

Abstract
Thymic stromal lymphopoietin (TSLP) is an IL-7 related cytokine, produced by epithelial cells, that has been linked to atopic dermatitis and asthma; however, it remains unclear how TSLP shapes the adaptive immune response that causes these allergic disorders. Here we demonstrate a role for TSLP in a Th2 model of contact hypersensitivity (CHS) in mice. TSLP is required for the development of Th2-type CHS induced by the hapten fluorescein isothiocyanate (FITC) in combination with the sensitizing agent dibutyl phthalate (DBP). TSLPR−/− mice exhibited a dramatically reduced response, including markedly reduced local infiltration by eosinophils, Th2 cytokine production, and serum IgE levels, following FITC sensitization and challenge. The reduced response by TSLPR−/− mice is likely due to decreased frequency, and reduced T cell stimulatory function, of skin-derived antigen-bearing FITC+CD11c+ dendritic cells (DCs) in draining lymph nodes following FITC sensitization. These data suggest that skin-derived DCs are direct or indirect targets of TSLP in the development of type-2 immune responses in the skin, where TSLP drives their maturation, accumulation in skin draining lymph nodes, and ability to induce proliferation of naïve allergen-specific T cells.
Keywords: Th1/Th2 Cells, Inflammation, Allergy, Dendritic Cells, Rodent
INTRODUCTION
Thymic stromal lymphopoietin (TSLP) is an epithelial-derived cytokine expressed primarily in the lung, skin, and intestine, in response to inflammation, tissue damage, or Toll-like receptor ligation (1–4). The TSLP receptor complex is composed of the IL-7Rα chain and a TSLP-specific receptor chain (referred to as TSLPR), and is expressed primarily on hematopoietic cells (1,5). Several cell types respond to TSLP in both humans and mice, including DCs, CD4 T cells, macrophages, and B cells (1,6,7). TSLP has been shown to act directly on immature DCs in vitro leading to maturation and the upregulation of costimulatory molecules, and the Th2-associated chemokines CCL17 and CCL22 (1,2). TSLP-treated human DCs induce a unique inflammatory Th2 cytokine profile in CD4 T cells, specifically production of IL-4, IL-5, IL-13 and TNFα (1,2). In addition, it has been shown that TSLP can act directly on CD4 T cells in vitro to drive IL-4 production (6,8). However, much of this data was generated from in vitro studies, and there is very little known regarding the in vivo cellular targets of TSLP.
Consistent with its ability to drive Th2-type responses in vitro, TSLP has been suggested to play a role in Th2-type allergic responses in humans. Specifically, TSLP has been found at elevated levels in the lesional skin of atopic dermatitis (AD) patients and the lungs of asthmatics (2,9). Furthermore, TSLP has been shown to be critical in a mouse model of antigen-driven airway inflammation (10,11), and constitutive overexpression of TSLP in the lung leads to a spontaneous airway inflammatory disease that closely resembles human asthma (10). In addition, we and others have demonstrated a role for TSLP in the development of a chronic form of dermatitis in mice, that closely resembles human atopic dermatitis (12,13). In these models, elevated TSLP due to overexpression of a keratinocyte specific TSLP transgene, or conditional ablation of RXRα and β in mouse epidermal keratinocytes, led to the development of spontaneous dermatitis characterized by mast cell hyperplasia, eosinophil, and lymphocyte infiltration, Th2 cytokine production, and increased serum IgE (12,13). Interestingly, T cells are not required for the development of AD when TSLP is conditionally overexpressed by keratinocytes, or expression is induced by topical treatment with vitamin D3 analogs, most likely due to chronic activation of innate immune cells by the constitutive presence of elevated TSLP (12,14). In contrast, it has recently been shown that intradermal administration of TSLP induces inflammation in a manner that requires an adaptive immune response, as RAG−/− mice are resistant to inflammation in this model (15). In addition, administration of exogenous TSLP and foreign antigen intranasally results in the development of asthma-like disease in a CD4 T cell dependent manner (16). However, the nature of the TSLP-responsive cell populations in each model required to induce Th2 inflammation remains unclear.
Contact hypersensitivity (CHS) develops as a consequence of epicutaneous contact with an allergen (sensitization) resulting in the development of immunologic memory and enhanced inflammatory skin responses at sites of re-exposure to allergen (17). Haptens, electrophilic small molecular compounds, and proteins serve as allergens in CHS assays. The type of CHS response that occurs, Th1- versus Th2-type, is allergen and adjuvant-dependent. The hapten fluorescein isothiocyanate (FITC), in combination with dibutyl phthalate (DBP), induces a Th2-type response in mice (18–20). FITC sensitized mice develop swelling at the site of challenge and increased serum IgE, both of which are STAT6 dependent (18,21). Ear swelling is largely dependent on mast cells, CD4 T cells, MHC class II, IL-4 and IL-5 and the cellular infiltrate is primarily eosinophilic (18).
AD in humans is considered an allergic disease, driven by initial sensitization to environmental antigens through skin with altered epidermal barrier function. Increased penetration of microbial products through barrier deficient skin in AD may activate innate receptors and induce proinflammatory signals contributing to sensitization. Alternatively, defective barrier function may directly lead to the activation of innate immune cells in the skin. This results in the development of Th2 effector cells, formation of immunologic memory, and an inflammatory cellular response with acute and chronic phases/relapses (22). These factors, together with the inability to clearly distinguish between sensitization and challenge phases, make it difficult to characterize the role that TSLP plays in the immune mechanisms underlying the initiation of AD. Because AD shares many features with CHS to FITC, understanding the role TSLP plays in this model of Th2-type CHS may elucidate features involved in the development of AD and other allergic and non-allergic Th2-type disorders.
The role of TSLP in asthma and chronic dermatitis models and its association with Th2 allergic diseases led us to hypothesize that it may also play a significant role in a Th2 model of CHS. Here we demonstrate that the presence of functional TSLP receptor (TSLPR) is required for the development of Th2-type CHS to the hapten FITC. This defect in the development of Th2-type CHS response in TSLPR deficient (TSLPR−/−) mice is due to multiple factors including decreased migration of skin-derived DCs to lymph nodes post-sensitization, as well as diminished ability of those DCs to drive antigen-specific T cell proliferation. This is likely the mechanism for the observed reduction in the overall CHS response to FITC by TSLPR−/− mice.
MATERIALS & METHODS
Mice
6–12 week old wild-type Balb/c mice were purchased from Taconic or Charles River (l’Arbresle, France). TSLPR−/− mice (23) were backcrossed to a Balb/c background for eight generations, or more. All animals were housed in the Benaroya Research Institute vivarium, the animal facility at the Centre Médicale Universitaire in Geneva, or the Amgen vivarium in specific pathogen free conditions, and all experiments were done with approval from the Benaroya Research Institute Institutional Animal Care Committee; the State Veterinarian’s Office (Geneva, Switzerland) and performed according to the Federal Law (Switzerland); or the Amgen Institutional Animal Care Committee.
Contact Hypersensitivity
CHS was induced using the hapten fluorescein isothiocyanate (FITC, Sigma Aldrich). FITC was resuspended in 1:1 acetone and DBP to a concentration of 0.5%. Mice were sensitized with 100 μl of 0.5% FITC or vehicle (1:1 DBP/acetone) on days 0 and 2, or days 0 and 1, on the shaven or unshaven back or abdomen. On day 6, baseline ear thickness was measured with calipers followed by challenging the mice on one ear with 20μl of 0.5% FITC on one ear and vehicle on the contralateral ear. On day 7, 24 H post challenge, ear thickness was measured to calculate the change in ear thickness.
Skin DC Migration
Skin-derived DC migration was measured using FITC or TRITC reagents. 0.5% FITC or 0.1% TRITC diluted in acetone/DBP was applied to shaved (FITC) or unshaven (TRITC) abdomen of Balb/c and TSLPR−/− mice. 24 H later mice were sacrificed and inguinal, axillary, brachial, and auricular lymph nodes were removed and processed using Collagenase D (Roche) or mechanically dissociated between frosted microscope slides. Low density cells were enriched for by running lymph node cell suspensions over a 60% Percoll (GE Healthcare) gradient. Cells were then stained for flow cytometric analysis.
TSLP Antibody Treatment
For CHS experiments, mice were treated with PBS, anti-TSLP antibody (clone M702), or isotype control (rat IgG2a) antibody 200 μg/mouse intravenously on days -1 and 6, and 100 μg/mouse IP on days 0 and 7. For migration experiments, Mice were treated with anti-TSLP antibody or isotype control antibody 200 μg/mouse IP 24 hours prior to FITC painting, at the time of FITC painting, and 8 hours after FITC painting.
Histology
Whole ears were excised at the base and fixed overnight in 10% formalin at room temperature, sectioned, and stained with hematoxylin and eosin.
