment of p-GSK-3β, activated by autophosphorylation at Tyr216 (Kozikowski et al., 2006; Duka et al., 2009). GSK-3β is a proline-directed serine/threonine kinase, ubiquitously expressed in mammalian tissues, and epitopes phosphorylated by GSK-3β are among the pathological phosphorylation sites of Tau seen in NFTs and paired helical filaments [PHFs] of Alzheimer’s disease, and GSK-3β is a major kinase implicated in Tau hyperphosphorylation (Baum et al., 1995).
To examine the clinical relevance of our previous findings, the current studies were undertaken, and are the first neurochemical examination of the α-Syn/p-Tau/GSK-3β pathway in human postmortem striata and IFG from PD and PD with dementia [PDD]. The results show higher neurodegeneration in PDD compared to PD, with tauopathy restricted to striata of both diseases, along with decreased proteasomal activity in the IFG of PDD. Our studies show that unlike Alzheimer’s disease where tauopathy is a more global event, tauopathy in brains of PD and PDD has a restricted expression and is limited to dopaminergic neurons of the nigrostriatal region, possibly due to increased dopamine-linked oxidative stress of the latter region.
MATERIALS AND METHODS
Materials
The antibodies used in this study are: anti-DAT, MAB369 and TAU MAB361 both from Millipore [Temecula, CA]; anti-α-Syn CAT# 610787, Anti-GSK-3β CAT# 612313 and anti-pGSK-3B – Purified Mouse Anti-GSK-3B (pY216) CAT # 612313, all from BD Transduction Labs [San Jose, CA]; Anti-parkin ab77924, Abcam Inc. [Cambridge, MA]; Anti-tyrosine hydroxylase sc-25269 and anti-β-actin SC-1616, both from Santa Cruz Biotechnology, Inc. [Santa Cruz, CA]. The CP-13 and PHF-1 antibodies [recognizing Tau-Ser202 and Tau-Ser396/404] were gifts from Dr. Peter Davies [New York]. Antibodies against the 19S S6′ and 20S alpha 5 proteasome subunits were catalog #s AP-111 and AP-120, respectively, from Boston Biochemicals [Boston, MA].
Postmortem tissue
Postmortem tissue was provided by the Sun Health Research Institute Brain and Body Donation Program (Sun City, AZ) and included samples from PD cases without dementia (and neuropathologically confirmed to be absent AD pathology or cortical Lewy Bodies), PDD cases that had clinically diagnosed dementia (and neuropathologically were confirmed to have cortical Lewy bodies and confirmed to be absent AD pathology), and well characterized, neurological and neuropathological normal controls. Clinical evaluation and neuropathological diagnosis of these cases have been published in greater detail elsewhere (Joyce et al., 2002; Adler et al., 2002). Since there were no differences between male and female tissues, all data were pooled together. In all, striata were used from 17 PD cases, 18 PDD cases, and 22 controls, while inferior frontal gyrus [IFG] was obtained from 9 PD cases, 7 PDD and 9 controls, with average postmortem interval of ~3 hours.
Tissue solubilization
A 100 mg of tissue was weighed and cold tissue homogenization solution (5M guanidine HCl, 50mM Tris-HCl pH 8.0) was added and thoroughly homogenized with a hand held electric homogenizer. Cold homogenization buffer was added to equal a total of 8X total vol. Samples were mixed for 3.5–4 hrs at room temperature using micro-magnets and stir plate. Samples were diluted (1:200) with cold BSAT-DPBS solution (0.2g/L KCl, 0.2g/L KH2PO4, 8.0g/L NaCl, 1.15g/L Na2HPO4, 5%BSA, 0.03% Tween–20) and adjusted to pH 7.4. Protease inhibitor cocktail was added to all solutions immediately before use. In some studies samples were centrifuged at 16,000 × g for 20 min. at 4 °C and supernatant collected and used immediately in ELISAs. In other studies, the homogenates were centrifuged at 1,500 × g to remove cellular debris and the supernatant was used for further studies in Western blots.
When smaller amounts of tissues were analyzed, the tissues were homogenized in buffer containing 80 mM Pipes (pH 6.8), 1mM MgCl2, 2 mM EGTA. 0.1 mM EDTA, 0.1% Triton X-100 and 30% glycerol. Lysates were incubated at 37 °C for 10 minutes prior to room temperature centrifugation at 14,000 × g to separate soluble and insoluble fractions. Insoluble fractions were re-suspended in 2% SDS, 5 mM EDTA, 5 mM EGTA, 10 % glycerol, 0.25 M Tris-HCl (pH 6.8) and sonicated with a Branson Sonifier. Protein concentrations for soluble and insoluble fractions were determined and equal volumes of each fraction were combined, diluted with Laemelli buffer containing 5% β-mercaptoethanol, 5% SDS and 1% sodium deoxycholate. Samples were heated at 65 °C for 1 hour and run on 12% SDS-PAGE gels.
Determination of 26S proteasome activity
Frozen striatum and inferior frontal gyrus (IFG) tissue samples were resuspended in 10X volume ice-cold extraction buffer (10mM Tris-HCl pH 7.4, 1mM ethylene-diamine-tetra-acetic acid, 4mM dithiothreitol, 20% glycerol) and disrupted by 50 strokes in a dounce homogenizer on ice. Lysates were cleared by centrifugation (20 min, 16,000 × g, 4°C). Soluble protein concentration was determined by Bio-Rad Protein Assay (Bio-Rad, Hercules, CA), and protein concentrations were equalized to 0.75mg/ml by dilution in extraction buffer. For determination of 26S proteasome activity, 10μl lysate was combined with 85μl reaction buffer (20mM Tris-HCl pH 7.4, 1mM ATP, 20% glycerol) plus 5μl 0.1mg/ml N-Succinyl-Leu-Leu-Val-Tyr-7-amino-4-methylcoumarin (suc-LLVY-AMC; Biomol, Plymouth Meeting, PA), and incubated at 37°C for 60 min. Reactions including 50μM clasto-lactacystin ß-lactone (lactacystin) 26S proteasome inhibitor were conducted in parallel. After incubation, reactions were transferred to opaque 96-well plates, and AMC fluorescence was measured on a Victor3V, 1420 Multi-label Counter (PerkinElmer, Waltham, MA) for 1 sec, using an excitation filter of 355 nm and an emission filter of 460 nm. Lactacystin-resistant activity was subtracted from total activity to determine 26S activity.
Enzyme-linked immunosorbent assays
ELISA’s were conducted using special 96-well plates with high binding capacity (Corning Costar). All samples were assayed in triplicate along with a known α-Syn standard ranging from 0–500pg/ml. Plates were coated and incubated overnight at 4°C using coating buffer (0.025 M sodium bicarbonate, 0.025 M sodium carbonate pH 9.7) prior to blocking plate. Rest of the methods were completed according to Alpha-Synuclein Immunoassay Kit (#KHB0061) from Biosource International Inc. Absorbance was taken at 450 nm using a Wallac 1420 Multilabel Counter within 30 min. of stopping the reaction with 1M HCl. The bottom of each 96-well plate was thoroughly wiped off with 70% ethanol before reading.
