

Abstract
Accurate and precise methods for estimating incorrect peptide and protein identifications are crucial for effective large-scale proteome analyses by tandem mass spectrometry. The target-decoy search strategy has emerged as a simple, effective tool for generating such estimations. This strategy is based on the premise that obvious, necessarily incorrect “decoy” sequences added to the search space will correspond with incorrect search results that might otherwise be deemed to be correct. With this knowledge, it is possible not only to estimate how many incorrect results are in a final data set but also to use decoy hits to guide the design of filtering criteria that sensitively partition a data set into correct and incorrect identifications.
Keywords: Proteomics, Target-decoy, False positive, False discovery, Mass spectrometry, Estimation
1. Introduction
Peptide and protein identifications made in most mass spectrometry-based proteomic work flows first involve acquiring a set of tandem mass (MS/MS) spectra and then interrogating each spectrum against spectra predicted from a list of protein sequences by search engines, such as SEQUEST (1), Mascot (2), OMSSA (3), and X!Tandem (4). The output of these programs indicates the best theoretical peptide matches to the input spectra, which are then used to infer the source protein that was present in the biological sample. Unfiltered sets of peptide identifications produced in this manner are necessarily imperfect for three reasons: (1) not all peptide species in a sample are represented in the search space; (2) spectra derived from background nonpeptide species will often be given a peptide assignment; and (3) incorrect candidate peptide sequences occasionally may outscore correct sequences. For many search engines, nearly all input MS/MS spectra will be assigned a peptide match if there are any that lie within the supplied mass tolerance. Thus, the primary task of proteomics researchers is to distinguish incorrect from correct peptide assignments.
When working with very small data sets, such as those produced from a single spot on a 2D gel or a gel band representing a component of an isolated protein complex, identifying correct peptide identifications is almost trivial: they are the ones with the highest scores and tend to map to the same protein. It is also reasonable and appropriate to manually examine individual peptide-spectrum matches (PSMs) to verify that they are correct. However, the increasingly large data sets created by modern tandem mass spectrometers in global proteomic efforts are not amenable to these strategies. Simple filtering criteria based on score magnitude or numbers of peptides per protein tend to be neither sensitive nor accurate (5), and the staggering amount of information that can be produced in a single experiment renders the manual validation of peptide assignments impractical. Consequently, high-throughput protein sequencing efforts must rely on methods for estimating the frequencies of incorrect peptide and protein identifications among correct ones. The “target-decoy” search strategy is a simple yet powerful way to deliver false positive estimations and can be applied to nearly any MS/MS workflow. Here, we present several methods for preparing decoy sequences and strategies for selecting correct peptide identifications.
2. Materials
2.1. MS/MS Spectra
MS/MS spectra can be acquired on any number of tandem mass spectrometers, including the LTQ family of ESI-ion trap instruments from ThermoFisher, the QSTAR from Applied Biosystems, and the FLEX family from Bruker Daltonics. Alternatively, several public sources of MS/MS spectra are freely available on the internet, including PeptideAtlas (6) and the Open Proteomics Database (7). It is recommended that the target-decoy approach be applied to data sets consisting of several thousand MS/MS spectra (see Note 1).
2.2. Protein Sequences
MS/MS spectra are generally searched against peptides predicted from FASTA-formatted protein sequence lists. Sequence lists should be chosen such that any peptide that may have given rise to an observed spectrum is represented. For example, if a mouse-derived sample was sequenced by MS/MS, the spectra should be searched against a list of all known mouse proteins. Protein lists can be downloaded from numerous sources, including the International Protein Index (8) and UniProt/SwissProt (9). It is useful to also include sequences of known contaminants, such as trypsin and human keratins.
2.3. Search Engine
Numerous MS/MS search engines are in common usage. Some are commercially available, for example:
SEQUEST (http://www.thermo.com/com/cda/product/detail/0,1055,22209,00.html)
Mascot (http://www.matrixscience.com)
SpectrumMil (http://www.chem.agilent.com/scripts/pds.asp?lpage=7771) Other search engines are freely-distributed via the internet:
X!Tandem (http://www.thegpm.org/tandem/)
All of these produce some form of a score indicating the degree to which observed and predicted MS/MS spectra agree. Several of these search engines’ scores may be probability-based. See refs. (10–13) for more detailed descriptions and comparisons of these search engines. One principle benefit to target-decoy searching is its applicability to data generated by any search engine.
