

Summary
Medicinal inorganic chemistry can exploit the unique properties of metal ions for the design of new drugs. This has, for instance, led to the clinical application of chemotherapeutic agents for cancer treatment, such as cisplatin. The use of cisplatin is, however, severely limited by its toxic side effects. This has spurred chemists to employ different strategies in the development of new metal-based anticancer agents with different mechanisms of action. Recent trends in the field are discussed in this review. These include the more selected delivery and/or activation of cisplatin-related prodrugs and the discovery of new non-covalent interactions with the classical target, DNA. The use of the metal as scaffold rather than reactive centre and the departure from the cisplatin paradigm of activity towards a more targeted, cancer cell-specific approach, a major trend, are discussed as well. All this, together with the observation that some of the new drugs are organometallic complexes, illustrates that exciting times lie ahead for those interested in ‘metals in medicine’.
Introduction
Medicinal inorganic chemistry [••1-3] is a field of increasing prominence as metal-based compounds offer possibilities for the design of therapeutic agents not readily available to organic compounds. The wide range of coordination numbers and geometries, accessible redox states, thermodynamic and kinetic characteristics, and the intrinsic properties of the cationic metal ion and ligand itself offer the medicinal chemist a wide spectrum of reactivities that can be exploited. Although metals have long been used for medicinal purposes in a more or less empirical fashion [4], the potential of metal-based anticancer agents has only been fully realised and explored since the landmark discovery of the biological activity of cisplatin [5]. To date, this prototypical anticancer drug remains one of the most effective chemotherapeutic agents in clinical use. It is particularly active against testicular cancer and, if tumours are discovered early, an impressive cure rate of nearly 100% is achieved. The clinical use of cisplatin against this and other malignancies is, however, severely limited by dose-limiting side-effects such as neuro-, hepato- and nephrotoxicity [5]. In addition to the high systemic toxicity, inherent or acquired resistance is a second problem often associated with platinum-based drugs, with further limits their clinical use. Much effort has been devoted to the development of new platinum drugs and the elucidation of cellular responses to them to alleviate these limitations [5,6]. These problems have also prompted chemists to develop alternative strategies based on different metals and aimed at different targets. We summarize here recent activities in the field of metal-based anticancer drugs. Space limitations mean that this overview is not comprehensive, but aims to highlight significant advances and illustrate emerging trends.
New modes of interaction with the classical target, DNA
In classical chemotherapy, anticancer agents target DNA directly according to the cisplatin paradigm to generate lesions which ultimately trigger cell death. Much effort has been directed towards combatting the high systemic toxicity of traditional platinum anticancer agents by designing drug delivery systems capable of delivering platinum to tumour cells only.
A recent example of the latter strategy is the encapsulation of cisplatin and carboplatin in the hollow protein cage of the iron storage protein ferritin, which can be internalized by some tumour tissues. Indeed, the drug-loaded protein showed cytotoxic activity against the rat pheochromocytoma cell line (PC12) [7]. In a different approach, minicells, bacterially-derived 400 nm anucleate particles, have been packed with chemotherapeutics, such as cisplatin, and labelled with bispecific antibodies. This resulted in endocytosis and ultimately drug release in cancer cells [8].
Platinum(iv) prodrugs can be used to overcome some of the problems associated with cisplatin and its analogues [9]. The high kinetic inertness of Pt(iv) complexes relative to their Pt(ii) analogues introduces drug stability and the two extra ligands on the octahedral metal centre offer many possibilities for modification of pharmacokinetic parameters. As intracellular reduction of platinum(iv) to platinum(ii) is usually essential for activation and subsequent cytotoxicity, these prodrugs essentially present better ways of delivering cisplatin (or its analogues) to the target tumour cell. Synthetic advances now allow the inclusion of various bioactive ligands in the axial positions, and, hence, targeting to specific types of cancer cells.
The observation that estrogen receptor-positive, ER(+), breast cancer cells treated with estrogen are sensitized to cisplatin, led Lippard et al. to synthesize the estrogen-tethered platinum(iv) complex 1 (see Figure 1a). Intracellular reduction releases one equivalent of cisplatin and two equivalents of estradiol. The latter induces up-regulation of high-mobility group (HMG) domain protein HMGB1, a protein that shields platinated DNA from nucleotide excision repair [10].
Figure 1.

Metal-based anticancer drugs that primarily target DNA. (a) Pt(iv) complexes that deliver cisplatin and two equivalents of estradiol (1) and the GST inhibitor ethacrynic acid (2) after activation. (b) Photolabile platinum diazide complexes 4 and 5 demonstrate good cytotoxicity upon irradiation. (c) Typical, cytotoxic examples of the ruthenium- and osmium-arene family of complexes.
Along a similar vein, but with the intention of overcoming platinum drug resistance rather than cell sensitization, Dyson et al. used the Pt(iv) complex 2 (Figure 1a) to target cytosolic glutathione-S-transferase (GST), which constitutes the main cellular defence against xenobiotics. Ethacraplatin (2) has the GST inhibitor ethacrynic acid, a diuretic in clinical use, attached. Reduction of ethacraplatin in the cell results in the release of two equivalents of a potent GST inhibitor together with one equivalent of the cytotoxic cisplatin [11].
Lippard et al. have tackled the problem of drug delivery by attaching a Pt(iv) prodrug to functionalized soluble single-walled carbon nanotubes (SWNT), highly effective carriers that can transport various cargos across the cell membrane through clathrin-dependent endocytosis [••12]. Platinum(iv) complex 3 has two different axial ligands (Figure 1a) and binds non-covalently to the nanotube surface and one SWNT longboat carries on average 65 platinum complexes. The conjugate shows a substantial increase in cytotoxicity compared to the untethered complex and to cisplatin.