Western Blot Analysis
Western blot analysis was performed as described previously (16), except that samples contained 5% SDS and 1% sodium deoxycholate, and were heated to 70 °C for 90–120 min to ensure maximal solubilization of aggregates of α-Syn and p-Tau. Samples were analyzed by Western blots with 12% SDS-PAGE and all blots were blocked with 5% milk. Primary antibodies used were: TAU-5 (phosphorylation-independent antibody; 1/1000; Chemicon), PHF-1 (pSer396/404; 1/000; provided by P. Davies, Albert Einstein College of Medicine, Bronx, NY), CP13 (pSer202; 1/1000; provided by P. Davies) and pSer262 (1/1000; Biosource); α-Syn (mouse mAb; 1/1000; BD Transduction Laboratories); DAT (1/1000, Chemicon); GSK-3β (mouse mAb; 1/1000; BD Transduction Laboratories); pY216-GSK-3β (mouse mAb; 1/1000; BD Transduction Laboratories); parkin (AbCam, 1/1000); 19S proteasome (Boston Biochemicals, 1/1000); 20S proteasome (Boston Biochemicals, 1/1000); β-actin (Santa Cruz Biotechnology, 1/1000). Equal protein loading was confirmed with anti-β-actin antibody (1/1000; Santa Cruz Biotechnology, sc-1616). Protein bands were scanned by Scanner EPSON Perfection 4870 Photo and quantitatively analyzed by Scion Image Program.
Statistics
Results were expressed as mean ± SEM and statistically analyzed by the t test between two groups and analysis of variance among multiple groups. Statistical significance was accepted at the p < 0.05 level.
RESULTS
Expression levels of tyrosine hydroxylase and dopamine transporter in human post mortem brains of PD, PDD, and age-matched controls
Since decreases in tyrosine hydroxylase [TH] and dopamine transporter [DAT] levels indicate the extent of loss of monoaminergic neurons and loss of dopaminergic nerve terminals, respectively, we examined expression levels of these proteins in post mortem striata [Fig. 1A] and inferior frontal gyri [IFG, Fig. 1B] from PD, PDD, and controls using Western blots. In striata [Fig.1A], TH levels were decreased by ~40% in PD and PDD cases [p<0.01]. In the IFG [Fig. 1B], TH levels were significantly decreased by ~40% only in the PDD cases compared to both PD and controls [p<0.01].
Figure 1. Western blot analyses of tyrosine hydroxylase [TH] and dopamine transporters [DAT] in striata [A] and inferior frontal gyri [IFG] [B] from control [Cont], PD and PDD groups.

Tissue lysates were prepared as described under Materials and Methods, and analyzed by Western blots using antibodies against TH and DAT. Blots are from representative experiments, while the graph is a summary of quantitation of protein levels normalized against β-actin from striata of 17 PD cases, 18 with PDD and 22 controls and while IFG are from 9 PD cases, 7 PDD and 9 controls. * denotes p<0.01 for PD and PDD groups compared to controls, while † denotes p<0.01 for PDD compared to PD.
Large decreases in DAT levels were observed in striata from PD and PDD [44 and 50% respectively, p<0.01] compared to age-matched controls [Fig. 1A]; there were no significant differences between the two disease groups. Compared to the age matched control group, DAT levels in the IFG of PD were decreased by 20% [p<0.01] and were decreased by 38% [p<0.01] in PDD [Fig. 1B]. Moreover, the decrease seen in IFG of PDD patients was significantly different than the decrease noted in PD patients [p<0.01, Fig. 1B].
Taken together, these combined data suggest that there is substantial and equal degeneration of dopaminergic neurons in striata of patients with PD and PDD of about ~40%, although there appears to be greater loss of dopaminergic terminals in PDD striata relative to PD striata. In the IFG, patients with PDD, however, appear to sustain a greater loss of dopaminergic neurons and nerve terminals [about ~40%], while PD patients have a loss of half as many dopaminergic neurons and terminals [~20%], as indicated by decreases in both TH and DAT immunoreactivity.
Measurement of soluble and insoluble α-synuclein levels in striata and IFG of PD, PDD, and age-matched controls
We measured both soluble and insoluble α-Syn in striata [Fig. 2A] and IFG [2B] by ELISA and Western blots, respectively, as described in Materials and Methods. Soluble levels of α-Syn were low in all tissues, ranging from 0.9–0.1.2 absorbance units, with no significant differences between the three groups. For insoluble α-Syn levels, however, control subjects had insoluble levels of α-Syn in striata that were 4.3-fold higher than soluble levels [Fig. 2A]. The ratio of insoluble/soluble α-Syn was also the same at 4.4-fold higher than soluble levels [Fig. 2A]. In PD striata, insoluble levels of α-Syn were12.4-fold higher than soluble levels and insoluble/soluble levels were also significantly increased [by ~17-fold]. In PDD striata, insoluble and insoluble/soluble levels of α-Syn levels were even higher [29- and 30-fold, respectively]. Moreover, these elevated levels of insoluble α-Syn seen in PDD brains were higher [by 141-76%] than that of PD brains, for insoluble and insoluble/soluble α-Syn, respectively, and the difference between the two groups was highly significant [p <0.01].
Figure 2. ELISA and western blot analyses of α-synuclein [α-Syn] in striata [A] and inferior frontal gyri [IFG] [B] from control, PD and PDD groups.
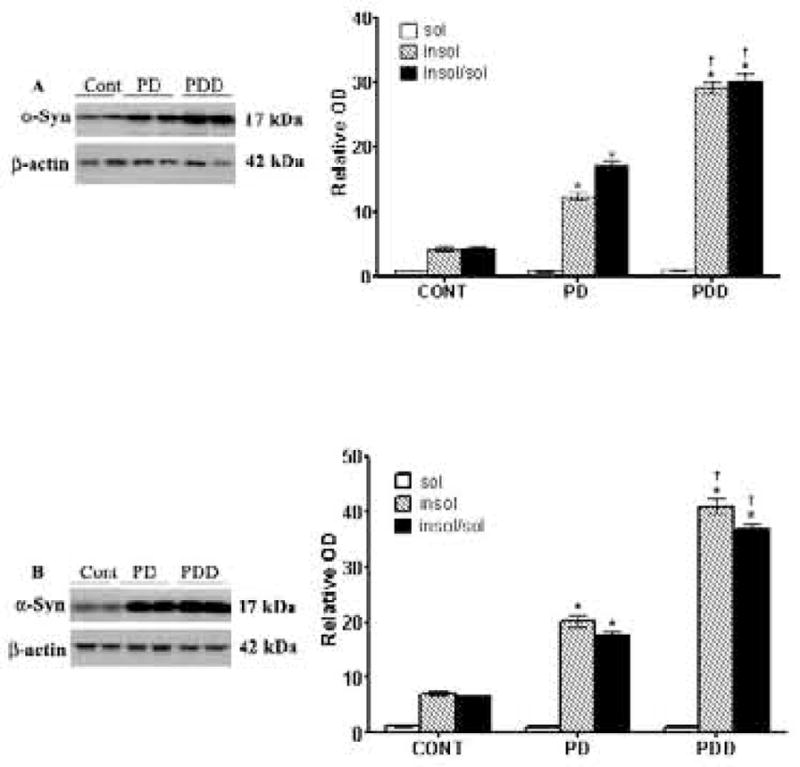
ELISA was conducted to measure soluble levels of α-Syn, while Western blots were conducted on insoluble α-Syn, as described in Materials and Methods. Blots are from representative experiments, while the graph is a summary of quantitation of studies from striata of 17 PD cases, 18 with PDD and 22 controls and IFG from 9 PD cases, 7 PDD and 9 controls. * denotes p<0.01 for PD and PDD groups compared to controls, while † denotes p<0.01 for PDD compared to PD.
