

Abstract
Adoptive cell transfer has been shown to significantly reduce established tumors in both experimental models and cancer patients. Due to the toleragenic nature of cancer, approaches that lead to durable maintenance of functional T cells in tumor-bearing hosts are needed to maximize tumor regression. In this study, we investigated strategies to augment CD8+ T cell (T-CD8)-mediated adoptive immunotherapy of mice bearing advanced-stage autochthonous brain tumors by targeting a weakly immunogenic epitope. We found that immunization enhanced the accumulation of adoptively transferred T-CD8 at the tumor site, but that the timing of immunization was critical for optimal T cell expansion. A more rapid accumulation of T-CD8 was achieved when mice were conditioned with agonist anti-CD40 antibody prior to adoptive transfer due to increased T cell activation against the endogenous tumor antigen. Both approaches led to an increase in the lifespan of SV11 mice due to decreased tumor progression. However, tumor-specific T-CD8 did not persist long-term at the tumor site following administration of either regimen. Importantly, the combination of anti-CD40 conditioning followed by optimally-timed immunization synergistically promoted long-term maintenance of T-CD8 in the brain and dramatically enhanced survival. A second round of combination immunotherapy resulted in a further increase in survival, suggesting long-term tumor sensitivity to CD8+ T cell-based immunotherapy. These results demonstrate that even a weak antigen can be effectively targeted for control of established tumors using a combined adoptive transfer plus immune modulation approach and suggest that similar strategies may translate to clinical practice.
Keywords: CD8+ T cell, CD40, Immunotherapy, SV40 T antigen transgenic mice, vaccination
Introduction
CD8+ T lymphocytes (T-CD8) have the capacity to destroy tumor cells through recognition of specific peptide/MHC class I complexes. The demonstration that tumor-reactive T-CD8 can be isolated from cancer patients (1, 2) has fueled the development of both vaccination and T cell-based adoptive immunotherapy approaches (3, 4). However, the finding that many tumor-associated antigens represent self antigens has raised concerns that immunological tolerance and other mechanisms that limit self-reactive immunity preclude effective T-CD8-based approaches to cancer therapy. Indeed, since the most stongly self-reactive T-CD8 are eliminated in the thymus during T cell development, the pool of self/tumor reactive T-CD8 is often comprised of low avidity T cells (5) or higher avidity T-CD8 specific for antigens expressed at low or undetectable levels during T cells development (6), including mutated antigens (7).
The use of adoptive immunotherapy approaches can overcome some of the constraints on the responder T cells through ex vivo generation of large numbers of tumor-responsive T-CD8 and selection of the most strongly reactive T cell clones. Once transferred into the tumor-bearing host however, these tumor-reactive T cells are often subject to a number of regulatory mechanims that prevent their expansion, migration into the tumor, effector function and persistence (8–10). Thus, a current challenge lies in the ability to direct transferred cells to the tumor site and maintain persistence of functional intratumoral cells at a level within the tumor-bearing host that produces a clinical effect (11, 12).
Mice that develop tumors due to transgenic expression of an oncoprotein provide a challenging setting for immunotherapy due to the autochthonous nature of the tumor and its associated self antigens. Mice of the SV11 (H-2b) lineage express the SV40 large T antigen (Tag) oncoprotein from the viral promoter, resulting in development of choroid plexus papillomas within the brain ventricles (13). Tumors are detectable by 40 days of age and progress rapidly and reproducibly until mice succumb to tumor burden at a mean age of 105 days (14). Tag is the target of a potent T-CD8 response in normal C57BL/6 mice, in which immunization with wild-type Tag induces T-CD8 specific for three dominant epitopes: epitope I (Tag residues 206-215), epitope II/III (residues 223-231), and epitope IV (residues 404-411) (15–17). In SV11 mice, Tag expression in the thymus induces central tolerance to all three dominant epitopes, but a residual population of Tag-specific T-CD8 specific for an immunorecessive epitope, designated Tag-V (residues 489-497), survive negative selection and persist in SV11 mice (18).
We recently investigated the fate of Tag-V specific T-CD8 in tumor-bearing SV11 mice using adoptive transfer of T-cell receptor transgenic T-CD8 specific for Tag-V (TCR-V cells) (19). TCR-V cells were activated in the tumor-draining lymph nodes within hours of transfer into tumor-bearing SV11 hosts and reached peak levels of accumulation by day 4. Immunization on the day of adoptive transfer led to only a two-fold increase in T cell numbers in SV11 mice and subsequent trafficking to the tumor site, but T cell persistence was short-lived and had no effect on tumor progression. In contrast, a booster immunization administered 7 days post transfer and primary immunization resulted in a dramatic increase in TCR-V T cell accumulation in the brain and was associated with a significant delay in tumor progression. These findings raised the possibility that the timing of immunization following adoptive immunotherapy may be critical for establishment of an effective anti-tumor T-CD8 response.
In the current study, we determined both the optimal time for immunization following adoptive transfer of TCR-V T cells into tumor-bearing mice and whether the initial response to endogenous tumor antigen could be enhanced. Our results indicate that both carefully timed immunization and conditioning with agonist anti-CD40 antibody can enhance initial T-CD8 accumulation at the tumor site and that the combination of these approaches prolongs T cell persistence and enhances control of established tumors.