Sadler et al. are using a strategy that relies on the photochemical activation of platinum(IV) drugs to release active antitumour agents, rather than spontaneous intracellular reduction. This can then provide localized treatment of cancers accessible to irradiation. The trans-dihydroxy platinum(iv) prodrugs are non-toxic in the dark and incorporate two azide ligands, either positioned cis (4) [•13] or trans (5) [14] to each other (Figure 1b). Irradiation results in growth inhibition of human bladder cancer cells (5) and cytotoxicity towards human skin cells (HaCaT keratinocytes) (4 and 5). The discovery that the trans-isomer, a potential precursor of the inactive transplatin, is as active as cisplatin is notable [14]. It is also notable that the cis-azide complex is not cross-resistant to cisplatin and different DNA platination pathways seem to be involved [•13]. Incorporation of a pyridine ligand into these complexes can greatly increase their potency (FS Mackay et al., unpublished).
The activity of the clinically ineffective transplatin itself is markedly enhanced upon irradiation [15]. This, together with the observed increase in cytotoxicity upon irradiation of dirhodium complexes [16], further illustrates the potential of photoactivation.
A variety of ruthenium complexes have been designed which interact specifically with the classical target, DNA [17,18]. A family of ruthenium(ii)-arene complexes developed by Sadler et al. [•19], for instance, exhibits high in vitro and in vivo anticancer activity [20]. For example, the direct coordinative binding of the monofunctional Ru-arene complex (6) (Figure 1c) to N7 of G bases in DNA is complemented by intercalative binding of the biphenyl ligand and specific hydrogen bonding interactions of the ethylenediamine NH2 groups with C6O of guanine. These additional interactions result in unique binding modes to duplex DNA and induce different structural distortions in DNA compared to cisplatin, which may explain why these complexes are not cross-resistant with cisplatin [18]. Interestingly, this chemistry has recently been extended to include osmium(ii)-arene analogues, such as 7 (Figure 1c), whose hydrolytic properties can be tuned to achieve promising activity against human A549 and A2780 ovarian cancer cells [•21].
Other ruthenium-based anticancer drugs, including the NAMI-A (9) and KP1019 (10) drugs which are under clinical evaluation, have different modes of action and specifically aim at non-classical targets such as gene products and cellular transduction pathways [22]. This shift in interest, which complements the classical approach, is one of the major trends in the field and is discussed in the later sections of this review.
Although much less studied than the metallodrug-DNA interactions, the interaction of metallodrugs with protein targets and the proteome deserves more attention, especially since such studies will not only shed light on the mechanisms of action, but also help to identify new targets for drug therapy [17,23]. Metallodrug-protein interactions studied by various advanced analytical techniques have been recently reviewed [23]. Recent characterization of protein adducts of platinum and ruthenium anticancer drugs by X-ray crystallography [24,25] or advanced mass spectrometry [26,27] show the timeliness of this approach.
Non-covalent interactions with DNA
Single-stranded ends of human telomeric DNA, which consist of guanine-rich TTAGGG repeats known to fold into G-quadruplex structures (Figure 2a), provide interesting targets for drug design. Telomeric DNA shortens after every cell division and after critical shortening of the telomeres, cells stop dividing and commit suicide. Telomerase, however, maintains the length of the telomeric DNA and overexpression of this enzyme endows the (cancer) cell with the ability to replicate indefinitely and thus proliferate. Since telomerase accepts only the single-stranded overhang, stabilization of the G-quadruplexes provides an attractive means of preventing telomerase from maintaining telomere length. The Ni(ii)-salphen complex (11) (Figure 2a) incorporates the main requirements for quadruplex-stabilizing molecules, i.e. a π-delocalized system prone to stacking on a G-quartet, a positive charge that is able to lie in the centre of the quartet, and finally, positively-charged substituents which can interact with the grooves and loops of the quadruplex. Indeed, 11 induces a high degree of quadruplex DNA stabilization and telomerase inhibition with telEC50 values in the range of 0.1 μM [28].
Figure 2.
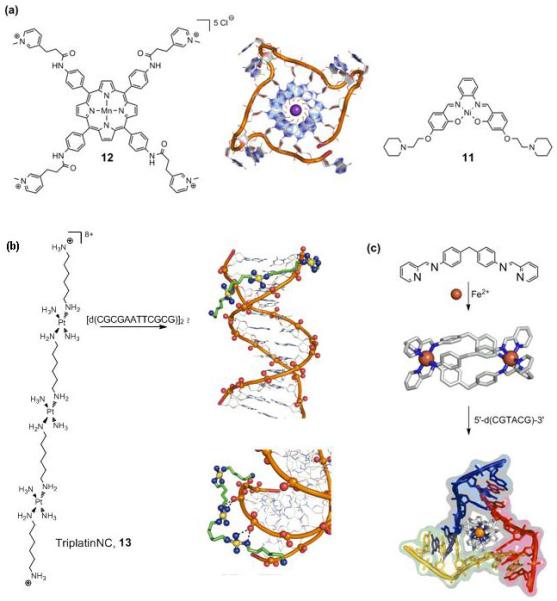
New (non-covalent) interactions with DNA. (a) G-quadruplex binding complexes 11 and 12 show high affinity and good (11) to exceptional (12) selectivity for telomeric DNA (centre, 1KF1.pdb). (b) The highly modular TriplatinNC (13) complex binds to DNA via so-called ‘phosphate clamps’ (2DYW.pdb). (c) The saturated iron triple helicate binds to a three-way DNA junction (2ET0.pdb).
An important challenge in this field is the design of complexes that bind selectively to quadruplex over duplex DNA. Whereas the Ni(ii)-salphen complex 11 shows selectivity of > 50-fold, the manganese porphyrin 12 (Figure 2a) which follows the same design criteria as mentioned previously, shows an exceptional 10,000-fold selectivity for quadruplex over duplex DNA (IC50 of around 0.6 μM) [•29]. These results illustrate the potential of metal complexes as telomere-targeted chemotherapeutics.
