

Abstract
We previously identified a lysine to arginine transition at residue 303 (K303R) in ERα in invasive breast cancers, which confers resistance to the aromatase inhibitor (AI) anastrozole (Ana) when expressed in MCF-7 breast cancer cells. Here we show that AI resistance arises through an enhanced cross-talk of the IGF-1R/IRS-1/Akt pathway with ERα, and the serine (S) residue 305 adjacent to the K303R mutation plays a key role in mediating this cross-talk. The ERα S305 residue is an important site that modifies response to tamoxifen; thus, we questioned whether this site could also influence AI response. We generated stable transfectants expressing wild-type (WT), K303R ERα, or a double K303R/S305A mutant receptor, and found that the AI-resistant phenotype associated with expression of the K303R mutation was dependent on activation of S305 within the receptor. Ana significantly reduced growth in K303R/S305A-expressing cells. Preventing S305 phosphorylation with a blocking peptide inhibited IGF-1R/IRS-1/Akt activation, and also restored AI sensitivity. Our data suggest that the K303R mutation and the S305 ERα residue may be a novel determinant of aromatase inhibitor response in breast cancer, and blockade of S305 phosphorylation represents a new therapeutic strategy for treating tumors resistant to hormone therapy.
Keywords: Breast Cancer, Estrogen receptor, K303R mutant ERα, Aromatase Inhibitors Resistance, S305 ERα phosphorylation, IGF-1 Signaling Pathway
Introduction
Aromatase inhibitors (AIs) have become the choice for first-line hormonal therapy ofestrogen receptor alpha (ERα)-positive breast cancer in postmenopausal women. However, many patients are initially refractory or acquire resistance to AI treatment. The mechanisms responsible for the development of resistance remain poorly defined. Several mechanisms have been proposed, including the loss of ERα expression or function (Encarnacion et al., 1993), alterations in the balance of regulatory cofactors, increased oncogenic kinase signaling (Blume-Jensen and Hunter, 2001), and deregulated cell proliferation (Evan et al., 1995). Resistance may also result from altered expression of growth factor signaling pathways, such as insulin-like growth factor receptor-1 (IGF-1R), and human epidermal growth factor receptor ERBB2, that serve to stimulate estrogen-independent growth (Martin et al., 2003; Sabnis et al., 2005; Schiff et al., 2004; Staka et al., 2005).
The IGF-1R is a receptor tyrosine kinase that is activated by its ligands, IGF-1, IGF-2, and insulin (Foulstone et al., 2005). Activation of kinase activity results in autophosphorylation of the receptor, phosphorylation of downstream substrates, such as insulin receptor substrate (IRS1-4) proteins, and consequent stimulation of signaling cascades, such as the MAPK and PI3K/Akt pathways (Backer et al., 1992; Skolnik et al., 1993). IGF-1R and IRS-1 are often coexpressed with ERα in hormone-dependent breast cancer cells, and epidemiological studies suggest a positive correlation between IGF-1 serum levels and breast cancer risk (Hankinson et al., 1998). Accordingly, a number of IGF-1R inhibitors have entered into human clinical trials (Sachdev and Yee, 2007).
We have previously identified a frequent somatic mutation at nucleotide 908 of ERα (A908G) in premalignant and invasive breast cancers (Fuqua et al., 2000; Herynk et al., 2007). This mutation introduces a lysine to arginine substitution at residue 303 (K303R) of ERα, and confers the ability for enhanced breast tumor cell growth in low estrogen levels, and resistance to the non-steroidal AI anastrozole and tamoxifen (Tam) (Barone et al., 2009; Giordano et al., 2009). The mutation resides at a region of major post-translational modifications (acetylation, ubiquitination, sumoylation, and metylation) adjacent to the S305 phosphorylation site. Several studies have identified the ERα S305 as an important phosphorylation site that modifies response to Tam in breast tumors (Holm et al., 2009; Michalides et al., 2004; Rayala and Kumar, 2007). Phosphorylation of S305 has also been implicated in ligand-independent ERα activation (Tharakan et al., 2008). The K303R ERα mutation is a more efficient substrate for phosphorylation of S305 by cAMP-dependent protein kinase (PKA), and its phosphorylation is a key determinant of enhanced estrogen hypersensitivity associated with this mutation (Cui et al., 2004).
In this report, the contribution of the ERα S305 site to AI response will be evaluated to determine if its site-specific phosphorylation can also result in escape from AI therapy. Our results suggest a dynamic interplay between the K303R ERα mutant, S305 phosphorylation, and IGF-1R signalling pathways resulting in enhanced cell growth and AI resistance (AIR).
Materials and Methods
Reagents, hormones and antibodies
17β-Estradiol, 4-Androstene-3, 17-dione and insulin-like growth factor-1 were purchased from Sigma (St. Louis, MO). Anastrozole and gefitinib were obtained from Astrazeneca (Macclesfield, England), and AG1024 from Calbiochem (Darmstadt, Germany). Antibodies used for immunoblotting were: ERα (Vector Laboratories, Burlingame, CA), Shc, Rho GDIα (Santa Cruz Biotechnology, Santa Cruz, CA), total IGF-1R, IRS-1, Akt, phosphorylated IGF-1R(Tyr1131), Akt(Ser473) (Cell Signaling Technology, Beverly, MA), and IRS-1(Tyr612) (Invitrogen, Carlsbad, CA), phospho-ER (Ser305) (Upstate, Temecula, CA). Goat anti-mouse or anti-rabbit secondary antibodies were from Amersham Bioscences (Piscataway, NJ).
Plasmids
Generation of the yellow-fluorescent protein (YFP)-tagged ERα expression constructs, YFP-WT, YFP-K303R, and YFP-K303R/S305A ERα, are previously described (Cui et al., 2004).
Cell culture
MCF-7 breast cancer cells were cultured as described (Cui et al., 2004). The aromatase-overexpressing cells MCF-7 Arom and K303R Arom, and CHO or MCF-7 Arom-expressing pools, stably transfected with YFP-WT, YFP-K303R, and YFPK303R/S305A ERα expression vectors were generated as described (Barone et al., 2009).
Expression microarray analysis and quantitative real-time RT-PCR analysis
Cells were starved in phenol red-free MEM with 5% charcoal-stripped FBS for 48h. RNA was extracted, and microarray analysis and real-time RT-PCR were performed as published in the supplemental information on the Oncogene website. A top table of expressed pathways is provided as supplementary Table 1, and a list of differentially expressed genes is shown in supplementary Table 2.
Immunoblot analysis
Cells were starved in serum-free MEM for 48h, and treated as indicated before lysis (Herynk et al., 2006). Equal amounts of cell extracts were subjected to SDS-PAGE, as described (Cui et al., 2004).
Blocking peptide delivery
A blocking peptide of 11 residues (298IKRSKKNSLAL308) surrounding the S305 residue (in bold) of the human ERα was transfected into cells as described (Giordano et al., 2009).
In vitro Akt kinase assay
Expression vectors for glutathione S-transferase (GST) fusion proteins of WT, K303R and K303R/S305A ERα hinge region (residues 253-310) were previously described (Cui et al., 2004). Kinase assays were performed as published in the supplemental information on the Oncogene website.
Cell proliferation assays
After 48h of starvation in 5% charcoal-stripped FBS phenol red-free MEM, cell proliferation was assessed using soft agar anchorage-independent or MTT growth assays as described (Barone et al., 2009; Giordano et al., 2009).
Statistical Analysis
Data were analyzed by Student’t-test using the GraphPAD Prism5 software (GraphPad Software, Inc., San Diego, CA).