We next examined soluble and insoluble α-Syn levels in the IFG [Fig. 2B]. Insoluble levels of α-Syn in control brains were 7-fold higher in the IFG, consistent with higher expression levels of α-Syn in this brain region (Wersinger et al., 2004), while insoluble/soluble levels of α-Syn remained the same. In PD IFG, insoluble levels of α-Syn were elevated 20-fold, while insoluble/soluble levels were elevated 18-fold [Fig. 2B]. In IFG of PDD, insoluble levels of α-Syn were increased 41-fold, while insoluble/soluble levels were also increased by 37-fold. The higher levels of insoluble α-Syn seen in PDD IFG compared to the PD IFG were highly significant [p<0.01].
Hyperphosphorylation of Tau in striata and IFG
Several protocols exist for solubilizing p-Tau for their analyses by Western blots, often giving varied results. In our hands, we found that when using large pieces of tissues are to be extracted [>100–300 mg wet weight], the guanidine HCl method gave satisfactory results. However, for small amounts of tissues [~20 mg], the PIPES/SDS method of extraction was far superior. Moreover, this protocol had the added advantage that it allowed for detection of other proteins. We examined p-Tau, hyperphosphorylated at pSer262 [seen as a doublet protein band with molecular weight of 64 and 66 kDa] in striata and found an increase of 34% [p<0.01] in striata of the PD and PDD groups, compared to the age-matched controls [Fig. 3A]. pSer262 levels were calculated relative to total Tau levels within the same samples [Fig. 3A] and there was no difference between PD and PDD. pSer396/404 Tau was increased by 81% [p<0.01] in PD striata and by 64% [p<0.01] in PDD striata compared to control subjects with no significant difference between the PD and PDD groups. A significant increase of 23% in pSer202 was seen in PD striata, while no change was observed for the PDD group compared to the control group.
Figure 3. Western blot analyses of p-Tau levels in striata [A] and inferior frontal gyri [IFG] [B] from control, PD and PDD groups.

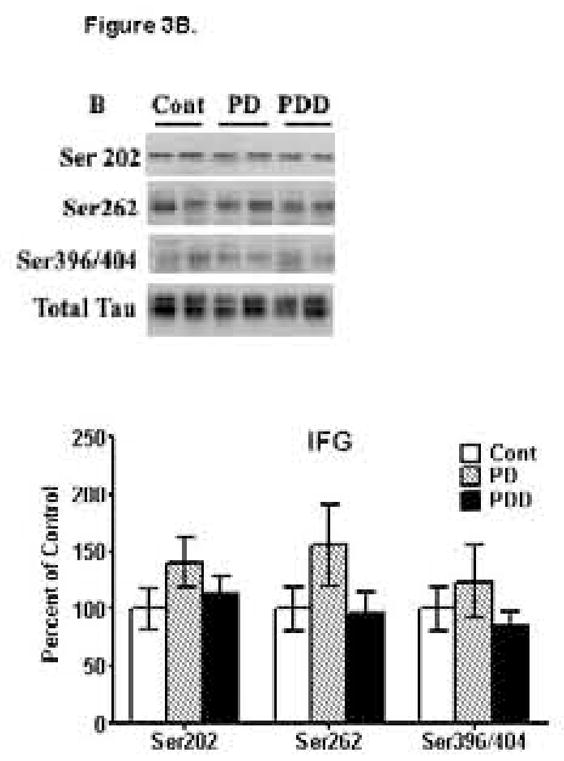
Western blot analyses of p-Tau levels were conducted using antibodies to detect hyperphosphorylation of Tau at Ser202 [CP-13 antibodies], Ser262 [anti-p-Tau-Ser262 antibodies] and Ser396/404 [PHF-1 antibodies]. Blots are from representative experiments, while the graph is a summary of quantitation of protein levels normalized against total Tau in samples, from striata of 17 PD cases, 18 with PDD and 22 controls and IFG from 9 PD cases, 7 PDD and 9 controls. * denotes p<0.01 for PD and PDD groups compared to controls.
We next examined p-Tau levels in IFG [Fig. 3B]. In both PD and PDD IFG, we were unable to detect any significant changes in p-Tau levels hyperphosphorylated at pSer202, pSer262 or pSer396/404, when compared to age-matched controls. Although there appeared to be an increase in pSer262 Tau in PD, this was not significant and was due to increases in a few of a samples, but was not uniformly observed in other tissues, and the increase was not significant (p>0.05). Thus, no increases in p-Tau were seen for either PD or PDD, despite repeating conducting these studies multiple times and under diverse methods of tissue extraction and solubilization. In all instances, we were completely unable to detect any increases in p-Tau levels in IFG of PD or PDD compared to controls. These combined data suggests a complete absence of tauopathy in the IFG of PD or PDD despite having observed tauopathy in striatum of the very same patients.
Measurement of GSK-3β and its activation in striata and IFG
pGSK-3β levels were examined by Western blots using an antibody that recognizes GSK-3β phosphorylated at Tyr216 [Fig. 4]. In both PD and PDD striata, pGSK-3β/GSK-3β levels were significantly [p<0.01] increased 4- and 4.6-fold, respectively, compared to the control group [Fig. 4A]; the difference between PD and PDD was not significant. In the IFG, however, pGSK-3β/GSK-3β levels were unchanged and similar levels of these proteins were seen in control, PD and PDD with no significant differences between the three groups [Fig. 4B].
Figure 4. Western blot analyses of p-GSK-3β levels in striata [A] and inferior frontal gyri [IFG] [B] from control, PD and PDD groups.
