

Abstract
Summary: METAL provides a computationally efficient tool for meta-analysis of genome-wide association scans, which is a commonly used approach for improving power complex traits gene mapping studies. METAL provides a rich scripting interface and implements efficient memory management to allow analyses of very large data sets and to support a variety of input file formats.
Availability and implementation: METAL, including source code, documentation, examples, and executables, is available at http://www.sph.umich.edu/csg/abecasis/metal/
Contact: goncalo@umich.edu
1 INTRODUCTION
Meta-analysis is becoming an increasingly important tool in genome-wide association studies (GWAS) of complex genetic diseases and traits (de Bakker et al., 2008). Meta-analysis provides an efficient and practical strategy for detecting variants with modest effect sizes (Skol et al., 2007). We, and others, have used METAL for performing meta-analysis of GWAS to identify loci reproducibly associated with a variety of traits, such as type 2 diabetes (Scott et al., 2007; Zeggini et al., 2008), lipid levels (Kathiresan et al., 2008, 2009; Willer et al., 2008), BMI (Willer et al., 2009), blood pressure (Newton-Cheh et al., 2009) and fasting glucose levels (Prokopenko et al., 2009).
Meta-analysis of genome-wide association summary statistics, in contrast to direct analysis of pooled individual-level data, alleviates common concerns with privacy of study participants and avoids cumbersome integration of genotype and phenotypic data from different studies. Meta-analysis allows for custom analyses of individual studies to conveniently account for population substructure, the presence of related individuals, study-specific covariates and many other ascertainment-related issues. It has been shown that meta-analysis of summary statistics is as efficient (in terms of statistical power) as pooling individual-level data across studies, but much less cumbersome (Lin and Zeng, 2009). Since GWAS routinely examine evidence for association at millions of directly genotyped and imputed SNPs across dozens or even hundreds of individual studies, it is important to use a fast and flexible tool to perform meta-analysis.
2 METHODS
The basic principle of meta-analysis is to combine the evidence for association from individual studies, using appropriate weights. METAL implements two approaches. The first approach converts the direction of effect and P-value observed in each study into a signed Z-score such that very negative Z-scores indicate a small P-value and an allele associated with lower disease risk or quantitative trait levels, whereas large positive Z-scores indicate a small P-value and an allele associated with higher disease risk or quantitative trait levels. Z-scores for each allele are combined across samples in a weighted sum, with weights proportional to the square-root of the sample size for each study (Stouffer et al., 1949). In a study with unequal numbers of cases and controls, we recommend that the effective sample size be provided in the input file, where Neff = 4/(1/Ncases+1/Nctrls). This approach is very flexible and allows results to be combined even when effect size estimates are not available or the β-coefficients and standard errors from individual studies are in different units. The second approach implemented in METAL weights the effect size estimates, or β-coefficients, by their estimated standard errors. This second approach requires effect size estimates and their standard errors to be in consistent units across studies. Asymptotically, the two approaches are equivalent when the trait distribution is identical across samples (such that standard errors are a predictable function of sample size). Key formulae for both approaches are in Table 1.
Table 1.
Formulae for meta-analysis
| Analytical strategy |
||
|---|---|---|
| Sample size based | Inverse variance based | |
| Inputs | Ni - sample size for study i | βi- effect size estimate for study i |
| Pi−P-value for study i | ||
| Δi - direction of effect for study i | sei - standard error for study i | |
| Intermediate Statistics | Zi = Φ−1(Pi/2) * sign(Δi) | wi = 1/SEi2 |
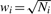 |
 |
|
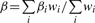 |
||
| Overall Z-Score | 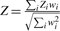 |
Z=β/SE |
| Overall P-value | P=2Φ(|−Z|) | |
3 RESULTS
3.1 Implementation
In implementing our software for meta-analysis, a primary consideration was to facilitate identification and resolution of common problems in meta-analysis. A secondary consideration was the ability to specify custom headers and delimiters so as to combine input files with varying formats generated from a variety of statistical packages. METAL tries to resolve or flag common problems that result from an inconsistent choice of allele labels or genomic strand across studies, or the presence of invalid P-values or test statistics at a subset of markers (due to numerical errors). METAL allows data to be filtered according to quality control measures, and can handle very large data sets (that typically total several GB in size) in workstations with a memory capacity not exceeding 2 GB.
