

Abstract
Motivation: A gene set test is a differential expression analysis in which a P-value is assigned to a set of genes as a unit. Gene set tests are valuable for increasing statistical power, organizing and interpreting results and for relating expression patterns across different experiments. Existing methods are based on permutation. Methods that rely on permutation of probes unrealistically assume independence of genes, while those that rely on permutation of sample are suitable only for two-group comparisons with a good number of replicates in each group.
Results: We present ROAST, a statistically rigorous gene set test that allows for gene-wise correlation while being applicable to almost any experimental design. Instead of permutation, ROAST uses rotation, a Monte Carlo technology for multivariate regression. Since the number of rotations does not depend on sample size, ROAST gives useful results even for experiments with minimal replication. ROAST allows for any experimental design that can be expressed as a linear model, and can also incorporate array weights and correlated samples. ROAST can be tuned for situations in which only a subset of the genes in the set are actively involved in the molecular pathway. ROAST can test for uni- or bi-direction regulation. Probes can also be weighted to allow for prior importance. The power and size of the ROAST procedure is demonstrated in a simulation study, and compared to that of a representative permutation method. Finally, ROAST is used to test the degree of transcriptional conservation between human and mouse mammary stems.
Availability: ROAST is implemented as a function in the Bioconductor package limma available from www.bioconductor.org
Contact: smyth@wehi.edu.au
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
A gene set test is a differential expression analysis in which a set of putatively co-regulated genes is treated as a unit. A single P-value is evaluated for the set rather than evaluating individual P-values for individual genes. Typically, the gene set is chosen to represent a particular molecular pathway. In this way, gene set testing can simplify and organize differential expression analyses by focusing attention on larger and more biologically meaningful processes than individual genes. Gene set tests are valuable for relating expression patterns across different studies, even across different platforms or species (Manoli et al., 2006). Gene set tests can be statistically more powerful than genewise tests as evidence is accumulated from many genes.
There are a number of gene set testing methodologies with different aims. This article is concerned with what might be called focused gene set testing, in which interest focuses on one or more gene sets of special relevant to the experiment at hand (Dinu et al., 2007; Goeman et al., 2004; Jiang and Gentleman, 2007; Tian et al., 2005). This approach contrasts with what might be called battery gene set testing, in which a large database of gene sets is evaluated on a microarray dataset, to see whether any sets stand out from the others (Dørum et al., 2009; Efron and Tibshirani, 2007; Saxena et al., 2006). Battery gene set testing was made popular by the Gene Set Enrichment Analysis (GSEA) method of Mootha et al. (2003) and Subramanian et al. (2005). In focused gene set testing, each set is evaluated on its own terms. In battery gene set testing, sets are evaluated relative to the other sets in the database. For this reason, methods designed for battery testing cannot be applied to individual gene sets.
Focused gene set tests can be classified into those which evaluate P-values by permuting samples and those which permute genes. Methods that permute genes are limited by the fact that they treat genes as if statistically independent. This assumption is usually suspect, particularly when the set is specifically chosen to contain co-regulated genes. Moreover, gene permutation P-values are very sensitive to inter-gene correlations, potentially leading to dangerously over-stated statistical significance (Dørum et al., 2009; Efron and Tibshirani, 2007). This has led some authors to conclude that statistically rigorous testing is only possible by permuting samples (Goeman and Bühlmann, 2007).
The need to permute samples to obtain P-values severely limits the experimental designs which can be analysed. Permutation is basically limited to two-group comparisons, with a moderate to large number of replicates in each group. Some authors have adapted permutation to the needs of one- or two-way ANOVA designs. Adewale et al. (2008) and Hummel et al. (2008) suggested permuting sample labels while holding all covariates fixed except the covariate of interest. Oron et al. (2008) permuted sample labels within each level of a blocking factor. However, these stratified permutation methods are still limited to special designs and moderate to large numbers of replicates.
Another possibility is to try to model inter-gene correlations explicitly using mixed linear models (Wang et al., 2008). As genes operate with complex covariance patterns, this will be a simplification of the truth and, again, the method is restricted to experimental designs with a special structure.
We present a statistically rigorous gene set test that fully allows for gene-wise correlations while being applicable to almost any experimental design. We have in mind the sort of small but complex experimental designs that arise frequently in experimental medicine, which may have many experimental factors but only a few biological replicates. Instead of permutation, we use rotation, a Monte Carlo simulation technology recently proposed for multivariate regression models (Langsrud, 2005). Rotation has also been used recently for a battery gene set testing (Dørum et al., 2009), but here we use it for focused testing. Rotation can be viewed as analogous to fractional permutation. Since there is no limit on the number of rotations which can be done, the problem of granularity of P-values in small sample sizes is avoided. Rotation can be applied to the residual space of a linear model, and so utilizes all the available degrees of freedom regardless of the experimental design.