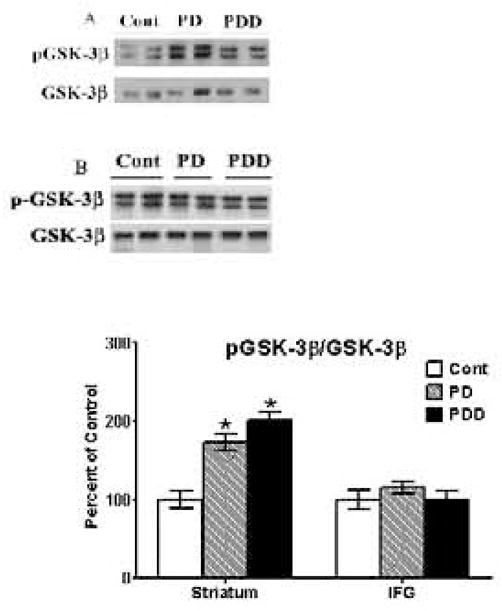
Western blot analyses of p-Tau levels were conducted using antibodies to detect phosphorylation of GSK-3β at Y216. Blots are from representative experiments, while the graph is a summary of quantitation of protein levels normalized against total GSK-3β [nonphosphorylated form] in samples, from striata of 17 PD cases, 18 with PDD and 22 controls and IFG from 9 PD cases, 7 PDD and 9 controls. * denotes p<0.01 for PD and PDD groups compared to controls, while † denotes p<0.01 for PDD compared to PD.
Examination of Parkin and the 26S Proteasome Activity in Striatum and IFG of PD and PDD Patients
In striatum, expression levels of parkin were unchanged [Fig. 5A] in PD and PDD, as compared to control. Similarly in the IFG, we failed to observe any significant changes in parkin levels between any of the three groups [Fig. 5B]. These data suggests an absence of a link between the E3 ubiquitin ligase enzyme, parkin, and disease pathology, especially tauopathy, seen in PD and PDD.
Figure 5. Western blot analyses of parkin levels in striata [A] and inferior frontal gyri [IFG] [B] from control, PD and PDD groups.
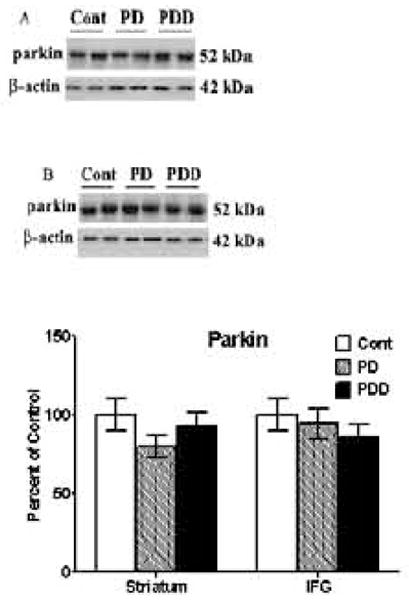
Western blot analyses of parkin levels were conducted using antibodies to detect parkin. Blots are from representative experiments, while the graph is a summary of quantitation of protein levels normalized against β-actin in samples, from striata of 17 PD cases, 18 with PDD and 22 controls and IFG from 9 PD cases, 7 PDD and 9 controls.
26S proteasome activity was measured in soluble lysates from striata of PD (n =15), PDD (n = 12), and controls (n = 16), and from IFG tissue of PD (n = 9), PDD (n = 7), and controls (n = 7). Lactacystin-sensitive digestion of the model substrate suc-LLVY-AMC was used to measure 26S proteasome activity; in this assay, cleavage of the LLVY peptide leads to an increase in AMC fluorescence. Greater than 80% of the total 26S proteasome activity in each sample was solubilized by our extraction procedure (data not shown), so only soluble 26S activity data is presented here. Striatum 26S proteasome activity was reduced by nearly 20% (not significant) in both PD and PDD samples relative to control (Fig. 6A). The reduction in 26S activity in the IFG was more dramatic, with PD samples showing a 30% decrease (p<0.05) and PDD samples a greater than 40% decrease (p<0.01) relative to controls (Fig. 6A). There were no significant differences between the PD and PDD samples in either striatum or IFG.
Figure 6. 26S Proteasome Activity in Striatum and IFG.
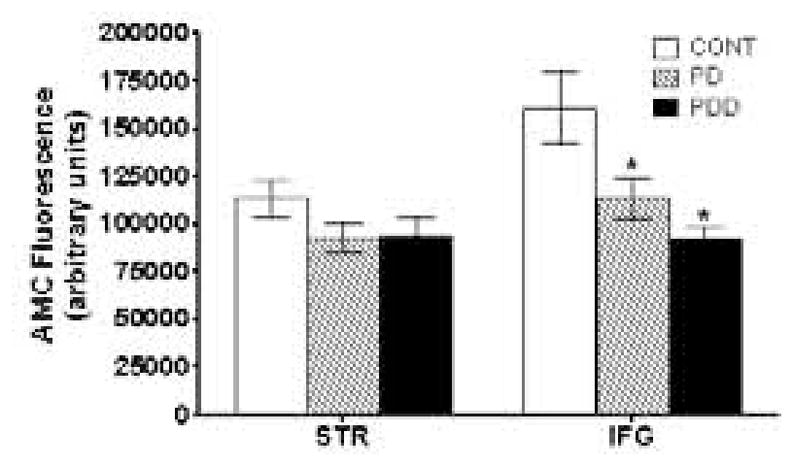
26S proteasome activity was determined as described in Materials and Methods from striatum and IFG of PD and PDD patients, and compared with age-matched controls. Activity is reported as AMC fluorescence in arbitrary units. Statistically significant decreases in activity relative to control are indicated by an (*).
We also examined the protein levels of representative subunits of the 19S and 20S components of the 26S proteasome by Western blot (Fig. 6B and C). There were no significant differences for the 19S S6′ subunit in striatum between the control and diseased groups (Fig. 6B). In the IFG, the 19S S6′ subunit was similar in control and PD patients and was significantly (p<0.05) decreased in PDD patients. The 20S alpha 5 subunit was decreased in both striatum (p<0.05) and in IFG (p<0.01), with larger decreases observed in IFG [of ~45%], as compared to control. Thus, reduced proteasomal activity in the IFG is likely due to reduction in proteasome levels.
DISCUSSION
We show here for the first time an increased state of tauopathy and an increase in associated proteins that modulate this pathway in Parkinson’s disease, α-Syn and p-GSK-3β, in postmortem striata of both PD and PDD patients. Our findings demonstrate a large increase in Tau abnormally hyperphosphorylated at Ser202, Ser262 and Ser396/404 in striatum of PD patients, with a similar increase seen in Ser262 and Ser396/404 in PDD patients; notably, Ser202 levels were not increased in striatum of PDD. The high levels of p-Tau hyperphosphorylated at these sites were also accompanied by elevated levels of insoluble α-Syn in striatum. Moreover, in PDD, insoluble levels of α-Syn were nearly 2-fold higher than the levels seen in PD in both striatum and IFG, suggesting that the increased accumulation and aggregation of this protein may lead to dementia.