Besides direct coordinative binding of metallo-agents to DNA bases, other potential DNA binding modes include intercalation and groove binding. The latter modes are generally non-covalent in nature.
Farrell et al. have described a discrete binding mode based on interactions that utilize exclusively backbone functional groups [•30]. The new binding mode was observed in the crystal structure of a double-stranded B-DNA dodecamer with TriplatinNC (13) (Figure 2b). This trinuclear Pt(ii) complex is related to the trinuclear trans-platinum drug BBR3464, but lacks the reactive chloride ligands. TriplatinNC displays micromolar activity against human ovarian cancer cell lines [31]. The phosphate-selective complex binds through a multitude of specific “phosphate clamps” (see Figure 2b), bifurcated ammine(NH)⋯phosphate(O)⋯amine hydrogen bonds. A series of such phosphate clamps with one strand of DNA results in so-called “backbone tracking”, and a combination of two interstrand clamps gives rise to (minor) “groove-spanning”. Both interactions may be present in solution [•30].
A X-ray crystallographic study of the adduct between triple helicate [Fe2L3]4+ (Figure 2c) and palindromic DNA 5′-d(CGTACG) by Hannon, Coll et al. reveals the metallosupramolecular helicate comfortably occupying the central hydrophobic cavity of a three-way (Y-shaped) junction (Figure 2c) [•32]. The positive charge of the helicate, together with the large hydrophobic surface of the aromatic rings (together with its π-stacking potential), are the driving forces behind this specific interaction. This unprecedented mode of non-covalent DNA recognition shows that three-way junctions, naturally occurring both in DNA and RNA, provide a new structural target for design of novel, highly specific drugs [•32]. In principle, the palindromic sequence allows for the formation of any oligomeric formulation through Watson-Crick hydrogen bonding, thus presenting a dynamic combinatorial library. Addition of the triple helices then drives the selective formation of the three-junction member of this library. The DNA junction recognition compliments the previously reported major groove binding of the iron metallohelicates, which led to remarkable intramolecular DNA coiling [33]. The ruthenium metallohelicate induces a similar bending/coiling effect, further illustrating that the cylinder is responsible for this effect [34]. The Ru triple helicate exhibits cytotoxicity towards human breast cancer HBL-100 and T47D cells, albeit with modest activity (2-5 fold less potent than cisplatin). Related unsaturated dinuclear ruthenium double helicates, capable of classic coordinative binding to DNA, show greatly improved cytotoxicity towards the same cell lines (30-fold more active than cisplatin). These complexes illustrate the many possibilities of using metallosupramolecular architectures in anticancer drug design [35].
These examples not only illustrate the role of non-covalent DNA interactions, but also the newly-emerging trend of using the metal in a scaffold, rather than as the reactive centre. In a highly modular approach, the use of a metal centre as a building block allows for the spatial orientation of other functionalities (as part of the ligands), which in turn interact favourably with the target via, for instance, hydrogen bonds (phosphate clamps) or π-stacking interactions (helicates). This trend will be discussed in the next section.
The metal as scaffold
Metals ions have been traditionally included in anticancer agents to exploit their reactivity and have been particularly attractive because of the exceptionally wide range of reactivities available. On the other hand, metals can also be used as building blocks for well-defined, three-dimensional constructs. In this way, the availability of many different coordination geometries allows for the synthesis of structures with unique stereochemistry and orientation of organic ligands and structures which are not accessible through purely organic, carbon-based compounds. The kinetic inertness of the coordination/organometallic bonds make these compounds in principle behave like organic compounds. This approach immensely expands our ability to chart biologically-relevant chemical space [••36]. The group of Meggers has pioneered this approach in their development of organometallic ruthenium complexes that mimic organic enzyme inhibitors. The natural product staurosporine, for instance, is a highly potent inhibitor for various kinases (Figure 3a). Meggers et al. replaced the carbohydrate unit with ruthenium fragments. Structural variation by simple substitution of the ligands on the metal to optimize the enzyme-inhibitor interactions has resulted in the discovery of nanomolar and even picomolar protein kinase inhibitors (Figure 3b) [37]. The co-crystal structures of Pim-1 with the organometallic complexes nicely illustrate all salient features of these potent kinase inhibitors (Figure 3c) [38]. The relevance of these organometallic inhibitors as anticancer agents has been demonstrated recently. They are highly cytotoxic towards human melanoma cells. The organometallic GSK-3β inhibitor DW1/2 is a potent activator of p53 and thus induces p53-activated apoptosis via the mitochondrial pathway in otherwise highly chemoresistant melanoma cells [39]. The anticancer agent DW1/2 works by specifically targeting a protein, rather than DNA. The development of novel drugs with non-classical protein targets is becoming a major new theme in metal-based drug development and will be discussed in the next section.
Figure 3.

The metal as scaffold. (a) Concept: mimicking the protein kinase inhibitor staurosporine with an octahedral ruthenium complex. (b) DW1 (the R enantiomer of the DW1/2 racemic mixture) activates p53 and induces apoptosis in human melanoma cells. (c) The concept is demonstrated by the crystal structure of with the protein kinase Pim-1. (d) The remarkably close match is illustrated by the superimposed cocrystal structures of the organometallic inhibitor (white, 2BZI.pdb) and staurosporine (purple, 1YHS.pdb).
Proteins and enzymes as non-classical targets
Traditional anticancer drugs that target DNA make use of the fact that malignant cells divide rapidly. A drawback of this strategy is that rapidly dividing healthy cells are affected as well, causing severe toxic side-effects. Alternatively, the design of novel agents that target cellular signalling pathways specific to cancer cells would therefore be preferred. As genomics and proteomics have resulted in an explosion of available information concerning the biology of cancer cells, such targeted therapies have come within reach and offer much potential. This current shift of focus will be illustrated with a few typical examples [17].