Flow Cytometry
Single cell suspensions were prepared from the cutaneous and control draining lymph nodes using collagenase D or mechanical dissociation. Cells were stained with the following fluorochrome conjugated antibodies (clone, manufacturer): anti-CD11c (N418, eBioscience) conjugated to Pacific Blue, anti-CD11b (M1/70, eBioscience) conjugated to allophyocyanin, anti-MHC II (M5/114.15.2, eBioscience) conjugated to Alexa Fluor 700, and anti-CD8 (53-6.7, BD Biosciences) conjugated to PE or FITC, anti-CD40 (1C10, eBioscience) conjugated to allophyocyanin, anti-CD80 (16-10A1, eBioscience) conjugated to PE, anti-CD86 (PO3, eBioscience) conjugated to PE, and anti-OX40L (RM134L, eBioscience) conjugated to PE. Cells were also stained for intracellular CD207 conjugated to AlexaFluor488 (Dendritics). Samples were analyzed with BD LSR II or BD FACSCalibur flow cytometry.
Real-Time PCR
Ears were painted with 20 μl of vehicle (acetone/DBP left ear), 0.5% FITC in acetone or 0.5% FITC in acetone/DBP on both the ventral and dorsal side of the ear or left untreated. At different time points mice were sacrificed and frozen ears were cut in thin slices using a cryostat. The slices were snap-frozen in TRIzol reagent (Gibco BRL) and total RNA was extracted with chloroform (Sigma Aldrich). RNA integrity and quantity were assessed using RNA 6000 nanochips with an Agilent 2100 bioanalyzer (Agilent Technologies). RT-PCR assays were performed with 1 μg total RNA. Total RNA was reverse transcribed using random hexamers and SuperScript II reverse transcriptase (Invitrogen Life Technologies) according to the manufacturer’s instructions, and 1/60 cDNA template was used as template for each PCR. cDNA was PCR amplified in a 7900HT Sequence Detection Systems (Applied Biosystems) using Power SYBR Green PCR master mix (Applied Biosystems). Raw threshold-cycle (Ct) values were obtained from Sequence Detection Systems 2.2 software (Applied Biosystems). Relative quantities (RQs) were calculated with the formula RQ=E−CT, using efficiencies calculated for each run with the Data Analysis for Real-Time PCR (DART-PCR) algorithm, as described (24). A mean quantity was calculated from triplicate PCR for each sample and this quantity was normalized to three similarly measured quantity of normalization genes (EEF1A1, HPRT and Trf1R) as described (25). Normalized quantities were averaged for three replicates for each data point. The highest normalized relative quantity was arbitrarily designated as value of 1.0. Fold changes were calculated from the quotient of means of these normalized quantities. PAM212 keratinocytes were seeded in tissue culture plates overnight to allow for adherence. DBP was added and cells were lysed at various time points post treatment. RNA was prepared with TRIzol (Invitrogen) following manufacturer’s protocol. cDNA was prepared using the SuperScript II reverse transcriptase kit (Invitrogen) from 1 μg of RNA. Relative TSLP expression was calculated using the delta delta Ct method, after normalization to GAPDH expression (26). Primer sequences for detecting TSLP expression by PAM212 cells, 5′-TGAGAGCAAGCCAGCTTGTC-3′ and 5′-GTGCCATTTCCTGAGTACCG-3′. Primer sequences for detecting GAPDH expression by PAM212 cells, 5′-TGCACCACCAACTGCTTAG-3′ and 5′-GGATGCAGGGATGATGTTC-3′. Primer sequences for detecting T helper cytokine expression: 5′-TTTCAGTGATGTGGACTTGGACTC-3′ and 5′-AAGAACACCACAGAGAGTGAGCTC-3′ for IL-4; 5′-AGCAAGAGATCCTGGTCCTGAA-3′ and 5′-CATCTTCTCGACCCTGAAAGTGA-3′ for IL-17A; and 5′-CTGCAGAGCCAGATTATCTC-3′ and 5′-CCTGTGGGTTGTTGACCTCA-3′ for IFNγ.
Tissue Lysate ELISA
Ears were treated with acetone, 1:1 acetone/DBP, FITC in 1:1 acetone/DBP, FITC in acetone, or left untreated. 24 H later mice were euthanized and ear tissue was excised and placed in T-PER tissue protein extraction reagent (Thermo Scientific). Tissue was homogenized and protein was quantified using a Nanodrop spectrophotometer at 280nm. 100 ug of total protein per sample was loaded per well using the R&D Systems murine TSLP ELISA kit following the manufacturer’s protocol. Each mouse was treated the same on both ears and TSLP expression in each ear was analyzed independently.
Serum IgE ELISA
Sera were obtained before sensitization and 2–3 days after challenge with FITC and stored at −20°C. MaxiSorp microtiter plates (NUNC) were coated with 100 ng/well anti-mouse IgE (BD Pharmingen) antibody during 3 H at 37°C. The plates were washed 3 times with PBS containing 0.5% Tween and blocked overnight at RT with 200 μl/well of a solution of polyvinylpyrrolidon K25 (PVP) (Fluka). Plates were washed again and purified IgE (BD Pharmingen) (standard curve) or sera diluted 1:10 in PVP were incubated in duplicates for 3 H at 37°C. Plates were washed and a biotinylated anti-mouse IgE diluted 1:1000 in PVP was added for 2 H at 37°C. Plates were washed and incubated for 1 h at 37°C in presence of streptavidin horseradish peroxidase (Sigma Aldrich). Finally, substrate (orthophenylenediamine) was added for 30 minutes at room temperature in the dark. The reaction was stopped with 2N H2SO4. Optical density was measured at 490nm.
EPO Activity
EPO activity was determined as described (27). Eight mm punch biopsies of vehicle or FITC treated ears were frozen in liquid nitrogen and ground to fine powder. This powder was homogenized in 0.05 M Tris HCl, pH 8 and 0.1% Triton X-100 using a sonicator. The samples were centrifuged and 20-fold diluted supernatants were incubated for 1 H at RT in the dark with 10mM o-phenyldiamine in 0.05 M Tris HCL and 4mM hydrogen peroxide. The reaction was stopped by addition of 2 N H2SO4. Optical density was measured at 490 nm.
Cytokine ELISPOT and ELISA
ELISPOT or ELISA plates (Millipore) were coated over night at 4°C with 500 ng/well purified anti-mouse IL-4, IL-5, IL-17, or IFNγ antibodies. The plates were washed and blocked for 1 H at 37°C with RPMI 1640 (Gibco/Invitrogen) containing 10% FBS and β-mercaptoethanol. Pooled superficial inguinal, axillary and brachial LN cells were resuspended in RPMI supplemented with penicillin, streptomycin, L-glutamine, polymyxin, β-mercaptoethanol and 10% FBS, added to the precoated wells and incubated for 4 days at 37°C in presence of 20 ng/ml PMA and 1μg/ml ionomycin or medium alone as negative control. For cytokine ELISAs day 3 supernatants from DC-T cell cocultures were added for 2 H at RT. Biotin-labelled antibodies against IL-4, IL-5, IL-17, or IFNγ at 0.1μg/well were added for overnight (1 H for ELISA) incubation at 4°C followed by streptavidin horseradish peroxidase 1:1000 for 2 hours (0.5 H for ELISA). 100μl/well amino-ethyl-carbazole was added for 20 minutes at room temperature in the dark and the spots were counted using the KS ELISPOT 4.2.1 Software (Zeiss) and expressed as cell-forming units (CFU) per 106 cells. 1-Step Ultra TMB-ELISA substrate (Pierce) was used to develop ELISAs.
Proliferation Assay
WT and TSLPR−/− mice were sensitized with 200 μl, or 20 μl 0.5% FITC 1:1 acetone/DBP on the shaven abdomen or ear, respectively. Mice were sacrificed 24 H post FITC treatment, inguinal, axillary, brachial, and retroauricular lymph nodes were harvested, digested, and enriched as discussed previously. Low density cells were stained with anti-CD11c conjugated to allophyocyanin and sorted on a BD FACSVantage cell sorter. 2×103/well of FITC+ or FITC-CD11c+ cells were cultured with 5 μg/ml class II restricted OVA323-339 peptide for 24 H at 37°C in 50 μl complete RPMI in a 96-well plate. 5×104 DO11.10 transgenic T cells in 50 μl of complete RPMI were added to each well. Additional conditions included FITC+ or FITC-CD11c+ DC with DO11.10 T cells in the absence of peptide, FITC+CD11c+ DCs alone, and DO11.10 T cells alone. After 72 H of culture, each well was pulsed with 1 μCi of tritiated thymidine for 12 H, followed by measurement of tritiated thymidine incorporation.
Statistical Analysis
Statistical analyses were carried out as noted within figure legends.