Materials and Methods
Mice
C57BL/6J (H-2b), B6.SJL-PtprcaPepcb/BoyJ (CD45.1+), and UBI-GFP/BL6 mice were purchased from the Jackson Laboratories (Bar Harbor, ME) and maintained at the animal facility of the Milton S. Hershey Medical Center. SV11 mice on the C57BL/6 background express full length SV40 Tag under the control of the SV40 promoter/enhancer (13) and were maintained by backcrossing Tag transgene positive males with C57BL/6J females. SV11 transgene positive mice were identified by PCR amplification of the transgene as previously described (20). Transgene positive (SV11) and negative (B6) littermates were used at 85 days of age. Line 459 mice (designated TCR-V) expressing the TCRα and TCRβ chains specific for Tag epitope V on the C57BL/6 background have been previously described (21). Line 459 mice were bred with B6.SJL-PtprcaPepcb/BoyJ mice in order to generate TCR-V/CD45.1 congenic mice, referred to as heterozygous (CD45.1/CD45.2) or homozygous (CD45.1/CD45.1) TCR-V mice. TCR-V transgene positive mice were identified by screening peripheral blood lymphocytes for TCRVβ7+ CD8+ cells by flow cytometry. All animal experiments were performed using protocols approved by the institutional animal care and use committee of the Pennsylvania State University College of Medicine and comply with federal guidelines.
Cell lines and media
B6/T116A1 (B6/V-only Tag) cells express a Tag variant in which epitopes I (residues 207-215) and II/III (residues 223-231) are deleted and epitope IV is inactivated by alanine substitution of residues 406, 408, and 411, but in which epitope V remains intact (22) and were propagated in vitro as previously described (19). Ex vivo lymphocytes were maintained in complete RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), 100U/mL of penicillin, 100 μg/mL streptomycin, 2 mM L-glutamine, and 50 μM 2-mercaptoethanol.
Synthetic peptides
Peptides were synthesized at the Macromolecular Core Facility of the Milton S. Hershey Medical Center by Fmoc chemistry using an automated peptide synthesizer (9050 MiliGen PepSynthesizer; Milipore). Peptides were solubilized in dimethyl sulfoxide and diluted to 5 μM in RPMI-1640 medium. Peptides used include SV40 Tag-V (QGINNLDNL; peptide V) and influenza virus (Flu) nucleoprotein (NP) 366-374 (ASNENMETM)(23).
Adoptive transfer, immunization and α-CD40 conditioning
Adoptive transfers were performed as described previously (19) by intravenous injection of 5×106 clonotypic naïve TCR-V cells per mouse. For immunization, 5×107 B6/V-only Tag cells were injected intraperitoneally in 0.5 mL of PBS. For in vivo conditioning with α-CD40 mice received 100 μg of agonistic α-CD40 mAb, clone FGK45 (a generous gift of Dr. Stephen Schoenberger, LaJolla Institute for Immunology), or polyclonal rat IgG (Sigma-Aldrich) by i.p. injection in 200 μl PBS the day prior to and the day after adoptive transfer of TCR-V cells. For combination immunotherapy, mice received 100 μg of purified α-CD40 or polyclonal rat IgG the day prior to and the day after adoptive transfer of 5×106 naïve clonotypic TCR-V cells followed seven days later by immunization with 5×107 B6/V-only Tag cells via intraperitoneal injection. For secondary combinatorial therapy, mice initially received combinatorial immunotherapy as just described at 85 days of age with adoptive transfer of homozygous CD45.1/CD45.1 TCR-V cells. At 125 days of age, a second round of combinatorial therapy was administered in which mice received 100 μg of purified α-CD40 or polyclonal rat IgG the day prior to and the day after adoptive transfer of 5×106 naïve clonotypic heterozygous CD45.1/CD45.2 TCR-V cells, followed seven days later by immunization with 5×107 B6/V-only Tag cells via intraperitoneal injection.
Lymphocyte isolation
Mice were anesthetized via intraperitoneal injection of sodium pentobarbital (70 mg/kg body weight) diluted in 10% ethanol and perfused transcardially with 10 ml PBS. Spleens, brains, cervical lymph nodes (CLN) and inguinal lymph nodes (ILN) were harvested into cold RPMI-1640. Spleens and lymph nodes (LN) were processed to single cell suspensions and spleens were depleted of RBCs using Tris NH4Cl as previously described (19). Lymphocytes were isolated from brains as described previously (19).
MHC tetramers and antibodies
MHC class I tetrameric complexes corresponding to the H-2Db/Tag epitope V (Tag-V tetramer) and H-2Db/influenza virus NP epitope 366-374 (Flu tetramer) conjugated with streptavidin-PE were prepared as previously described (22). Purified anti-CD16/CD32 was purchased from BD-Pharmingen. The following antibodies were purchased from eBioscience: PE-Cy5-labeled anti-mouse CD8a (clone 53-6-7); FITC-labeled anti-mouse CD44 (clone IM7), FITC-labeled anti-mouse CD45.1 (clone A20), biotin-labeled anti-mouse CD45.1 (clone A20), FITC-labeled anti-mouse CD45.2 (clone 104), PercP-Cy5 labeled anti-mouse CD8a (clone 53-6-7), FITC-labeled anti-BrdU (clone PRB-1), PE-and FITC-conjugated anti-mouse IFNγ (clone XMG1.2). Unlabeled streptavidin and streptavidin-Alexa-647 were purchased from Molecular Probes.