Human thioredoxin reductase (hTrxR) is associated with many cellular processes such as antioxidant defence and redox homeostasis. hTrxR is found at elevated levels in human tumour cell lines. A strong connection with the apoptosis regulator protein p53 has been established and it is strongly associated with tumour proliferation, making hTrxR an interesting target for anticancer drugs [40]. Gold(i) complexes are amongst the most potent inhibitors of hTrxR, a feature attributable to the high electrophilicity of Au(i) and its preference for the selenocysteine residue of hTrxR. For example, phosphole-gold(i) complexes (14, Figure 4a) are highly potent, nanomolar inhibitors of hTrxR and the related human glutathione reductase (hGR) [41]. A crystal structure of 14 with hGR shows, surprisingly, the coordinative binding of one phosphole-gold unit to an exposed cysteine and a second gold atom that has lost both its chloride and phosphole ligand to form a linear S-Au-S adduct at the active site (Figure 4a). IC50 values for gliobastoma cells are in the 5-15 μM range.
Figure 4.
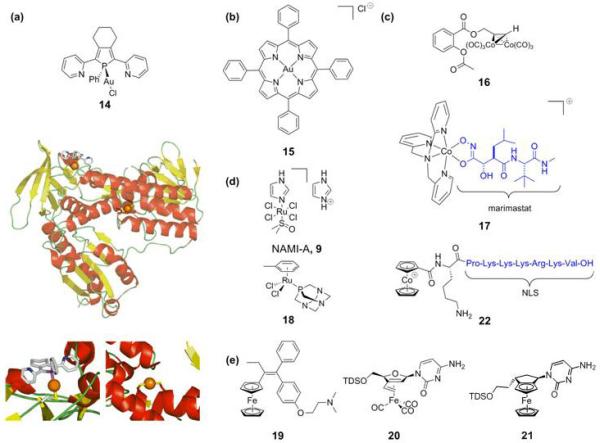
Proteins and enzymes as non-classical targets. This cross-section illustrates the diversity in structure of the complexes and the many organometallic agents being studied. (a) The Au(i)-phosphole complex 14 inhibits hTrxR. The crystal structure shows the two gold binding sites (insets, gold atoms as orange spheres, 2AAQ.pdb). (b) Gold(iii)-porphyrin anticancer agent 15. (c) The cobalt-alkyne (16) and cobalt-marimastat chaperone (17) complexes inhibit COX and MMP, respectively. The cobaltocenium complex (22) carries a nuclear localization signal for directed nuclear delivery. (d) NAMI-A (9) and the RAPTA-ruthenium complexes (18) show antimetastatic activity. (e) Ferrocifen (19) constitutes a prototypical example of a bioorganometallic drug. The nucleotide-appended organometallic iron complexes 20 and 21 show pronounced cytotoxicity.
This is one example from the active field of gold anticancer drugs [42], many of which target mitochondria, increasingly recognized as a regulator of cell death. Another promising group of gold-containing anticancer agents are the gold(iii) porphyrins (15) studied by Che et al. (Figure 4b). These complexes exhibit potent in vitro and in vivo anticancer activity towards hepatocellular and nasopharyngeal carcinoma [•43]. A functional proteomics study indeed suggests involvement of the mitochondria in the induced apoptosis [•44]. Finally, gold(iii) thiocarbamates are more cytotoxic in vitro than cisplatin, including intrinsically-resistant cell lines. The primary target is thought to be the proteasome, inhibition of which results in induction of apoptosis [45].
Other metal-containing anticancer agents are known to target specific enzymes. The cobalt-alkyne analogue 16 of the non-steroidal anti-inflammatory drug aspirin (acetylsalicylic acid) (Figure 4c) exhibits high cytotoxicity in breast cancer cell lines. The cytotoxicity correlates with cyclooxygenase (COX) inhibition. COX is involved in eicosanoid metabolism and interference with this pathway is a promising strategy for the development of new cytostatics [46].
Hambley et al. have explored the use of cobalt-containing compounds for the selective inhibition of enzymes involved in the process of tumour metastasis [47]. Their interesting strategy focuses on the selective delivery of the established maxtrix metalloproteinase (MMP) inhibitor marimastat by complexing it to a ‘chaperone’ Co(iii)-complex (17, Figure 4c). MMPs are overexpressed in tumour cells and high levels of MMPs in cancer patients correlate with poor prognosis. The Co(iii) carrier provides an inert framework for the transportation of the inhibitor and the prodrug is activated by a bioreduction pathway generating the more labile Co(ii)-complex, which leads to inhibitor release. Back-oxidation to the prodrug would be prevented by the hypoxic nature of tumour tissue, thus achieving selective release in targeted cells only. showed that Prodrug 17 shows significantly more growth inhibition towards 4T1.2 tumours in vivo in mice than marimastat alone, but also, unexpectedly, that both the inhibitor and the prodrug complex potentiates metastasis [47].
Inspired by the remarkable properties of NAMI-A (9), a compound devoid of in vitro cytotoxicity but capable of in vivo metastasis inhibition, metastasis was identified as a primary new target [•48] and a number of other ruthenium(iii) complexes have been studied for such potential activity [•48]. Remarkably, the lead complexes of the RAPTA family of ruthenium(ii)-arene compounds (18, Figure 4d) developed by Dyson et al. also showed the same low in vitro cytotoxicity but in vivo inhibition of lung metastases in CBA mice bearing MCa mammary carcinoma [49].
These examples illustrate the need to develop new assays that look beyond traditional in vitro cytotoxicity tests [22].