RESULTS
Dibutyl Phthalate Induces TSLP Expression
TSLP is expressed by epithelial cells, in an NFκB-dependent manner, following exposure to various stimuli, including tissue injury or disruption, TLR ligation, and inflammatory cytokines (1–4,28). Given that the hapten FITC in combination with DBP initiates a Th2-type CHS response, and that TSLP has been shown to be an important factor in type-2 immune responses, we initially measured TSLP expression in the skin after sensitization with FITC. Epicutaneous application of FITC (in 1:1 acetone/DBP) or vehicle alone (1:1 acetone/DBP) resulted in significant upregulation of TSLP mRNA and protein in the skin compared with controls (acetone alone or FITC in acetone) (Fig. 1A and B). These data demonstrate that DBP alone is capable of inducing expression of TSLP in the skin in the absence of FITC. However, FITC appears to have an additive effect on TSLP protein levels when DBP is present, as treatment with FITC in combination with vehicle induced significantly greater levels of TSLP protein than vehicle alone or FITC in acetone (Fig. 1B). Different phthalate esters variably mobilize DCs to draining lymph nodes correlating with their capacity to sensitize to FITC, while FITC in acetone alone does not mobilize DCs or induce CHS (29). This demonstrates that DBP is sufficient for TSLP upregulation in skin and suggests that TSLP is one of the endogenous mediators of the adjuvant effect of DBP. It is, however, not excluded that DBP also acts directly on skin-resident DCs (30).
Figure 1. DBP induces expression of TSLP in vivo and in vitro by a keratinocyte cell line.

(a) Relative TSLP mRNA expression in ears following sensitization with 0.5% FITC in vehicle (acetone/dibutyl phthalate), vehicle alone, 0.5% FITC in acetone, acetone alone, or untreated ears. The leftmost two columns show TSLP mRNA induction after treatment of ears with acetone alone (gray) or 0.5% FITC in acetone (black). Significant upregulation of TSLP message 24 H post 1:1 acetone/DBP (vehicle) or FITC (diluted in vehicle) treatment relative to untreated or FITC in acetone control (**P<0.01, Wilcoxon test, n≥3 mice per group). (b) TSLP protein levels 24 H post treatment with acetone, 0.5% FITC in acetone, 0.5% FITC in vehicle, vehicle alone, or left untreated as detected by loading 100 ug of total protein per sample in a tissue lysate ELISA (***P<0.001, one-way ANOVA, n≥5 ears per group). (c) TSLP expression by DBP treated PAM212 keratinocytes. PAM212 cells were cultured in the presence of DBP for the time shown. Expression is relative to the 2H untreated sample and is normalized to GAPDH expression (representative of three independent experiments).
It has previously been shown that keratinocyte-derived TSLP is required for the development of an AD-like disease in mice (31). Therefore, we sought to determine whether DBP could directly induce TSLP expression by keratinocytes by treating the PAM212 murine keratinocyte cell line with DBP in vitro. We found that treatment of the PAM212 cells with DBP resulted in the induction of TSLP expression (Fig. 1D), suggesting DBP acts directly on keratinocytes in the skin to induce TSLP expression. Since TSLP has been shown to play a role in Th2 allergic responses, we sought to determine whether there was a requirement for the cytokine during a Th2 response in the skin.
TSLP-TSLPR Interactions are Required for Th2-type Contact Hypersensitivity
The finding that TSLP expression was induced by DBP suggested the cytokine may play a role in the Th2-type CHS response to FITC (always diluted in 1:1 acetone DBP, unless otherwise stated). To address this question wild-type (WT) and TSLPR−/− mice were sensitized and challenged with FITC or vehicle. WT mice displayed a significantly greater increase in ear thickness compared to all controls (Fig. 2A). Vehicle sensitized animals did not experience a significant increase in ear thickness (data not shown). Swelling was observed as early as 12 H after challenge, and was sustained up to 48 H post challenge (Fig. 2A). In contrast, TSLPR−/−animals sensitized and challenged with FITC exhibited markedly reduced ear swelling (Fig. 2A). These data demonstrate a requirement for TSLP responsiveness for the CHS response to FITC.
Figure 2. Th2-Type contact hypersensitivity is significantly reduced in TSLPR−/− mice.

(a) Ear swelling time course of FITC sensitized WT and TSLPR−/− mice challenged with either FITC or vehicle. Mice were sensitized with 0.5% FITC on days 0 and 2, baseline ear thickness was measured on day 6 followed by a challenge with 0.5% FITC on the right ear and vehicle on the left. Change in ear thickness was calculated by subtracting baseline ear thickness from the ear thickness measured at each time point post challenge. FITC sensitized and challenged WT mice experience significantly greater ear swelling starting at twelve hours up to forty-eight hours compared to TSLPR−/− mice under the same treatment (*P<0.05, **P<0.01, ***P<0.001, two-way ANOVA test, n=3 mice per group). (b and c) Hematoxylin and eosin stained ear sections fixed twenty-four hours post challenge from WT and TSLPR−/− mice sensitized and challenged with FITC. (b) Cellular infiltration of FITC challenged ear of FITC-sensitized WT and TSLPR−/−mice. (c) Epidermal lesions are a hallmark of FITC CHS observed in WT mice (left panel) and absent in TSLPR−/− mice. (d) Significantly reduced EPO activity in vehicle and FITC challenged TSLPR−/− ears twenty-four hours post challenge of FITC sensitized mice. EPO activity of WT mice sensitized and challenged with FITC set to 100% (*P<0.05, **P<0.01, Wilcoxon test).
When examined histologically, ears from FITC sensitized and challenged WT mice displayed a significant cellular infiltrate consisting primarily of eosinophils that was dramatically reduced in ears from similarly treated TSLPR−/− mice (Fig 2B and C). FITC sensitized and challenged WT mice exhibited pronounced epidermal lesions and thickening which were substantially reduced in TSLPR−/− mice (Fig. 2B and C). These data were further supported by the significant reduction in eosinophil peroxidase activity measured in the ears of TSLPR−/−, as compared with WT mice, after FITC sensitization and challenge (Fig. 2D). However, neutrophil infiltration, as measured by myeloperoxidase activity, was equivalent between WT and TSLPR−/− mice (data not shown). Cellular infiltration was absent in vehicle sensitized or challenged mice of both genotypes (data not shown). Taken together, these data demonstrate a requirement for TSLP signals in the development of a Th2-type CHS inflammatory response.
Diminished Th2 Responses in FITC Sensitized TSLPR−/− Mice
Consistent with the reduced infiltration and inflammatory response, the number of IL-4 and IL-5 producing cells was significantly reduced in the draining lymph nodes of the FITC sensitized TSLPR−/− mice, as compared to WT controls (Fig. 3A). FITC sensitized WT mice also displayed significantly higher serum IgE levels compared to TSLPR−/− mice when measured after FITC challenge, as well as reduced baseline levels (Fig. 3B). Decreased levels of IL-4 and IL-5 observed in TSLPR−/− mice may be responsible for the reduction of serum IgE and eosinophil infiltration, as IL-4 is important for antibody isotype switch to IgE, and IL-5 is important for eosinophil recruitment and activation (32,33). Thus, TSLPR−/− mice displayed an overall reduction in Th2-type phenotypic changes typically seen in WT mice sensitized and challenged with FITC. However, it is not clear if the reduced Th2 cytokine production observed is a T cell intrinsic defect, or if there is a defect upstream of T cell activation. TSLP appeared to have no effect on the development of Th1 and Th17 cells in this model as the number of IFN-γ and IL-17 producing T cells in the draining lymph nodes 1 week after FITC sensitization remained unchanged (data not shown).
Figure 3. Reduced features of a Th2 allergic response in FITC sensitized TSLPR−/− mice.

(a) Cytokine secretion by PMA/ionomycin stimulated skin-draining lymph node cells one week following sensitization of WT and TSLPR−/− mice with 0.5% FITC in acetone/dibutyl phthalate. The results are expressed as percent of WT cytokine forming spots/106 cells from three independent experiments (**P < 0.01, Wilcoxon test, n=15). (b) Serum IgE levels in untreated or FITC sensitized and challenged WT or TSLPR−/− mice. WT mice have significantly higher basal and post-challenge serum IgE levels (**P < 0.01, Wilcoxon test, n≥9 for each group).
TSLPR−/− Mice Exhibit Impaired Skin-Derived DC Migration after Epicutaneous Hapten Sensitization
TSLP treatment of DCs in vitro has been shown to induce increased expression of co-stimulatory molecules, and to generate DCs capable of promoting Th2 cell differentiation (2,10). However, very little is known about the effect of TSLP on DCs during an immune response in vivo. Based on in vitro data, and the finding that human epidermal Langerhans cells have been shown to express TSLPR and respond to TSLP treatment (34), we hypothesized that skin-resident DCs would be a target of TSLP during the sensitization phase of a CHS response. To initiate an adaptive immune response tissue-resident DCs undergo a series of processes that include antigen uptake, maturation, migration to draining lymph nodes, and presentation of antigen to T cells (35). To determine if the failure of TSLPR−/− mice to develop a CHS response to FITC was due to a deficit in DC function, we first assessed the migration of DCs to draining lymph nodes following FITC sensitization (Fig. 4A). Skin draining inguinal and axillary lymph node cells from FITC-sensitized WT and TSLPR−/− mice were analyzed for the relative frequency and absolute numbers of FITC+CD11c+ cells (Fig. 4B and C). There was a significant reduction in frequency and number of FITC+CD11c+ DCs in the draining lymph nodes of TSLPR−/− mice 24 H post sensitization (Fig. 4B and C). As shown previously, we observed a requirement for DBP in driving the migration of skin-resident DCs to the draining LN, as there were essentially no FITC+CD11c+ cells after treatment with FITC in acetone alone (data not shown) (29).