Although we have previously demonstrated tauopathy induced by α-Syn, through activation of p-GSK-3β, our studies were done in toxin [MPTP/MPP+] in vivo and in vitro models of PD (Duka et al., 2006; Duka and Sidhu 2006; Duka et al., 2009). In those studies, we found Tau to be hyperphosphorylated at Ser262 and Ser396/404, but not at Ser202. However, in striata of PD patients, our current studies show that Tau is hyperphosphorylated at Ser202, indicating that this site may also be involved in the disease pathology of PD. Hyperphosphorylation of Tau in synaptic-enriched fractions in the frontal cortex of PD patients has been previously demonstrated (Mutane et al., 2008); however, these studies found Tau to be hyperphosphorylated only at Ser396/404. This study contrasts with ours, where we were unable to find hyperphosphorylation of Tau in the IFG of either PD or PDD patients. Part of this discrepancy may be related to the fact that we examined a specific region of the frontal cortex, the IFG, whereas in the other study the entire frontal cortex was used.
The pathological hyperphosphorylation of Tau at specific sites is well-related to neurodegenerative changes. In a mouse model of AD, neurons expressing Tau hyperphosphorylated at Ser202/Thr205 were more prone to undergo degeneration (Schindowski et al., 2006). Hyperphosphorylation at both Ser262 and Ser396/404 has been shown to lead to reduced binding of Tau to microtubules (Zhong et al., 1999), resulting in increased destabilization of the microtubules (Zhong et al., 1999) and ultimately leading to collapse of the cytoskeleton and neurodegeneration. The neurodegenerative scenarios presented by Tau hyperphosphorylated at Ser202, Ser262 and Ser396/404 are consistent with the degenerative changes that accompany dopaminergic neurons innervating the striatum in PD, which we have shown earlier in a previous study (Duka et al., 2006).
Our studies also show that parkin levels are unchanged and are expressed to similar levels in striatum of PD and PDD patients compared to controls. In the IFG as well, there were no changes in levels of parkin in PD or PDD patients compared to the control group. Thus, in both striata and IFG, parkin, the E3 ubiquitin ligase enzyme, is likely ubiqutinating and targeting proteins for ultimate degradation by the 26S proteasomes normally, and there does not appear to be any evidence that it has a role in pathological accumulations of p-Tau and α-Syn in these diseases. Thus, despite several in vitro studies suggesting that parkin overexpression can rescue cells from neurodegeneration, our findings of normal parkin levels in PD and PDD do not support a role for this protein in the sporadic human disease brain.
Studies have suggested an impairment in the 26S proteasomal pathway in Substantia nigra of PD (McNaught et al., 2003) which may account for accumulation of both α-Syn and p-Tau in striatum, and in PD, the 20S proteasome protein is colocalized with α-Syn in Lewy bodies (Lindersson et al., 2004). When we examined the 26S proteasomal activity in striata there was a small decrease in proteasomal activity in PD and PDD compared to controls, but the decrease was not significant. This is consistent with previous studies, which also did not show any significant changes in proteasomal activity in PD striata compared to controls, but where lower activity was detected only in the Substantia nigra (McNaught et al., 2003). In the IFG, however, we found a clear and significant decrease in 26S proteasomal activity in both PD and PDD compared to controls. This is the first time that decreased proteasomal activity has been detected in this brain region. It is interesting to note that previously, no differences were found in the frontal cortex of PD patients compared to controls (McNaught et al., 2003). However, in that study (McNaught et al., 2003), the superior frontal gyrus was used, which is anatomically distinct from the inferior frontal gyrus used in our studies. Interestingly, the decrease in proteasomal activity in the both the striata IFG of PD and PDD was accompanied by significant decreases in levels of the 20S alpha 5 subunit, and the decrease was larger in the IFG as compared to the striata. In addition, the 19S S6′ subunit was as also reduced in the IFG of both PD and PDD, although it was significant for only PDD; lower, nonsignificant reductions in 19S S6′ levels were also seen in striatum. Together, the reduced levels of subunits from both the 19S and 20S components of the proteasomes may account for the overall significantly lower level of proteasomal activity seen in the IFG of PD and PDD.
This is the first evidence of reduced levels of 19 and 20S subunits of the proteasome in PD and PDD brains and is likely to account for the abnormal accumulation of α-Syn seen in these diseases, as well as of p-Tau. Previously, reduction in proteasomal activity has been implicated in the accumulation and aggregation of hyperphosphorylated Tau in AD brains (Keck et al., 2003). Moreover, it has been suggested that proteasome dysfunction in AD brain may result from the inhibitory binding of hyperphosphorylated Tau to the proteasomes themselves (Keck et al., 2003). In particular, it was found that Tau that was hyperphosphorylated by GSK-3β was especially potent in inhibiting proteasomal activity in vitro (Poppek et al., 2006). However, since there were no increases in p-Tau levels in the IFG of either PD or PDD groups, it is unlikely that 26S proteasomal activity is inhibited by p-Tau. Studies have also shown that α-Syn aggregates can also directly bind to the 20S proteasome, in a non-competitive manner, resulting in inhibition of the 20S activity (Lindersson et al., 2004). Therefore, in PD and PDD brains, it is more likely that binding of α-Syn aggregates to the 20S proteasomes occurs in the IFG of both PD and PDD, causing accumulation of α-Syn resulting in decreased activity of the 26S proteasome in the IFG. This is particularly important in view of the fact that we were unable to detect any changes in p-GSK-3β [or p-Tau] in IFG that could account for the increases and accumulation of α-Syn in IFG.
The most interesting aspect of our studies was the distribution of the tauopathy seen in PD and PDD brains, where p-Tau changes were observed only in striatum and not in IFG. Tauopathy correlated very well with increased levels of both α-Syn and p-GSK-3β, and in this regard, it should be noted that this is also the first report of elevated levels of p-GSK-3β in PD or PDD brains. Interestingly, a recent report demonstrated strong linkage of two single nucleotide polymorphisms in the GSK-3β gene to PD (Kwok et al, 2005), implicating participation of this kinase in the genesis of this disease. Moreover, the lack of tauopathy in IFG, despite seeing an increase in α-Syn, can clearly be attributed to lack of p-GSK-3β activation, which further suggests that increases in this enzyme are essential for observing changes in p-Tau levels. Indeed, in our previous studies using cultured co-transfected SH-SY5Y neuronal cells, we had shown early accumulations of both α-Syn and p-GSK-3β activation preceded increases in p-Tau levels by at least 16 h, and such increases were sustained throughout production of p-Tau (Duka et al, 2009). In addition to increases in both α-Syn and p-GSK-3β, earlier studies from our laboratory have also shown an absolute requirement for oxidative stress in the α-Syn-dependent induction of p-Tau formation, and in the absence of external stressors, we have failed to observe any tauopathy in neuronal cells (Duka et al, 2006; Duka & Sidhu., 2006; Duka et al, 2009). In striatum, which is comprised of dopaminergic neurons and terminals, oxidative stress through autoxidation of dopamine under pathological conditions is likely to be high, which in turn promotes formation of p-Tau through α-Syn/p-GSK-3β dependent pathways. Thus, although the pathomechanisms of tauopathy in both PD and AD appear to be similar, our data suggests that tauopathy in PD may be restricted to dopaminergic neurons and that additional underlying mechanisms may come into play that are unique to PD.