3.2 Usage
METAL has been used extensively by many groups since its initial release in January 2008. This field testing enabled not only thorough debugging but improvements in error-detection methods. METAL can be run interactively or with a command script as input. Input files are processed one at a time and used to update intermediate statistics stored in memory. METAL implements Cochran's Q-test for heterogeneity (Cochran, 1954) and the appropriate statistics can be calculated if requested by the user. METAL was designed for flexible formatting of input files, and allows users to customize labels for key columns, input field delimiters and other characteristics of each input file. Information on genomic strand is used, if available, and—when it is unavailable—METAL automatically resolves strand mismatches for markers where strand is obvious (e.g. all SNPs except those with A/T and C/G alleles). METAL has an option to estimate a genomic control parameter (Devlin and Roeder, 1999) for each input file and apply an appropriate genomic control correction to input statistics prior to performing meta-analysis. To facilitate the detection of allele labels that may have been mis-specified by the user, which is critical for the correct determination of the direction of effect, METAL implements an option to output the mean, variance and minimum and maximum allele frequencies for each marker. METAL will track custom statistics, such as cumulative sample size, even when the standard error-weighted meta-analysis was performed. METAL can read gzipped files to allow for efficient use of disk space and optionally allows for subsets of markers to be analyzed. Full documentation of all options is available at http://www.sph.umich.edu/csg/abecasis/metal/.
3.3 Performance
METAL was written in C++ and is freely available for download. METAL compiles and runs on most Unix and Linux systems, and on Windows and Mac workstations. We recently performed a meta-analysis of GWAS for BMI (Willer et al., 2009). The analysis included 15 studies, each with association statistics at 2.2–2.5 million SNPs (average file size 225 MB), for a total of 36 million association statistics and a set of input files totaling 3.4 GB. This analysis required <6 min computing time and 790 MB of memory on a 2.83 GHz Intel processor. Runtime scales linearly with the number of studies examined—a meta-analysis including 74 input files (each with >2.5 m SNPs) took 36 min and 1 GB of memory.
ACKNOWLEDGEMENTS
The authors thank Michael Boehnke, Hyun Min Kang and Anne Jackson for reviewing early versions of this article. We are also grateful to numerous collaborators in the GIANT Consortium, the Global Lipids Genetic Consortium and the DIAGRAM Consortium for testing METAL and providing many useful suggestions.
Funding: G.R.A. was supported in part by the National Human Genome Research Institute (HG0002651 and HG0005214) and the National Institute of Mental Health (MH084698). C.J.W. was supported by a Pathway to Independence Award from the National Heart, Lung and Blood Institute (K99HL094535). Y.L. was supported by the National Institute for Diabetes and Digestive and Kidney Diseases (DK078150-03, PI Mohlke) and the National Cancer Institute (CA082659-11S1, PI Lin).
Conflict of Interest: none declared.
REFERENCES
- Cochran WG. The combination of estimates from different experiments. Biometrics. 1954;10:101–129. [Google Scholar]
- de Bakker PI, et al. Practical aspects of imputation-driven meta-analysis of genome-wide association studies. Hum. Mol. Genet. 2008;17:R122–R128. doi: 10.1093/hmg/ddn288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devlin B, Roeder K. Genomic control for association studies. Biometrics. 1999;55:997–1004. doi: 10.1111/j.0006-341x.1999.00997.x. [DOI] [PubMed] [Google Scholar]
- Kathiresan S, et al. Six new loci associated with blood low-density lipoprotein cholesterol, high-density lipoprotein cholesterol or triglycerides in humans. Nat. Genet. 2008;40:189–197. doi: 10.1038/ng.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kathiresan S, et al. Common variants at 30 loci contribute to polygenic dyslipidemia. Nat. Genet. 2009;41:56–65. doi: 10.1038/ng.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DY, Zeng D. Meta-analysis of genome-wide association studies: no efficiency gain in using individual participant data. Genet. Epidemiol. 2009;34:60–66. doi: 10.1002/gepi.20435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton-Cheh C, et al. Genome-wide association study identifies eight loci associated with blood pressure. Nat. Genet. 2009;41:666–676. doi: 10.1038/ng.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prokopenko I, et al. Variants in MTNR1B influence fasting glucose levels. Nat. Genet. 2009;41:77–81. doi: 10.1038/ng.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott LJ, et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 2007;316:1341–1345. doi: 10.1126/science.1142382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skol AD, et al. Optimal designs for two-stage genome-wide association studies. Genet. Epidemiol. 2007;31:776–788. doi: 10.1002/gepi.20240. [DOI] [PubMed] [Google Scholar]
- Stouffer SA, et al. Adjustment During Army Life. Princeton, NJ: Princeton University Press; 1949. [Google Scholar]
- Willer CJ, et al. Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat. Genet. 2008;40:161–169. doi: 10.1038/ng.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willer CJ, et al. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat. Genet. 2009;41:25–34. doi: 10.1038/ng.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeggini E, et al. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat. Genet. 2008;40:638–645. doi: 10.1038/ng.120. [DOI] [PMC free article] [PubMed] [Google Scholar]