3. Methods
One deceptively simple way to estimate false positives is to manufacture “decoy” sequences that do not exist in nature, and then allow the search engine to consider these alongside “target” sequences derived from the organism being studied. Necessarily, incorrect decoy hits should be similar to incorrect but unknown hits derived from target sequences in terms of length, amino acid composition, mass accuracy, and search engine-assigned scores. Therefore, knowing the proportion of decoy versus target sequences in the search space allows one to estimate the number of incorrect target sequences in a reasonably large collection of PSMs. More than providing a means to estimate the number of incorrect target hits in a collection of PSMs, decoy hits can be used to guide researchers in the design of sensitive filtering criteria to precisely distinguish correct from incorrect PSMs.
Target-decoy searching is usually performed in the following steps:
Construct a concatenated target-decoy sequence list, marking decoy sequences with a text flag in their annotation.
Use a MS/MS search engine to interpret input MS/MS spectra using target-decoy sequence list.
Evaluate the relative proportion of target and decoy sequences in the search space to derive the multiplicative factor required to estimate false positives, if necessary.
Estimate false positive-related statistics.
Use decoy hits to guide the establishment of filtering criteria.
Report statistics for filtered data set.
Each of these steps will be discussed in further details below.
3.1. Decoy Sequence Construction
Several methods for creating decoy sequences have been described (14–16). Each has varying advantages and disadvantages, and it must be stressed that no single decoy type is perfect. Ideal decoy sequences should have the following characteristics:
Similar amino acid distributions as target protein sequences.
Similar protein length distribution as target protein sequence list.
Similar numbers of proteins as target protein list.
Similar numbers of predicted peptides as target protein list.
No predicted peptides in common between target and decoy sequence lists.
If each of these conditions are reasonably met, one can safely assume that decoy sequence selected by the search engine are incorrect, and that there is a one-to-one correspondence between incorrect target hits and decoy hits. By design or as a consequence of the decoy sequence construction method, conditions 3 or 4 may not be met. In this case, one should take into account the discrepancy between target and decoy sequences (see Subheading 3.3). This is particularly true when using stochastic means to generate decoy sequences based on target sequences demonstrating substantial amounts or repetition or homology.
3.1.1. Reversed Proteins
Protein reversal is by far the simplest and most widely used method for creating decoy sequences (see Note 2 for a simple Perl script to create a concatenated target-decoy sequence list based on an input target sequence list) (17, 18). By switching the amino-carboxyl orientation of a protein’s amino acids, a negligible number of peptide sequences are preserved, particularly when imposing in silico digestion constraints with proteases like trypsin. Protein reversal has two main advantages: First, because it preserves the general features of the target sequence list, reversed protein sequences will share the same degree of interprotein redundancy as the input target sequences; Second, since it is a defined transformation, multiple research groups can generate the same decoy sequences. The main disadvantage to protein reversal is that it is not a random transformation as some may prefer. Consequently, it can be argued that it does not strictly represent a null random distribution, and for certain types of peptides (e.g., palendromic or low sequence complexity), it may not be possible to create a suitable decoy counterpart. In practice, however, protein reversal stands up to the five conditions listed above (14), and can therefore be used to faithfully estimate the occurrences of incorrect identifications.
3.1.2. Shuffled Proteins
Protein shuffling is another method used for creating decoy sequences (16) in which the amino acids of each input target protein are randomly rearranged to yield a new decoy protein. Like protein reversal, shuffling is fairly simple to implement programmatically, and it preserves both the amino acid composition and length of each input target protein sequence. Unlike sequence reversal, this transformation has desired stochastic properties. As is true of most random transformations though, redundancies and homologies between protein entries will not be preserved, resulting in a greater number of decoy peptides than originally present in the target sequence list. This imbalance must be measured and then taken into account when generating estimations of false positives.
3.1.3. Random Proteins
Proteins can also be generated in a completely random fashion. This is the method internally implemented by some search engines, such as Mascot, for performing target-decoy analyses. Ideally, randomized sequences should have the same amino acid biases and protein length distribution as an input target sequence list. One way to do this is to first evaluate the target sequence list to generate a frequency matrix of amino acids and a histogram of protein lengths. Decoy proteins are then constructed by randomly selecting amino acids according to the frequency matrix, and adding these to the growing decoy protein until it reaches a specified length, randomly determined from the length histogram.
Rather than relying on a simple amino acid frequency matrix, one can construct a Markov chain model of amino acid frequencies to better replicate small scale patterns found in the target sequence list, such as single or double amino acid repeats or highly basic or acidic regions. Essentially, this is done by generating a frequency matrix reflecting the likelihood of observing a particular amino acid given the preceding n amino acids (14). Another frequency matrix should be constructed consisting of only the n amino acids that initiate the protein sequence. After randomly selecting from the initiating sequence frequency matrix, the protein can be extended by randomly selecting from the conditional frequency matrix until the protein achieves a specified length.