From inorganic to bioorganometallic drugs
The fact that many of the compounds mentioned above are organometallic complexes illustrates the emergence of the field of medicinal organometallic chemistry. More generally, bioorganometallic chemistry is relatively new, but has already led to exciting developments [50]. A prototypical example of a promising organometallic (pro)drug is the now well-established ferrocifen (19, Figure 4e) system. Ferrocifen, in contrast to its parent tamoxifen, is active against both ER(+) and ER(−) human breast cancer cell lines. The antiproliferative effect arises from the anti-estrogenic effect of the tamoxifen-like unit combined with cytotoxicity caused by the redox properties of the attached ferrocene group. Electrochemical studies on a variety of ferrocifen-like compounds have revealed a structure-activity relationship and thus established minimal structural requirements for cytotoxic effect [••51]. Organometallic iron complexes with nucleosides appended to either a ferrocenyl [52] (21) or an η4-butadiene-Fe(CO)3 group[53] (20) also show pronounced cytotoxic activity resulting induction of apoptosis.
Metzler-Nolte et al. have reported the directed nuclear delivery of an organometallic cobalt compound (22). To achieve this, the SV4-40 T antigen nuclear localization signal (NLS) was modified with a cobaltocenium cation and other groups. These cobaltocenium-NLS-bioconjugates present an intriguing opportunity for the targeted delivery of therapeutics to the cell nucleus [•54].
The large diversity in recently-reported organometallic anticancer complexes, illustrate that the full arsenal of synthetic organometallic chemistry, is now available to the medicinal chemist. Many more exciting developments can therefore be expected.
Conclusions
This survey of recent literature illustrates that many different new creative approaches are being taken towards the design of innovative metal-based anticancer drugs. The clinical success of cisplatin remains a stimulus for the development of new complexes that address the downsides associated with cisplatin, especially the systemic toxicity and (acquired) resistance. Targeted delivery and/or controlled prodrug activation, be it by light, intracellular reduction or other means, hold the promise of more selective and effective drug administration. The field of classical chemotherapy with DNA as the established target continues to produce interesting discoveries. A clearly discernible, emerging trend, however, is the departure from the cisplatin paradigm of activity. The newly discovered, mainly non-covalent DNA interactions offer a glimpse of the rich chemistry that remains to be discovered, most possibly with applications reaching further than medicinal chemistry. The concept of the metal as scaffold for the construction of unique, yet well-defined three-dimensional structures, rather than reactive centre, holds much promise. This highly modular approach, combined with currently available combinatorial techniques and knowledge of supramolecular chemistry, yields a very powerful method for optimizing drug interactions with carefully selected targets.
Future clinical success will benefit from targets which are highly specific for cancer cells. The rapidly expanding knowledge of their cellular characteristics offers many new opportunities for drugs that show low systemic toxicity and efficiently tackle the problem of drug resistance. The different examples mentioned in this review offer a promising start. Finally, the advent of medicinal bioorganometallic chemistry has further expanded the toolbox of the medicinal inorganic chemist. The nature of the research will rely ever more heavily on interdisciplinary collaboration, but many exciting discoveries and applications almost certainly lie ahead.
Acknowledgements
PCAB thanks the Netherlands Organisation for Scientific Research (NWO) for financial support through a Rubicon Scholarship. We thank the EPSRC, BBSRC, Royal Society, Wellcome Trust, EC (Marie Curie and COST), Scottish Enterprise, and Oncosense Ltd for support of our recent research on therapeutic metal complexes.
The University of Edinburgh (former employer of PJS) has filed patent applications relating to the ruthenium arene and platinum diazido complexes under study in the PJS laboratory.
References and recommended reading
Papers of particular interest, published within the annual period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- ••1.Hambley TW. Developing new metal-based therapeutics: challenges and opportunities. Dalton Trans. 2007:4929–4937. doi: 10.1039/b706075k. This recent perspective provides an excellent introduction to the present status of the field of medicinal inorganic chemistry and discusses the difficulties that are currently faced in the development of new metal-based therapeutics
- 2.Orvig C, Abrams MJ. Medicinal inorganic chemistry: introduction. Chem Rev. 1999;99:2201–2203. doi: 10.1021/cr980419w. [DOI] [PubMed] [Google Scholar]
- 3.Guo Z, Sadler PJ. Metals in Medicine. Angew Chem Int Ed. 1999;38:1512–1531. doi: 10.1002/(SICI)1521-3773(19990601)38:11<1512::AID-ANIE1512>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 4.Thompson KH, Orvig C. Metal complexes in medicinal chemistry: new vistas and challenges in drug design. Dalton Trans. 2006:761–764. doi: 10.1039/b513476e. [DOI] [PubMed] [Google Scholar]
- 5.Jung Y, Lippard SJ. Direct cellular responses to platinum-induced DNA damage. Chem Rev. 2007;107:1387–1407. doi: 10.1021/cr068207j. [DOI] [PubMed] [Google Scholar]
- 6.van Zutphen S, Reedijk J. Targeting platinum anti-tumour drugs: Overview of strategies employed to reduce systemic toxicity. Coord Chem Rev. 2005;249:2845–2853. [Google Scholar]
- 7.Yang Z, Wang XY, Diao HJ, Zhang JF, Li HY, Sun HZ, Guo ZJ. Encapsulation of platinum anticancer drugs by apoferritin. Chem Commun. 2007:3453–3455. doi: 10.1039/b705326f. [DOI] [PubMed] [Google Scholar]
- 8.MacDiarmid JA, Mugridge NB, Weiss JC, Phillips L, Burn AL, Paulin RP, Haasdyk JE, Dickson KA, Brahmbhatt VN, Pattison ST, et al. Bacterially derived 400 nm particles for encapsulation and cancer cell targeting of chemotherapeutics. Cancer Cell. 2007;11:431–445. doi: 10.1016/j.ccr.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 9.Hall MD, Mellor HR, Callaghan R, Hambley TW. Basis for design and development of platinum(IV) anticancer complexes. J Med Chem. 2007;50:3403–3411. doi: 10.1021/jm070280u. [DOI] [PubMed] [Google Scholar]
- 10.Barnes KR, Kutikov A, Lippard SJ. Synthesis, characterization, and cytotoxicity of a series of estrogen-tethered platinum(IV) complexes. Chem Biol. 2004;11:557–564. doi: 10.1016/j.chembiol.2004.03.024. [DOI] [PubMed] [Google Scholar]
- 11.Ang WH, Khalaila I, Allardyce CS, Juillerat-Jeanneret L, Dyson PJ. Rational design of platinum(IV) compounds to overcome glutathione-S-transferase mediated drug resistance. J Am Chem Soc. 2005;127:1382–1383. doi: 10.1021/ja0432618. [DOI] [PubMed] [Google Scholar]
- ••12.Feazell RP, Nakayama-Ratchford N, Dai H, Lippard SJ. Soluble single-walled carbon nanotubes as longboat delivery systems for platinum(IV) anticancer drug design. J Am Chem Soc. 2007;129:8438–8439. doi: 10.1021/ja073231f. A dual strategy against platinum drug inactivation is presented by attaching a Pt(iv) prodrug to a carbon nanotube delivery system. The conjugate showed increased cellular uptake and displayed high cytotoxicity. The ability to attach other additional compounds to the platinated longboats provides an interesting opportunity for targeted delivery.