Figure 4. Reduced accumulation of FITC+CD11c+ cells in skin draining lymph nodes of TSLPR−/− mice 24 H post-sensitization with FITC in acetone/DBP.

(a) Representative FACS plots from skin-draining inguinal and axillary lymph node cells from WT and TSLPR−/− mice 24 H post-sensitization on the abdomen with 0.5% FITC, after Percoll enrichment for low density cells. Values represent frequency of FITC+CD11c+ cells within the live cell gate. (b) Frequency and (c) absolute number of FITC+CD11c+ cells found in inguinal and axillary LN of WT and TSLPR−/− mice 24 H post FITC sensitization. TSLPR−/− FITC+CD11c+ cell frequency and number are significantly reduced compared to WT. (*P<0.05, Students T-test, results pooled from two independent experiments, n=7 mice per group).
Selective Reduction of Dermis-Derived DC after TRITC Sensitization of TSLPR−/− Mice
To confirm and extend these data, mice were sensitized with a second fluorescent hapten, TRITC, in DBP and acetone (Fig. 5A). As was found for FITC painted animals, the TSLPR−/−mice displayed a marked reduction in TRITC+ cells in the skin draining lymph nodes following treatment (Fig. 5A and B). Specifically, the skin-derived TRITC+CD11c+CD207+ and TRITC+CD11bintCD11cint subsets were significantly reduced, whereas the blood-derived TRITC+CD11c+CD8+ subset was similar between WT and TSLPR−/− mice (Fig. 5A and 5B). This demonstrates a selective reduction in the skin-derived TRITC+ DC subsets in the TSLPR−/−mice, while blood-derived DC subsets were unaffected. The skin-derived DC subsets are most likely of dermal origin as the analysis was performed 24 H following sensitization, and epidermal Langerhans cells arrive in draining lymph nodes 72–96 H post sensitization (36). These findings suggest that TSLP acts locally on skin-resident DCs during sensitization, resulting in accumulation in skin-draining lymph nodes, and the ability to drive proliferation of hapten-specific naïve CD4 T cells. This phenotype is not due to a deficit in skin-resident DCs in the TSLPR−/− mice as they have comparable numbers of epidermal Langerhans cells and dermal DCs to WT mice (data not shown).
Figure 5. Decreased accumulation of TRITC+ dermis-derived dendritic cells in draining lymph nodes 24 H post sensitization.
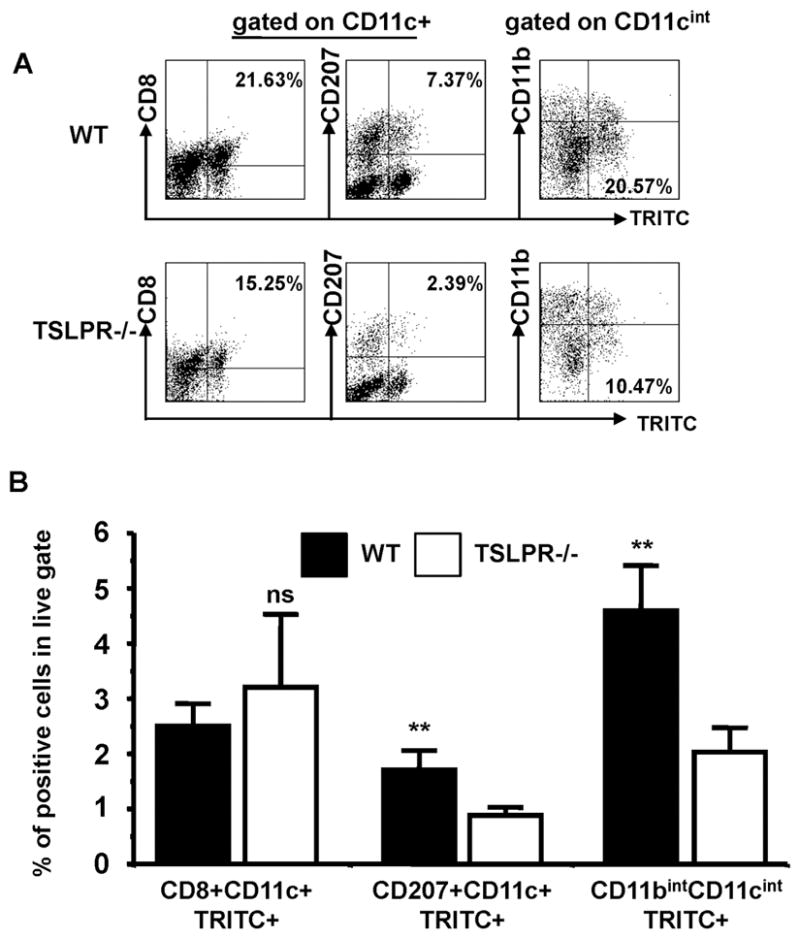
(a) WT and TSLPR−/− mice were sensitized with 0.1% TRITC diluted in acetone and DBP on the abdomen. Plots in the left two columns are gated on CD11c+ cells, and plots in the rightmost column are gated on CD11cint cells. The percentage represent frequency within the CD11c+ (left and middle columns) or CD11cint (right column) populations. (b) Frequency of blood-derived (left column, CD8+CD11c+TRITC+), and skin-derived (middle and right columns) TRITC+ dendritic cells in the lymph nodes from TRITC sensitized mice. Inguinal, axillary and brachial lymph nodes were pooled. TSLPR−/− have reduced frequencies of TRITC+ skin-derived DCs accumulating in skin-draining lymph nodes after TRITC sensitization, while blood-derived DCs are similar between TSLPR−/− and WT (** P < 0.01, * P < 0.05, Wilcoxon test, representative of three independent experiments, n≥3 mice per group)
Acute Blockade of TSLP Abrogates FITC-Induced Contact Hypersensitivity
The data presented above is consistent with an essential role for TSLP signaling in a Th2 CHS response. However, as the studies were performed using mice genetically deficient for the TSLP receptor, it is formally possible that, in the absence of TSLP signals there is a developmental defect resulting in reduced Th2 responses. As TSLP expression is seen most prominently at barrier surfaces (skin, gut, and lung) under steady-state conditions, the loss of a constant, albeit low level TSLP signal may lead to loss of some intrinsic DC function. To address this issue, WT mice were treated with a TSLP neutralizing antibody (M702) (37), control antibody, or PBS prior to FITC sensitization and challenge, and ear swelling was measured 24 H after challenge. Mice that were treated with M702 displayed significantly reduced ear swelling upon FITC sensitization and challenge, as compared with control antibody or PBS treated WT mice (Fig. 6A). These data, along with those generated in the TSLPR−/− mice, demonstrate that TSLP signals are required for Th2-type CHS, further supporting the role of TSLP as a mediator of this type of Th2 response.
Figure 6. TSLP blockade inhibits Th2-type CHS and DC accumulation in skin draining lymph nodes.

(a) WT mice were treated with TSLP blocking antibody (clone M702) on the day before (200 μg IV) and the day of (100 μg IP) FITC sensitization. Mice received similar anti-TSLP treatments on days 6 and 7, followed by FITC challenge on day 7. Ear swelling was measured 24 H post challenge (*P<0.05, **P<0.01, one-way ANOVA, n=5 mice per group). (b) To determine the effect of anti-TSLP treatment on FITC+ DC accumulation in the skin draining lymph nodes, a similar sensitization and antibody treatment was used, with an additional anti-TSLP treatment at 8 H post sensitization. 24 H post sensitization mice were sacrificed and FITC+ DC populations were analyzed in the skin draining lymph nodes. (c) Frequency of FITC+CD11c+ cells in the skin draining lymph nodes of anti-TSLP treated WT mice 24 H post FITC sensitization.
Furthermore, as seen in the TSLPR−/− mice, M702 treatment also led to a diminished frequency of FITC+CD11c+ DCs in the skin draining lymph nodes 24 H post FITC sensitization (Fig. 6B and C), These data demonstrate that the DC migration defect seen in the TSLPR−/− mice is not developmental, and that TSLP responsiveness at the time of antigen acquisition determines the nature of the adaptive immune response.