Section 2 of this article describes our statistical model for microarray data. Our implementation of ROAST (rotation gene set testing) allows for any experimental design, which can be expressed as a linear model, and also accommodates array quality weights and correlations between samples. To ensure good behaviour in very small samples, ROAST uses empirical Bayes t-statistics for gene-level differential expression (Smyth, 2004). Then our approach to gene set testing is described. Alternative hypotheses are considered for gene sets in which all genes are expected to be regulated in the same direction and for those in which genes may vary in both directions. Scenarios are considered in which only a subset of the genes in the set actively contribute to the overall result. Different gene set summary statistics are developed that are appropriate for detecting different proportions of active genes.
Next, the numerical implementation of the ROAST algorithm is outlined. The power and size of the ROAST procedure is demonstrated in a simulation study, and compared to a representative permutation method. Finally, ROAST is used to test the degree of transcriptional conservation between human and mouse mammary stems
2 THE STATISTICAL MODEL
2.1 Data and gene set
We assume that an experiment or observational study has been conducted resulting in expression data on G probes in each of n target RNA samples. Different treatments, phenotypes or other characteristics are associated with the n samples, and we are interested in finding genes that are differentially expressed (DE) between the samples in some particular way. For example, the samples might be in two or more groups, and we want to find genes DE between two specified groups. Or we might want to find genes that show an interaction between two treatments. Or we might want to find genes that show a trend in a time-course experiments. Our aim is to accommodate quite general and arbitrarily complex experiments, so there is no limit on the number of treatment factors associated with the experiment, provided of course there is enough data available to estimate all the effects.
We assume that a particular set of probes or genes is of prior interest. This a priori specified gene set might represent a molecular pathway believed to be relevant to the experiment, or it might be a gene list from a previous microarray experiment hypothesized to be related to the current experiment. We want to test whether the gene set contains any probes that are DE. In some cases, the expected direction and magnitude of change may be specified in advance for individual genes. We are particularly interested in detecting sets that contain a good proportion, say 25–50% or more, of co-regulated DE genes. Sets that contain a small proportion of DE genes are of a lower level of interest.
2.2 The linear model
To be as general as possible, we use a linear model representation for the experiment, as described previously (Smyth, 2004). Write ygi for the log2-expression value for sample i and probe g. We assume that the expression values have already been background corrected, normalized and, perhaps, filtered in a way appropriate for the microarray or expression platform. Write yg = (yg1,…, ygn)T for the vector of expression values for probe g. We assume
where X is an n× p design matrix of full column rank and αg an unknown coefficient vector of length p. The matrix X represents the design of the experiment, and describes how the different treatment factors are assigned to the RNA samples. The coefficients αgj, which make up αg, represent the treatment effects or differences associated with probe g. We also assume
where σ2g is the unknown probewise variance and W a positive-definite matrix of weights. The weight matrix W provides the ability to incorporate array weights if desired (Ritchie et al., 2006) or to incorporate correlations between samples (Smyth et al., 2005). If W is unknown, then it is estimated from the expression data on all G probes, as described in the papers just cited. After this step, W is treated as known.
2.3 Distributional assumptions across genes
We assume that the ygi are multivariate normal, at least for the probes in our gene set, with a general but unknown correlation structure between the probes. Multivariate normality is theoretically a strong assumption, but we show later that our test procedure is robust against departures from normality.
To borrow information across probes when estimating standard errors, we assume a hierarchical model for the probewise variances, as described previously (Lonnstedt and Speed, 2002; Smyth, 2004; Wright and Simon, 2003). We assume that the probewise variances are sampled from an inverse-χ2 distribution
where s20 is the prior variance and d0 the prior degrees of freedom. The prior variance represents typical variability and the prior degrees of freedom control how consistent the variability is across probes.
2.4 Probe level tests
A particular contrast of the coefficients, represented by βg =cTαg, is assumed to be of interest. We want to find genes for which βg is non-zero. The linear model described above is fitted to the expression data for each probe. The t-statistic for testing βg = 0 is
where 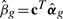 is the least squares estimator of βg, sg the residual SD for probe g and v = cT(XTWX)−1c the unscaled SD of
is the least squares estimator of βg, sg the residual SD for probe g and v = cT(XTWX)−1c the unscaled SD of 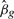 . Under the null hypothesis, tg follows a t-distribution on d = n − p degrees of freedom.
. Under the null hypothesis, tg follows a t-distribution on d = n − p degrees of freedom.
Following Wright and Simon (2003) and Smyth (2004), an improved test can be obtained by computing the posterior variances
| (1) |
and moderated t-statistics
Under the null hypothesis, 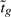 follows a t-distribution on d0+d degrees of freedom. The gain in degrees of freedom of the moderated over the ordinary t-statistic reflects the information that is borrowed from other probes when making inferences about an individual probe. The moderated t-test has been shown to be superior to other tests in comparative studies (Diboun et al., 2006; Kooperberg et al., 2005; Murie et al., 2009).
follows a t-distribution on d0+d degrees of freedom. The gain in degrees of freedom of the moderated over the ordinary t-statistic reflects the information that is borrowed from other probes when making inferences about an individual probe. The moderated t-test has been shown to be superior to other tests in comparative studies (Diboun et al., 2006; Kooperberg et al., 2005; Murie et al., 2009).