- •13.Bednarski PJ, Grunert R, Zielzki M, Wellner A, Mackay FS, Sadler PJ. Light-activated destruction of cancer cell nuclei by platinum diazide complexes. Chem Biol. 2006;13:61–67. doi: 10.1016/j.chembiol.2005.10.011. Photolabile platinum(iv) diazide complexes are nontoxic in the dark, but demonstrate good cytotoxicity upon irradiation with light against human bladder cancer cells. No crossresistance with cisplatin is observed. The photoactivation pathway does not rely on oxygen, unlike current photodynamic therapy. See also [14].
- 14.Mackay FS, Woods JA, Moseley H, Ferguson J, Dawson A, Parsons S, Sadler PJ. A photoactivated trans-diammine platinum complex as cytotoxic as cisplatin. Chem Eur J. 2006;12:3155–3161. doi: 10.1002/chem.200501601. [DOI] [PubMed] [Google Scholar]
- 15.Heringova P, Woods J, Mackay FS, Kasparkova J, Sadler PJ, Brabec V. Transplatin is cytotoxic when photoactivated: Enhanced formation of DNA cross-links. J Med Chem. 2006;49:7792–7798. doi: 10.1021/jm0606692. [DOI] [PubMed] [Google Scholar]
- 16.Lutterman DA, Fu PKL, Turro C. cis-[Rh2(μ-O2CCH3)2(CH3CN)6]2+ as a photoactivated cisplatin analog. J Am Chem Soc. 2006;128:738–739. doi: 10.1021/ja057620q. [DOI] [PubMed] [Google Scholar]
- 17.Ang WH, Dyson PJ. Classical and non-classical ruthenium-based anticancer drugs: Towards targeted chemotherapy. Eur J Inorg Chem. 2006:4003–4018. [Google Scholar]
- 18.Liu HK, Berners-Price SJ, Wang FY, Parkinson JA, Xu JJ, Bella J, Sadler PJ. Diversity in guanine-selective DNA binding modes for an organometallic ruthenium arene complex. Angew Chem Int Ed. 2006;45:8153–8156. doi: 10.1002/anie.200602873. [DOI] [PubMed] [Google Scholar]
- •19.Habtemariam A, Melchart M, Fernandez R, Parsons S, Oswald IDH, Parkin A, Fabbiani FPA, Davidson JE, Dawson A, Aird RE, et al. Structure-Activity Relationships for Cytotoxic Ruthenium(II) Arene Complexes Containing N,N-, N,O-, and O,O-Chelating Ligands. J Med Chem. 2006;49:6858–6868. doi: 10.1021/jm060596m. Systematic ligand variation resulted in organometallic ruthenium-arene complexes as cytotoxic as cisplatin and established a structure-activity relationship for cytotoxicity. No cross-resistance with cisplatin was observed .
- 20.Yan YK, Melchart M, Habtemariam A, Sadler PJ. Organometallic chemistry, biology and medicine: ruthenium arene anticancer complexes. Chem Commun. 2005:4764–4776. doi: 10.1039/b508531b. [DOI] [PubMed] [Google Scholar]
- •21.Peacock AFA, Parsons S, Sadler PJ. Tuning the hydrolytic aqueous chemistry of osmium arene complexes with N,O-chelating ligands to achieve cancer cell cytotoxicity. J Am Chem Soc. 2007;129:3348–3357. doi: 10.1021/ja068335p. The aqueous and hydrolytic chemistry of osmium analogues of the half-sandwich ruthenium(ii) complexes were studied by systematic variation of the chelating ligand on osmium. The rational control of chemical reactivity resulted in cytotoxic osmium-arene complexes.