Results
Gene transcription patterns of K303R ERα-overexpressing cells
We have previously described a model of ERα-positive MCF-7 breast cancer cells that overexpress the K303R ERα mutant receptor along with aromatase, and reported that expression of the mutant conferred resistance to the AI anastrozole (Barone et al., 2009). As shown in Figure 1a, cells expressing the K303R receptor expressed a 66kDa endogenous ERα protein, along with a 96kDa receptor representing the exogenously-added mutant tagged with YFP. Increased aromatase protein expression was observed in MCF-7 Arom 1 (one clone stably expressing aromatase) and K303R Arom 1 (one clone co-expressing the YFP-K303R mutant and aromatase) cells. To identify genes whose expression were associated with the development of AIR, we compared RNA isolated from K303R Arom 1-expressing cells with WT-expressing cells using expression microarray analysis. Gene expression analyses showed marked changes in the expression of insulin/IGF family members between the two cell lines based on pathway analysis (Figure 1b and Table 1). We found that K303R ERα mutant expression induced genes that positively regulate IGF signaling (insulin-like growth factor-1: IGF-1, insulin receptor: INSR, insulin receptor substrate-1 and -2: IRS-1 and IRS-2), and suppressed genes that negatively regulate this pathway (insulin-like growth factor binding protein 3 and 5, IGFBP3-5) (McGuire et al., 1992; Salerno et al., 1999; Umayahara et al., 1994). We also observed increased expression of JAK2 kinase, and the trascritption factors fos and STAT1. The two clones had equivalent levels of ERα RNA. These data suggest increased activation of the IGF signaling pathway in mutant-expressing cells that may be related to increased transcriptional activity of the mutant receptor (Barone et al., 2009).
Figure 1.
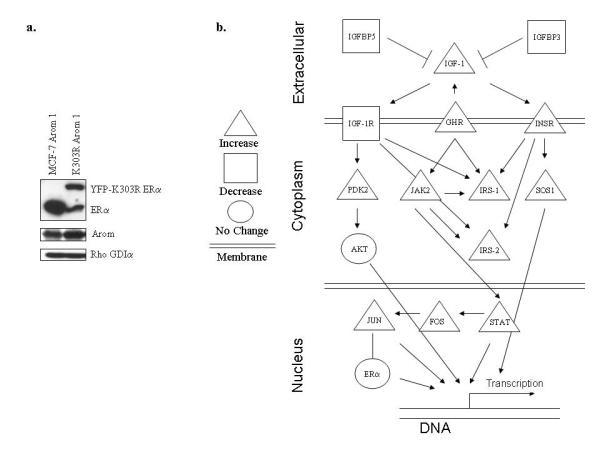
(a) Immunoblot analysis for ERα and aromatase (Arom) expression in MCF-7 Arom 1 and K303R Arom 1-expressing cells. Rho GDIα was used as a control for equal loading and transfer. (b) Schematic representation of gene expression changes of insulin/IGF family members of K303R-expressing cells (n=4) compared to WT cells (n=4) studied by microarray analysis. The genes that were induced are represented in triangles; those that were repressed are indicated with a square, and unchanged genes are shown with a circle.
Table 1.
Genes overexpressed in mutant cells.
| Gene Name | Gene Symbol |
Parametric P-value |
Fold Change K303R Arom 1 vs MCF-7 Arom 1 |
|---|---|---|---|
| Insulin-like Growth Factor Binding Protein 3 |
IGFBP 3 |
5e-07 | 0.49 |
| Insulin-like Growth Factor Binding Protein 5 |
IGFBP 5 |
5e-07 | 0.17 |
| Insulin Receptor Substrate 1 | IRS1 | <1E-07 | 2,9 |
| Insulin Receptor Substrate 2 | IRS2 | <1E-07 | 3,6 |
| Janus Kinase 2 | JAK2 | <1E-07 | 3,7 |
| Signal Transducer and Activator of Transcription 1 |
STAT1 | <1E-07 | 15,1 |
| v-fos FBJ murine osteosarcoma viral oncogene homolog |
FOS | 4e-07 | 5,2 |
Representative probesets from pathway analysis showing gene expression changes in the insulin signaling along with the p-value and the fold change of K303R-expressing cells compared to WT cells. Only genes at a fold change greater than 2 are showed. In cases where the same genes were deemed significant across multiple probesets, only one is shown.
IGF-1 signaling pathway activation in K303R ERα-overexpressing cells
To validate the gene expression profile identified in the microarray study, specific transcript levels were examined using quantitative real-time PCR, choosing to validate genes based on their potential regulatory role in mediating IGF signaling. For instance, IRS-1 is the predominant molecule activated in response to IGF-1 stimulation, and it has been shown that downregulation of IGF-binding proteins is a mechanism by which estrogen can increase IGF responses. We found a significant increase in IRS-1 mRNA, and a significant decrease in IGFBP3 mRNA in K303R Arom 1-expressing cells (Figure 2a).
Figure 2.
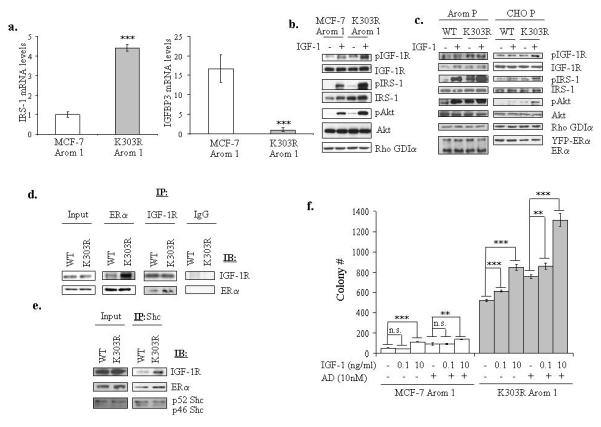
The K303R ERα mutant cells exhibited increased activation of the IGF-1 signaling pathway. (a) IRS-1 and IGFBP3 mRNA expression was assessed by quantitative real-time RT-PCR analysis. Each bar represents mRNA expression relative to β-actin mRNA (endogenous control gene) ± SD (average of four samples). ***P<0.0001 K303R-Arom 1 mRNA levels compared to MCF-7 Arom 1 (b, c) Serum-deprived MCF-7 Arom 1 and K303R Arom 1 cells (b) or MCF-7 WT, K303R P and CHO WT, K303R P (c) pools of stably transfected and overexpressing cells were treated with vehicle or IGF-1 10ng/ml for 5min. Total cellular extracts were analyzed for phosphorylation (p) and expression of IGF-1R, IRS-1 and Akt by immunoblot analysis. Rho GDIα was used as a control for equal loading and transfer. Immunoblots show a single representative of three separate experiments. (d, e) Lysates from CHO cells transiently transfected with WT or K303R ERα were immunoprecipitated (IP) with anti-ERα or IGF-1R or IgG (negative control) (d) or Shc (e) antibodies and immunoblotted for IGF-1R, ERα or Shc. (f) Cells were plated in soft agar, and then treated with vehicle (−) or increasing amounts of IGF-1 (0.1 and 10ng/ml) ± AD 10nM. Cells were allowed to grow for 14 days, and the number of colonies >50μm were quantified, and the results were graphed. Data are the mean colony number of three plates and representative of two independent experiments. Bars, SD. n.s.=nonsignificant, **P<0.01, ***P<0.0005.