TSLPR−/− DCs are Defective at Inducing CD4 T Cell Proliferation
The data presented above demonstrates that TSLP provides a maturation signal to skin resident DCs resulting in migration to draining lymph nodes following encounter with antigen. To determine whether DCs from TSLPR−/− mice were also functionally defective, FITC+ and FITC-CD11c+ DCs were isolated from skin draining lymph nodes 24 H after FITC sensitization. These cells were pulsed with peptide antigen (OVA321-339), and then co-cultured with OVA-specific DO11.10 CD4 T for 3 days. FITC+CD11c+ DCs from WT mice effectively drove proliferation of DO11.10 CD4 T cells, whereas TSLPR−/− FITC+ DCs were significantly impaired in their ability to drive proliferation of naïve CD4 T cells (Fig. 7A). These data are consistent with a defect in DC maturation in the absence of a TSLP signal at the time of antigen acquisition, and emphasize a major role for TSLP in the sensitization phase of FITC induced CHS.
Figure 7. FITC+ DC from TSLPR−/− are defective in their ability to induce proliferation of naïve CD4 T cells.

FITC+ (a) and FITC− (c) CD11c+ DC from WT and TSLPR−/− mice were FACS sorted from FITC-sensitized mice and co-cultured at a 25:1 T cell: DC ratio with OVA-specific DO11.10 transgenic T cells in the presence of 5 μg/ml OVA323-339 peptide. Tritiated thymidine uptake (12 H pulse, 1 μCi/well) was measured on day 3. TSLPR−/− FITC+ DCs are reduced in their ability to drive proliferation of DO11.10 CD4 T cells (***P<0.001, one-way ANOVA, representative of three independent experiments). (b) Representative histograms and combined mean fluorescence intensities depicting expression of the costimulatory molecules CD86 and OX40L by WT and TSLPR−/− FITC+CD11c+ DCs 24 H post-sensitization. TSLPR−/−FITC+CD11c+ DCs express reduced levels of CD86 (*P<0.05, Student’s T-test, n≥4 mice per group).
Consistent with reduced T cell proliferation in the presence of TSLPR−/− FITC+DC, we observed reduced CD86 expression by TSLPR−/− FITC+ DCs (Fig. 7B), suggesting the FITC+TSLPR−/− DC may be defective in providing costimulation to naïve CD4 T cells. Interestingly, there was no difference in expression of the putative TSLP target OX40L, as well as CD80 and CD40, between WT and TSLPR−/− FITC+DC (Fig. 7B and data not shown). Taken together, these data suggest the defective Th2 CHS response observed in TSLPR−/− mice is multifaceted, including a deficiency in FITC+DC accumulation in skin draining lymph nodes and a reduced ability to induce proliferation of naïve CD4 T cells, potentially due to reduced expression of costimulatory molecules on FITC+ DCs. In addition, there was no difference in the ability of WT and TSLPR−/− FITC-CD11c+ DCs to drive proliferation of naïve CD4 T cells (Fig. 7C), demonstrating that the deficit in the FITC+ DCs from TSLPR−/− mice was due to lack of an acute TSLP signal, and not a developmental defect in the TSLPR−/− mice. Taken as a whole, these data show that TSLP acts in the periphery on tissue resident DCs, inducing a mature phenotype.
FITC Sensitization and Challenge of TSLPR−/− mice does not affect Th1 or Th17 Responses
One possibility for the reduced response to FITC sensitization and challenge experienced by TSLPR−/− mice is the induction of an antagonistic Th1 or Th17 response. Others have shown that TSLPR−/− mice experience an aberrant Th1 response to infection with the intestinal helminth Trichuris muris rather than the normal Th2 response (38). In order to determine whether the reduced FITC CHS response experienced by TSLPR−/− mice was due to aberrant Th1 or Th17 responses we sensitized and challenged WT and TSLPR−/− with FITC and measured IL-4, IL-17, and IFNγ expression in the inflamed skin. Consistent with the ELISpot data in Figure 3, we found significantly greater IL-4 expression in the FITC challenged skin of WT mice compared to TSLPR−/− mice. However, we observed no difference in IFNγ or IL-17 expression, suggesting TSLPR−/− mice experience a reduced Th2 response, rather than an antagonistic Th1 or Th17 response to FITC (Fig. 8A).
Figure 8. TSLPR−/− do not experience an aberrant Th1 or Th17 response to FITC sensitization and challenge.

(a) IL-4, IL-17, and IFNγ mRNA expression in the vehicle and FITC-challenged ears of FITC-sensitized WT and TSLPR−/− mice. Mice were sensitized with FITC on days 0 and 2, and challenged with FITC or vehicle on the contralateral ear on day 6. Ear tissue was excised and processed 24 H post challenge. Fold induction is relative to the WT vehicle challenged samples and normalized to GAPDH. (**P<0.01, one-way ANOVA, n≥3 mice per group, representative of two independent experiments). (b) IL-4, IL-17 and IFNγ expression by DO11.10 CD4 T cells induced by FITC+ or FITC- DCs from FITC sensitized WT or TSLPR−/− mice. WT and TSLPR−/− mice were sensitized with FITC and skin draining lymph nodes were harvested 24 H post sensitization. CD11c+FITC+ and CD11c+FITC− DCs were sorted and co-cultured at a 25:1 (T:DC) ratio with DO11.10 CD4 T cells in the presence of 5ug/ml OVA peptide (323-339). Supernatants were collected on day 3 for cytokine ELISAs (*P<0.05, ***P<0.001, one-way ANOVA).
In addition, we wanted to determine whether DCs from TSLPR−/− preferentially drove Th1 or Th17 responses after FITC sensitization. In order to test this we sorted FITC+ and FITC-DCs from FITC sensitized WT and TSLPR−/− mice and co-cultured them with DO11.10 OVA-specific CD4 T cells. While we observed a significant reduction in IL-4 produced by T cells co-cultured with TSLPR−/− FITC+ DCs, there was no difference in IFNγ production induced by WT and TSLPR−/− FITC+ DCs (Fig. 8B). Interestingly, WT FITC+ DCs induced significantly greater IL-17 production by DO11.10 CD4 T cells, suggesting there may be a previously unappreciated Th17 component to FITC CHS response (Fig. 8B). Taken together, these data suggest that the reduced response observed in TSLPR−/− and anti-TSLP treated mice is not due to an antagonistic Th1 or TH17 response to FITC sensitization and challenge.
DISCUSSION
Several lines of evidence demonstrate a role for TSLP in regulating Th2 responses in allergic disease. First, in both humans and mice, TSLP treated DCs can prime CD4 T cells to take on a Th2 phenotype (1,2). Second, TSLP expression is markedly elevated in the lesional skin of individuals with AD and in the lungs of asthmatics (1,2,9). Likewise, in mice, TSLP expression in the lung is increased in a model of antigen-induced airway inflammation, whereas TSLPR−/− mice are resistant to disease development (10,11). Finally, overexpression of TSLP specifically in the skin or lung leads to development of spontaneous disease that closely resembles human AD or asthma, respectively (10,12). We have now expanded these observations to an antigen-driven model of skin inflammation in mice, CHS, which is dependent on the adaptive immune system. We show that, in the absence of a TSLP response (either through genetic ablation or antibody blockade), mice fail to mount a response to the Th2-eliciting FITC/DBP combination. This defective CHS response correlated with decreased number and function of antigen-positive DCs in skin draining lymph nodes following sensitization, and reduced Th2 cytokine producing cells in skin draining lymph nodes (Figs. 3 and 4). These data support a model whereby DBP-induced TSLP controls the response to antigens encountered in the skin through local stimulation of resident DCs after sensitization.
In addition to our studies, recent work has also demonstrated a role for TSLP in a delayed type hypersensitivity model. He et al., using a model involving sensitization with ovalbumin, demonstrated impaired allergic skin inflammation in TSLPR−/− mice (39). However, the mechanism they describe for this defect is distinct from the mechanisms described here. He et al. used epicutaneous sensitization with protein antigen and did not observe striking deficits in the sensitization phase in the absence of TSLP receptor (39). Thus, the relative TSLP-independence of the sensitization phase in their experimental system was not suited to show an effect of this cytokine during this phase of the response. However, in the FITC CHS model we have clearly demonstrated induction of TSLP in the skin during the sensitization phase. Thus the FITC CHS model is more suitable for determining a role for TSLP during sensitization, while the ovalbumin model used by He et al. has proven suitable to demonstrate a role for TSLP during the challenge phase. In addition, He et al. suggest that TSLPR−/− DCs are not defective in the priming of DO11.10 T cells. However, in this study FITC+CD11c+ DCs were co-cultured with DO11.10 T cells at a 1:10 (DC:T) ratio, potentially masking any defects due to the high DC:T ratio (39). We observed significant differences between WT and TSLPR−/− FITC+CD11c+ DCs ability to induce proliferation of DO11.10 T cells at 1:25 DC:T ratio (Figure 7A), which is likely a more physiologically relevant ratio. In addition, we observed significantly reduced expression of the costimulatory molecule CD86 on FITC+DC from TSLPR−/− mice, a possible mechanism for reduced ability to drive proliferation of CD4 T cells (Fig 7B). OX40L has been hypothesized to be directly downstream of TSLP and critically involved in driving the differentiation of inflammatory Th2 cells by TSLP treated DC in human systems (40). However, there was no detectable difference in OX40L expression by FITC+DC in our model (Fig. 7B), suggesting that TSLP-dependent CHS responses are independent of OX40L induction by TSLP. Furthermore, this elucidates a potential distinction in the TSLP pathway between murine and human systems. Our data, in combination with the data from He et al., suggests TSLP has pleiotropic effects in driving allergic responses in the skin. We propose a model in which keratinocyte-derived TSLP acts directly or indirectly on skin-resident DCs resulting in maturation and homing to skin draining lymph nodes, where the TSLP-programmed DCs drive proliferation of naïve CD4 T cells. Interestingly, we only observed a modest reduction in the ability of TSLPR−/− FITC+ DCs ability to induce IL-4 production by DO11.10 CD4 T cells, suggesting there may be other roles for TSLP in driving Th2 responses in vivo, distinct from their action on DCs, such as direct action on CD4 T cells. Furthermore, we do not exclude a potential role for basophils in this response, as it was recently demonstrated that basophils are involved in the early development of Th2-type responses (41–44).