Flow cytometric analysis
For ex vivo characterization of isolated T cells, lymphocytes were resuspended at 2×107/ml in FACS buffer (PBS containing 2%FBS/0.01% NaN3), and incubated in the presence of anti-CD16/CD32 and unconjugated streptavidin for 30 minutes on ice. Cells were washed in FACS buffer and resuspended in a cocktail of the indicated fluorochrome-conjugated antibody and tetramer. PE-conjugated tetramers were diluted 1:200; FITC-conjugated mAbs (1:50); all other mAbs were diluted 1:100. Cells were stained in the dark on ice for 1 hour. In the case of biotin-conjugated mAb-containing cocktails, cells were washed 3 times in FACS buffer and resuspended in streptavidin-conjugated Alexa 647 (Molecular Probes) at 1:500 dilution for 30 minutes on ice. Cells were washed 3 times in FACS buffer, and fixed with 2% paraformaldehyde/PBS and analyzed using a FACScan or FACSCalibur flow cytometer (BD Biosciences, San Jose, CA). Data were analyzed using FlowJo software (Tree Star, San Carlos, CA). The percentage of Tag-V tetramer+ CD8+ cells was calculated by subtracting the percentage of CD8+ cells that stained with the control Flu tetramer on parallel samples.
In vivo proliferation assay (CFSE)
RBC-depleted lymphocytes derived from spleens and LNs of TCR-V transgenic mice were resuspended at 1×107/mL in PBS/0.2% bovine serum albumin (BSA) and labeled with 5 μM carboxy fluorescein diacetate succinimidyl ester (CFSE) (Molecular Probes) for 10 minutes at 37°C. Cells were washed three times with PBS/0.2% BSA, resuspended in Hank’s balanced salt solution (HBSS), filtered through a cell strainer (Falcon), and injected i.v. at a dose of 5×106 clonotypic TCR-V cells per mouse in 0.4 ml. After 7 days the dilution of CFSE was determined on Tag-V tetramer+, CD8+ lymphocytes.
Intracellular cytokine assay
In vitro stimulation of lymphocytes ex vivo and subsequent staining for intracellular IFNγ production was performed as described previously (19) and analyzed by flow cytometry as described above. The percentage of CD8+ cells that stained specifically for intracellular IFNγ following stimulation with Tag-V peptide was determined by subtracting the percentage of CD8+ cells that stained for IFNγ in the presence of the control Flu NP peptide.
In vivo labeling with bromodeoxyuridine (BrdU)
Mice were injected intraperitoneally every 12 hours for 48 hours with 1 ml of 1 mg/mL BrdU (Sigma) diluted in PBS two days prior to sacrifice in order to label proliferating cells. Mice were sacrificed 12 hours after the fourth dose of BrdU was administered and lymphocytes from the brain, spleen, and CLN were prepared as described above for lymphocyte isolation. Single cell suspensions were surface stained with Tag-V tetramer and anti-CD8 as described above and then fixed and permeabilized in 200 μL 1% paraformaldehyde/0.01% Tween-20 (Sigma) overnight at 4°C. The following day, cells were washed and incubated in 200 μL of bovine pancreatic DNAse I (50 Kunitz Units/mL = 0.1 mg/mL) (Sigma) prepared in PBS containing calcium and magnesium for 45 minutes at 37°C. Cells were washed twice and resuspended in 150 μl 0.5% Tween-20 diluted in PBS supplemented with 10% FBS. 20 μl of FITC-conjugated anti-BrdU mAb (or isotype control Ab) was added to each well and stained for 45 min on ice. Cells were washed twice and resuspended for flow cytometric analysis.
Lifespan analysis
SV11 mice were monitored for the development of hydrocephalus, indicative of end-stage choroid plexus tumors. Mice were euthanized following the development of neurological symptoms and the presence of tumors confirmed by gross examination. In some cases, spleens and brains were processed and stained for flow cytometric detection of TCR-V cells. Survival curves were constructed by the Kaplan-Meier method using GraphPad Prism software (GrafPad Software, Inc). Significance was determined by single-factor ANOVA, and validated using the log-rank test. p values of <0.05 were considered significant.