We next determined whether this altered gene expression resulted in increased phosphorylation, and activation of IGF signaling. Cells were maintained under estrogen-depleted conditions, treated with IGF-1, and analyzed for phosphorylation of IGF-1R and IRS-1 (Figure 2b). MCF-7 Arom 1-expressing cells showed low basal levels of pIGF-1R and pIRS-1 that were increased with IGF-1 treatment. In contrast, K303R-expressing cells showed elevated constitutive phosphorylation of IGF-1R and IRS-1 further increased with IGF-1. The increase in IGF-1R/IRS-1 phosphorylation resulted in increased phosphorylation of downstream Akt. Since expression of exogenous ERα alone might contribute to the increase in IGF activation, we also stably transfected MCF-7 Arom 1-expressing cells or ERα-negative, aromatase-positive CHO cells with an expression vector for YFP-WT ERα. Pools expressing exogenous WT or mutant receptor were evaluated for IGF-1 growth factor signaling activation. Our results demonstrate that the expression of the mutant receptor in different backgrounds and at differing levels of receptor induced elevated constitutive and IGF-1-mediated phosphorylation of IGF-1R/IRS-1/Akt signaling (Figure 2c).
ERα can bind to IGF-1R (Song et al., 2004). We have previously shown that the mutant receptor exhibited altered binding with several regulatory proteins, such as the TIF-2 coactivator, the p85α regulatory subunit of PI3K, and the ERBB2 receptor compared with WT ERα (Barone et al., 2009; Fuqua et al., 2000; Giordano et al., 2009). To examine whether the mutation might alter binding with the IGF-1R, we transiently transfected CHO cells with YFP-tagged ERs, and coimmunoprecipitation studies were performed. Enhanced binding of IGF-1R to the K303R ERα was observed in the absence of estrogen (Figure 2d). We also confirmed this enhanced binding by immunoprecipitation of Shc, a key component in mediating ERα-IGF-1R interaction (Song et al., 2004) (Figure 2e).
We next evaluated whether activation of the IGF pathway by the K303R mutation could enhance the effects of IGF-1 on growth using anchorage-independent growth assays (Figure 2f). Cells were plated in soft agar, and treated with IGF-1 (0.1 or 10ng/ml) and the aromatase substrate androstenedione (AD). As previously demonstrated (Barone et al., 2009), control basal growth of mutant-expressing cells was significantly elevated compared to WT-expressing cells; IGF-1 treatment at 10ng/ml, and AD treatment increased the number of colonies in both cells. However, growth stimulation was not observed with low IGF-1 treatment (0.1ng/ml) in WT cells, while IGF-1 at this concentration significantly enhanced mutant growth. Estrogen and IGF-1 are potent mitogens for ERα-positive breast cancer cells, and addition of both may result in additive or synergistic effects (Lee et al., 1999). In WT-expressing cells only AD+IGF-1 at 10ng/ml increased colony numbers, compared to AD- or IGF-1 alone-stimulated growth. In contrast, in mutant-expressing cells, the combination of AD and IGF-1 at both concentrations increased growth which was greater than either ligand alone. Therefore, K303R mutant expression was associated with increased IGF signaling activation, and increased sensitivity to IGF-1 stimulation.
AIR and the IGF-1R pathway
Previous studies have proposed a dynamic interplay between ERα and several signal transduction pathways, such as IGF-1R and ERBB2, as mechanisms responsible for acquired resistance in breast cancer cells (long-term estrogen-deprived cells, tamoxifen or letrozole-resistant cells) (Jelovac et al., 2005; Martin et al., 2003; Schiff et al., 2004; Staka et al., 2005). Therefore we addressed whether the activated IGF signaling we observed in our model may represent a potential mechanism of AIR. First, we examined the activation of IGF-1R/IRS-1/Akt in cells treated with AD and Ana (Figure 3a, left panel). WT ERα-expressing cells showed increased pIGF-1R/pIRS-1/pAkt levels following AD treatment that was reduced with Ana cotreatment. In contrast, the mutant-expressing cells showed elevated constitutive IGF-1R/IRS-1/Akt phosphorylation that was not affected by AD and Ana treatments. Similar results were obtained in stably transfected pools (Figure 3a, right panel) and transiently transfected (Supplemental Figure 1) MCF-7 Arom 1 cells expressing equal amount of exogenous WT or mutant receptor.
Figure 3.
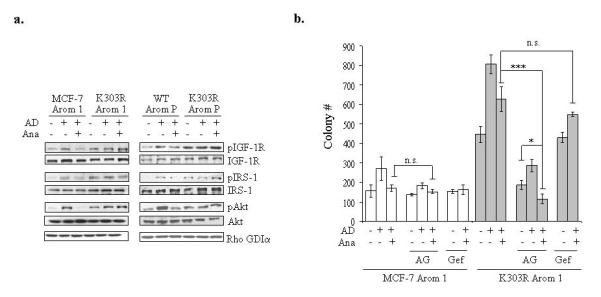
Inhibition of the IGF-1R pathway reversed AIR. (a) MCF-7 Arom 1 and K303R Arom 1 cells or MCF-7 WT, K303R P pools of stably transfected and overexpressing cells (WT Arom P and K303R Arom P) were treated with vehicle (C), AD 10nM ± anastrozole (Ana 1μM) for 1h. Levels of phosphorylated (p) IGF-1R, IRS-1 and Akt and total non-phosphorylated proteins were measured using immunoblot analysis. Rho GDIα was used as a control for equal loading and transfer. Immunoblots shown are a single representative blot of two separate experiments. (b) Cells were plated in soft agar and then treated with vehicle, AD 10nM, Ana 1μM with or without the IGF-1R inhibitor AG1024 (AG 10μM) or the EGFR inhibitor Gefitinib (Gef, 1μM). The number of colonies >50μm were quantified, and data are the mean colony number of three plates and representative of three independent experiments. Bars, SD. n.s.=nonsignificant, *P=0.02, ***P<0.0005.
To further confirm the role of IGF signaling in the AIR phenotype associated with mutant ERα expression, we performed soft agar assays in the presence of the IGF-1R inhibitor AG1024 (Figure 3b). AG treatment induced only a slight reduction in the relative growth of MCF-7 Arom 1-expressing cells. In contrast, AG completely suppressed the proliferation of mutant-expressing cells, and the combination of Ana and AG resulted in a higher inhibition of mutant-expressing cell growth. To emphasize the specific involvement of the IGF pathway in AIR, we used a clinically useful EGFR inhibitor (gefitinib [Gef], Figure 3b). Gefitinib was unable to reverse the AIR phenotype in mutant-expressing cells. Our results suggest that enhanced IGF signaling may contribute to the AIR phenotype associated with K303R ERα mutant expression. These results also question how constitutively activated IGF signaling may affect mutant ERα’s function?
Phosphorylation at serine 305 of the K303R ERα mutant and IGF-1R cross-talk
Post-translational modifications of ERα, such as phosphorylation, are important regulatory components of cross-talk between different signaling networks. For instance, phosphorylation at S305 by PKA and PAK1 has been implicated in ligand-independent ERα activation, and in Tam-resistance of breast tumors (Michalides et al., 2004; Rayala and Kumar, 2007b; Tharakan et al., 2008). Furthermore, the K303R ERα mutant is a more efficient substrate for phosphorylation by PKA at S305, which enhanced hormone sensitivity and Tam-resistant cell growth (Cui et al., 2004; Giordano et al., 2009).
We first evaluated the phosphorylation status of the S305 residue in either WT or K303R ERα-expressing cells using immunoblot analysis. As previously demonstrated (Giordano et al., 2009), we found a costitutively higher phosphorylation of S305 in the mutant receptor (Figure 4a). We then mutated the predicted S305 phosphorylation site to alanine (A) in the K303R ERα construct (K303R/S305A) to eliminate potential phosphorylation at this site, and found that S305A mutation completely abrogated this phosphorylation.
Figure 4.