We have also shown that DBP is sufficient to induce TSLP expression, and the adjuvant effect of DBP during the sensitization phase of CHS may in part be mediated by TSLP. Consistent with our data, Boehme et al. recently showed that treatment with FITC in acetone and DBP, or acetone and DBP alone, resulted in increased levels of TSLP protein in the skin (45). We have extended this study to show that FITC alone (diluted in acetone) is not sufficient to induce TSLP expression (Fig. 1A and B). Interestingly, FITC in the presence of DBP appears to have an additive effect on the induction of TSLP protein but not mRNA (Fig. 1A and B). These data are in agreement with Boehme et al., as they observed an increase in TSLP protein in samples treated with FITC in acetone/DBP over samples treated with acetone/DBP alone (45). This suggests that the presence of antigen enhances TSLP production, potentially by inducing the recruitment of adaptive immune cells producing cytokines that are involved in a positive feedback loop for TSLP expression. In addition, we have shown that DBP induces TSLP expression by the PAM212 keratinocyte cell line, suggesting that DBP acts directly on keratinocytes to drive TSLP expression in the skin. Previous CHS studies using FITC have shown that phthalate esters such as DBP are required for many aspects of the CHS response, including inflammation, trafficking of FITC+ cells to draining lymph nodes, and the subsequent generation of Th2 cells (29,30). The data presented in our study further elucidates the mechanism by which DBP, and other sensitizing phthalate esters, drive Th2 responses following skin exposure, via induction of TSLP gene expression. The mechanism of DBP-induced TSLP gene expression remains to be elucidated. Previous work has shown that combinations of inflammatory cytokines (TNFα and IL-1β) and Th2 cytokines (IL-4 and IL-13) synergize to induce TSLP gene expression by human keratinocytes (46). There is no data available on the regulation of TSLP gene expression in mouse keratinocytes, but NFκB has been shown to be critical for TSLP gene induction in airway and colonic epithelium (28,47). Whether DBP utilizes similar pathways to induce TSLP gene expression remains to be determined.
Recently, Boehme et al. demonstrated that treatment of FITC-sensitized mice with an anti-TSLP antibody during FITC challenge resulted in a modest reduction (less than two-fold) in ear swelling compared to isotype treated controls (45). These data, coupled with the data presented here that TSLP blockade at both sensitization and challenge resulted in a significant decrease in inflammation, suggests that TSLP plays a role at both phases. The fact that treatment at both sensitization and challenge had a greater effect suggests that TSLP plays a more important role during sensitization phase than at challenge.
The critical role of TSLP in promoting inflammatory Th2 responses in a Th2 model of CHS supports a role for epidermal TSLP expression in the immunopathogenesis of AD and underscores FITC CHS as an animal model of AD. AD is characterized by TSLP expression in the epidermis and deficient epidermal barrier function. There is a significant association between AD and mutations in the gene encoding filaggrin, resulting in abnormal epidermal differentiation and epidermal barrier dysfunction (49,50). Induction of TSLP in skin has been reported in an animal model of genetically disrupted epidermal barrier function, rendering the association of barrier deficiency and TSLP induction very striking (51,52). Moreover, there is evidence that abnormal keratinocyte differentiation is linked to NFκB dependent induction of TSLP in these cells (51). While current animal models of AD do not incorporate mutations of genes involved in epidermal structure and barrier function, induction of TSLP in skin by DBP in the FITC CHS model substitutes for any genetically determined cutaneous TSLP induction. It remains to be shown whether DBP induces abnormal epidermal differentiation, defective skin barrier function and NFκB activation in vivo.
Our results demonstrate that in order to generate a response to FITC, DCs require a TSLP-mediated signal. The lack of this signal results in a series of maturational and functional deficits, including decreased migration and diminished capability for T cell activation. Taken together, these deficiencies result in a dampened adaptive immune response to FITC. Previous studies have demonstrated a requirement for CCR7 and its ligands CCL19 and CCL21 for optimal migration of skin-derived DCs after FITC sensitization (53–56). We observed no difference in WT and TSLPR−/− bone marrow-derived DC migration towards CCR7 ligands in vitro (data not shown), suggesting TSLPR−/− DCs are capable of upregulating CCR7 sufficiently in an in vitro setting. However, other chemokine receptors and ligands have been shown to play a role in DC migration from the skin to draining lymph nodes, such as CXCR4 and CXCL12, which we have not investigated (57). The reduced accumulation of FITC+ DCs in draining lymph nodes following FITC sensitization in TSLPR−/− mice included the CD207+ (Langerin; Fig. 5) subset. Recently a novel subset of dermal DCs expressing CD207 was identified and found to be involved in CHS responses (58). Therefore, it is conceivable that the reduced accumulation of this subset in draining lymph nodes following sensitization contributed to the diminished CHS responses to FITC in TSLPR−/− mice.
TSLP has been shown to inhibit Th1 and Th17 responses in the intestine (38,47). Therefore, it is feasible that the reduced FITC CHS response observed in TSLPR−/− mice is due to the development of an antagonistic Th1 or Th17 response. However, we did not observe increased IFNγ or IL-17 in the inflamed tissue of FITC sensitized TSLPR−/− mice (Fig. 8A), nor did we observe increased IFNγ or IL-17 production by DO11.10 CD4 T cells cocultured with TSLPR−/− FITC+ DCs (Fig. 8B). This suggests that an aberrant Th1 or Th17 response is not occurring in FITC-sensitized and challenged TSLPR−/− mice, and is most likely not the reason for the significantly reduced response observed in these mice. In contrast, we observed a selective reduction in IL-4 expression in the skin of TSLPR−/− mice (Fig. 8A); suggesting Th2 cells mediate the significant response observed in WT mice.
Taken as a whole these data suggest that TSLP regulates responses to allergens at barrier surfaces through promotion of Th2 responses. The presence of elevated TSLP at sites of allergic inflammation also suggests that TSLP is a prime therapeutic target for these diseases. The fact that the induction of Th1 and Th17 responses in skin remained unaffected in the absence of TSLP signaling and that TSLP induction in skin may be involved in early steps of the immunopathogenesis of AD as shown here, in addition to late steps, make it a particularly attractive therapeutic target.
Acknowledgments
We would like to thank Drs. Jessica Hamerman, Daniel Campbell, and Hai Chon Lee, and other members of the Benaroya Research Institute Immunology Program, for helpful discussion and critical reading of the manuscript. We also thank Matt Warren for assistance with preparing the manuscript.
Footnotes
The work was supported in part from NIH grants AI44259, AI68731, AR56113, and AR55695 to S.F.Z, and a grant from the SNF (310000-109716) to C.H. R.P.L. was supported by training grant CA009537, T32-GM007270, and the Howard Hughes Institute Med into Grad initiative through a grant to UW.