Results
Timing of immunization is critical for optimal expansion of TCR-V cells and accumulation at the tumor site
We determined whether a delay in immunization relative to the time of adoptive transfer might increase expansion of adoptively transferred T cells in vivo. We examined the efficiency of T cell accumulation following adoptive transfer of CFSE-labeled TCR-V cells into tumor-bearing SV11 or control B6 mice and subsequent immunization with B6/V-only Tag cells at increasing times post transfer. Spleens cells were analyzed seven days post immunization to determine the frequency of TCR-V T cells by staining with Db/epitope V tetramer (Fig. 1A, dot plots). Groups of unimmunized mice were analyzed at 7, 14 and 21 days post adoptive transfer to determine the baseline frequency of TCR-V cells prior to immunization (Fig. 1A, parentheses). Immunization of SV11 mice at day 0 induced high level expression of CD44 on TCR-V cells, indicative of antigen recognition, and accumulation of cells that had undergone multiple rounds of proliferation as indicated by loss of CFSE (Fig. 1A, histograms). Consistent with previous results (19), immunization at day 0 resulted in a 2-fold increase in TCR-V cells in the spleen. In contrast, TCR-V cell frequency increased 10-fold in SV11 mice following immunization on day 7 post transfer (30% of CD8+ splenocytes). Immunization at later time points failed to induce this same level of accumulation, indicating that TCR-V T cells became resistant to antigenic challenge after extended exposure to the endogenous Tag. In B6 mice, the highest level of TCR-V T cell accumulation also was observed when immunization was delivered on day 7 post transfer (25% of CD8+ splenocytes) (Fig. 1A). However, similar frequencies were achieved when immunization was delivered at later time points. A comparable trend was observed when these results were analyzed as total number of TCR-V splenocytes (Fig. 1B). Thus, similar numbers of TCR-V T cells accumulate in the spleens of both SV11 and B6 mice following immunization at day 7, but not at day 0 or later time points.
Figure 1.

Immunization on day 7 post adoptive transfer promotes optimal accumulation of TCR-V T cells in SV11 mice. (A–B) TCR-V cells were labeled with CFSE and adoptively transferred into 85 day-old SV11 or B6 mice. Groups of three mice were immunized with B6/Tag-V only cells at weekly intervals, including day 0, 7, 14, or 21 following adoptive transfer. A control group received no immunization (none). Mice from each group were analyzed 7 days post-immunization by staining spleen cells with anti-CD8a and Tag-V tetramer (Tet-V). (A) The percentage of CD8+ cells that stained specifically with Tet-V are indicated in the dot plots. Gated populations of CD8+Tet-V+ cells were assessed for activation status (CD44) and proliferation (CFSE). Separate groups of unimmunized mice were analyzed at day 7, 14 and 21 to determine baseline frequency of TCR-V cells at the time of immunization (percentage indicated within parentheses). (B) The total number of CD8+ Tet-V+ cells per spleen for each experimental group in (A). Error bars indicate standard error of the mean. (C) TCR-V cells were adoptively transferred into 85 day-old SV11 mice. Groups of three mice received immunization at various time points post-transfer: 0, 2, 4, 5, 7, 9, 12, and 14 days. Splenocytes were isolated and assessed 7 days following immunization to determine the frequency of TCR-V cells by staining of CD8+ cells with Tag-V tetramer. Error bars indicate standard error of the mean. (D) TCR-V cells were adoptively transferred into 85 day-old SV11 mice. Groups of three mice received no immunization (none); immunization on the day of transfer (Day 0); or immunization on day 7 post transfer (Day 7). Splenocytes and brain lymphocytes were isolated and assessed 7 days later for TCR-V cell frequency in the spleen and brain by staining of CD8+ cells with Tag-V tetramer and for production of IFNγ in response to Tag-V peptide stimulation (% = % of tetramer-V+ or IFNγ+/CD8+ cells).
To determine more specifically the time when immunization induces optimal accumulation of TCR-V T cells, we immunized groups of SV11 mice at 0, 2, 4, 5, 7, 9, 12 and 14 days post-transfer of naïve TCR-V T cells. The results in figure 1C demonstrate that immunization as early as day 4 post adoptive transfer enhanced T cell accumulation in the spleen relative to day 0 immunization, but that immunization at day 7 promoted the highest level of accumulating TCR-V T cells. This response was slightly decreased, although still substantial, following immunization at day 9 and dramatically decreased by day 12. These data indicate that immunization at day 7 promotes optimal accumulation of TCR-V cells in tumor-bearing mice and suggest that prolonged exposure to endogenous Tag ultimately tolerizes TCR-V cells.
We tested the functional capacity of the responding TCR-V T cells and determined whether immunization at day 7 promoted TCR-V cell accumulation in the brain of tumor-bearing mice. Groups of mice received (i) no immunization; (ii) immunization on the day of transfer (day 0); or (iii) immunization on day 7 post adoptive transfer. The frequency of TCR-V cells in spleen and brain was determined by MHC tetramer analysis and by intracellular cytokine assay in parallel to estimate the proportion of epitope V-specific T cells that could produce IFNγ. The results indicate that immunization at day 7, but not day 0, promoted high level infiltration and accumulation of TCR-V cells in the brain (Fig. 1D; 50% of CD8+ cells). Furthermore, a population of T-CD8 in both the spleen (20%) and brain (32%) produced Tag-V-specific IFNγ, indicating that a significant proportion of TCR-V cells that accumulate in SV11 mice following day 7 immunization are functional. These data demonstrate that adoptive transfer followed by day 7 immunization promotes accumulation of functional TCR-V T cells at the tumor site without the need for a booster immunization.