With either randomization method, it is possible to modulate the number of decoy sequences with respect to the number of target sequences considered. This has been done to examine the effects of interrogating a set of MS/MS spectra against search spaces of varying sizes (19). As with shuffled decoy proteins, random proteins do not preserve redundancies and homologies, so care must be taken to measure the relative proportion of target and decoy sequences, and then account for any observed bias when generating false positive estimations (see Subheading 3.3).
3.1.4. Decoy Peptides
Rather than generating entire decoy proteins from which decoy peptides will be derived according to in silico enzymatic digestion rules, one can instead generate decoy peptides directly by altering each peptide sequence derived from the target sequence list. Alterations can take the form of reversals or shuffling. This procedure has the advantage of creating decoy peptides exactly matching the masses of all target peptides considered by the search engine. If reversal or nonrandom shuffling was the transformation applied, the number of target and decoy sequences will match exactly both in number and in mass distributions. Otherwise, decoy peptides may outnumber target peptides, as with stochastically created proteins. Since in silico digestion is usually performed by the search algorithm prior to querying observed spectra, the generation of decoy peptides directly is typically performed within the search algorithm. An example of a search engine with this feature is the Sorcerer-SEQUEST platform from SAGE-N.
3.2. Spectrum Search
Once a target-decoy sequence list has been generated, the analysis of a set of MS/MS spectra can begin. The generally accepted means to do this is to supply the search engine with a single protein sequence list consisting of both target and decoy sequences. For each spectrum, the search engine must then choose between target and decoy sequences. Correctly-identified peptides will exclusively be selected from target protein sequences, while incorrect peptide matches will be randomly drawn from target and decoy sequences. If the number of target and decoy sequences considered by the search engine are equal, there should be a one-to-one correlation between target and decoy sequences among incorrect identifications. If the number of target and decoy sequences are unequal, the correlation between target and decoy sequences should reflect this bias. It should be noted that some groups advocate searching target sequences separately from decoy sequences. For a variety of reasons, this procedure can lead to an overly conservative interpretation of search results (14) (see Note 3).
3.3. Measuring Decoy Bias
In order to properly estimate the number of false positive identifications in a set of peptide identifications, it is essential that one first knows the relative proportion of decoy to target hits in the search space. For reversed-decoy databases, it can generally be assumed that there is a 1:1 correlation between target and decoy sequences (14). For decoy sequence lists generated with a stochastic component, there are usually more decoy sequences than target sequences, particularly when there is a substantial degree of homology or redundancy among target sequences. One computational approach for measuring this proportion is to create in silico digests of each target and decoy component, and then ask how many peptides from each component are within a specified tolerance near a given mass. For example, one would determine how many target and decoy peptides are within 1.0 Da surrounding a mass of 1,000 Da. The proportion of target and decoy peptides should be consistent across all masses in the range of peptides one might consider (e.g., 600–5,000 Da).
More simply, one can examine the frequency with which a search engine returns target and decoy hits for incorrect identifications. Since correct peptide identifications usually achieve the top-ranked hit for a given MS/MS spectrum, it can be usually assumed that lower ranked peptide hits are incorrect (14, 20, 21). Alternatively, if one shifts the precursor masses of input MS/MS spectra outside of the specified mass tolerance, they cannot be correctly matched (14, 20). Comparing the frequencies of target and decoy hits for incorrect spectra reveals the effective proportion of target and decoy sequences in the search space and therefore the factor one should use to estimate the number of hidden incorrect target hits, given the observed decoy hits (Fig. 1) (14).
Fig. 1.
