

Abstract
We introduce a novel experimental methodology for the reverse-phase protein microarray platform which reduces the typical measurement CV as much as 70%. The methodology, referred to as array microenvironment normalization, increases the statistical power of the platform. In the experiment, it enabled the detection of a 1.1-fold shift in prostate specific antigen concentration using approximately six technical replicates rather than the 37 replicates previously required. The improved reproducibility and statistical power should facilitate clinical implementation of the platform.
Keywords: Microarrays, Protein arrays, Reproducibility
Immunoassays such as Western blots and ELISA are a primary source of quantitative information in the fields of clinical chemistry and proteomics. In addition to these benchmark methods, researchers have begun developing new assays to meet the high-throughput challenges of 21st century biology. The level of imprecision (CV) is one of the main criteria used to evaluate the effectiveness and ultimate utility of these assays [1, 2]. Here, we describe a novel method for improving the reproducibility of measurements made by the developing platform known as reverse-phase protein microarrays (RPMAs). We refer to this new method as “Array Microenvironment Normalization” (AMN) and experiments indicate that it reduces measurement CV as much as 70%.
RPMAs are unique in that they immobilize protein extracts from many samples onto a planar substratum (typically nitrocellulose) via robotic printing and probe all extracts concomitantly with a well-validated antibody directed against a target protein. A detailed protocol for an iteration of the assay can be found in Spurrier et al. [3]. Typical within-array CV for the RPMA assay is approximately 15% in the linear range of detection while across-array variability is somewhat greater [4]. The variability can be broken into two types: random and systematic. Random variability arises from unknown and unpredictable sources and is accounted for by increasing the number of sample replicates. Systematic variability is due to identifiable imperfections in the assay itself and increasing the number of replicates may not account for the bias introduced. Potential sources of systematic error in RPMAs, analogous to many cDNA microarray sources [5–7], include but are not limited to the following:
Spatial inconsistencies in the planar substratum (nitrocellulose) density and permeability.
Non-uniform reagent and antibody incubation.
Pin-head inconsistency in dimensions and printing properties.
Evaporation of lysate buffer from the source plate throughout the robotic deposition process.
Humidity and temperature differences that affect the nitrocellulose-protein and antibody-protein binding kinetics, as well as the spread of reagents during incubation.
Differences in reagent lot and diluted antibody concentration from incubation to incubation.
Image collection and analysis process.
The simultaneous contribution of all sources of systematic error cause the measured variability to be spatially heterogeneous within arrays and unpredictable across arrays. Each spot on every array experiences a unique microenvironment of conditions (i–vii above) that influence the magnitude of variation in measured intensity. Thus, any viable variability reduction strategy must attempt to correct for the local microenvironment effects.
Total protein normalization (TPN) is used to account for systematic errors that result in total protein deposition differences across replicate spots within an array. TPN is also vital to enable the comparison of target protein signal across independent samples that have total protein concentration differences [8]. Using a fluorescent reporter based approach [9], a SYPRO ruby (Invitrogen) stain is first carried out per manufacturer’s instructions to yield the total protein signal (scanned at 532 nm). Subsequently, within the same array and after total protein values are collected, the target protein signal is detected using IRDye680 (LiCor, Lincoln, NE, USA) (scanned at 635 nm). The ratio of target to total protein signal for each corresponding spot is calculated to normalize expression to the signal per unit protein. While TPN accounts for total protein deposition differences, it ignores and potentially contributes to within-array spatial measurement variability and across-array scaling artifacts.
AMN was developed to correct for the remaining spatial and scaling variability. Figure 1 displays the array design that facilitates the correction for microenvironment measurement effects. The layout can be described as an alternating (checkerboard) pattern of common control and experimental samples on the array. Using 185 μm diameter pins, approximately 1500 spots composed of samples of interest and 1500 spots of control sample can be printed per array. The control sample should be carefully selected to have a total protein concentration similar to that of the study sample population and a target protein concentration within the linear range of detection. The ratio of each sample TPN signal to that of its surrounding controls is used as the feature of interest
Figure 1.

RPMA with AMN experimental design. Top is the array 1 total protein stain. Bottom is a rendering of the “checkerboard” control layout used to carry out AMN for each sample spot.
| (1) |
where indices i and j are the array row and column positions respectively, s is the TPN sample signal, and c is the TPN control signal.