Control of tumor progression in SV11 mice is associated with high level TCR-V T cell persistence in the brain
We next assessed the potential for day 7 immunization to enhance survival of 85 day-old tumor-bearing SV11 mice. Adoptive transfer of TCR-V cells + day 7 immunization significantly enhanced SV11 survival compared to mice receiving no treatment, adoptive transfer alone or adoptive transfer plus day 0 immunization (Fig. 2), with a median lifespan of 122 days. These data suggest that the high-level accumulation of TCR-V cells in the brain resulting from day 7 immunization delays progression of established tumors in SV11 mice.
Figure 2.
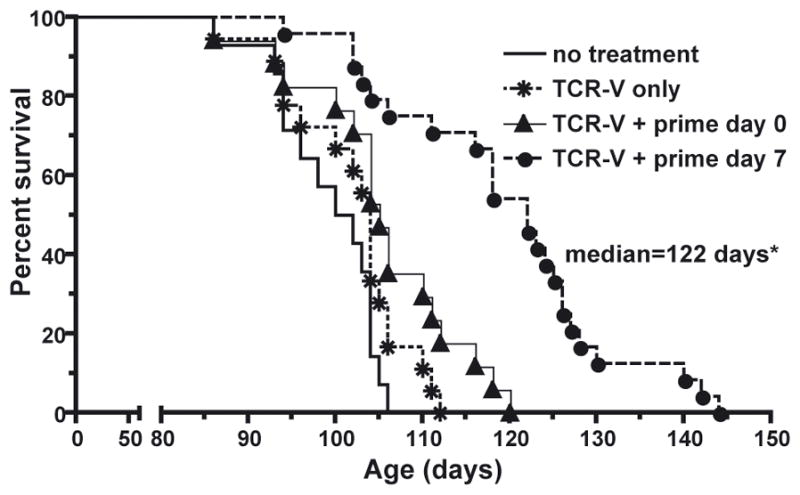
Immunization on Day 7 Post Adoptive Transfer of TCR-V Cells Enhances Survival of SV11 Mice. Cohorts of 85-day old SV11 mice (16 mice/group) received the following treatments: (i) no treatment; (ii)TCR-V cell adoptive transfer alone; (iii) TCR-V cells + Tag-V only immunization on the day of transfer (day 0); or (iv) TCR-V cells + immunization with B6/V-only Tag cells on day 7 post transfer (day 7). Mice were euthanized following the development of neurological symptoms. The presence of tumors was confirmed by gross examination and percent survival calculated using the Kaplan-Meir method. *p<0.0001 vs. “no treatment” by logrank.
Since all mice eventually succumbed to tumor progression despite enhanced survival following day 7 immunization, we determined how long TCR-V cells persisted in SV11 mice following this treatment regimen. SV11 or control B6 mice were assessed at various time points post immunization to measure the frequency of TCR-V cells in the spleen, CLN, and brain (Fig. 3A). Each group of mice was injected with BrdU over a two day period prior to analysis in order to determine the percentage of TCR-V cells that had divided during the past 48 hours (Fig. 3B).
Figure 3.

Day 7 immunization induces high level accumulation and proliferation of TCR-V cells in the brain. (A–C) Naïve TCR-V cells were adoptively transferred into 85 day-old SV11 or B6 mice (three mice/group) followed by immunization with B6/V-only Tag cells seven days later. Beginning at 2 days prior to sacrifice, mice were injected every 12 hours with 1 mg BrdU in order to label proliferating cells. Groups of three mice were analyzed at days 2, 4, 7, 14, and 21 post immunization to determine the total number of TCR-V cells in the spleen, CLN, and brain by staining with anti-CD8 and Tag-V tetramer (A). Cells were additionally stained with anti-BrdU to determine the percentage of CD8+/Tag-V tetramer+ cells that had undergone proliferation in the past 48 hours (B). An additional group of mice was analyzed prior to immunization in order to determine the baseline frequency of TCR-V cells (day 0). Error bars indicate standard deviation from the mean. (C) SV11 mice treated as in A and B were analyzed 33 days following immunization, at 125 days of age, to determine the frequency of CD8+ Tag-V tetramer+ cells in the spleen and brain. Dot plots show data for a representative mouse from a group of six, indicating both the frequency of CD8+ cells that stained positive with Tag-V tetramer and the absolute number of CD8+/Tag-V tetramer+ cells per organ (in parentheses).
The kinetics of TCR-V cell expansion and contraction was similar in the spleens of SV11 and B6 mice following immunization, although the frequency in the spleens of SV11 mice remained at 2-fold higher levels by day 21 (Fig. 3A). The highest percentage of dividing cells in the spleen was detected at day 4 for both strains of mice (Fig. 3B). In the CLN of SV11 mice, TCR-V cells were already detectable at the time of immunization (Fig. 3A), and 20% of these TCR-V cells were proliferating, presumably due to prior activation by endogenous Tag (Fig. 3B). This frequency decreased approximately 4-fold by day 21 post-immunization (Fig. 3B). In contrast, the kinetics of TCR-V cell accumulation and contraction in the CLN of B6 mice mimicked that observed in the spleen, although only a low percentage of cells were actively dividing (Fig. 3B). Thus, these T cells may have migrated from another site following their initial proliferation. The kinetics of TCR-V cell contraction in the CLN of SV11 mice corresponded with a sharp increase in TCR-V cell accumulation in the brain between day 2 and day 4 post-immunization, which remained at high levels even at day 21 post-immunization (Fig. 3A). A high proportion of TCR-V cells detected in the brains of SV11 mice were proliferating up to 14 days post immunization (Fig. 3B), but this proportion decreased by day 21 corresponding to a slight decrease in total TCR-V cells in the brain by this time point (Fig. 3A). These results suggest that active T cell proliferation was required to maintain high levels of TCR-V cells in the brains of SV11 mice.