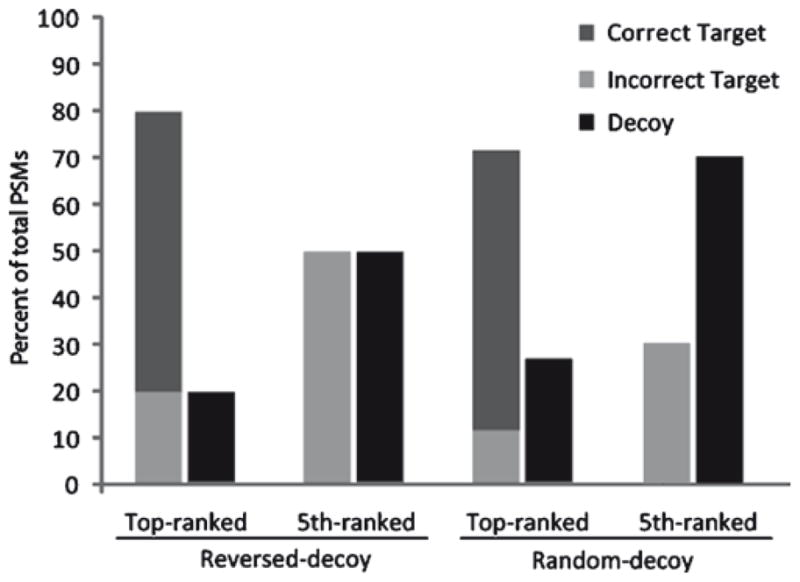
Decoy PSMs indicate incorrect target PSMs, depending on the underlying proportion of target and decoy sequences. Under the reversed-decoy model, the proportion of target and decoy peptides considered are approximately equal (5th-ranked, reversed-decoy). Thus, the proportion of decoy PSMs observed in the presence of correct identifications equals the proportion of target PSMs that are incorrect (Top-ranked, reversed-decoy). When the underlying proportion of target and decoy sequences are not equal, as is usually the case with randomly created protein sequence lists, one must first measure this proportion (5th-ranked, random-decoy), and then apply it to the condition containing correct identifications (top-ranked, random-decoy). See ref. 14 for further details
Once the background frequencies of target and decoy hits are determined (t, d), one can determine the multiplicative factor (f) used to estimate the total (target + decoy) number of incorrect identifications:
| (1) |
where d = 1 − t. For reversed decoy sequences in which target and decoy search spaces are nearly equal, it can be assumed that t and d are both equal to 0.5, and f is therefore equal to 2. One can then estimate the total number of incorrect peptides by doubling the number of observed decoy hits. If t and d are determined to be 0.37 and 0.63, respectively, as can be the case for randomly-created decoy sequences (14), then f should be 1.6.
3.4. False Positive Statistics
In order to fairly compare data sets collected in different laboratories, acquired on different instruments, searched with different search engines, and representing different biological samples, it is crucial that they meet similar false positive-related constraints. The first step in this process is to estimate the total number of correct PSMs in the entire data set. One way to do this is as follows:
Sort all peptide hits by score, descending.
Count how many target hits are greater than or equal to a given score
Count how many decoy hits are greater than or equal to a given score
- Estimate the number of correct hits (true positive, TP) from total (T) and decoy hits (d) greater than or equal to a given score:
(2) Estimate the total number of correct hits in the data set from the maximum value of TP observed across all score thresholds.
Given the total number of correct identifications in the data set, the number of identifications being considered, and how many of these are incorrect, one can populate the Venn diagram shown in Fig. 2. Given estimations of false positives (FP), true positives (TP), false negatives (FN), and true negatives (TN), one can generate the measurements shown in Table 1. Of these, precision and sensitivity are often the most useful for evaluating and comparing MS/MS data sets.
Fig. 2.
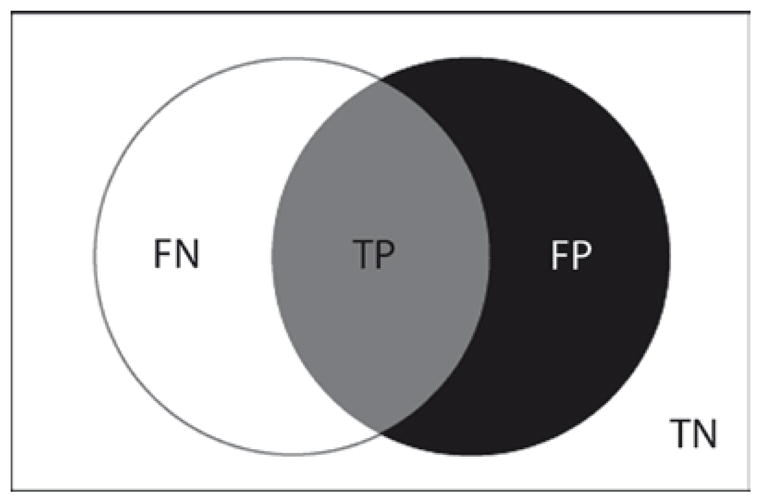
Venn diagram of basic measurements related to estimated false positive identifications. The total number of identifications are contained within the rectangle. All correct identifications are contained within the white circle. All identifications passing a given set of selection criteria (positive identifications) are contained within the black circle. The overlap between these circles are true positives (TP). False positive identifications (FP) are the remaining positive identifications, and false negative identifications (FN) are the remaining correct identifications that do not meet the selection criteria. True negatives (TN) are the incorrect identifications that are correctly classified as such by the selection criteria. This Venn diagram scheme is elaborated in Fig. 3
Table 1.