The assumption is made that the density of target protein per unit total protein is equal for all controls regardless of the position on the array. Due to this equivalence, the variation in the control TPN measured intensities can be attributed to variability in the assay. Each control experiences nearly identical nitrocellulose properties, staining conditions, and even image focus plane to that of its adjacent samples. Any systematic variability caused by the microenvironment conditions affects each sample and its surrounding controls congruently, causing a highly correlated variation in measured intensity. The ratio of each sample to its local controls (Eqn. 1) thus removes (reduces) the correlated systematic variability. AMN also scales across-array measurements identically as all reported intensities are relative to the same control. Lastly, the layout of uniformly distributed controls on the array can provide a robust quality control construct for the assay. AMN has strong parallels to the internal control methodology developed by Alban et al. for 2-D DIGE as well as the two-dye common reference design of cDNA microarray studies, which have each been widely accepted to reduce systematic variability [10, 11].
To test the efficacy of AMN, experimental samples were created by pipetting a calculated amount of purified prostate specific antigen (PSA) (Calbiochem, San Diego, CA, USA) (stock solution concentration verified by Immulite 2000, Siemens) into a 250 μg/mL HeLa cell lysate (BD Biosciences, San Jose, CA, USA). HeLa cell lysate was chosen as it contains no endogenous PSA and represents a “real world” lysate with a physiological matrix of proteins. Five samples were created to have 1.4-fold differences in PSA concentration per sample: 50, 34.76, 24.16, 16.8, and 11.7 ng/mL as well as five samples with 1.1-fold differences: 28, 25.45, 23.14, 21.04, and 19.12 ng/mL. The sample containing 24.16 ng/mL PSA was selected as the control for this experiment. Using the layout in Fig. 1, 32 replicates of each sample were printed (spatially randomized) across each array surface. Four arrays (FAST Slides Whatman, Florham Park, NJ, USA) were printed with an Aushon 2470 arrayer equipped with 185-μm pins (Aushon Biosystems, Billerica, MA, USA).
Three arrays were stained for both total protein (SYPRO) and PSA. The PSA target protein stain used an a-PSA antibody from Dako diluted 1:5000 in antibody diluent. The fourth array underwent the exact same protocol but the a-PSA antibody was omitted as a negative control. Imaging was carried out using the Vidar Revolution™ 4200 laser scanner (Vidar Systems, Herndon, VA, USA). All array tiff images (10 μm resolution) were uploaded into MicroVigene (Vigene Tech, Carlisle, MA, USA) for signal quantification. Text files for each array were exported from MicroVigene and imported into R to implement AMN (www.R-Project.org)[12]. Each array was analyzed once utilizing only TPN and then a second time using AMN. The signal from the negative control stain was not significant, confirming the specificity of the staining process to the primary antibody. All array images, raw data, and AMN data are provided: http://gforge.icm.jhu.edu/gf/project/rpma_locl_cntrl/docman/.
The improvement in reproducibility and linearity for all arrays is exemplified by Fig. 2A and B, which displays scatter plots of measured relative intensity versus PSA concentration before and after AMN for array 1. Figure 2C and D displays scatter plots of the AMN relative intensity versus the TPN relative intensity for each sample of array 1 in order to visualize the difference in variability. AMN shows significant improvement in separation of sample measurements based on PSA concentration. Along the AMN dimension (y-axis), cutoff intensities can be established that completely separate the samples which differ by 1.4-fold in PSA concentration (Fig. 2C), and near complete separation exists for the samples with 1.1-fold differences (Fig. 2D). Separation of the data based on PSA concentration is not feasible without AMN (x-axis of Fig. 2C and D) demonstrating its utility.
Figure 2.

Linearity and reproducibility improvement with AMN demonstrated for array 1. (A) Scatter plot of TPN relative intensity (no AMN) for each sample of array 1 versus the known PSA concentration. (B) Scatter plot of AMN relative intensity for each sample of array 1 versus the known PSA concentration. (C) Scatter plot of the AMN relative intensity versus TPN relative intensity for samples of array 1 which differed in PSA concentration by 1.4-fold. (D) Same as (C) except displays samples differing in PSA concentration by 1.1-fold for array 1. The spread in the data is always much greater before AMN is implemented.
The improved resolution of measurements in Fig. 2C and D through the implementation of AMN can be quantified as a reduction in measurement CV for all three arrays (Fig. 3A). CVs were calculated for each of the ten samples based on their 32 replicates, the average of the CVs was taken as the within-array CV. Using the TPN relative intensities, the average CVs for arrays 1, 2, and 3 were 17, 11, and 33%, respectively. Implementing AMN, the CV dropped to 4, 4, and 8% for an average relative reduction of 72±6%. Similarly, the average across-array CV dropped by 68%. The significant improvement in both within and across-array variability supports the hypothesis that AMN controls for systematic variability as described. The higher CV of the third array was due to an artifact (region of low signal possibly caused by systemic error sources (i) and/or (ii) in the staining that can be seen by downloading the image. The artifact may not have been identifiable without the alternating control layout and demonstrates AMN’s additional utility for quality control.
Figure 3.