Since only one mouse in the day 7 treatment group had died by 21 days post immunization (Fig. 2; 106 days of age), we examined TCR-V cell frequency at 33 days post-immunization when approximately 50% of mice in this group had succumbed to tumor burden. By this time point, TCR-V cell frequency had declined approximately 2-fold in both the spleen and brain compared to day 21 (compare Fig. 3C with Fig. 3A). Taken together, these results indicate that day 7 immunization prolonged high-level TCR-V cell accumulation in the brain for at least 2 weeks and was associated with increased SV11 survival.
Pre-conditioning with anti-CD40 followed by day 7 immunization prolongs tumor-specific T cell accumulation at the tumor site and enhances survival of tumor-bearing mice
We considered that improving the initial response of adoptively transferred TCR-V cells against the endogenous Tag might enhance control of tumor progression. A possible explanation for the meager response induced by the endogenous Tag is that T cells encounter antigen presenting cells (APCs) that induce tolerance rather than immunity (24). This tolerizing signal can be overcome by administration of agonistic antibody directed against CD40 (α-CD40), resulting in APC activation and subverting the need for CD4+ T cell help to initiate T-CD8 responses (25).
TCR-V cells were adoptively transferred into 85 day-old SV11 or B6 mice that received either α-CD40 or control IgG on the day before and after transfer. Another group of mice additionally received immunization at day 7 post-adoptive transfer. Mice that received α-CD40 plus TCR-V cell transfer were analyzed at 7 and 14 days post-adoptive transfer to determine the frequency and function of the recovered TCR-V cells. Administration of α-CD40 induced a dramatic response against the endogenous Tag by day 7 in both the spleens and brains of SV11 mice compared to mice that received control rat IgG (Fig. 4A). A proportion of cells at both sites were functional as indicated by peptide-induced IFNγ production. In contrast, α-CD40 conditioning had no effect on the TCR-V T cells transferred into B6 mice, which remained CD44lo. Despite the initial high level T cell accumulation in α-CD40 conditioned SV11 mice, TCR-V cell frequency declined 3- to 4-fold in the spleen and brain, respectively between days 7 and 14 post transfer (Fig. 4B, left panels and Fig. 4C). This rapid attrition of TCR-V cells was prevented by immunization at day 7 (Fig. 4B, right panels and Fig. 4C), resulting in high level TCR-V cell maintenance in both the spleen and brain at day 14 post-adoptive transfer. Over half of these T cells were able to produce IFNγ in response to Tag V peptide (Fig. 4B and 4C). Thus, administration of anti-CD40 dramatically enhanced the response of TCR-V T cells toward the endogenous Tag in SV11 mice, but this effect was short-lived. However, the addition of day 7 immunization to the treatment regimen prolonged high level T cell accumulation within the brain and lymphoid organs.
Figure 4.

Pre-conditioning with α-CD40 enhances early accumulation of functional TCR-V cells in the brains of SV11 mice. (A and B) Naïve TCR-V cells were adoptively transferred into 85 day-old SV11 or control B6 mice. Some mice received α-CD40 (or control IgG) via the intraperitoneal (i.p.) route on the day before (day -1) and after (day +1) transfer. (A) Groups of mice from each treatment condition (3 mice/group) were assessed on day 7 post transfer to determine frequency and activation of TCR-V cells in the spleen and brain by staining with anti-CD8, Tag-V tetramer and anti-CD44, and function by IFNγ production in response to Tag-V peptide. Representative data are shown with the percentage of CD8+ cells that stained with Tag-V tetramer or for IFNγ production indicated. The level of CD44 expression on CD8+, Tag-V tetramer+ cells is shown. (B) Groups of three SV11 mice that received treatments at the same time as those in (A) were assessed on day 14 post transfer to determine frequency of TCR-V cells in the spleen and brain by staining with anti-CD8 and Tag-V tetramer or for Tag-V peptide-induced IFNγ production (No Immunization). Some mice also received immunization with B6/V-only Tag cells 7 days post adoptive transfer and were analyzed simultaneously (Day 7 immunization). (C) Data from A and B are summarized as the mean percentage of CD8+ T cells specific for epitope V +/− the standard error with the data representing three mice per group.