The hyper-parameters s0 and d0 in the prior distribution for σ2g are estimated from the expression data on all G probes as described by Smyth (2004). After this step, s0 and d0 are treated as known.
For the calculations in this article, the moderated t-statistics are transformed to equivalent standard normal random variables
| (2) |
where F and Ft are the cumulative distribution functions of the standard normal and td0+d distributions, respectively.
3 APPROACH TO GENE SET TESTING
3.1 Gene set hypotheses
Let 𝒮 be the set of indices of the probes in our gene set of interest. We want to test the null hypothesis H0 that βg = 0 for all g ∈ 𝒮. The alternative hypothesis depends on whether the DE genes in 𝒮 are expected to change in the same direction or not, and whether this direction is specified in advance. We consider three possible alternative hypotheses. The up hypothesis Hu is that βg > 0 for at least one g ∈ 𝒮. The down hypothesis Hd is that βg < 0 for at least one g ∈ 𝒮. The mixed hypothesis Hm is that βg≠0 for at least one g ∈ 𝒮, i.e. the genes can change in mixed (up or down) directions. Clearly, it is possible for more than one of the alternative hypotheses Hm, Hu or Hd to be true for the same gene set.
3.2 Gene weights
In some cases, there are prior reasons to give more weight to some genes in the gene set than others; for example, genes that are more highly expressed might be of more interest. We, therefore, allow the possibility of gene weights ag, which are used to weight the zg when the gene set summary statistic T is computed, similar to a suggestion of Jiang and Gentleman (2007).
Positive or negative gene weights can be used to reflect the expected direction of change of genes in the gene set. For example, the gene weights might be ±1 depending on whether the genes are known to be up- or down-regulated in a particular pathway, or the gene weights might be set equal to the log-fold changes for these genes in a prior experiment.
3.3 Gene set statistics
A test statistic T for the gene set 𝒮 is constructed from the moderated t-statistics for probes in the set. Several gene set level summary statistics have been proposed in previous research on gene set testing (Ackermann and Strimmer, 2009; Jiang and Gentleman, 2007). We propose a number of new summary statistics that have a good power for different gene set scenarios. Our statistics are computed in terms of the z-scores zg.
When all genes in the set 𝒮 are DE by about the same amount, the weighted mean of the genewise statistics is a logical gene set statistic. Write A = ∑g∈𝒮|ag| for the sum of absolute gene weights of genes in the set. To test the directional hypotheses Hu or Hd, we define Tmean = (∑g∈𝒮 agzg)/A. To test Hm, agzg is replaced by |agzg|.
When only a few genes in the set are DE, or if some log-fold-changes are much larger than others, the mean of the squared genewise statistics is a more sensitive measure. To test the mixed hypothesis Hm, we define Tmsq = (∑g∈𝒮 |ag|zg2)/A. To test Hu, the sum in the numerator of the statistic is taken only over those agzg that are positive. To test Hd, the sum in the numerator of the statistic is taken only over those agzg that are negative.
We define two more gene set statistics that are designed to be sensitive to gene sets in which about half of the genes are DE. The first is the mean-50 statistic. Let h be the smallest integer greater than or equal to half the number of genes in the set, i.e. h = ⌈(m+1)/2⌉, where m is the number of genes in the gene set. The mean50 statistic Tmean50 is the weighted mean of the top h most significant z-statistics. To test Hm, the mean50 statistic is the mean of the h largest absolute agzg values. To test Hu, the mean50 statistic is the mean of the h largest agzg values. To test Hd, the mean50 statistic is the mean of the h smallest agzg values.
The last statistic is inspired by the max–mean statistic of Efron and Tibshirani (2007). We call it floor-mean because it applies a floor value to the z-statistics. To test Hu, we compute the floored genewise statistics fg = max(zg, 0). To test Hd, the floored statistics are fg = min(agzg, 0). To test Hm, the floored statistics are fg = max(|zg|, 0.67), where 0.67 is the square-root of the median of the χ21 distribution. In each case, Tfloormean is computed as for Tmean but with fg in place of zg. The floor-mean statistic behaves similarly to the mean50 statistic, but is faster to compute.
3.4 Assigning P-values
We now seek to assign a P-value to the gene set statistic T. The distribution of T is unknown, because the correlation of expression scores between probes is unknown, so a resampling method must be used to assign the P-value. We avoid permuting genes because this would destroy the inter-probe dependence (Goeman and Bühlmann, 2007). We also avoid permuting samples, for several reasons. First, permutation requires a large number of replicate samples in order to provide a reliable P-value estimate, whereas we wish to analyse experiments with small numbers of replicates. Second, permutation does not have the ability to test general linear model hypotheses, such as we have specified. Third, permutation assumes samples to be identically distributed and exchangeable, whereas we wish to accommodate various types of weighting and correlation structures.