Reference List
- 1.Liu YJ, Soumelis V, Watanabe N, Ito T, Wang YH, Malefyt RW, Omori M, Zhou B, Ziegler SF. TSLP: an epithelial cell cytokine that regulates T cell differentiation by conditioning dendritic cell maturation. Annu Rev Immunol. 2007;25:193–219. doi: 10.1146/annurev.immunol.25.022106.141718. [DOI] [PubMed] [Google Scholar]
- 2.Soumelis V, Reche PA, Kanzler H, Yuan W, Edward G, Homey B, Gilliet M, Ho S, Antonenko S, Lauerma A, Smith K, Gorman D, Zurawski S, Abrams J, Menon S, McClanahan T, Waal-Malefyt RR, Bazan F, Kastelein RA, Liu YJ. Human epithelial cells trigger dendritic cell mediated allergic inflammation by producing TSLP. Nat Immunol. 2002;3:673–680. doi: 10.1038/ni805. [DOI] [PubMed] [Google Scholar]
- 3.Ziegler SF, Liu YJ. Thymic stromal lymphopoietin in normal and pathogenic T cell development and function. Nat Immunol. 2006;7:709–714. doi: 10.1038/ni1360. [DOI] [PubMed] [Google Scholar]
- 4.Allakhverdi Z, Comeau MR, Jessup HK, Yoon BR, Brewer A, Chartier S, Paquette N, Ziegler SF, Sarfati M, Delespesse G. Thymic stromal lymphopoietin is released by human epithelial cells in response to microbes, trauma, or inflammation and potently activates mast cells. J Exp Med. 2007;204:253–258. doi: 10.1084/jem.20062211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Park LS, Martin U, Garka K, Gliniak B, Di Santo JP, Muller W, Largaespada DA, Copeland NG, Jenkins NA, Farr AG, Ziegler SF, Morrissey PJ, Paxton R, Sims JE. Cloning of the murine thymic stromal lymphopoietin (TSLP) receptor: Formation of a functional heteromeric complex requires interleukin 7 receptor. J Exp Med. 2000;192:659–670. doi: 10.1084/jem.192.5.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Omori M, Ziegler S. Induction of IL-4 expression in CD4(+) T cells by thymic stromal lymphopoietin. J Immunol. 2007;178:1396–1404. doi: 10.4049/jimmunol.178.3.1396. [DOI] [PubMed] [Google Scholar]
- 7.Pandey A, Ozaki K, Baumann H, Levin SD, Puel A, Farr AG, Ziegler SF, Leonard WJ, Lodish HF. Cloning of a receptor subunit required for signaling by thymic stromal lymphopoietin. Nat Immunol. 2000;1:59–64. doi: 10.1038/76923. [DOI] [PubMed] [Google Scholar]
- 8.Rochman I, Watanabe N, Arima K, Liu YJ, Leonard WJ. Cutting edge: direct action of thymic stromal lymphopoietin on activated human CD4+ T cells. J Immunol. 2007;178:6720–6724. doi: 10.4049/jimmunol.178.11.6720. [DOI] [PubMed] [Google Scholar]
- 9.Ying S, O’Connor B, Ratoff J, Meng Q, Mallett K, Cousins D, Robinson D, Zhang G, Zhao J, Lee TH, Corrigan C. Thymic stromal lymphopoietin expression is increased in asthmatic airways and correlates with expression of Th2-attracting chemokines and disease severity. J Immunol. 2005;174:8183–8190. doi: 10.4049/jimmunol.174.12.8183. [DOI] [PubMed] [Google Scholar]
- 10.Zhou B, Comeau MR, De Smedt T, Liggitt HD, Dahl ME, Lewis DB, Gyarmati D, Aye T, Campbell DJ, Ziegler SF. Thymic stromal lymphopoietin as a key initiator of allergic airway inflammation in mice. Nat Immunol. 2005;6:1047–1053. doi: 10.1038/ni1247. [DOI] [PubMed] [Google Scholar]
- 11.Al Shami A, Spolski R, Kelly J, Keane-Myers A, Leonard WJ. A role for TSLP in the development of inflammation in an asthma model. J Exp Med. 2005;202:829–839. doi: 10.1084/jem.20050199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yoo J, Omori M, Gyarmati D, Zhou B, Aye T, Brewer A, Comeau MR, Campbell DJ, Ziegler SF. Spontaneous atopic dermatitis in mice expressing an inducible thymic stromal lymphopoietin transgene specifically in the skin. J Exp Med. 2005;202:541–549. doi: 10.1084/jem.20041503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li M, Messaddeq N, Teletin M, Pasquali JL, Metzger D, Chambon P. Retinoid X receptor ablation in adult mouse keratinocytes generates an atopic dermatitis triggered by thymic stromal lymphopoietin. Proc Natl Acad Sci U S A. 2005;102:14795–14800. doi: 10.1073/pnas.0507385102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li M, Hener P, Zhang Z, Kato S, Metzger D, Chambon P. Topical vitamin D3 and low-calcemic analogs induce thymic stromal lymphopoietin in mouse keratinocytes and trigger an atopic dermatitis. Proc Natl Acad Sci U S A. 2006;103:11736–11741. doi: 10.1073/pnas.0604575103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jessup HK, Brewer AW, Omori M, Rickel EA, Budelsky AL, Yoon BR, Ziegler SF, Comeau MR. Intradermal administration of thymic stromal lymphopoietin induces a T cell- and eosinophil-dependent systemic Th2 inflammatory response. J Immunol. 2008;181:4311–4319. doi: 10.4049/jimmunol.181.6.4311. [DOI] [PubMed] [Google Scholar]
- 16.Headley MB, Zhou B, Shih WX, Aye T, Comeau MR, Ziegler SF. TSLP conditions the lung immune environment for the generation of pathogenic innate and antigen-specific adaptive immune responses. J Immunol. 2009;182:1641–1647. doi: 10.4049/jimmunol.182.3.1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Watanabe H, Unger M, Tuvel B, Wang B, Sauder DN. Contact hypersensitivity: the mechanism of immune responses and T cell balance. J Interferon Cytokine Res. 2002;22:407–412. doi: 10.1089/10799900252952181. [DOI] [PubMed] [Google Scholar]
- 18.Takeshita K, Yamasaki T, Akira S, Gantner F, Bacon KB. Essential role of MHC II-independent CD4+ T cells, IL-4 and STAT6 in contact hypersensitivity induced by fluorescein isothiocyanate in the mouse. Int Immunol. 2004;16:685–695. doi: 10.1093/intimm/dxh073. [DOI] [PubMed] [Google Scholar]
- 19.Tang A, Judge TA, Nickoloff BJ, Turka LA. Suppression of murine allergic contact dermatitis by CTLA4Ig. Tolerance induction of Th2 responses requires additional blockade of CD40-ligand. J Immunol. 1996;157:117–125. [PubMed] [Google Scholar]
- 20.Kehren J, Desvignes C, Krasteva M, Ducluzeau MT, Assossou O, Horand F, Hahne M, Kagi D, Kaiserlian D, Nicolas JF. Cytotoxicity is mandatory for CD8(+) T cell-mediated contact hypersensitivity. J Exp Med. 1999;189:779–786. doi: 10.1084/jem.189.5.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dearman RJ, Kimber I. Role of CD4(+) T helper 2-type cells in cutaneous inflammatory responses induced by fluorescein isothiocyanate. Immunology. 2000;101:442–451. doi: 10.1046/j.1365-2567.2000.01126.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bieber T. Atopic dermatitis. N Engl J Med. 2008;358:1483–1494. doi: 10.1056/NEJMra074081. [DOI] [PubMed] [Google Scholar]
- 23.Carpino N, Thierfelder WE, Chang MS, Saris C, Turner SJ, Ziegler SF, Ihle JN. Absence of an essential role for thymic stromal lymphopoietin receptor in murine B-cell development. Mol Cell Biol. 2004;24:2584–2592. doi: 10.1128/MCB.24.6.2584-2592.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peirson SN, Butler JN, Foster RG. Experimental validation of novel and conventional approaches to quantitative real-time PCR data analysis. Nucleic Acids Res. 2003;31:e73. doi: 10.1093/nar/gng073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3:RESEARCH0034. doi: 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 27.Takeshita K, Yamasaki T, Nagao K, Sugimoto H, Shichijo M, Gantner F, Bacon KB. CRTH2 is a prominent effector in contact hypersensitivity-induced neutrophil inflammation. Int Immunol. 2004;16:947–959. doi: 10.1093/intimm/dxh096. [DOI] [PubMed] [Google Scholar]
- 28.Lee HC, Ziegler SF. Inducible expression of the proallergic cytokine thymic stromal lymphopoietin in airway epithelial cells is controlled by NFkappaB. Proc Natl Acad Sci U S A. 2007;104:914–919. doi: 10.1073/pnas.0607305104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Imai Y, Kondo A, Iizuka H, Maruyama T, Kurohane K. Effects of phthalate esters on the sensitization phase of contact hypersensitivity induced by fluorescein isothiocyanate. Clin Exp Allergy. 2006;36:1462–1468. doi: 10.1111/j.1365-2222.2006.02574.x. [DOI] [PubMed] [Google Scholar]
- 30.Maruyama T, Shiba T, Iizuka H, Matsuda T, Kurohane K, Imai Y. Effects of phthalate esters on dendritic cell subsets and interleukin-4 production in fluorescein isothiocyanate-induced contact hypersensitivity. Microbiol Immunol. 2007;51:321–326. doi: 10.1111/j.1348-0421.2007.tb03914.x. [DOI] [PubMed] [Google Scholar]