- 22.Dyson PJ, Sava G. Metal-based antitumour drugs in the post genomic era. Dalton Trans. 2006:1929–1933. doi: 10.1039/b601840h. [DOI] [PubMed] [Google Scholar]
- 23.Timerbaev AR, Hartinger CG, Aleksenko SS, Keppler BK. Interactions of antitumor metallodrugs with serum proteins: Advances in characterization using modern analytical methodology. Chem Rev. 2006;106:2224–2248. doi: 10.1021/cr040704h. [DOI] [PubMed] [Google Scholar]
- 24.Calderone V, Casini A, Mangani S, Messori L, Orioli PL. Structural investigation of cisplatin-protein interactions: Selective platination of His19 in a cuprozinc superoxide dismutase. Angew Chem Int Ed. 2006;45:1267–1269. doi: 10.1002/anie.200502599. [DOI] [PubMed] [Google Scholar]
- 25.McNae IW, Fishburne K, Habtemariam A, Hunter TM, Melchart M, Wang FY, Walkinshaw MD, Sadler PJ. Half-sandwich arene ruthenium(II)-enzyme complex. Chem Commun. 2004:1786–1787. doi: 10.1039/b408141b. [DOI] [PubMed] [Google Scholar]
- 26.Casini A, Mastrobuoni G, Ang WH, Gabbiani C, Pieraccini G, Moneti G, Dyson PJ, Messori L. ESI-MS characterisation of protein adducts of anticancer ruthenium(II)-arene PTA (RAPTA) complexes. ChemMedChem. 2007;2:631–635. doi: 10.1002/cmdc.200600258. [DOI] [PubMed] [Google Scholar]
- 27.Weidt SK, Mackay CL, Langridge-Smith PRR, Sadler PJ. Platination of superoxide dismutase with cisplatin: tracking the ammonia ligands using Fourier transform ion cyclotron resonance mass spectrometry (FT-ICR MS) Chem Commun. 2007:1719–1721. doi: 10.1039/b701903c. [DOI] [PubMed] [Google Scholar]
- 28.Reed JE, Arnal AA, Neidle S, Vilar R. Stabilization of G-quadruplex DNA and inhibition of telomerase activity by square-planar nickel(II) complexes. J Am Chem Soc. 2006;128:5992–5993. doi: 10.1021/ja058509n. [DOI] [PubMed] [Google Scholar]
- •29.Dixon IM, Lopez F, Tejera AM, Esteve JP, Blasco MA, Pratviel G, Meunier B. A G-quadruplex ligand with 10000-fold selectivity over duplex DNA. J Am Chem Soc. 2007;129:1502–1503. doi: 10.1021/ja065591t. A metalloporphyrin is reported that shows both a very high affinity and an excellent selectivity for G-quadruplex DNA over GC-rich or AT-rich DNA.
- •30.Komeda S, Moulaei T, Woods KK, Chikuma M, Farrell NP, Williams LD. A third mode of DNA binding: Phosphate clamps by a polynuclear platinum complex. J Am Chem Soc. 2006;128:16092–16103. doi: 10.1021/ja062851y. The polynuclear platinum complex TriplatinNC binds to DNA through specific hydrogen bonding interactions called phosphate clamps, presenting a new DNA binding mode. The modularity of TriplatinNC holds promise for the assembly of drugs that selectively bind to specific DNA forms.
- 31.Harris AL, Yang X, Hegmans A, Povirk L, Ryan JJ, Kelland L, Farrell NP. Synthesis, characterization, and cytotoxicity of a novel highly charged trinuclear platinum compound. Enhancement of cellular uptake with charge. Inorg Chem. 2005;44:9598–9600. doi: 10.1021/ic051390z. [DOI] [PubMed] [Google Scholar]
- •32.Oleksi A, Blanco AG, Boer R, Uson I, Aymami J, Rodger A, Hannon MJ, Coll M. Molecular recognition of a three-way DNA junction by a metallosupramolecular helicate. Angew Chem Int Ed. 2006;45:1227–1231. doi: 10.1002/anie.200503822. The crystal structure of a supramolecular cylinder with a palindromic hexanucleotide revealed a new DNA binding mode. The iron-containing triple helicate was found at the heart of a three-way DNA junction.
- 33.Hannon MJ. Supramolecular DNA recognition. Chem Soc Rev. 2007;36:280–295. doi: 10.1039/b606046n. [DOI] [PubMed] [Google Scholar]
- 34.Pascu GI, Hotze ACG, Sanchez-Cano C, Kariuki BM, Hannon MJ. Dinuclear ruthenium(II) triple-stranded helicates: Luminescent supramolecular cylinders that bind and coil DNA and exhibit activity against cancer cell lines. Angew Chem Int Ed. 2007;46:4374–4378. doi: 10.1002/anie.200700656. [DOI] [PubMed] [Google Scholar]
- 35.Hotze ACG, Kariuki BM, Hannon MJ. Dinuclear double-stranded metallosupramolecular ruthenium complexes: Potential anticancer drugs. Angew Chem Int Ed. 2006;45:4839–4842. doi: 10.1002/anie.200601351. [DOI] [PubMed] [Google Scholar]
- ••36.Meggers E, Atilla-Gokcumen GE, Bregman H, Maksimoska J, Mulcahy SP, Pagano N, Williams DS. Exploring chemical space with organometallics: Ruthenium complexes as protein kinase inhibitors. Synlett. 2007:1177–1189. This account discusses the concept of exploration of chemical space with organometallic ruthenium complexes and gives an overview of the impressive results obtained by the Meggers group thus far. See also references [37] and [38].