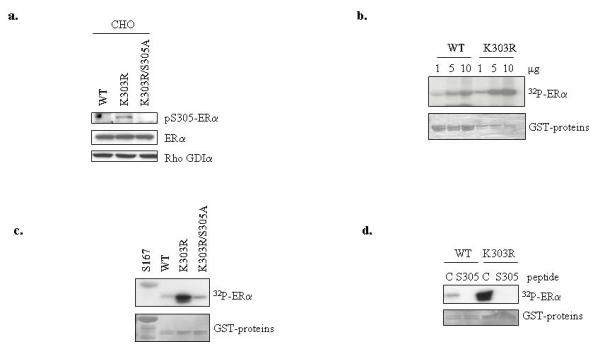
The S305 residue of the K303R mutant receptor is a novel Akt phosphorylation site. (a) Lysates from CHO cells transiently transfected with WT, K303R ERα or K303R/S305A ERα plasmids were analyzed for phosphorylation levels of S305 YFP ERα (pS305) and total non-phosphorylated YFP- ERα using immunoblot analysis. Rho GDIα was used as a control for equal loading and transfer. (b) In vitro Akt kinase assays using increasing concentrations (1, 5, 10μg) of WT and K303R ERα GST-fusion hinge fragments (residues 253-310) were performed in kinase buffer. Reactions products were analyzed using SDS-PAGE, and phosphorylated ERα bands were visualized by autoradiography. (c) In vitro Akt kinase assays using WT, K303R and K303R/S305A ERα GST-fusion fragments. The ERα fragment containing the known S167 site was used as positive control in the reaction. (d) In vitro Akt kinase assays using WT, K303R ERα GST-fusion fragments were performed in the presence of the S305 blocking or a control (C) peptide. The input GST proteins were visualized with Ponceau S staining (bottom panels).
Then, we examined whether there were kinase-specific consensus phosphorylation sites in ER‘s hinge domain using publicly available software (NETPHOSK1.0 server) from the Center for Biological Sequence Analysis (http://www.cbs.dtu.dk). Our search suggested that the S305 ERα site was a potential Akt phosphorylation site, with a low consensus score but only in the mutant; also as expected, S305 was identified as a potential consensus PKA site with a high potential score in both WT and K303R ERα receptors (data not shown). Because we have shown enhanced Akt signaling in mutant-expressing cells (Barone et al., 2009), we initially tested whether the mutant receptor could be phosphorylated by recombinant Akt using an in vitro kinase assay with GST-ERα hinge fragments deleting the known S167 Akt consensus sequence. Using increasing amounts of the GST-ERα constructs as phosphorylation substrates, we discovered that the K303R ERα mutant was more efficiently phosphorylated in a dose-dependent manner compared to WT receptor and S305A mutation completely prevented in vitro phosphorylation by Akt (Figure 4c). A receptor fragment containing the S167 Akt site was used as positive control. To evaluate the specificity of the S305 phosphorylation by Akt kinase, we also performed kinase assays using a selective ERα blocking peptide of 11 amino acids corresponding to the sequence surrounding S305, along with a negative control peptide (Figure 4d). Akt kinase failed to phosphorylate the GST-ERα constructs in the presence of the S305 peptide. These findings suggest that the K303R ERα mutation may generate a super-active substrate for Akt kinase phosphorylation at S305, a site which we have previously shown to be critically involved in the estrogen hypersensitivity of K303R ERα mutant (Cui et al., 2004).
We next addressed whether S305 phosphorylation could be a determinant of the increased sensitivity to IGF-1 stimulation associated with K303R mutant expression. We generated pools of stable transfectants expressing exogenous WT, K303R, or K303R/S305A receptors in MCF-7 Arom 1-expressing cells, and evaluated for the effects of increasing concentrations of IGF-1 in soft agar assays (Figure 5a). The basal non-stimulated growth of K303R Arom pools was higher than WT ERα-Arom pools, and expression of the K303R/S305A receptor resulted in a reduction in colony number. IGF-1 10ng/ml increased anchorage-independent growth in all three clones, while IGF-1 treatment at the lowest concentration (0.1ng/ml) enhanced growth only in the K303R-expressing cells. Accordingly, expression of the K303R/S305A double mutant reduced the activation induced by AD+IGF 10ng/ml and inhibited the increase on growth mediated by AD+IGF 0.1ng/ml when compared to K303R ERα expression (supplemental Figure 2). To extend these results, we also generated stable transfected pools of YFP-WT, YFP-K303R and YFP-K303R/S305A ERα in CHO cells (Figure 5b). Again, the K303R/S305A double ERα mutant-expressing cells exhibited growth similar to WT cells, showing a significant reduction in colony number compared to K303R-expressing cells, and no increase in growth with low doses of IGF-1.
Figure 5.
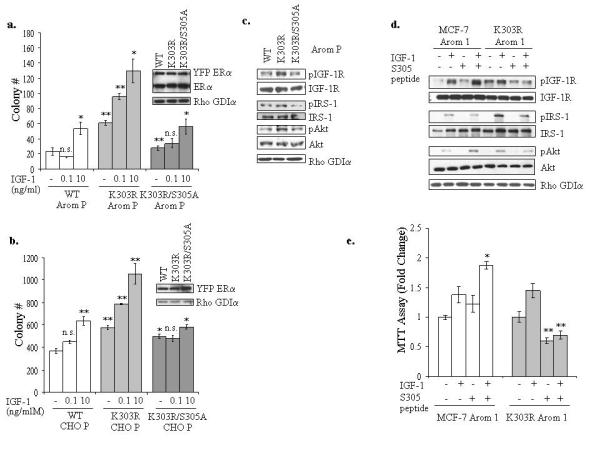
Inhibition of serine 305 ERα phosphorylation blocked IGF-1R and mutant ERα cross-talk. (a, b) MCF-7 WT, K303R and K303R/S305A Arom P (a) and CHO WT, K303R and K303R/S305A P (b) pools of stably transfected and overexpressing cells were plated in soft agar and then treated with vehicle, IGF-1 0.1 or 10ng/ml for 14 days. The number of colonies >50μm were quantified and data are the mean colony number of three plates and representative of two independent experiments. Bars, SD. n.s.=nonsignificant, *P<0.01, **P<0.005. Equal expression of protein was determined by immunoblot with anti-ERα and Rho GDIα antibodies (right panel for a and b). (c) Cellular extracts from serum-deprived MCF-7 Arom WT, K303R K303R/S305A P pools of stably transfected and overexpressing cells were analyzed for phosphorylation (p) and expression of IGF-1R, IRS-1 and Akt by immunoblot analysis. Rho GDIα was used as a control for equal loading and transfer. (d) Cells were incubated with the S305 peptide (4μg/well) for 4h, and then treated with or without IGF-1 (10ng/ml) for 5min. Levels of phosphorylated (p) IGF-1R, IRS-1 and Akt, and total non-phosphorylated proteins were measured in cellular extracts by immunoblot analysis. Blots are representative of three separate experiments. Rho GDIα was used as a control for equal loading and transfer. (e) MTT growth assays in MCF-7 Arom 1 and K303R Arom 1 cells treated for 3 days with vehicle, IGF-1 (10ng/ml) and/or the S305 peptide (0.6μg/well). Cell proliferation is expressed as fold change relative to vehicle-treated cells. The data are representative of four independent experiments, each performed in triplicate. Columns, mean. Bars, SD. *P<0.005 compared to IGF-1 treatment in MCF-7 Arom 1-cells, **P<0.001 compared to vehicle and IGF-1 treatments in K303R Arom1-expressing cells.
To confirm the role of S305 phosphorylation in IGF-1R signaling activation, pools of MCF-7 Arom1 cells expressing YFP-WT, YFP-K303R and YFP-K303R/S305A receptors were analyzed for IGF-1R/IRS-1/Akt phosphorylation. The exogenous expression of the K303R/S305A mutant was associated with a reduction in pIGF-1R/pIRS-1/pAkt activation compared to K303R receptor (Figure 5c).