At the present time, the precise nature of the degenerative change taking place in the IFG in PD or PDD is unclear, but our data suggests that they are not tauopathic in nature, even though tauopathy appears to have a role in striatum. PDD is distinguished from PD by the additional presence of Lewy bodies [LBs] in the frontal gyrus (Mattila et al., 2000), which are primarily composed of α-Syn inclusions, and aggregation of the latter is thought to be a key pathogenic event in formation of LBs (Beyer et al., 2009). In addition to aggregation of α-Syn, thought to the first step in formation of LBs, impairment of protein degradative pathways, including both the ubiquitin-proteasome system and the autophagy-lysosomal pathway, are also believed to play an important role during the development of LBs (Beyer et al., 2009). In IFG of both PD and PDD, the diminished proteasomal activity we observe as a consequence of decreased 19S and 20S subunits of the proteasome, may lead to increased accumulation of α-Syn, but which in the absence of oxidative stress and p-GSK-3β, does not lead to any Tauopathic changes.
Given the restricted expression of tauopathy in PD and PDD, strategies aimed at targeting GSK-3β to prevent its activation holds promise in the development of novel therapeutic strategies aimed at treating this disease as well as other synucleopathies.
Figure 7. Western blot analyses of 19S and 20S levels in striata [A] and inferior frontal gyri [IFG] [B] from control, PD and PDD groups.
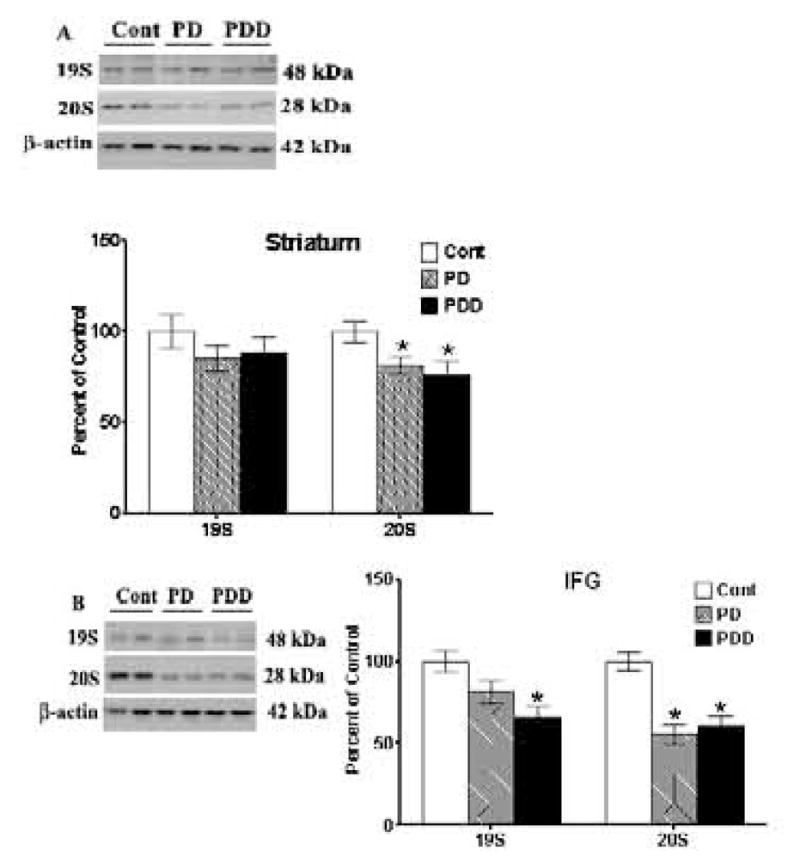
Western blot analyses of 19S and 20S proteasomal subunits were conducted using antibodies to detect these proteins. Blots are from representative experiments, while the graph is a summary of quantitation of protein levels normalized against β-actin in samples, from striata of 17 PD cases, 18 with PDD and 22 controls and IFG from 9 PD cases, 7 PDD and 9 controls.
Acknowledgments
We are grateful to Peter Davies [Albert Einstein College of Medicine, NY] for the generous gift of the CP-13 and PHF-1 antibodies. We are grateful to Dr. Dianca Graham for work on unpublished studies that enabled us to form some of our conclusions. We are grateful to the families of the many patients for their generosity in donating the organs that made this study possible and to Sun Health Research Institute Brain Donation Program of Sun City, Arizona for the provision of human brain tissue. The Brain Donation Program is supported by the National Institute on Aging (P30 AG19610 Arizona Alzheimer’s Disease Core Center), the Arizona Department of Health Services (contract 211002, Arizona Alzheimer’s Research Center), the Arizona Biomedical Research Commission (contracts 4001, 0011, 05-901 and 1001 to the Arizona Parkinson’s Disease Consortium) and the Prescott Family Initiative of the Michael J. Fox Foundation for Parkinson’s Research. This study was supported in part by grants from NIAID, R01 AI062854 [J.J.] and NIA, R01 NIA AGO28108 [A.S.].