We instead adapt the idea of rotation tests from Langsrud (2005). Rotation tests use a type of simulation to generate P-values. The first step is to remove the nuisance parameters in the linear model, all the αgj other than the contrast of interest βg, by projecting the data for each probe onto the d+1 dimensional residual space orthogonal to them. This yields a set of d+1 independent residuals, such that the t-statistic 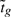 can be computed from the first residual. This step allows us to test βg = 0 without making assumptions about the other coefficients in the linear model. The second step randomly rotates the residuals in d+1 dimensional space. For each rotation, the gene set statistic T is re-computed, and compared to the observed value. The final P-value is p = (b + 1)/(B + 1), where B is the total number of rotations and b the number that yield a rotation statistic at least as extreme as that observed. This is an exact P-value (Barnard, 1963).
can be computed from the first residual. This step allows us to test βg = 0 without making assumptions about the other coefficients in the linear model. The second step randomly rotates the residuals in d+1 dimensional space. For each rotation, the gene set statistic T is re-computed, and compared to the observed value. The final P-value is p = (b + 1)/(B + 1), where B is the total number of rotations and b the number that yield a rotation statistic at least as extreme as that observed. This is an exact P-value (Barnard, 1963).
3.5 Estimating the active proportion
The above procedure attaches a P-value to the gene set. When the P-value is statistically significant, it is of interest to know how many genes in the set are contributing to this result. We consider a gene to be active in the result if 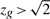 (for Hu) or
(for Hu) or 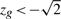 (for Hd) or
(for Hd) or 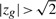 for (for Hm). The threshold of
for (for Hm). The threshold of 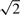 is somewhat arbitrary but is motivated by Akaike's information criterion in the model selection theory, wherein the addition of one parameter to a model is considered worthwhile if it improves the χ2 goodness of fit by two or greater.
is somewhat arbitrary but is motivated by Akaike's information criterion in the model selection theory, wherein the addition of one parameter to a model is considered worthwhile if it improves the χ2 goodness of fit by two or greater.
4 NUMERICAL IMPLEMENTATION
4.1 Independent residual effects
For the statistical model considered here, we are able to substantially simplify the rotation P-value computations outlined by Langsrud (2005), resulting in a very fast algorithm. All the computations below are for genes g in 𝒮 only. Other genes can be ignored.
The first step is to remove the nuisance parameters, i.e. the contrasts not of interest, from the linear model. This is done by projecting each yg onto the space orthogonal to the columns of X not involved in the null hypothesis.
Let C be an invertible p × p matrix with last column equal to c, and write βg = CTαg. Note
and that βg, as defined in Section 2.4, is the last element of βg. In this way, the linear model is re-parameterized so that the null hypothesis concerns the last regression coefficient.
The projection is obtained as a byproduct of the usual QR decomposition used in the numerical calculations for fitting the linear model,
We use the full QR-decomposition in which Q is n×n. Let Q2 be Q with the first p − 1 columns removed. Then the nuisance parameters are removed by the projection
for each g in 𝒮. In the actual numerical computations, the vector ug can be obtained from the QR-decomposition without forming Q or Q2 explicitly.
Under the null hypothesis, the elements ug1,…, ug,d+1 of ug are independent and identically N(0, σ2g). We have
then the posterior variance is computed from (1), and finally the moderated t-statistics are
4.2 Rotation
Let ρ2g = ugTug. The rotation test method rotates the vector of residuals ug to a random point ug* on the d + 1-sphere of radius ρg. For every rotation, the moderated t-statistics and the overall gene set statistic T are recomputed. After a large number of rotations, the Monte Carlo P-value is computed as above.
It is actually only necessary to randomly generate the first element u1* of u*g, because the residual variances for the rotated data can be computed from sg*2 = (ρg2 − u1*2)/d. The computation is extremely efficient. A random rotation vector r is generated satisfying rTr = 1. Then u1* is generated for all probes in the gene set by
where U is the m × (d + 1) matrix with rows ugT for g in 𝒮. This yields rotated t-statistics and z-statistics for each gene in the gene set, and hence to a null distribution value T* of the gene set statistic. New rotation vectors r and T* are generated large number of times, typically B = 10 000.
5 SIMULATION STUDY
5.1 Scenarios
Four multi-group experimental designs were simulated. The first design (D1–3) was a three-group experiment with n1 = n2 = 3 replicate samples in the first two groups and n3 = 20 replicate samples in the third. Interest is in differential expression between the first two groups. The second design (D1–5) was the same but with n1 = n2 = 5 replicate samples in the first two groups. The third (D2–3) and fourth (D2–5) designs were two-way factorial designs with two levels per factor, with three samples and five samples per group, respectively. For the factorial designs, interest was in differential expression for the first factor, the other being a blocking factor.