Immunization at day 7 promotes long-term TCR-V cell persistence in the brain of α-CD40 conditioned mice, prolonging control of tumor progression
We next determined whether the high level TCR-V T cell response induced by the combination of α-CD40 conditioning and day 7 immunization was maintained long-term. Groups of SV11 mice received adoptive transfer with TCR-V T cells plus either day 7 immunization, conditioning with α-CD40 or both treatments. The results in figure 5A demonstrate that TCR-V cells persisted at high levels in the brains of SV11 mice at 40 days post adoptive transfer following combination immunotherapy, but not in mice that received either therapy administered individually. In addition, approximately half of these cells were able to produce IFNγ ex vivo (Fig. 5B), demonstrating that functional T-CD8 were present at this late time point. Interestingly, although TCR-V cells were maintained at high levels in the brain, combined α-CD40 + immunization did not prolong high level TCR-V cell accumulation in the spleen of SV11 mice compared to that observed initially after combinatorial therapy (Fig. 4B and C). While the basis for this finding remains unknown, activated TCR-V cells may preferentially accumulate at the site of Tag expression while TCR-V cells that remain in the spleen may undergo a normal contraction phase following immunization. Thus, immunization at day 7 post-transfer is capable of sustaining the dramatic accumulation of TCR-V cells at the tumor site achieved by α-CD40 conditioning.
Figure 5.
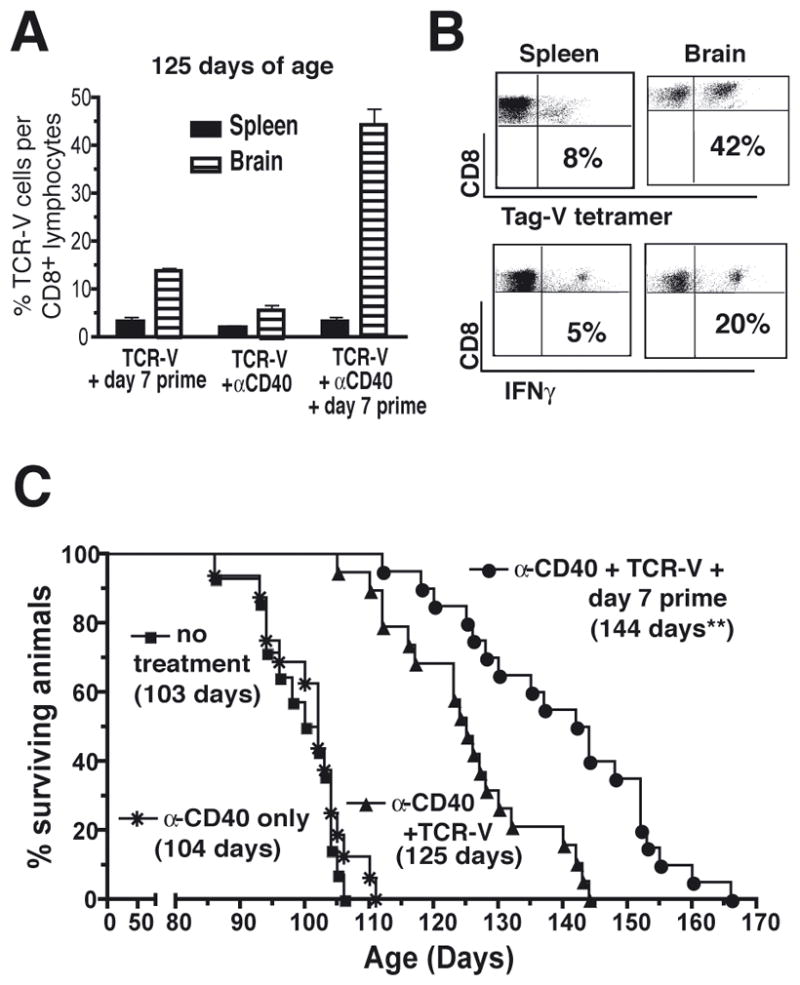
Combination immunotherapy promotes long-term TCR-V cell persistence at the tumor site and enhances survival of SV11 mice. (A) Naïve TCR-V cells were adoptively transferred into groups of 85 day-old SV11 mice (six per group) that were administered one of three treatments: (i) α-CD40 the day before and after transfer; (ii) immunization with B6/V-only Tag cells 7 days post transfer; or (iii) α-CD40 the day before and after transfer plus immunization 7 days post transfer. Mice were analyzed at 125 days of age (40 days following initiation of immunotherapy) to assess the frequency of TCR-V cells that persisted in the spleen and brain by quantitation of CD8+, Tag-V tetramer+ cells. Data are presented as frequency of Tag-V tetramer+ cells/CD8+ cells with error bars indicating standard error of the mean. (B) The persisting population of TCR-V cells in 125 day-old SV11 mice was assessed for function and proliferative capacity, determined by the production of IFNγ. The percentage of CD8+ cells from spleen or brain that produced IFNγ in response to epitope V is indicated. MHC tetramer staining of CD8+ cells from parallel samples is shown for comparison. (C) Cohorts of 85-day old SV11 mice (16/group) received the following treatments: (i) α-CD40 administration alone; (ii) α-CD40 + TCR-V cell adoptive transfer; (iii) α-CD40 + TCR-V cell adoptive transfer + day 7 immunization with B6/V-only Tag cells; or (iv) no treatment. Mice were euthanized following the development of neurological symptoms. The presence of tumors was confirmed by gross examination and percent survival calculated using the Kaplan-Meir method. The median survival age is indicated in parentheses for each group. **p< 0.0001 vs. “α-CD40 only” by logrank.