To investigate the role of the S305 residue in K303R ERα’s cross-talk with IGF-1R, we delivered the S305 blocking peptide to cells, and after IGF-1 treatment, downstream growth factor signaling activation was analyzed (Figure 5d). Blockade of phosphorylation at S305 resulted in specific inhibition IGF-1R/IRS-1/Akt phosphorylation in mutant-expressing cells, while in WT cells the S305 peptide slightly enhanced IGF-1-mediated activation of pIGF1-R and pAkt. We next examined the effects of the S305 blocking peptide on cell growth using MTT assays (Figure 5e). Cells were treated with IGF-1 in the presence or absence of the S305 blocking peptide; IGF-1 treatment enhanced cell growth in both cells. Blockade of S305 resulted in a significant reduction in basal and IGF-1-stimulated growth only in mutant cells, while in WT ERα Arom cells the S305 peptide was unable to reduce growth. As seen with immunoblot analysis, we actually observed that treatment with IGF-1 plus the S305 peptide stimulated growth compared to IGF-1 treatment alone in WT cells. The reasons of this increase are not presently understood, although the influence of S305 phosphorylation on methylation at K302, and thus stability of the receptor (Subramanian et al., 2008) or protein acetylation (Cui et al., 2004) are possibilities. These collective data indicate that phosphorylation of the S305 ERα residue may represent a significant downstream signaling nodule of cross-talk between IGF-1R and the mutant receptor.
K303R/ S305 ERα residues and AIR
We next questioned whether the K303R/S305 site could influence AI response. Stable pools of WT and mutant receptors were evaluated for the effects of AD and Ana treatments on anchorage-independent growth (Figure 6a). Inhibition of aromatase activity by Ana caused a significant reduction in AD-stimulated growth in WT ERα Arom P cells, while in K303R-expressing cells Ana was unable to reduce AD-stimulated growth. In the K303R Arom pool we observed that AD+Ana stimulated growth compared to AD treatment alone (P<0.01), but this increase may be a variable clonal effect in this pool. Expression of the K303R/S305A receptor restored sensitivity to the inhibitory effects of Ana on growth, thus mimicking the same response profile of WT ERα clones. We also obtained similar results in CHO pools stably expressing WT and mutant receptors (Figure 6b).
Figure 6.
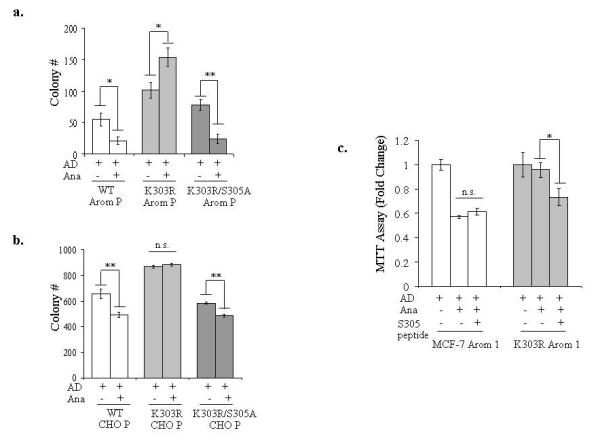
The S305 residue in the K303R ERα mutant is a determinant of AIR. (a, b) MCF-7 WT, K303R and K303R S305A Arom P (a) and CHO WT, K303R and K303R S305A P (b) pools of stably transfected cells were plated in soft agar and then treated with AD (10nM) ± Ana (1μM) for 14 days. The number of colonies >50μm were quantified, and data shown are the mean colony number of two plates and representative of three independent experiments. Bars, SD. n.s.= nonsignificant, *P<0.01, **P<0.005. (c) MTT growth assay in MCF-7 Arom 1 and K303R Arom 1 cells treated for 6 days with AD (10nM), Ana (1μM) and/or the S305 peptide (0.6μg/well). Cell proliferation is expressed as fold change relative to AD-treated cells. The data are representative of three independent experiments, each performed in triplicate. Columns, mean. Bars, SD. n.s.=nonsignificant, *P=0.01 AD+Ana+S305 peptide compared to Ana+AD.
Finally, we delivered the S305 blocking peptide to cells, and evaluated its effect on AI response using growth assays (Figure 6c). Ana treatment decreased AD-stimulated growth in WT cells, but had no effect in mutant-expressing cells. Blockade of S305 phosphorylation using the peptide inhibited Ana+AD-mediated growth only in mutant-expressing cells. These data suggest that enhanced phosphorylation at S305 within the hinge domain of the receptor might specifically mediate enhanced cell growth and the AIR phenotype associated with K303R mutant expression, and that blockade of ERα phosphorylation may be an effective strategy to reverse resistance to hormone therapy in tumors bearing the ERα mutation.
Discussion
Despite the efficacy of AIs in the treatment of breast cancer, tumors can develop resistance to estrogen deprivation and relapse with recurrent disease. To explore mechanisms involved in the development of AIR, we recently discovered that expression of a naturally-occurring K303R ERα mutation in MCF-7 breast cancer cells conferred resistance to the non-steroidal AI anastrozole (Barone et al., 2009). This somatic mutation (A908G) has been identified in about a third of premalignant lesions, and one-half of invasive breast tumors (Fuqua et al., 2000; Herynk et al., 2007), although using another detection method the mutation was identified in only 6% of tumors (Conway et al., 2005). This transition introduces a lysine to arginine substitution at residue 303 (K303R) within the hinge domain. The region between the DNA-binding and the ligand-binding domains, known as the hinge, has long been considered to simply serve as a flexible linker to orient these functional domains. However, it has now shown that this region is a multifunctional domain that binds co-regulatory proteins, participates in DNA binding, and undergoes post-translational modifications (Cui et al., 2004; Haelens et al., 2007; Kim et al., 2006). Naturally-occurring mutations have been also identified in the hinge of the androgen receptor in prostate cancer, and the progesterone receptor in ovarian cancer. These mutations affect acetylation or phosphorylation of the hinge, and result in hyper-active receptors with enhanced hormone sensitivity (Agoulnik et al., 2004; Deeb et al., 2008; Romano et al., 2006). Our studies also suggest a new critical role for the hinge domain of the K303R ERα mutant receptor. We reported that overexpression of K303R mutation in MCF-7 breast cancer cells conferred increased sensitivity to subphysiological levels of estrogen, and decreased sensitivity to Tam when engaged in cross-talk with growth factor receptor signaling (Fuqua et al., 2000; Giordano et al., 2009). We also demonstrated that K303R ERα expression conferred resistance to the AI anastrozole in ERα-positive cells also co-expressing human aromatase; resistance occurred via activation of the PI3K/Akt prosurvival signaling pathway to which the mutant cells had become “addicted” for maintenance of growth (Barone et al., 2009). In this study, we investigated the role of a post-translational modification of the hinge region of the K303R mutant receptor as a determinant of resistance to AI, and as a possible therapeutic target to reverse resistance.
Additional biomarkers to identify patients who are resistant to AIs are lacking in clinical practice. Thus, a better understanding of resistance mechanisms may enable accurate prediction of response, which is relevant since alternative therapeutic strategies are available. To identify genes whose expression were associated with the development of AIR, we performed expression microarray analyses, and found increased expression of several IGF activators, such as IRS-1, and decreased expression of specific IGF inhibitors, such as IGFBP3 in the mutant cells. This altered gene expression resulted in enhanced phosphorylation and activation of IGF signaling molecules, and increased sensitivity to IGF-1 stimulation. We also found an enhanced association between the mutant receptor and IGF-1R, consistent with our earlier observations of enhanced binding of the mutant receptor to other regulatory molecules. This enhanced binding along with the increased ligand-independent transcriptional activity of the mutant we previously reported may help to sustain enhanced bidirectional cross-talk between IGF-1R and K303R ERα, further augmenting signaling between these important growth pathways.