Measurements derived from target-decoy estimations of FP, TP, FN, TN
| Measurement | Formula | Description | |
|---|---|---|---|
| Precision |
|
Proportion of assignments passing selection criteria that are correct | |
| False discovery rate (FDR) | , 1 - precision | Proportion of assignments passing selection criteria that are incorrect | |
| Sensitivity |
|
Proportion of correct assignments passing selection criteria | |
| Specificity |
|
Proportion of all incorrect assignments excluded by selection criteria | |
| Accuracy |
|
Proportion of all assignments correctly classified by selection criteria |
3.5. Designing Filtering Criteria
For several years, large MS/MS data sets were subject to predefined, general filtering constraints to attempt to separate correct from incorrect peptide identifications. Often, these constraints were learned from a training data set consisting of known proteins, and then applied to experimental data sets that were often orders of magnitude larger than the training data set. Through target-decoy searching, it was determined that the proportion of false positive identifications that surpass standard criteria varies with individual data sets, as does the proportion of correct identifications that fail to meet them (i.e., false negatives). Thus, application of identical filtering criteria across multiple data sets does not necessarily yield data sets with comparable sensitivity or precision rates. It is often desirable, therefore, to design filtering criteria that can accommodate the diversity of LC-MS/MS analyses while yielding optimized, comparable error profiles.
Since decoy peptide matches and incorrect target matches have similar properties, one can examine decoy hits to learn how all incorrect hits can be segregated from correct hits in a sensitive and precise manner. This is fairly easy to accomplish when one considers a single monotonic score provided by the search engines, such as SEQUEST’s XCorr, Mascot’s Ion Score, and the E-value from OMSSA and X!Tandem, or composite scores, such as the Discriminant Score, returned by Peptide Prophet’s linear discriminant function (5):
Sort all peptide hits by score, descending.
Count how many target hits are greater than or equal to a given score
Count how many decoy hits are greater than or equal to a given score
- Estimate the total number of incorrect hits (false positive, FP) from observed decoy hits (d) greater than or equal to a given score:
(3) Calculate statistics related to FP for each given score threshold (see Subheading 3.5).
Select score threshold based on a desired statistic threshold.
Single scores are generally less able to sensitively separate correct from incorrect hits than consideration of multiple peptide measurements, such as mass accuracy, enzyme specificity, and alternate scoring methods. Composite scores are therefore superior to single scores, since they can incorporate these multiple lines of evidence that influence the likelihood that a peptide is correct. Another approach is to use the target-decoy strategy to examine multiple peptide measurements in a holistic fashion without condensing them into a single composite score. This is done by seeking an optimal (or several optimal) threshold combination(s) that maximizes the number of peptide identifications while minimizing the number of false positive identifications, or at least restricting them to a specified proportion of all positive identifications (Fig. 3). Evaluating and optimizing multiple candidate score threshold combinations can be tedious to perform manually; computational approaches for doing this have been described, however (22, 23).
Fig. 3.

Considering multiple selection criteria enhances accuracy. Selection criteria applied to score distributions (left) determine the form of the Venn diagrams (right). Venn diagram shapes and colors correspond with those in Fig. 2. (a) Distribution of FP and TP hits sorted by an arbitrary score. When no score criteria are applied, all selected correct identifications are denoted in grey circle, and all selected incorrect identifications are denoted in black rectangle. (b) Application of a single score threshold, which excludes most incorrect identifications (lighter region), can yield an acceptable precision rate, but yields sub-optimal sensitivity. (c) Considering two scores allows for greater separation between correct and incorrect identifications. The distribution of incorrect identifications is indicated by the distribution of decoy hits. Application of global criteria that excludes most decoy hits in two score dimensions (lighter region) provides greater sensitivity than one score alone. (d) Designing selection criteria that take into account numerous peptide measurements, such as mass accuracy, charge, enzymatic specificity, and peptides per protein, can yield far greater sensitivity while maintaining acceptable precision
3.6. Report Statistics for Filtered Data Set
Increasingly, journals are requiring an assessment of data quality when publishing MS/MS results (24–26). As previously stated, the most useful measurements are usually precision (or FDR) and sensitivity. Although it is convenient to include decoy hits in a data set during analysis (see Note 4), decoy hits should not contribute to the final tally of incorrect hits since they can be easily recognized and removed. Thus, the reported number of FP and corresponding precision rate should be:
| (4) |
| (5) |
It must be stressed that the above calculations apply to the aggregate of all identifications that meet or exceed a given set of filtering criteria. The final precision rate represents the proportion of the final data set that is likely to be correct; it does not indicate the likelihood of any particular identification of being correct (see Note 5).