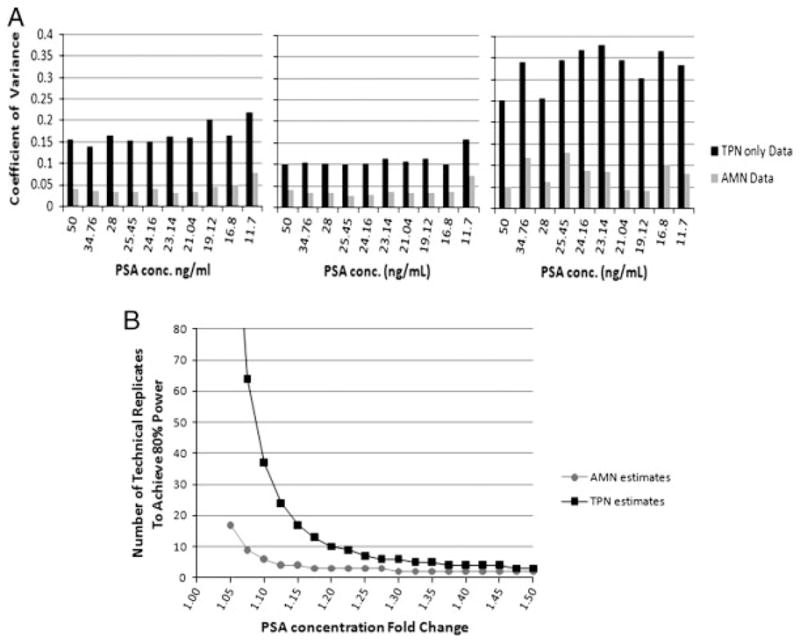
AMN improves the average coefficient of variance by 72% which results in an increased power to detect shifts in expression with fewer replicates. (A) Bar plots of measured coefficient of variance for the 32 replicates of each sample containing the specified amount of PSA for arrays 1, 2 and 3 both with and without AMN. (B) Power curve displaying the minimum number of technical replicates needed to detect the fold change specified in the x-axis with p<0.05 and power >0.80 using a t-test when the CV=5 or 15% (representative of the CV with and without AMN).
AMN’s significant reduction in measurement CV results in an ability to detect smaller shifts in true target protein concentration using fewer technical replicates, known as an increase in the statistical power. Figure 3B shows the estimated number of replicates needed to detect shifts in PSA concentration of 1.05- to 1.5-fold when the CV is 15 versus 5%, representing the approximate CV reduction by AMN. Each point in Fig. 3B is the calculated number of replicates needed to detect the specified fold shift in true PSA concentration with a p-value <0.05 and power >0.80 using a two-sided t-test. In the experiment, measurements were made using the arrays to detect PSA concentration shifts of both 1.1 and 1.4-fold. In Fig. 3B, before AMN was implemented, we estimated that detecting the shifts in PSA concentration of 1.1 and 1.4-fold would require 37 and 4 replicates, respectively. After AMN implementation, the shifts were predicted to be detectable with just six and two replicates.
These estimates were verifiable using the collected measurements from the arrays. The first step was to set the number of technical replicates (2–32) to be used in the power calculation. Then, the PSA samples had the specified number of replicates selected at random from among their 32 total reps on the array. Using the selected replicate measurements, a t-test was applied for each of the 1.1 and 1.4-fold comparisons to determine if the null hypothesis was rejected (cutoff p<0.05). For each number of replicates, random selection and the t-test significance calculation were repeated 100 times. The percentage of comparisons that produced a significant p-value was used as the estimate of the power. To achieve a power of 80% with AMN, arrays 1–3 needed to use 4, 4, and 8 replicates to detect a 1.1-fold change in PSA and 3, 2, and 3 replicates to detect a 1.4-fold shift. Using TPN only, the number of replicates required to detect the 1.1-fold shift were greater than 32, 16, and greater than 32 as arrays 1 and 3 could not achieve 80% power with all 32 replicates. To detect the 1.4 PSA shift, the number of replicates required were 4, 3, and 12. The calculated number of replicates for each array agree well with the number estimated in Fig. 3B, demonstrating that the theoretical increase in statistical power by AMN implementation translates to the physical measurements.
Overall, the Pearson correlation coefficient (R2) of measured intensity to PSA concentration (for arrays 1–3, respectively) increased from (0.92, 0.96, and 0.76) to (0.99, 0.99, and 0.975) when using AMN. The across-array correlation (array 1 to 2, 2 to 3, and 1 to 3) increased from R2=(0.98, 0.84, and 0.84) to (0.99, 0.99, and 0.99), while the slope of the linear fit improved from (0.56, 0.57, and 0.99) to (1.02, 1.02, and 0.998). A perfect across-array intensity correlation with no variability would result in a slope of 1 and R2=1, which the data begins to approach when AMN is implemented. The linear fit approaching a slope of 1 indicates that the array measurements are scaled identically as predicted because the intensities are all relative to the same control.