- 37.Bregman H, Carroll PJ, Meggers E. Rapid access to unexplored chemical space by ligand scanning around a ruthenium center: discovery of potent and selective protein kinase inhibitors. J Am Chem Soc. 2006;128:877–884. doi: 10.1021/ja055523r. [DOI] [PubMed] [Google Scholar]
- 38.Debreczeni JE, Bullock AN, Atilla GE, Williams DS, Bregman H, Knapp S, Meggers E. Ruthenium half-sandwich complexes bound to protein kinase Pim-1. Angew Chem Int Ed. 2006;45:1580–1585. doi: 10.1002/anie.200503468. [DOI] [PubMed] [Google Scholar]
- 39.Smalley KSM, Contractor R, Haass NK, Kulp AN, Atilla-Gokcumen GE, Williams DS, Bregman H, Flaherty KT, Soengas MS, Meggers E, et al. An organometallic protein kinase inhibitor pharmacologically activates p53 and induces apoptosis in human melanoma cells. Cancer Res. 2007;67:209–217. doi: 10.1158/0008-5472.CAN-06-1538. [DOI] [PubMed] [Google Scholar]
- 40.Urig S, Becker K. On the potential of thioredoxin reductase inhibitors for cancer therapy. Semin Cancer Biol. 2006;16:452–465. doi: 10.1016/j.semcancer.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 41.Urig S, Fritz-Wolf K, Reau R, Herold-Mende C, Toth K, Davioud-Charvet E, Becker K. Undressing of phosphine gold(I) complexes as irreversible inhibitors of human disulfide reductases. Angew Chem Int Ed. 2006;45:1881–1886. doi: 10.1002/anie.200502756. [DOI] [PubMed] [Google Scholar]
- 42.Barnard PJ, Berners-Price SJ. Targeting the mitochondrial cell death pathway with gold compounds. Coord Chem Rev. 2007;251:1889–1902. [Google Scholar]
- •43.Sun RWY, Ma DL, Wong ELM, Che CM. Some use of transition metal complexes as anti-cancer and anti-HIV agents. Dalton Trans. 2007:4884–4892. doi: 10.1039/b705079h. This perspective covers the recent contributions of the Che laboratory to the development of transition-metal-based anticancer complexes, including gold, platinum, ruthenium and iron complexes.
- •44.Wang Y, He QY, Che CM, Chiu JF. Proteomic characterization of the cytotoxic mechanism of gold(III) porphyrin 1a, a potential anticancer drug. Proteomics. 2006;6:131–142. doi: 10.1002/pmic.200402027. A functional proteomics study on the cytotoxic mechanism of a gold(iii) porphyrin anticancer drug was conducted to identify the proteins involved in apoptosis pathways. Multiple factors were identified, suggestive of mitochondria being centrally involved in cell death.
- 45.Milacic V, Chen D, Ronconi L, Landis-Piwowar KR, Fregona D, Dou QP. A novel anticancer gold(III) dithiocarbamate compound inhibits the activity of a purified 20S proteasome and 26S proteasome in human breast cancer cell cultures and xenografts. Cancer Res. 2006;66:10478–10486. doi: 10.1158/0008-5472.CAN-06-3017. [DOI] [PubMed] [Google Scholar]
- 46.Ott I, Schmidt K, Kircher B, Schumacher P, Wiglenda T, Gust R. Antitumor-active cobalt-alkyne complexes derived from acetylsalicylic acid: Studies on the mode of drug action. J Med Chem. 2005;48:622–629. doi: 10.1021/jm049326z. [DOI] [PubMed] [Google Scholar]
- 47.Failes TW, Cullinane C, Diakos CI, Yamamoto N, Lyons JG, Hambley TW. Studies of a cobalt(III) complex of the MMP inhibitor marimastat: A potential hypoxia-activated prodrug. Chem Eur J. 2007;13:2974–2982. doi: 10.1002/chem.200601137. [DOI] [PubMed] [Google Scholar]
- •48.Bergamo A, Sava G. Ruthenium complexes can target determinants of tumour malignancy. Dalton Trans. 2007:1267–1272. doi: 10.1039/b617769g. The authors argue a case for a change in strategy for the development of new metallotherapeutics. Metastasis is identified as the primary target for drug therapy. See also reference [22].
- 49.Scolaro C, Bergamo A, Brescacin L, Delfino R, Cocchietto M, Laurenczy G, Geldbach TJ, Sava G, Dyson PJ. In Vitro and in Vivo Evaluation of Ruthenium(II)-Arene PTA Complexes. J Med Chem. 2005;48:4161–4171. doi: 10.1021/jm050015d. [DOI] [PubMed] [Google Scholar]
- 50.Schatzschneider U, Metzler-Nolte N. New Principles in Medicinal Organometallic Chemistry. Angew Chem Int Ed. 2006;45:1504–1507. doi: 10.1002/anie.200504604. [DOI] [PubMed] [Google Scholar]
- ••51.Hillard E, Vessieres A, Thouin L, Jaouen G, Amatore C. Ferrocene-mediated proton-coupled electron transfer in a series of ferrocifen-type breast-cancer drug candidates. Angew Chem Int Ed. 2006;45:285–290. doi: 10.1002/anie.200502925. Electrochemical studies on a series of ferrocifen-type complexes revealed a structure-activity relationship and thus established minimal structural requirements for cytotoxic effect.
- 52.James P, Neudorfl J, Eissmann M, Jesse P, Prokop A, Schmalz HG. Enantioselective synthesis of ferrocenyl nucleoside analogues with apoptosis-inducing activity. Org Lett. 2006;8:2763–2766. doi: 10.1021/ol060868f. [DOI] [PubMed] [Google Scholar]
- 53.Schlawe D, Majdalani A, Velcicky J, Hessler E, Wieder T, Prokop A, Schmalz HG. Iron-containing nucleoside analogues with pronounced apoptosis-inducing activity. Angew Chem Int Ed. 2004;43:1731–1734. doi: 10.1002/anie.200353132. [DOI] [PubMed] [Google Scholar]
- •54.Noor F, Wustholz A, Kinscherf R, Metzler-Nolte N. A cobaltocenium-peptide bioconjugate shows enhanced cellular uptake and directed nuclear delivery. Angew Chem Int Ed. 2005;44:2429–2432. doi: 10.1002/anie.200462519. Attaching a nuclear localization signal peptide to a cobaltocenium group resulted in significant accumulation of the conjugate in the nucleus of HepG2 cells. In addition to the peptide-mediated nuclear delivery, a metal-enhanced cellular uptake was observed as the organometallic group was essential for active endocytosis.