These statistics may also be applied at the protein level. However, protein inference from multiple peptides poses additional challenges beyond the scope of this chapter (see Note 6). Protein precision is often worse than the precision measured from PSMs. This usually can be attributed to proteins that are incorrectly identified by just one peptide. In contrast, proteins identified by multiple peptides are usually correct. Thus, correct peptide identifications map to fewer proteins than incorrect peptides, reducing the final protein precision. This situation can be addressed by paying specific attention to single peptide identifications (see Note 7).
Acknowledgments
This work was supported in part by National Institutes of Health (NIH) GM67945 and HG00041 (S.P.G.).
Footnotes
| (6) |
| (7) |
| (8) |
A simple Perl script for generating a target-reversed decoy sequence list:
$NUM_COL = 80; ## set the column width of output file
$infile = shift; ## grab input sequence file name from command line
$outfile = “REV”. $infile; ## name output file, prepend with “REV”
open (IN, $infile);
open (OUT, >$outfile);
$/ = undef; ## allow entire input sequence file to be read into memory
my $text = <IN>; ## read input sequence file into memory
print OUT $text; ## output sequence file into new decoy sequence file
my @proteins = split (/>/, $text); ## put all input sequences into an array
for my $protein (@proteins) { ## evaluate each input sequence individually
$protein =~ s/(^.*)\n//m; ## match and remove the first descriptive line of
## the FATA-formatted protein
my $name = $1; ## remember the name of the input sequence
print OUT “>#REV#$name\n”; ## prepend with #REV#; a # will help make the
## protein stand out in a list
$protein =~ s/\n//gm; ## remove newline characters from sequence
$protein = reverse($protein); ## reverse the sequence
while (length ($protein) > $NUM_C0L) { ## loop to print sequence with set number of cols
## per line
$protein =~ s/(.{$NUM_C0L})//;
my $line = $1;
print OUT “$line\n”;
}
print OUT “$protein\n”; ## print last portion of reversed protein
}
close (IN);
close (OUT);
print “done\n”;
Several groups recommend first searching MS/MS spectra against decoy sequences to derive a null distribution of scores, and then basing filtering criteria on the null distribution. Furthermore, by restricting the target database search to just target sequences, scores that are dependent on the search space will often be greater for correct identifications in comparison to the combined target-decoy search. While the practice of separate searches is reasonable in principle, it creates a variety of situations that must be accounted for in the final analysis. These include, but are not limited to:
(a) Correct/incorrect PSM noncompetition: A high-quality MS/MS spectrum will often receive an elevated score compared to a low-quality spectrum, even if both corresponding PSMs are incorrect. When searching against a concatenated target-decoy sequence list, a correct target PSM necessarily competes with an incorrect decoy PSM, and is then returned by the algorithm. Under the separate searches paradigm, high-scoring decoy PSMs will indicate setting an exceptionally stringent filtering threshold that undermines sensitivity, unless these PSMs are secondarily compared to their target PSM counterparts following the search.
(b) Imbalanced incorrect target and decoy numbers: Typical search results consist of a mixture of correct and incorrect PSMs. Under the concatenated target-decoy paradigm, incorrect PSMs are distributed between target and decoy sequences according to their background frequency (i.e., 1:1 for reversed sequences). When searching target and decoy sequences separately, decoy PSMs will necessarily outnumber incorrect PSMs, since spectra that can be correctly assigned to target sequences will be matched to decoy sequences. For example, if 20% of all spectra are correctly assigned, the proportion of incorrect target to incorrect decoy will be 0.8:1, even if the underlying target and decoy sequences were equal in number. Further complicating matters, the larger decoy distribution presents the opportunity for them to achieve a wider range of scores, inappropriately suggesting more stringent filtering thresholds.
Even after a set of filtering criteria have been arrived at, it is often useful to leave decoy PSMs mixed among the target ones. Should one choose to revisit the data analysis, one can derive further filtering/selection criteria involving additional parameters not considered in the original analysis.