Each simulated dataset had G = 10 000 probes per array. Genes were divided either into 250 gene sets of 40 genes each, or into 10 gene sets of 1000 genes each. In each case, only the first gene set was simulated to contain DE genes. Genewise variances were simulated according to the model described in Section 2 with d0 = 4 and s0 = 0.25.
For each design, datasets were simulated in 10 scenarios with different proportions of up- and down-regulated genes in the set, and either independent or correlated expression profiles. In each scenario, the DE genes share the same fold-change. The fold-change was chosen for each scenario to make the highest powers around 80%. Table 1 shows the scenarios for designs D1–3 and D1–5. Smaller fold-changes were simulated with 1000 genes in each set than with 40 so as to keep the power about the same in each case (Table 1).
Table 1.
Ten simulated scenarios for designs D1–3 and D1–5
| Scenario | Proportion | Proportion | Correlation | Log-fold | Hypothesis |
|---|---|---|---|---|---|
| up | down | change | tested | ||
| 1 | 1.00 | 0.00 | 0.0 | 0.1 (0.02) | up |
| 2 | 1.00 | 0.00 | 0.1 | 0.2 (0.2) | up |
| 3 | 0.25 | 0.00 | 0.0 | 0.3 (0.07) | up |
| 4 | 0.25 | 0.00 | 0.1 | 0.4 (0.2) | up |
| 5 | 0.50 | 0.50 | 0.0 | 0.2 (0.07) | mixed |
| 6 | 0.50 | 0.50 | 0.1 | 0.2 (0.2) | mixed |
| 7 | 0.20 | 0.20 | 0.0 | 0.3 (0.1) | mixed |
| 8 | 0.20 | 0.20 | 0.1 | 0.4 (0.2) | mixed |
| 9 | 0.00 | 0.00 | 0.0 | 0.0 (0.0) | up/mix |
| 10 | 0.00 | 0.00 | 0.1 | 0.0 (0.0) | up/mix |
Fold-changes are for 40 genes per set (or 1000 genes per set). Genes regulated in the same direction are positively correlated, those in opposite directions are negatively correlated. In Scenario 10, the correlation applies to all genes in the set. Scenarios differ according to the proportion of up- and down-regulated genes in the set and the intergene correlation.
5.2 Size
The most important property of a statistical test is that its size (Type I error rate) is controlled correctly. It follows from the theory of rotation tests that ROAST must hold its size correctly if the data is normally distributed, and this was confirmed by simulations (Supplementary Tables S1 and S4).
To explore the robustness of ROAST to non-normal data, we simulated expression data to be exponentially distributed. The exponential distribution is highly right skew, far more non-normal than would be seen in any real microarray experiment. To simulate correlated exponential random variables, data was first simulated as for the normal simulations (Table 1, Scenarios 9 and 10), then transformed to be exponentially distributed, via the appropriate normal and exponential cumulative distribution functions. Even in this extreme situation, ROAST continued to hold its power correctly when used with the mean, mean50 or floor-mean gene set statistics (Supplementary Table S2). This was not quite true with the msq gene summary statistic but, even then, the true test sizes were only slightly higher than nominal sizes. We conclude that ROAST is likely to be robust to non-normality in realistic data situations.
5.3 Power
ROAST was found to have good power to detect sets containing DE genes with very modest fold-changes, across a range of scenarios (Supplementary Tables S3 and S5). The mean set statistic was the best of the statistics for detecting scenarios with all genes changing (Supplementary Tables S3 and S5, Scenarios 1, 2, 5 and 6). The msq set statistic was the best of the statistics for detecting scenarios with only a minority of genes changing (Supplementary Tables S3 and S5, Scenarios 3, 4, 7 and 8). The mean50 and floor-mean statistics were intermediate between the mean and msq statistics in both scenarios. The statistics can be ordered as mean, floor-mean, mean50, then msq, in terms of increasing sensitivity to a subset of DE genes and decreasing sensitivity to all genes changing by the same amount. In all cases, power was reduced for a given fold-change when the genes in set had correlated expression values. More genes in the gene set increased power, but this increase was not so apparent when the genes were correlated (Supplementary Tables S3 and S5).
5.4 Comparison with permutation tests
The key advantage of ROAST is the ability to handle situations for which no other gene set test is suitable. It is of interest, however, to see how ROAST compares with other methods when conditions are suitable for them. We compared ROAST to GSEAlm (Oron et al., 2008). We chose GSEAlm because it is a high-quality representative of permutation methods, and because it has more flexible options that most permutation software, being able to handle one-way ANOVA or block designs.
No permutation algorithm can be expected to work well on design D1–3, with only three arrays per group. Indeed, we found that GSEAlm failed to hold its size when the number of available permutations was not large (data not shown). This can be traced to the way in which the permutation P-values are computed. Whereas ROAST computes an exact P-value, GSEAlm, like all permutation software that we know of, computes an estimate 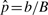 of the P-value, where B is the number of permutations and b the number of permuted statistics as extreme as that observed. This estimated P-value can often be zero if the number of permutations is modest, resulting in an over-statement of statistical significance (Ernst, 2004).
of the P-value, where B is the number of permutations and b the number of permuted statistics as extreme as that observed. This estimated P-value can often be zero if the number of permutations is modest, resulting in an over-statement of statistical significance (Ernst, 2004).