Abbreviations
- PD
Parkinson’s disease
- PDD
Parkinson’s disease with dementia
- AD
Alzheimer’s disease
- α-Syn
α-synuclein
- LBs
Lewy bodies
- p-Tau
hyperphosphorylated Tau
- GSK-3β
glycogen synthase kinase 3β
- p-GSK-3β
GSK-3β phosphorylated at Tyr216
- IFG
inferior frontal gyrus or gyri
- PHFs
paired helical filaments
- NFT
neurofibrillary tangle
- TH
tyrosine hydroxylase
- DAT
dopamine transporter
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adler CH, Hentz JG, Joyce JN, Beach T, Caviness JN. Motor Impairment in Normal Aging, Clinically Possible Parkinson’s Disease, and Clinically Probable Parkinson’s Disease: Longitudinal Evaluation of a Cohort of Prospective Brain Donors. Parkinsonism and Related Disorders. 2002;9:103–110. doi: 10.1016/s1353-8020(02)00012-3. [DOI] [PubMed] [Google Scholar]
- Arai Y, Yamazaki M, Mori O, Muramatsu H, Asano G, Katayama Y. Alpha-synuclein-positive structures in cases with sporadic Alzheimer’s disease: morphology and its relationship to tau aggregation. Brain Res. 2001;888:287–296. doi: 10.1016/s0006-8993(00)03082-1. [DOI] [PubMed] [Google Scholar]
- Baum L, Seger R, Woodgett J, Kawabata S, Maruyama K, Koyama M, Silver J, Saitoh T. Overexpressed tau protein in cultured cells is phosphorylated without formation of PHF: implication of phosphoprotein phosphatase involvement. Mol Brain Res. 1995;34:1–17. doi: 10.1016/0169-328x(95)00111-5. [DOI] [PubMed] [Google Scholar]
- Beyer K, Domingo-Sàbat M, Ariza A. Molecular pathology of lewy body diseases. Int J Mol Sci. 2009;10:724–45. doi: 10.3390/ijms10030724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns J, Galvin JE, Roe CM, Morris JC, McKeel DW. The pathology of the substantia nigra in Alzheimer disease with extrapyramidal signs. Neurology. 2005;64:1397–403. doi: 10.1212/01.WNL.0000158423.05224.7F. [DOI] [PubMed] [Google Scholar]
- Duda J, Giasson BI, Mabon ME, Miller DC, Golbe LI, Lee VM, Trojanowski JQ. Concurrence of alpha-synuclein and tau brain pathology in the Contursi kindred. Acta Neuropathol (Berl) 2002;104:7–11. doi: 10.1007/s00401-002-0563-3. [DOI] [PubMed] [Google Scholar]
- Duka T, Drolet R, Duka V, Wersinger C, Goudreau JL, Sidhu A. Alpha-synuclein induces hyperphosphorylation of TAU in the MPTP model of Parkinsonism. FASEB J. 2006;20:2302–2312. doi: 10.1096/fj.06-6092com. [DOI] [PubMed] [Google Scholar]
- Duka T, Sidhu A. The Neurotoxin MPP induces hyperphosphorylation of Tau in the presence of alpha-synuclein in SHSY-5Y neuroblastoma cells. Neurotox Res. 2006;10:1–10. doi: 10.1007/BF03033329. [DOI] [PubMed] [Google Scholar]
- Duka T, Duka V, Joyce JN, Sidhu A. Alpha-Synuclein contributes to GSK-3beta-catalyzed Tau phosphorylation in Parkinson’s disease models. FASEB J. 2009;23:2820–2830. doi: 10.1096/fj.08-120410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman M, Schmidt ML, Kasturi S, Perl DP, Lee VM, Trojanowski JQ. Tau and alpha-synuclein pathology in amygdala of Parkinsonism dementia complex patients of Guam. Am J Pathol. 2002;160:1725–1731. doi: 10.1016/s0002-9440(10)61119-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M, Spillantini M. Lewy body diseases and multiple system atrophy as alpha-synucleinopathies. Mol Psychiatry. 1998;3:462–465. doi: 10.1038/sj.mp.4000458. [DOI] [PubMed] [Google Scholar]
- Griffin W, Liu L, Li Y, Mrak R, Barger S. Interleukin-1 mediates Alzheimer and Lewy body pathologies. J Neuroinflammation. 2006;3:5. doi: 10.1186/1742-2094-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hishikawa N, Hashizume Y, Ujihira N, Okada Y, Yoshida M, Sobue G. Alpha-synuclein-positive structures in association with diffuse neurofibrillary tangles with calcification. Neuropathol Appl Neurobiol. 2003;29:280–287. doi: 10.1046/j.1365-2990.2003.00470.x. [DOI] [PubMed] [Google Scholar]
- Hofer A, Berg D, Asmus F, Niwar M, Ransmayr G, Riemenschneider M, Bonelli S, Steffelbauer M, Ceballos-Baumann A, Haussermann P, Behnke S, Kruger R, Prestel J, Sharma M, Zimprich A, Riess O, Gasser T. The role of alpha-synuclein gene multiplications in early-onset Parkinson’s disease and dementia with Lewy bodies. J Neural Transm. 2004;2:1249–54. doi: 10.1007/s00702-004-0263-3. [DOI] [PubMed] [Google Scholar]
- Iseki E, Marui W, Kosaka K, Uéda K. Frequent coexistence of Lewy bodies and neurofibrillary tangles in the same neurons of patients with diffuse Lewy body disease. Neurosci Lett. 1999;265:9–12. doi: 10.1016/s0304-3940(99)00178-0. [DOI] [PubMed] [Google Scholar]
- Joachim C, Morris JH, Kosik KS, Selkoe DJ. Tau antisera recognize neurofibrillary tangles in a range of neurodegenerative disorders. Ann Neurol. 1987;22:514–520. doi: 10.1002/ana.410220411. [DOI] [PubMed] [Google Scholar]
- Joyce JN, Ryoo HL, Beach TB, Caviness JN, Stacy M, Gurevich EV, Reiser M, Adler CH. Loss of response to levodopa in Parkinson’s disease and co-occurrence with dementia: role of D3 and not D2 receptors. Brain Res. 2002;955:138–52. doi: 10.1016/s0006-8993(02)03396-6. [DOI] [PubMed] [Google Scholar]
- Keck S, Nitsch R, Grune T, Ullrich O. Proteasome inhibition by paired helical filament-tau in brains of patients with Alzheimer’s disease. J Neurochem. 2003;85:115–122. doi: 10.1046/j.1471-4159.2003.01642.x. [DOI] [PubMed] [Google Scholar]
- Kotzbauer PT, Giasson BI, Kravitz AV, Golbe LI, Mark MH, Trojanowski JQ, Lee VM. Fibrillization of alpha-synuclein and tau in familial Parkinson’s disease caused by the A53T alpha-synuclein mutation. Exp Neurol. 2004;187:279–288. doi: 10.1016/j.expneurol.2004.01.007. [DOI] [PubMed] [Google Scholar]
- Kozikowski A, Gaisina I, Petukhov P, Sridhar J, King L, Blond S, Duka T, Rusnak M, Sidhu A. Highly Potent and Specific GSK-3 Inhibitors That Block Tau Phosphorylation and Decrease Alpha-Synuclein Protein Expression in a Cellular Model of Parkinson’s Disease. ChemMedChem. 2006;1:256–266. doi: 10.1002/cmdc.200500039. [DOI] [PubMed] [Google Scholar]