False positive statistics applied to entire data sets can obscure scoring data, which indicate that some PSMs are assigned with greater confidence than others. Particularly with very large data sets with a set precision threshold, it is possible that a small number of PSMs with a very low likelihood of being correct will be included. Recently, it has been proposed to restrict PSM selection based on the likelihood that a particular identification is correct (16, 23, 28, 29). This can be a highly useful practice, particularly when there is little tolerance for error, such as the submission of PSMs to a reference data set. However, many research applications are tolerant of some error, since it can allow for much greater sensitivity. A data set composed of PSMs with a minimum likelihood of being correct of 0.99, for example, may have an overall precision rate of 0.999, but nearly half the sensitivity of a data set restricted to have a precision of 0.99.
Although peptide identifications can be correct, it is possible to incorrectly infer the proteins that gave rise to them, due to sequence homologies. These proteins should be considered to be false positives, since the identified proteins were not actually present in the experimental sample. The target-decoy system cannot be used to estimate this source of error. Programs, such as Protein Prophet (30), can be used to formally identify the protein(s) that are most likely given the observed peptides. However, it is worth noting that despite some protein ambiguity, often, peptides restrict the protein identifications to a narrow group, often consisting of highly related isoforms.
Proteins identified by single peptides (“one-hit-wonders”) represent a special class of peptide and protein identifications. It is generally true that the vast majority of incorrect peptide identifications are in this category. As a result, the precision rate measured at the protein level is usually less than that observed at the protein level. It is often tempting, therefore to remove single peptide identifications from a final data set. While this practice certainly improves the precision rate at the protein level, it is usually accompanied by a substantial loss in sensitivity. Often, more than half of all correct peptide identifications fall into the one-hit-wonder category. Rather than removing these PSMs from the final data set, a more measured approach would be to apply filtering criteria tailored to just this subset.
References
- 1.Eng JK, McCormack AL, Yates JRI. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom. 1994;5:976–89. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 2.Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–67. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 3.Geer LY, Markey SP, Kowalak JA, Wagner L, Xu M, Maynard DM, Yang X, Shi W, Bryant SH. Open mass spectrometry search algorithm. Proteome Res. 2004;3:958–64. doi: 10.1021/pr0499491. [DOI] [PubMed] [Google Scholar]
- 4.Craig R, Beavis RC. TANDEM: matching proteins with tandem mass spectra. Bioinformatics. 2004;20:1466–7. doi: 10.1093/bioinformatics/bth092. [DOI] [PubMed] [Google Scholar]
- 5.Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem. 2002;74:5383–92. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 6.Deutsch EW, Lam H, Aebersold R. PeptideAtlas: a resource for target selection for emerging targeted proteomics workflows. EMBO Rep. 2008;9:429–34. doi: 10.1038/embor.2008.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prince JT, Carlson MW, Wang R, Lu P, Marcotte EM. The need for a public proteomics repository. Nat Biotechnol. 2004;22:471–2. doi: 10.1038/nbt0404-471. [DOI] [PubMed] [Google Scholar]
- 8.Kersey PJ, Duarte J, Williams A, Karavidopoulou Y, Birney E, Apweiler R. The International Protein Index: an integrated database for proteomics experi ments. Proteomics. 2004;4:1985–8. doi: 10.1002/pmic.200300721. [DOI] [PubMed] [Google Scholar]
- 9.The universal protein resource (UniProt) Nucleic Acids Res. 2008;36:D190–5. doi: 10.1093/nar/gkm895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bakalarski CE, Haas W, Dephoure NE, Gygi SP. The effects of mass accuracy, data acquisition speed, and search algorithm choice on peptide identification rates in phosphoproteomics. Anal Bioanal Chem. 2007;389:1409–19. doi: 10.1007/s00216-007-1563-x. [DOI] [PubMed] [Google Scholar]