GSEAlm was compared with ROAST on designs D1–5, D2–3 and D2–5. GSEAlm is computationally time consuming, so only 100 datasets were simulated for each scenario for each design. On design D1–5, GSEAlm is somewhat less powerful than ROAST, regardless of scenario and regardless of ROAST set statistic (Supplementary Table S6). This was presumably because ROAST is able to make use of residual degrees of freedom from the 20 arrays in the third group that is not involved in the hypothesis test. GSEAlm was not tested on the mixed scenarios, as it is not able to test bi-directional hypotheses. On the two-factor designs D2–3 and D2–5, for testing directional hypotheses, GSEAlm is similar in power to ROAST with the mean gene set statistic (Supplementary Tables S7 and S8).
6 MAMMARY STEM CELLS
Mammary epithelial cells can be sorted into a family tree of cell populations, including a mammary stem cell-enriched population (MaSC), luminal progenitor cells (LP) and mature luminal cells (ML) (Visvader and Lindeman, 2006). This family tree is of tremendous interest for many reasons, for example, because mammary stem cells or luminal progenitor cells may represent the cell of origin for different types of breast cancer (Lim et al., 2009). Most experimental research on mammary cells is undertaken using mouse as the model organism. Hence it is critically important to establish that results observed for mice will remain valid for humans.
Gene set testing provides a powerful way to relate expression profiles across different platforms or different species. In this example, we use gene set tests to demonstrate that the transcriptional differences between MaSC, LP and ML cells are broadly conserved between mouse and human. We focus particularly on stem cells. We define a gene set associated with stem cells in mouse, then examine the profile of this set in the human data. This allows us to confirm conclusions from Lim et al. (2010), but here we use ROAST to formally take account of inter-gene correlation in evaluating statistical significance, which we were not able to do in the earlier publication.
Mammary epithelial and stroma cells were sorted from breast tissue samples from three human patients. RNA was profiled on two Illumina HumanWG-6 V3 BeadChips, comprising 12 arrays (Table 2). A similar experiment was undertaken using mouse samples and Illumina mouse WG-6 V2 BeadChips. The microarray data was normalized and annotated as previously described (Lim et al., 2010). The data is available as GEO database series GSE16997 and GSE19446 for human and mouse, respectively.
Table 2.
Human mammary cell populations profiled
| Array | Cell population | Patient | BeadChip | Block |
|---|---|---|---|---|
| 1 | MaSC enriched | A | 4380071023 | 1 |
| 2 | Stroma | A | 4380071023 | 1 |
| 3 | ML | A | 4380071023 | 1 |
| 4 | LP | A | 4380071023 | 1 |
| 5 | MaSC enriched | B | 4380071023 | 2 |
| 6 | Stroma | B | 4380071023 | 2 |
| 7 | ML | B | 4380071027 | 3 |
| 8 | LP | B | 4380071027 | 3 |
| 9 | MaSC enriched | C | 4380071027 | 4 |
| 10 | Stroma | C | 4380071027 | 4 |
| 11 | ML | C | 4380071027 | 4 |
| 12 | LP | C | 4380071027 | 4 |
Block represents unique patient–BeadChip combinations.
A set of genes was selected to represent the transcriptional signature of MaSC cells in mouse, as previously described (Lim et al., 2010). Briefly, we identified signature probes as those significantly up- or down-regulated in MaSC cells versus both of the other two mammary epithelial populations, LP and ML. This yielded 2616 up- and 2305 down-regulated MaSC signature probes in mouse, corresponding to 3410 unique gene symbols, of which 2754 had human orthologs. This defined then a set of 2754 human gene symbols. We set gene weights ai equal to the log-fold-change observed for that gene in the mouse date, specifically the average of the log-fold-changes observed for MaSC versus LP and MaSC versus ML. Hence genes were weighted according to the strength and direction of their regulation in MaSC cells.
It is important to account for biological and technical correlations when analysing microarray data, as this improves the precision and reliability of the results, even with high-quality data such as we analyse here. For the human samples described in Table 2, it is essential to account for correlations between samples taken from the same patient. Also to be expected are more or less subtle batch effects between BeadChips. Our linear modelling approach permits us to adjust for patient and BeadChip effects in a variety of ways. We could include patient as a fixed effect in our linear model, when testing for differential expression between the cell populations. Alternatively, we could include patient as a random effect, with BeadChip as a fixed effect. The effects were relatively subtle, so we combined patient and BeadChip into one blocking factor (Table 2), which was included as a random effect. In this approach, samples are treated as correlated only if they corresponded to the same patient and the same BeadChip. Using the method of Smyth et al. (2005), the intra-block correlation was estimated to be 0.08, a small but detectable positive correlation between samples in the same block. Our weight matrix W, therefore, was made up of correlations ρij, where ρij was 1 if i = j, 0.08 if arrays i and j are in the same block and 0 otherwise.