Studies have shown that increased signaling via IGF-1R with consequent activation of IRS-1 and the PI3K/Akt survival pathway leads to resistance to Tam and AI treatments (Campbell et al., 2001; Santen et al., 2005; Wiseman et al., 1993). Indeed, IGF-1R inhibitors have been used to inhibit breast tumor growth and metastasis (Arteaga and Osborne, 1989; Burtrum et al., 2003; Sachdev et al., 2004). We found that an IGF-1R inhibitor, AG1024, completely suppressed the proliferation of mutant-expressing cells, and the combination of an AI with AG resulted in a greater growth inhibition. These results suggest that the persistence of active IGF-1R signaling may be a functional mechanism of resistance to AI therapies in cells expressing the mutant receptor.
ERα is known to be a target of several post-translational modifications (Faus and Haendler, 2006). ERα phosphorylation results from the activation of various cellular kinases and substantially alters its function. For example, ERα can be differentially phosphorylated by Akt and extracellular regulated kinase (Erk1/2) MAPK resulting in diverse responses to ligand (Likhite et al., 2006). Recently, we and others have shown that PKA and PAK1-mediated phosphorylation of ERα S305 impacted both estrogen hypersensitivity and Tam response (Cui et al., 2004; Michalides et al., 2004; Rayala and Kumar, 2007a). Our search for kinase consensus sequences identified the S305 site as a novel Akt phosphorylation site in the mutant receptor. Although the target site sequence around S305 is not a perfect match to the consensus target site for Akt (R-x-R-x-x-S/T) (Manning and Cantley, 2002), it is known for other protein kinases that the inclusion of additional amino acids of the same charge in this target site sequence enhanced substrate activity (Brenet et al., 2009). In addition, Akt may phosphorylate substrates efficiently which conform less rigorously to the requirement for arginine at positions n-3 and n-5 (Ksiezak-Reding et al., 2003). In vitro Akt kinase assays demonstrated that the K303R ERα mutant was more efficiently phosphorylated compared to WT ERα, and phosphorylation of the K303R mutant receptor at S305 was comparable with S167 phosphorylation. Both mutation of S305 to A to remove the phosphorylation site, or the use of a selective blocking peptide mimicking the sequence of ERα surrounding S305, prevented in vitro phosphorylation by Akt. We previously showed that the mutation leads to enhanced constitutive phosphorylation of Akt, and subsequent phosphorylation of the Akt S167 site within ER (Barone et al., 2009). Here we show that activated Akt can also elicit effects on phosphorylation of S305 in the mutant receptor, and that this phosphorylation increased its ligand-independent activity. To study the involvement of S305 phosphorylation in the AIR phenotype associated with the K303R mutation, we generated stable transfectants expressing exogenous K303R/S305A receptor. Double mutant-expressing cells exhibited soft agar growth similar to WT cells, showing a significant reduction of the colony numbers compared to K303R-expressing cells, no increase in anchorage-independent growth under low doses of IGF-1, and a marked sensitivity to anastrozole. These results suggested a prominent role for the S305 residue in mediating the mutant-resistant phenotype.
Others have synthesized small antagonistic peptides capable of interfering with signaling pathway activation (Fontenot et al., 2007; Rayala et al., 2006; Varricchio et al., 2007) Nakajima et al. demonstrated that a five amino acid peptide inhibited ERBB2 signaling, and proliferation equivalent to treatment with the ERBB2-antagonist trastuzumab (Nakajima et al., 2008). The S305 peptide used in this study blocked heregulin-induced S305 ERα phosphorylation, and prevented downstream phosphorylation events in K303R-expressing cells (Giordano et al., 2009). Thus, the contribution of the S305 site to the AIR phenotype associated with the K303R mutation was also evaluated using the S305 blocking peptide. We found specific inhibition of IGF-1R/IRS-1/Akt phosphorylation, and a significant reduction in basal and IGF-1-stimulated growth in the presence of the S305 peptide in mutant-expressing cells. Importantly, inhibition of IGF-1R and the mutant receptor cross-talk by blocking S305 phosphorylation reversed resistance in the mutant cells. Our studies suggest that the K303R/S305 ERα residues play a crucial role in ERα action in breast cancer by enhancing cross-talk between IGF-1R and the mutant receptor which leads to AIR. We propose a model in which the mutation enhances the ability of the receptor to bind to IGF-1R and activates IRS-1/Akt downstream signaling pathway. Activated Akt in turn phosphorylates ERα at S305, and increases the transcription of IGF signaling molecules, thus establishing a positive feedback loop between growth factor signaling and ERα to bypass normal endocrine controls and responsiveness (Figure 7).
Figure 7.
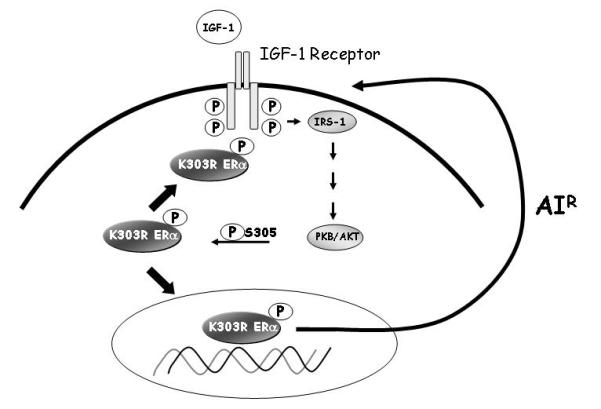
Model depicting potential cross-talk between the ERα and IGF-signaling components in breast cancer cells expressing the mutation. K303R ERα mutant receptor binds to IGF-1R and activates the IGF-1R signaling cascades, including IRS-1 and Akt pathways. Activated Akt promotes the phosphorylation of the K303R ERα mutant at the S305 residue, leading to an increase in the transcription of IGF signaling molecules, thus establishing a positive feedback loop between growth factor and steroid receptors that leads to AIR.
The role of S305 phosphorylation in AIR represents a novel mechanism. We speculate that blockade of this phosphorylation site may be a new therapeutic strategy to block ERα hyperactivation and overcome resistance in mutation-positive patients that are resistant to aromatase inhibitor therapy.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
This work was supported by NCI RO1 CA72038 to SAWF. The authors would like to thanks Mrs. Robin Sample for excellent administrative assistance.