The advantages to using RPMAs with AMN have been demonstrated by the results presented herein. The novel method greatly improves the linearity and reproducibility of quantification by controlling for the local microenvironment experienced by each sample on every array. Figure 2 demonstrates that the improvement in reproducibility makes it feasible to establish cutoff intensities that could discriminate between sample populations, which would yield more sensitive and specific ROC curves. Figure 3 shows that, when AMN is implemented, there is an average reduction in CV of 72% resulting in an increase in the statistical power of the assay to detect shifts in expression. An important current use of the RPMAs is the measurement of signaling protein activation for patient selection and stratification for targeted therapeutics. Given the importance that this application would have in deciding if a patient receives a therapeutic or not, the ability to establish the most reproducible cut-points for selection is critical. Overall, we believe the AMN strategy will bring improvement to data reproducibility across all antibodies and RPMA experiments as it reduces systematic variability. However, the improved reproducibility should be balanced against the potential increased cost and reduced number of independent samples per array for each unique RPMA use. When precision and accuracy are vitally important, AMN should be used. When a study can sacrifice some precision to increase throughput and decrease cost, AMN may not be necessary. Work is ongoing to establish an AMN control sample that will be independent of the study samples and applicable for any experiment.
Abbreviations
- AMN
array microenvironment normalization
- PSA
prostate specific antigen
- RPMA
reverse phase microarray
- TPN
total protein normalization
Footnotes
Conflict of interest: Authors (excluding Dr. Raimond Winslow) have affiliation with Theranostic Health Inc. which is commercializing the RPMA technology.
References
- 1.Kricka LJ, Master SR. Validation and quality control of protein microarray-based analytical methods. Mol Biotechnol. 2008;38:19–31. doi: 10.1007/s12033-007-0066-5. [DOI] [PubMed] [Google Scholar]
- 2.Reed GF, Lynn F, Meade BD. Use of coefficient of variation in assessing variability of quantitative assays. Clin Diagn Lab Immunol. 2002;9:1235–1239. doi: 10.1128/CDLI.9.6.1235-1239.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Spurrier B, Ramalingam S, Nishizuka S. Reverse-phase protein lysate microarrays for cell signaling analysis. Nat Protoc. 2008;3:1796–1808. doi: 10.1038/nprot.2008.179. [DOI] [PubMed] [Google Scholar]
- 4.Nishizuka S, Charboneau L, Young L, Major S, et al. Proteomic profiling of the NCI-60 cancer cell lines using new high-density reverse-phase lysate microarrays. Proc Natl Acad Sci USA. 2003;100:14229–14234. doi: 10.1073/pnas.2331323100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schuchhardt J, Beule D, Malik A, Wolski E, et al. Normalization strategies for cDNA microarrays. Nucleic Acids Res. 2000;28:E47. doi: 10.1093/nar/28.10.e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mary-Huard T, Daudin JJ, Robin S, Bitton F, et al. Spotting effect in microarray experiments. BMC Bioinformatics. 2004;5:63. doi: 10.1186/1471-2105-5-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Daly TM, Dumaual CM, Dotson CA, Farmen MW, et al. Precision profiling and components of variability analysis for Affymetrix microarray assays run in a clinical context. J Mol Diagn. 2005;7:404–412. doi: 10.1016/S1525-1578(10)60570-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.VanMeter AJ, Rodriguez AS, Bowman ED, Jen J, et al. Laser capture microdissection and protein microarray analysis of human non-small cell lung cancer: differential epidermal growth factor receptor (EGFR) phosphorylation events associated with mutated EGFR compared with wild type. Mol Cell Proteomics. 2008;7:1902–1924. doi: 10.1074/mcp.M800204-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Calvert VS, Tang Y, Boveia V, Wulfkuhle JD, et al. Development of multiplexed protein profiling and detection using near infrared detection of reverse-phase protein microarrays. Clin Proteomics J. 2004:1. [Google Scholar]
- 10.Alban A, David SO, Bjorkesten L, Andersson C, et al. A novel experimental design for comparative two-dimensional gel analysis: two-dimensional difference gel electrophoresis incorporating a pooled internal standard. Proteomics. 2003;3:36–44. doi: 10.1002/pmic.200390006. [DOI] [PubMed] [Google Scholar]
- 11.Dobbin K, Simon R. Comparison of microarray designs for class comparison and class discovery. Bioinformatics. 2002;18:1438–1445. doi: 10.1093/bioinformatics/18.11.1438. [DOI] [PubMed] [Google Scholar]
- 12.R Core Development Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2005. [Google Scholar]