- 11.Balgley BM, Laudeman T, Yang L, Song T, Lee CS. Comparative evalu ation of tandem MS search algorithms using target-decoy search strategy. Mol Cell Proteomics. 2007;6:1599–608. doi: 10.1074/mcp.M600469-MCP200. [DOI] [PubMed] [Google Scholar]
- 12.Elias JE, Haas W, Faherty BK, Gygi SP. Comparative evaluation of mass spectrometry platforms used in large scale proteomics investigations. Nat Methods. 2005;2:667–75. doi: 10.1038/nmeth785. [DOI] [PubMed] [Google Scholar]
- 13.Sadygov RG, Cociorva D, Yates JR., III Large-scale database searching using tandem mass spectra: looking up the answer in the back of the book. Nat Methods. 2004;1:195–202. doi: 10.1038/nmeth725. [DOI] [PubMed] [Google Scholar]
- 14.Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods. 2007;4:207–14. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- 15.Higdon R, Hogan JM, Van Belle G, Kolker E. Randomized sequence databases for tandem mass spectrometry peptide and protein identification. OMICS. 2005;9:364–79. doi: 10.1089/omi.2005.9.364. [DOI] [PubMed] [Google Scholar]
- 16.Kall L, Storey JD, MacCoss MJ, Noble WS. Assigning significance to peptides identified by tandem mass spectrometry using decoy databases. J Proteome Res. 2008;7:29–34. doi: 10.1021/pr700600n. [DOI] [PubMed] [Google Scholar]
- 17.Moore RE, Young MK, Lee TD. Qscore: an algorithm for evaluating SEQUEST database search results. J Am Soc Mass Spectrom. 2002;13:378–86. doi: 10.1016/S1044-0305(02)00352-5. [DOI] [PubMed] [Google Scholar]
- 18.Peng J, Elias JE, Thoreen CC, Licklider LJ, Gygi SP. Evaluation of multidimensional chromatography coupled with tandem mass spectrometry (LC/LC-MS/MS) for large-scale protein analysis: the yeast proteome. J Proteome Res. 2003;2:43–50. doi: 10.1021/pr025556v. [DOI] [PubMed] [Google Scholar]
- 19.Haas W, Faherty BK, Gerber SA, Elias JE, Beausoleil SA, Bakalarski CE, Li X, Villen J, Gygi SP. Optimization and use of peptide mass measurement accuracy in shotgun proteomics. Mol Cell Proteomics. 2006;5:1326–37. doi: 10.1074/mcp.M500339-MCP200. [DOI] [PubMed] [Google Scholar]
- 20.Beausoleil SA, Villen J, Gerber SA, Rush J, Gygi SP. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat Biotechnol. 2006;24:1285–92. doi: 10.1038/nbt1240. [DOI] [PubMed] [Google Scholar]
- 21.Elias JE, Gibbons FD, King OD, Roth FP, Gygi SP. Intensity-based protein identification by machine learning from a library of tandem mass spectra. Nat Biotechnol. 2004;22:214–19. doi: 10.1038/nbt930. [DOI] [PubMed] [Google Scholar]
- 22.Jiang X, Han G, Ye M, Zou H. Optimization of filtering criterion for SEQUEST database searching to improve proteome coverage in shotgun proteomics. BMC Bioinformatics. 2007;8:323. doi: 10.1186/1471-2105-8-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kall L, Canterbury JD, Weston J, Noble WS, MacCoss MJ. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat Methods. 2007;4:923–5. doi: 10.1038/nmeth1113. [DOI] [PubMed] [Google Scholar]
- 24.Binz PA, Barkovich R, Beavis RC, Creasy D, Horn DM, Julian RK, Jr, Seymour SL, Taylor CF, Vandenbrouck Y. Guidelines for reporting the use of mass spectrometry informatics in proteomics. Nat Biotechnol. 2008;26:862. doi: 10.1038/nbt0808-862. [DOI] [PubMed] [Google Scholar]
- 25.Bradshaw RA, Burlingame AL, Carr S, Aebersold R. Reporting protein identification data: the next generation of guidelines. Mol Cell Proteomics. 2006;5:787–8. doi: 10.1074/mcp.E600005-MCP200. [DOI] [PubMed] [Google Scholar]
- 26.Taylor CF. Minimum reporting requirements for proteomics: a MIAPE primer. Proteomics. 2006;6(Suppl 2):39–44. doi: 10.1002/pmic.200600549. [DOI] [PubMed] [Google Scholar]
- 27.Huttlin EL, Hegeman AD, Harms AC, Sussman MR. Prediction of error associated with false-positive rate determination for peptide identification in large-scale proteomics experiments using a combined reverse and forward peptide sequence database strategy. J Proteome Res. 2007;6:392–98. doi: 10.1021/pr0603194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kall L, Storey JD, MacCoss MJ, Noble WS. Posterior error probabilities and false discovery rates: two sides of the same coin. J Proteome Res. 2008;7:40–4. doi: 10.1021/pr700739d. [DOI] [PubMed] [Google Scholar]
- 29.Tang WH, Shilov IV, Seymour SL. Nonlinear fitting method for determining local false discovery rates from decoy database searches. J Proteome Res. 2008;7(9):3661–7. doi: 10.1021/pr070492f. [DOI] [PubMed] [Google Scholar]
- 30.Nesvizhskii AI, Keller A, Kolker E, Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem. 2003;75:4646–58. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]