5Financial Support: NCI RO1 CA72038 to SAWF
References
- Agoulnik IU, Tong XW, Fischer DC, Korner K, Atkinson NE, Edwards DP, et al. A germline variation in the progesterone receptor gene increases transcriptional activity and may modify ovarian cancer risk. J Clin Endocrinol Metab. 2004;89:6340–7. doi: 10.1210/jc.2004-0114. [DOI] [PubMed] [Google Scholar]
- Arteaga CL, Osborne CK. Growth inhibition of human breast cancer cells in vitro with an antibody against the type I somatomedin receptor. Cancer Res. 1989;49:6237–41. [PubMed] [Google Scholar]
- Backer JM, Myers MG, Jr., Shoelson SE, Chin DJ, Sun XJ, Miralpeix M, et al. Phosphatidylinositol 3′-kinase is activated by association with IRS-1 during insulin stimulation. EMBO J. 1992;11:3469–79. doi: 10.1002/j.1460-2075.1992.tb05426.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barone I, Cui Y, Herynk MH, Corona-Rodriguez A, Giordano C, Selever J, et al. Expression of the K303R estrogen receptor-alpha breast cancer mutation induces resistance to an aromatase inhibitor via addiction to the PI3K/Akt kinase pathway. Cancer Res. 2009;69:4724–32. doi: 10.1158/0008-5472.CAN-08-4194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001;411:355–65. doi: 10.1038/35077225. [DOI] [PubMed] [Google Scholar]
- Brenet F, Socci ND, Sonenberg N, Holland EC. Akt phosphorylation of La regulates specific mRNA translation in glial progenitors. Oncogene. 2009;28:128–39. doi: 10.1038/onc.2008.376. [DOI] [PubMed] [Google Scholar]
- Burtrum D, Zhu Z, Lu D, Anderson DM, Prewett M, Pereira DS, et al. A fully human monoclonal antibody to the insulin-like growth factor I receptor blocks ligand-dependent signaling and inhibits human tumor growth in vivo. Cancer Res. 2003;63:8912–21. [PubMed] [Google Scholar]
- Campbell RA, Bhat-Nakshatri P, Patel NM, Constantinidou D, Ali S, Nakshatri H. Phosphatidylinositol 3-kinase/AKT-mediated activation of estrogen receptor alpha: a new model for anti-estrogen resistance. J Biol Chem. 2001;276:9817–24. doi: 10.1074/jbc.M010840200. [DOI] [PubMed] [Google Scholar]
- Conway K, Parrish E, Edmiston SN, Tolbert D, Tse CK, Geradts J, et al. The estrogen receptor-alpha A908G (K303R) mutation occurs at a low frequency in invasive breast tumors: results from a population-based study. Breast Cancer Res. 2005;7:R871–80. doi: 10.1186/bcr1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y, Zhang M, Pestell R, Curran EM, Welshons WV, Fuqua SA. Phosphorylation of estrogen receptor alpha blocks its acetylation and regulates estrogen sensitivity. Cancer Res. 2004;64:9199–208. doi: 10.1158/0008-5472.CAN-04-2126. [DOI] [PubMed] [Google Scholar]
- Deeb A, Jaaskelainen J, Dattani M, Whitaker HC, Costigan C, Hughes IA. A novel mutation in the human androgen receptor suggests a regulatory role for the hinge region in amino-terminal and carboxy-terminal interactions. J Clin Endocrinol Metab. 2008;93:3691–6. doi: 10.1210/jc.2008-0737. [DOI] [PubMed] [Google Scholar]
- Encarnacion CA, Ciocca DR, McGuire WL, Clark GM, Fuqua SA, Osborne CK. Measurement of steroid hormone receptors in breast cancer patients on tamoxifen. Breast Cancer Res Treat. 1993;26:237–46. doi: 10.1007/BF00665801. [DOI] [PubMed] [Google Scholar]
- Evan GI, Brown L, Whyte M, Harrington E. Apoptosis and the cell cycle. Curr Opin Cell Biol. 1995;7:825–34. doi: 10.1016/0955-0674(95)80066-2. [DOI] [PubMed] [Google Scholar]
- Faus H, Haendler B. Post-translational modifications of steroid receptors. Biomed Pharmacother. 2006;60:520–8. doi: 10.1016/j.biopha.2006.07.082. [DOI] [PubMed] [Google Scholar]
- Fontenot DR, den Hollander P, Vela EM, Newman R, Sastry JK, Kumar R. Dynein light chain 1 peptide inhibits human immunodeficiency virus infection in eukaryotic cells. Biochem Biophys Res Commun. 2007;363:901–7. doi: 10.1016/j.bbrc.2007.09.046. [DOI] [PubMed] [Google Scholar]
- Foulstone E, Prince S, Zaccheo O, Burns JL, Harper J, Jacobs C, et al. Insulin-like growth factor ligands, receptors, and binding proteins in cancer. J Pathol. 2005;205:145–53. doi: 10.1002/path.1712. [DOI] [PubMed] [Google Scholar]
- Fuqua SA, Wiltschke C, Zhang QX, Borg A, Castles CG, Friedrichs WE, et al. A hypersensitive estrogen receptor-alpha mutation in premalignant breast lesions. Cancer Res. 2000;60:4026–9. [PubMed] [Google Scholar]
- Giordano C, Cui Y, Barone I, Ando S, Mancini MA, Berno V, et al. Growth factor-induced resistance to tamoxifen is associated with a mutation of estrogen receptor alpha and its phosphorylation at serine 305. Breast Cancer Res Treat. 2009 doi: 10.1007/s10549-009-0334-0. (Feb 11, Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haelens A, Tanner T, Denayer S, Callewaert L, Claessens F. The hinge region regulates DNA binding, nuclear translocation, and transactivation of the androgen receptor. Cancer Res. 2007;67:4514–23. doi: 10.1158/0008-5472.CAN-06-1701. [DOI] [PubMed] [Google Scholar]
- Hankinson SE, Willett WC, Colditz GA, Hunter DJ, Michaud DS, Deroo B, et al. Circulating concentrations of insulin-like growth factor-I and risk of breast cancer. Lancet. 1998;351:1393–6. doi: 10.1016/S0140-6736(97)10384-1. [DOI] [PubMed] [Google Scholar]
- Herynk MH, Beyer AR, Cui Y, Weiss H, Anderson E, Green TP, et al. Cooperative action of tamoxifen and c-Src inhibition in preventing the growth of estrogen receptor-positive human breast cancer cells. Mol Cancer Ther. 2006;5:3023–31. doi: 10.1158/1535-7163.MCT-06-0394. [DOI] [PubMed] [Google Scholar]
- Herynk MH, Parra I, Cui Y, Beyer A, Wu MF, Hilsenbeck SG, et al. Association between the estrogen receptor alpha A908G mutation and outcomes in invasive breast cancer. Clin Cancer Res. 2007;13:3235–43. doi: 10.1158/1078-0432.CCR-06-2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jelovac D, Sabnis G, Long BJ, Macedo L, Goloubeva OG, Brodie AM. Activation of mitogen-activated protein kinase in xenografts and cells during prolonged treatment with aromatase inhibitor letrozole. Cancer Res. 2005;65:5380–9. doi: 10.1158/0008-5472.CAN-04-4502. [DOI] [PubMed] [Google Scholar]
- Kim MY, Woo EM, Chong YT, Homenko DR, Kraus WL. Acetylation of estrogen receptor alpha by p300 at lysines 266 and 268 enhances the deoxyribonucleic acid binding and transactivation activities of the receptor. Mol Endocrinol. 2006;20:1479–93. doi: 10.1210/me.2005-0531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ksiezak-Reding H, Pyo HK, Feinstein B, Pasinetti GM. Akt/PKB kinase phosphorylates separately Thr212 and Ser214 of tau protein in vitro. Biochim Biophys Acta. 2003;1639:159–68. doi: 10.1016/j.bbadis.2003.09.001. [DOI] [PubMed] [Google Scholar]