ROAST was used to test whether the mouse MaSC signature gene set was able to distinguish human MaSC from LP cells, and human MaSC from ML (Table 3). Genes were weighted in the test by their direction of change in mouse, hence the alternative hypothesis Hu corresponds to genes changing in the same direction in human as they did in mouse, whereas Hd represents change in opposite directions. The highly significant results for Hu show that the main body of genes change in the same direction in human as in mouse. The non-significant P-values for Hd for the mean, mean50 and floormean statistics deny any sizeable group of genes changing in the opposite direction. The msq statistic is highly sensitive to even a small number of genes, and gives suggestive P-values for Hd. Close inspection of individual genes shows that a small number of the signature genes do indeed move in the reverse directions in human and mouse, explaining the msq result. Nevertheless, the high degree of conservation across species supports mouse as a model system for the study of mammary gland development.
Table 3.
ROAST P-values for distinguishing human cell populations using the mouse MaSC signature gene set
| MaSC−LP |
MaSC−ML |
|||
|---|---|---|---|---|
| Summary statistic | Hu | Hd | Hu | Hd |
| mean | 0.001 | 1.000 | 0.001 | 1.000 |
| mean50 | 0.001 | 0.397 | 0.001 | 0.396 |
| floormean | 0.001 | 0.345 | 0.001 | 0.318 |
| msq | 0.001 | 0.074 | 0.001 | 0.048 |
P-values based on 999 rotations.
7 DISCUSSION
ROAST is the first focused gene set test that correctly allows for inter-gene correlation and gives statistically rigorous P-values for small or complex microarray experimental designs. Like Dørum et al. (2009), we use rotation to evaluate P-values, but our methodology is for self-contained tests with specially targeted gene sets, whereas they considered competitive tests between a large database of gene sets. ROAST is the only existing software that could have been used for our mammary stem cell data example.
The use of rotation offers many advantages over permutation. It is computationally much faster. It yields exact P-values, so that ROAST holds its nominal size correctly even for small samples. The number of rotations can be chosen large enough to avoid any problem with granularity of P-values. Dørum et al. (2009) considered one or two-group t-tests, whereas we take full advantage of the possibilities of linear models. ROAST is applicable to any experiment that can be analysed using an linear model, i.e. to arbitrarily complex experiments. The gene set can be tested in any contrast between the coefficients, not necessarily a simple comparison between two groups. Indeed, our development could easily be extended to test gene sets in two or more contrasts simultaneously. In that case, we would replace our moderated t-statistics with F-statistics. Our implementation allows for correlations between RNA samples and for array quality weights, assuming that the correlations or weights can be estimated from the entire ensemble of probes on the arrays. In all cases, the test takes advantage of all available residual degrees of freedom. Our implementation uses empirical Bayes test statistics, which should add additional stability in small samples, especially when the gene set contains only a few genes.
Rotation theoretically depends on multivariate normality, but simulations with grossly non-normal data shows that ROAST is insensitive to departures from normality, agreeing with analogous results of Dørum et al. (2009).
We propose a number of new set summary test statistics. Like Jiang and Gentleman (2007), but unlike Ackermann and Strimmer (2009), we find that the choice of summary statistic does affect performance. Our summary statistics are designed to vary in their sensitivity to gene sets that contain a small proportion of DE genes. No statistic is universally better than the others in all situations. When the mean statistic is used, a gene set can be significant for the up or down hypothesis, but not both. With the other set statistics, a gene set can be judged as significant in both directions, if there is a subset of genes that are up-regulated and a subset that are down-regulated. Therefore, floormean, mean50 and especially msq give higher resolution results, more nuanced but potentially less clear cut. To help make judgements in this respect, ROAST gives an estimate of the proportion of genes that actively contribute to a significant result. The simulations presented in this article assumed a uniform fold-change for all DE genes. Simulations with random fold-changes tend to improve the performance of msq, and also of mean50 and floormean, relative to that of the mean statistic (data not shown). We suggest mean50 as a good compromise in many biological situations.
Potential applications for ROAST include those where the set might not be made up of genes. We have for example used it in exon-level expression analyses to test whether any exon of a given gene is DE.
Funding: Australian Postgraduate Award (to D.W.); National Health and Medical Research Council (grants to G.K.S. and J.E.V.).
Conflict of Interest: none declared.