- Krüger R, Kuhn W, Müller T, Woitalla D, Graeber M, Kösel S, Przuntek H, Epplen JT, Schöls L, Riess O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet. 1998;18:106–8. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- Kwok JB, et al. GSK3B polymorphisms alter transcription and splicing in Parkinson’s disease. Ann Neurol. 2005;58(6):829–39. doi: 10.1002/ana.20691. [DOI] [PubMed] [Google Scholar]
- Lindersson E, Beedholm R, Højrup P, Moos T, Gai W, Hendil KB, Jensen PH. Proteasomal inhibition by alpha-synuclein filaments and oligomers. J Biol Chem. 2004;279:12924–12934. doi: 10.1074/jbc.M306390200. [DOI] [PubMed] [Google Scholar]
- Lippa CF, Fujiwara H, Mann DM, Giasson B, Baba M, Schmidt ML, Nee LE, O’Connell B, Pollen DA, St George-Hyslop P, Ghetti B, Nochlin D, Bird TD, Cairns NJ, Lee VM, Iwatsubo T, Trojanowski JQ. Lewy bodies contain altered alpha-synuclein in brains of many familial Alzheimer’s disease patients with mutations in presenilin and amyloid precursor protein genes. Am J Pathol. 1998;153:1365–1370. doi: 10.1016/s0002-9440(10)65722-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattila PM, Rinne JO, Helenius H, Dickson DW, Roytta M. Alpha-synuclein-immunoreactive cortical Lewy bodies are associated with cognitive impairment in Parkinson’s disease. Acta Neuropathol. 2000;100:285–290. doi: 10.1007/s004019900168. [DOI] [PubMed] [Google Scholar]
- McNaught KS, Belizaire R, Jenner P, Olanow CW, Isacson O. Selective loss of 20S proteasome alpha-subunits in the substantia nigra pars compacta in Parkinson’s disease. Neurosci Lett. 2002;326:155–158. doi: 10.1016/s0304-3940(02)00296-3. [DOI] [PubMed] [Google Scholar]
- McNaught K, Belizaire R, Isacson O, Jenner P, Olanow CW. Altered Proteasomal Function in Sporadic Parkinson’s Disease. Experimental Neurology. 2003;179:38–46. doi: 10.1006/exnr.2002.8050. [DOI] [PubMed] [Google Scholar]
- Mori H, Oda M, Komori T, Arai N, Takanashi M, Mizutani T, Hirai S, Mizuno Y. Lewy bodies in progressive supranuclear palsy. Acta Neuropathol (Berl) 2002;104:273–278. doi: 10.1007/s00401-002-0555-3. [DOI] [PubMed] [Google Scholar]
- Murphy DD, Rueter SM, Trojanowski JQ, Lee VM. Synucleins are developmentally expressed, and α-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J Neurosci. 2000;20:3214–3220. doi: 10.1523/JNEUROSCI.20-09-03214.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muntané G, Dalfó E, Martinez A, Ferrer I. Phosphorylation of tau and alpha-synuclein in synaptic-enriched fractions of the frontal cortex in Alzheimer’s disease, and in Parkinson’s disease and related alpha-synucleinopathies. Neuroscience. 2008;152:913–923. doi: 10.1016/j.neuroscience.2008.01.030. [DOI] [PubMed] [Google Scholar]
- Nemes Z, Devreese B, Steinert PM, Van Beeumen J, Fésüs L. Cross-linking of ubiquitin, HSP27, parkin, and alpha-synuclein by gamma-glutamyl-epsilon-lysine bonds in Alzheimer’s neurofibrillary tangles. FASEB J. 2004;18:1135–1137. doi: 10.1096/fj.04-1493fje. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- Poppek D, Keck S, Emark G, Jung T, Stolzing A, Ullrich O, Davies KJ, Grune T. Phosphorylation inhibits turnover of the tau protein by the proteasome: influence of RCAN1 and oxidative stress. Biochem J. 2006;200:511–520. doi: 10.1042/BJ20060463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindowski K, Bretteville A, Leroy K, Bégard S, Brion JP, Hamdane M, Buée L. Alzheimer’s disease-like tau neuropathology leads to memory deficits and loss of functional synapses in a novel mutated tau transgenic mouse without any motor deficits. Am J Pathol. 2006;169:599–616. doi: 10.2353/ajpath.2006.060002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szpak GM, Lewandowska E, Lechowicz W, Bertrand E, Wierzba-Bobrowicz T, Gwiazda E, Pasennik E, Kosno-Kruszewska E, Lipczyńska-Lojkowska W, Bochyńska A, Fiszer U. Lewy body variant of Alzheimer’s disease and Alzheimer’s disease: a comparative immunohistochemical study. Folia Neuropathol. 2001;39:63–71. [PubMed] [Google Scholar]
- Wersinger C, Banta M, Sidhu A. Comparative analyses of alpha synuclein expression levels in rat brain tissue and transfected cells. Neurosci Lett. 2004;358:95–98. doi: 10.1016/j.neulet.2003.12.118. [DOI] [PubMed] [Google Scholar]
- Wersinger C, Sidhu A. Disruption of the interaction of alpha-synuclein with microtubules enhances cell surface recruitment of the dopamine transporter. Biochemistry. 2005;44:13612–24. doi: 10.1021/bi050402p. [DOI] [PubMed] [Google Scholar]
- Wersinger C, Rusnak M, Sidhu A. Modulation of the trafficking of the human serotonin transporter by human alpha-synuclein. Eur J Neurosci. 2006a;24:55–64. doi: 10.1111/j.1460-9568.2006.04900.x. [DOI] [PubMed] [Google Scholar]
- Wersinger C, Jeannotte A, Sidhu A. Attenuation of the norepinephrine transporter activity and trafficking via interactions with alpha-synuclein. Eur J Neurosci. 2006b;24:3141–52. doi: 10.1111/j.1460-9568.2006.05181.x. [DOI] [PubMed] [Google Scholar]
- Yamaguchi K, Cochran EJ, Murrell JR, Polymeropoulos MH, Shannon KM, Crowther RA, Goedert M, Ghetti B. Abundant neuritic inclusions and microvacuolar changes in a case of diffuse Lewy body disease with the A53T mutation in the alpha-synuclein gene. Acta Neuropathol (Berl) 2005;110:298–305. doi: 10.1007/s00401-005-1042-4. [DOI] [PubMed] [Google Scholar]
- Yamazaki M, Arai Y, Baba M, Iwatsubo T, Mori O, Katayama Y, Oyanagi K. Alpha-synuclein inclusions in amygdala in the brains of patients with the parkinsonism-dementia complex of Guam. J Neuropathol Exp Neurol. 2000;59:585–591. doi: 10.1093/jnen/59.7.585. [DOI] [PubMed] [Google Scholar]
- Yancopoulou D, Xuereb JH, Crowther RA, Hodges JR, Spillantini MG. Tau and alpha-synuclein inclusions in a case of familial frontotemporal dementia and progressive aphasia. J Neuropathol Exp Neurol. 2005;64:245–253. doi: 10.1093/jnen/64.3.245. [DOI] [PubMed] [Google Scholar]
- Zhong J, Iqbal K, Grundke-Iqbal I. Hyperphosphorylated tau in SY5Y cells: similarities and dissimilarities to abnormally hyperphosphorylated tau from Alzheimer disease brain. FEBS Lett. 1999;453:224–228. doi: 10.1016/s0014-5793(99)00715-2. [DOI] [PubMed] [Google Scholar]