- Lee AV, Jackson JG, Gooch JL, Hilsenbeck SG, Coronado-Heinsohn E, Osborne CK, et al. Enhancement of insulin-like growth factor signaling in human breast cancer: estrogen regulation of insulin receptor substrate-1 expression in vitro and in vivo. Mol Endocrinol. 1999;13:787–96. doi: 10.1210/mend.13.5.0274. [DOI] [PubMed] [Google Scholar]
- Likhite VS, Stossi F, Kim K, Katzenellenbogen BS, Katzenellenbogen JA. Kinase-specific phosphorylation of the estrogen receptor changes receptor interactions with ligand, deoxyribonucleic acid, and coregulators associated with alterations in estrogen and tamoxifen activity. Mol Endocrinol. 2006;20:3120–32. doi: 10.1210/me.2006-0068. [DOI] [PubMed] [Google Scholar]
- Manning BD, Cantley LC. Hitting the target: emerging technologies in the search for kinase substrates. Sci STKE. 2002;2002:PE49. doi: 10.1126/stke.2002.162.pe49. [DOI] [PubMed] [Google Scholar]
- Martin LA, Farmer I, Johnston SR, Ali S, Marshall C, Dowsett M. Enhanced estrogen receptor (ER) alpha, ERBB2, and MAPK signal transduction pathways operate during the adaptation of MCF-7 cells to long term estrogen deprivation. J Biol Chem. 2003;278:30458–68. doi: 10.1074/jbc.M305226200. [DOI] [PubMed] [Google Scholar]
- McGuire WL, Jr., Jackson JG, Figueroa JA, Shimasaki S, Powell DR, Yee D. Regulation of insulin-like growth factor-binding protein (IGFBP) expression by breast cancer cells: use of IGFBP-1 as an inhibitor of insulin-like growth factor action. J Natl Cancer Inst. 1992;84:1336–41. doi: 10.1093/jnci/84.17.1336. [DOI] [PubMed] [Google Scholar]
- Michalides R, Griekspoor A, Balkenende A, Verwoerd D, Janssen L, Jalink K, et al. Tamoxifen resistance by a conformational arrest of the estrogen receptor alpha after PKA activation in breast cancer. Cancer Cell. 2004;5:597–605. doi: 10.1016/j.ccr.2004.05.016. [DOI] [PubMed] [Google Scholar]
- Nakajima H, Mizuta N, Sakaguchi K, Fujiwara I, Yoshimori A, Takahashi S, et al. Development of HER2-antagonistic peptides as novel anti-breast cancer drugs by in silico methods. Breast Cancer. 2008;15:65–72. doi: 10.1007/s12282-007-0018-8. [DOI] [PubMed] [Google Scholar]
- Rayala SK, Kumar R. Sliding p21-activated kinase 1 to nucleus impacts tamoxifen sensitivity. Biomed Pharmacother. 2007a;61:408–11. doi: 10.1016/j.biopha.2007.05.006. [DOI] [PubMed] [Google Scholar]
- Rayala SK, Talukder AH, Balasenthil S, Tharakan R, Barnes CJ, Wang RA, et al. P21-activated kinase 1 regulation of estrogen receptor-alpha activation involves serine 305 activation linked with serine 118 phosphorylation. Cancer Res. 2006;66:1694–701. doi: 10.1158/0008-5472.CAN-05-2922. [DOI] [PubMed] [Google Scholar]
- Romano A, Lindsey PJ, Fischer DC, Delvoux B, Paulussen AD, Janssen RG, et al. Two functionally relevant polymorphisms in the human progesterone receptor gene (+331 G/A and progins) and the predisposition for breast and/or ovarian cancer. Gynecol Oncol. 2006;101:287–95. doi: 10.1016/j.ygyno.2005.10.040. [DOI] [PubMed] [Google Scholar]
- Sabnis GJ, Jelovac D, Long B, Brodie A. The role of growth factor receptor pathways in human breast cancer cells adapted to long-term estrogen deprivation. Cancer Res. 2005;65:3903–10. doi: 10.1158/0008-5472.CAN-04-4092. [DOI] [PubMed] [Google Scholar]
- Sachdev D, Hartell JS, Lee AV, Zhang X, Yee D. A dominant negative type I insulin-like growth factor receptor inhibits metastasis of human cancer cells. J Biol Chem. 2004;279:5017–24. doi: 10.1074/jbc.M305403200. [DOI] [PubMed] [Google Scholar]
- Sachdev D, Yee D. Disrupting insulin-like growth factor signaling as a potential cancer therapy. Mol Cancer Ther. 2007;6:1–12. doi: 10.1158/1535-7163.MCT-06-0080. [DOI] [PubMed] [Google Scholar]
- Salerno M, Sisci D, Mauro L, Guvakova MA, Ando S, Surmacz E. Insulin receptor substrate 1 is a target for the pure antiestrogen ICI 182,780 in breast cancer cells. Int J Cancer. 1999;81:299–304. doi: 10.1002/(SICI)1097-0215(19990412)81:2<299::AID-IJC21>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Santen RJ, Song RX, Zhang Z, Kumar R, Jeng MH, Masamura S, et al. Adaptive hypersensitivity to estrogen: mechanisms and clinical relevance to aromatase inhibitor therapy in breast cancer treatment. J Steroid Biochem Mol Biol. 2005;95:155–65. doi: 10.1016/j.jsbmb.2005.04.025. [DOI] [PubMed] [Google Scholar]
- Schiff R, Massarweh SA, Shou J, Bharwani L, Mohsin SK, Osborne CK. Cross-talk between estrogen receptor and growth factor pathways as a molecular target for overcoming endocrine resistance. Clin Cancer Res. 2004;10:331S–6S. doi: 10.1158/1078-0432.ccr-031212. [DOI] [PubMed] [Google Scholar]
- Skolnik EY, Lee CH, Batzer A, Vicentini LM, Zhou M, Daly R, et al. The SH2/SH3 domain-containing protein GRB2 interacts with tyrosine-phosphorylated IRS1 and Shc: implications for insulin control of ras signalling. EMBO J. 1993;12:1929–36. doi: 10.1002/j.1460-2075.1993.tb05842.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song RX, Barnes CJ, Zhang Z, Bao Y, Kumar R, Santen RJ. The role of Shc and insulin-like growth factor 1 receptor in mediating the translocation of estrogen receptor alpha to the plasma membrane. Proc Natl Acad Sci U S A. 2004;101:2076–81. doi: 10.1073/pnas.0308334100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staka CM, Nicholson RI, Gee JM. Acquired resistance to oestrogen deprivation: role for growth factor signalling kinases/oestrogen receptor cross-talk revealed in new MCF-7X model. Endocr Relat Cancer. 2005;12(Suppl 1):S85–97. doi: 10.1677/erc.1.01006. [DOI] [PubMed] [Google Scholar]
- Subramanian K, Jia D, Kapoor-Vazirani P, Powell DR, Collins RE, Sharma D, et al. Regulation of estrogen receptor alpha by the SET7 lysine methyltransferase. Mol Cell. 2008;30:336–47. doi: 10.1016/j.molcel.2008.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tharakan R, Lepont P, Singleton D, Kumar R, Khan S. Phosphorylation of estrogen receptor alpha, serine residue 305 enhances activity. Mol Cell Endocrinol. 2008;295:70–8. doi: 10.1016/j.mce.2008.07.018. [DOI] [PubMed] [Google Scholar]
- Umayahara Y, Kawamori R, Watada H, Imano E, Iwama N, Morishima T, et al. Estrogen regulation of the insulin-like growth factor I gene transcription involves an AP-1 enhancer. J Biol Chem. 1994;269:16433–42. [PubMed] [Google Scholar]
- Varricchio L, Migliaccio A, Castoria G, Yamaguchi H, de Falco A, Di Domenico M, et al. Inhibition of estradiol receptor/Src association and cell growth by an estradiol receptor alpha tyrosine-phosphorylated peptide. Mol Cancer Res. 2007;5:1213–21. doi: 10.1158/1541-7786.MCR-07-0150. [DOI] [PubMed] [Google Scholar]
- Wiseman LR, Johnson MD, Wakeling AE, Lykkesfeldt AE, May FE, Westley BR. Type I IGF receptor and acquired tamoxifen resistance in oestrogen-responsive human breast cancer cells. Eur J Cancer. 1993;29A:2256–64. doi: 10.1016/0959-8049(93)90218-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.