Supplementary Material
REFERENCES
- Ackermann M, Strimmer K. A general modular framework for gene set enrichment analysis. BMC Bioinformatics. 2009;10:47. doi: 10.1186/1471-2105-10-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adewale AJ, et al. Pathway analysis of microarray data via regression. J. Comput. Biol. 2008;15:269–277. doi: 10.1089/cmb.2008.0002. [DOI] [PubMed] [Google Scholar]
- Barnard GA. Discussion of the spectral analysis of point processes (by MS Bartlett) J. R. Stat. Soc. 1963;25:294. [Google Scholar]
- Diboun I, et al. Microarray analysis after RNA amplification can detect pronounced differences in gene expression using limma. BMC Genomics. 2006;7:252. doi: 10.1186/1471-2164-7-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinu I, et al. Improving gene set analysis of microarray data by SAM-GS. BMC Bioinformatics. 2007;8:242. doi: 10.1186/1471-2105-8-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dørum G, et al. Rotation testing in gene set enrichment analysis for small direct comparison experiments. Stat. Appl. Genet. Mol. Biol. 2009;8 doi: 10.2202/1544-6115.1418. Article 34. [DOI] [PubMed] [Google Scholar]
- Efron B, Tibshirani R. On testing the significance of sets of genes. Ann. Appl. Stat. 2007;1:107–129. [Google Scholar]
- Ernst M. Permutation methods: a basis for exact inference. Stat. Sci. 2004;19:686–696. [Google Scholar]
- Goeman JJ, Bühlmann P. Analyzing gene expression data in terms of gene sets: methodological issues. Bioinformatics. 2007;23:980–987. doi: 10.1093/bioinformatics/btm051. [DOI] [PubMed] [Google Scholar]
- Goeman JJ, et al. A global test for groups of genes: testing association with a clinical outcome. Bioinformatics. 2004;20:93–99. doi: 10.1093/bioinformatics/btg382. [DOI] [PubMed] [Google Scholar]
- Hummel M, et al. GlobalANCOVA: exploration and assessment of gene group effects. Bioinformatics. 2008;24:78–85. doi: 10.1093/bioinformatics/btm531. [DOI] [PubMed] [Google Scholar]
- Jiang Z, Gentleman R. Extensions to gene set enrichment. Bioinformatics. 2007;23:306–313. doi: 10.1093/bioinformatics/btl599. [DOI] [PubMed] [Google Scholar]
- Kooperberg C, et al. Significance testing for small microarray experiments. Stat. Med. 2005;24:2281–2298. doi: 10.1002/sim.2109. [DOI] [PubMed] [Google Scholar]
- Langsrud Ø. Rotation tests. Stat. Comput. 2005;15:53–60. [Google Scholar]
- Lim E, et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in brca1 mutation carriers. Nat. Med. 2009;15:907–913. doi: 10.1038/nm.2000. [DOI] [PubMed] [Google Scholar]
- Lim E, et al. Transcriptome analyses of mouse and human mammary cell subpopulations reveals multiple conserved genes and pathways. Breast Cancer Res. 2010;12:R21. doi: 10.1186/bcr2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonnstedt I, Speed T. Replicated microarray data. Stat. Sin. 2002;12:31–46. [Google Scholar]
- Manoli T, et al. Group testing for pathway analysis improves comparability of different microarray datasets. Bioinformatics. 2006;22:2500–2506. doi: 10.1093/bioinformatics/btl424. [DOI] [PubMed] [Google Scholar]
- Mootha VK, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- Murie C, et al. Comparison of small n statistical tests of differential expression applied to microarrays. BMC Bioinformatics. 2009;10:45. doi: 10.1186/1471-2105-10-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oron AP, et al. Gene set enrichment analysis using linear models and diagnostics. Bioinformatics. 2008;24:2586–2591. doi: 10.1093/bioinformatics/btn465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, et al. Empirical array quality weights in the analysis of microarray data. BMC Bioinformatics. 2006;7:261. doi: 10.1186/1471-2105-7-261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena V, et al. Absolute enrichment: gene set enrichment analysis for homeostatic systems. Nucleic Acids Res. 2006;34:e151. doi: 10.1093/nar/gkl766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth GK. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004;3 doi: 10.2202/1544-6115.1027. Article3. [DOI] [PubMed] [Google Scholar]
- Smyth GK, et al. Use of within-array replicate spots for assessing differential expression in microarray experiments. Bioinformatics. 2005;21:2067–2075. doi: 10.1093/bioinformatics/bti270. [DOI] [PubMed] [Google Scholar]
- Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian L, et al. Discovering statistically significant pathways in expression profiling studies. Proc. Natl Acad. Sci. USA. 2005;102:13544–13549. doi: 10.1073/pnas.0506577102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visvader JE, Lindeman GJ. Mammary stem cells and mammopoiesis. Cancer Res. 2006;66:9798–9801. doi: 10.1158/0008-5472.CAN-06-2254. [DOI] [PubMed] [Google Scholar]
- Wang L, et al. An integrated approach for the analysis of biological pathways using mixed models. PLoS Genet. 2008;4:e1000115. doi: 10.1371/journal.pgen.1000115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright GW, Simon RM. A random variance model for detection of differential gene expression in small microarray experiments. Bioinformatics. 2003;19:2448–2455. doi: 10.1093/bioinformatics/btg345. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.