- 31.Li M, Hener P, Zhang Z, Ganti KP, Metzger D, Chambon P. Induction of thymic stromal lymphopoietin expression in keratinocytes is necessary for generating an atopic dermatitis upon application of the active vitamin D3 analogue MC903 on mouse skin. J Invest Dermatol. 2009;129:498–502. doi: 10.1038/jid.2008.232. [DOI] [PubMed] [Google Scholar]
- 32.Yamaguchi Y, Hayashi Y, Sugama Y, Miura Y, Kasahara T, Kitamura S, Torisu M, Mita S, Tominaga A, Takatsu K. Highly purified murine interleukin 5 (IL-5) stimulates eosinophil function and prolongs in vitro survival. IL-5 as an eosinophil chemotactic factor. J Exp Med. 1988;167:1737–1742. doi: 10.1084/jem.167.5.1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Poulsen LK, Hummelshoj L. Triggers of IgE class switching and allergy development. Ann Med. 2007;39:440–456. doi: 10.1080/07853890701449354. [DOI] [PubMed] [Google Scholar]
- 34.Ebner S, V, Nguyen A, Forstner M, Wang YH, Wolfram D, Liu YJ, Romani N. Thymic stromal lymphopoietin converts human epidermal Langerhans cells into antigen-presenting cells that induce proallergic T cells. J Allergy Clin Immunol. 2007;119:982–990. doi: 10.1016/j.jaci.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 35.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 36.Kissenpfennig A, Henri S, Dubois B, Laplace-Builhe C, Perrin P, Romani N, Tripp CH, Douillard P, Leserman L, Kaiserlian D, Saeland S, Davoust J, Malissen B. Dynamics and function of Langerhans cells in vivo: dermal dendritic cells colonize lymph node areas distinct from slower migrating Langerhans cells. Immunity. 2005;22:643–654. doi: 10.1016/j.immuni.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 37.Zhou B, Headley MB, Aye T, Tocker J, Comeau MR, Ziegler SF. Reversal of thymic stromal lymphopoietin-induced airway inflammation through inhibition of Th2 responses. J Immunol. 2008;181:6557–6562. doi: 10.4049/jimmunol.181.9.6557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taylor BC, Zaph C, Troy AE, Du Y, Guild KJ, Comeau MR, Artis D. TSLP regulates intestinal immunity and inflammation in mouse models of helminth infection and colitis. J Exp Med. 2009;206:655–667. doi: 10.1084/jem.20081499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.He R, Oyoshi MK, Garibyan L, Kumar L, Ziegler SF, Geha RS. TSLP acts on infiltrating effector T cells to drive allergic skin inflammation. Proc Natl Acad Sci U S A. 2008;105:11875–11880. doi: 10.1073/pnas.0801532105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ito T, Wang YH, Duramad O, Hori T, Delespesse GJ, Watanabe N, Qin FX, Yao Z, Cao W, Liu YJ. TSLP-activated dendritic cells induce an inflammatory T helper type 2 cell response through OX40 ligand. J Exp Med. 2005;202:1213–1223. doi: 10.1084/jem.20051135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sokol CL, Barton GM, Farr AG, Medzhitov R. A mechanism for the initiation of allergen-induced T helper type 2 responses. Nat Immunol. 2008;9(3):318–318. doi: 10.1038/ni1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yoshimoto T, Yasuda K, Tanaka H, Nakahira M, Imai Y, Fujimori Y, Nakanishi K. Basophils contribute to T(H)2-IgE responses in vivo via IL-4 production and presentation of peptide-MHC class II complexes to CD4+ T cells. Nat Immunol. 2009;10:706–712. doi: 10.1038/ni.1737. [DOI] [PubMed] [Google Scholar]
- 43.Perrigoue JG, Saenz SA, Siracusa MC, Allenspach EJ, Taylor BC, Giacomin PR, Nair MG, Du Y, Zaph C, van Rooijen N, Comeau MR, Pearce EJ, Laufer TM, Artis D. MHC class II-dependent basophil-CD4+ T cell interactions promote T(H)2 cytokine-dependent immunity. Nat Immunol. 2009;10:697–705. doi: 10.1038/ni.1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sokol CL, Chu NQ, Yu S, Nish SA, Laufer TM, Medzhitov R. Basophils function as antigen-presenting cells for an allergen-induced T helper type 2 response. Nat Immunol. 2009;10:713–720. doi: 10.1038/ni.1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boehme SA, Franz-Bacon K, Chen EP, Sasik R, Sprague LJ, Ly TW, Hardiman G, Bacon KB. A small molecule CRTH2 antagonist inhibits FITC-induced allergic cutaneous inflammation. Int Immunol. 2009;21:81–93. doi: 10.1093/intimm/dxn127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bogiatzi SI, Fernandez I, Bichet JC, Marloie-Provost MA, Volpe E, Sastre X, Soumelis V. Cutting Edge: Proinflammatory and Th2 cytokines synergize to induce thymic stromal lymphopoietin production by human skin keratinocytes. J Immunol. 2007;178:3373–3377. doi: 10.4049/jimmunol.178.6.3373. [DOI] [PubMed] [Google Scholar]
- 47.Zaph C, Troy AE, Taylor BC, Berman-Booty LD, Guild KJ, Du Y, Yost EA, Gruber AD, May MJ, Greten FR, Eckmann L, Karin M, Artis D. Epithelial-cell-intrinsic IKK-beta expression regulates intestinal immune homeostasis. Nature. 2007;446:552–556. doi: 10.1038/nature05590. [DOI] [PubMed] [Google Scholar]
- 48.Gilliet M, Soumelis V, Watanabe N, Hanabuchi S, Antonenko S, Waal-Malefyt R, Liu YJ. Human dendritic cells activated by TSLP and CD40L induce proallergic cytotoxic T cells. J Exp Med. 2003;197:1059–1063. doi: 10.1084/jem.20030240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Palmer CN, Irvine AD, Terron-Kwiatkowski A, Zhao Y, Liao H, Lee SP, Goudie DR, Sandilands A, Campbell LE, Smith FJ, O’Regan GM, Watson RM, Cecil JE, Bale SJ, Compton JG, DiGiovanna JJ, Fleckman P, Lewis-Jones S, Arseculeratne G, Sergeant A, Munro CS, El Houate B, McElreavey K, Halkjaer LB, Bisgaard H, Mukhopadhyay S, McLean WH. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat Genet. 2006;38:441–446. doi: 10.1038/ng1767. [DOI] [PubMed] [Google Scholar]
- 50.Fallon PG, Sasaki T, Sandilands A, Campbell LE, Saunders SP, Mangan NE, Callanan JJ, Kawasaki H, Shiohama A, Kubo A, Sundberg JP, Presland RB, Fleckman P, Shimizu N, Kudoh J, Irvine AD, Amagai M, McLean WH. A homozygous frameshift mutation in the mouse Flg gene facilitates enhanced percutaneous allergen priming. Nat Genet. 2009;41:602–608. doi: 10.1038/ng.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Demehri S, Liu Z, Lee J, Lin MH, Crosby SD, Roberts CJ, Grigsby PW, Miner JH, Farr AG, Kopan R. Notch-deficient skin induces a lethal systemic B-lymphoproliferative disorder by secreting TSLP, a sentinel for epidermal integrity. PLoS Biol. 2008;6:e123. doi: 10.1371/journal.pbio.0060123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Segre JA. Epidermal barrier formation and recovery in skin disorders. J Clin Invest. 2006;116:1150–1158. doi: 10.1172/JCI28521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Forster R, Schubel A, Breitfeld D, Kremmer E, Renner-Muller I, Wolf E, Lipp M. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell. 1999;99:23–33. doi: 10.1016/s0092-8674(00)80059-8. [DOI] [PubMed] [Google Scholar]
- 54.Ohl L, Mohaupt M, Czeloth N, Hintzen G, Kiafard Z, Zwirner J, Blankenstein T, Henning G, Forster R. CCR7 governs skin dendritic cell migration under inflammatory and steady-state conditions. Immunity. 2004;21:279–288. doi: 10.1016/j.immuni.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 55.Engeman TM, Gorbachev AV, Gladue RP, Heeger PS, Fairchild RL. Inhibition of functional T cell priming and contact hypersensitivity responses by treatment with anti-secondary lymphoid chemokine antibody during hapten sensitization. J Immunol. 2000;164:5207–5214. doi: 10.4049/jimmunol.164.10.5207. [DOI] [PubMed] [Google Scholar]
- 56.Gunn MD, Kyuwa S, Tam C, Kakiuchi T, Matsuzawa A, Williams LT, Nakano H. Mice lacking expression of secondary lymphoid organ chemokine have defects in lymphocyte homing and dendritic cell localization. J Exp Med. 1999;189:451–460. doi: 10.1084/jem.189.3.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kabashima K, Shiraishi N, Sugita K, Mori T, Onoue A, Kobayashi M, Sakabe J, Yoshiki R, Tamamura H, Fujii N, Inaba K, Tokura Y. CXCL12-CXCR4 engagement is required for migration of cutaneous dendritic cells. Am J Pathol. 2007;171:1249–1257. doi: 10.2353/ajpath.2007.070225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bursch LS, Wang L, Igyarto B, Kissenpfennig A, Malissen B, Kaplan DH, Hogquist KA. Identification of a novel population of Langerin+ dendritic cells. J Exp Med. 2007;204:3147–3156. doi: 10.1084/jem.20071966. [DOI] [PMC free article] [PubMed] [Google Scholar]