We determined whether addition of α-CD40 to the treatment regimen would alter control of tumor progression in SV11 mice. Cohorts of 85-day old SV11 mice received the following treatments: (i) α-CD40 monoclonal antibody (mAb) alone; (ii.) α-CD40 + TCR-V cell adoptive transfer; (iii.) α-CD40 + TCR-V cell adoptive transfer + day 7 immunization; or (iv) no treatment. Consistent with the high level influx of TCR-V T cells into the brains of SV11 mice following TCR-V cell transfer coupled with α-CD40, this treatment significantly increased SV11 survival to a median age of 125 days (Fig. 5C), while administration of α-CD40 alone had no therapeutic value. The combination of day 7 immunization and α-CD40 treatment further enhanced SV11 survival to a median lifespan of 144 days (Fig. 5C), which was statistically significant compared to mice that received only adoptive transfer and α-CD40. Thus, the combination of α-CD40 conditioning + day 7 immunization promoted not only long-term persistence of brain-infiltrating TCR-V cells but significantly extended SV11 survival, suggesting that maintenance of high levels of functional TCR-V cells at the tumor site is important for prolonged tumor control.
Administration of a second round of combination therapy dramatically increases SV11 survival
We investigated the possibility that SV11 tumors develop resistance to TCR-V-mediated immunotherapy following combination therapy since treated mice eventually succumb to tumors despite a significant increase in survival. Mice that received an initial course of combination therapy beginning at 85 days of age received a second round of combination therapy (fresh transfer of TCR-V cells + α-CD40 + day 7 immunization) at 125 days of age – 40 days following the original adoptive transfer. TCR-V cells for the second transfer expressed both CD45.1 and CD45.2, enabling these cells to be distinguished from TCR-V cells from the first transfer (CD45.1 only). A second round of combination immunotherapy resulted in a dramatic increase in SV11 survival (181 day median) (Fig. 6). Administration of either α-CD40 alone or α-CD40 plus day 7 immunization without the second dose of TCR-V cells did not result in any improvement in control of tumor progression compared to mice given a single round of combination immunotherapy. This result demonstrates that SV11 tumors remain sensitive to epitope V-targeted immunotherapy at late time points.
Figure 6.

Administration of a second round of combination adoptive immunotherapy dramatically enhances control of tumor progression. (A) At 85 days of age, SV11 mice were administered combinatorial therapy (TCR-V cells expressing CD45.1, α-CD40 antibody + day 7 immunization with B6/V-only Tag cells). A second round of combinatorial therapy (fresh transfer of CD45.1/CD45.2 TCR-V cells + α-CD40 mAb + day 7 immunization) was delivered at 125 days of age – 40 days following the original adoptive transfer. Control groups included SV11 mice that received the initial round of TCR-V mediated immunotherapy + one or the following treatments at 125 days of age (i) a second dose of α-CD40 mAb only; (ii) a second dose of α-CD40 mAb + day 7 immunization; or (iii) no treatment (none). Ten mice were included in each group. Mice were euthanized following the development of neurological symptoms. The presence of tumors was confirmed by gross examination and percent survival calculated using the Kaplan-Meir method. ***p< 0.0001 by logrank compared to control groups (i) and (ii).
To determine whether TCR-V cells from the second adoptive transfer accumulated in SV11 mice, representative mice that received the second adoptive transfer and which showed a significant further increase in survival were sacrificed at days 140, 160 and 190 days age. TCR-V cells from the second adoptive transfer (CD45.1+/CD45.2+) were detected in both spleen and brain in all three mice, with high levels of total TCR-V T cells maintained in the brain even at 190 days of age (Fig. 7A). While the proportion of TCR-V T cells derived from each transfer was variable, TCR-V cells from the initial bolus (CD45.1 only) continued to represent the largest proportion within the brain. Evaluation of the IFNγ-producing potential of the recovered TCR-V T cells revealed that the ratio of IFNγ+ to Tag-V tetramer+ cells was higher for TCR-V cells derived from the second transfer compared to those derived from the first transfer at each time point, particularly in the brain (Fig. 7B). This result suggests that the majority of brain-infiltrating TCR-V cells from the first transfer are functionally compromised by the 140 day timepoint. This proportion decreased further by 160 and 190 days of age, although we were unable to determine whether this decrease is significant since only individual mice were analyzed.
Figure 7.

Donor TCR-V cells from both the first and second adoptive transfers persist in SV11 mice. (A) Representative animals taken from the groups of SV11 mice shown in figure 6 were analyzed at 140, 160 and 190 days of age for the presence of TCR-V T cells derived from the first (CD45.1+/CD45.2−) or second (CD45.1+/CD45.2+) adoptive transfers. The percentage of Tag-V tetramer+ cells of CD8+ cells is indicated in the dot plots in the left columns and the percentage of CD8+/Tag-V tetramer+ cells that stain for CD45.1 and/or CD45.2 is indicated in the right columns for lymphocytes isolated from the spleens and brains. (B) The same representative mice from A were assessed for function of TCR-V cells derived from each transfer in the spleen and brain at 140, 160, and 190 days of age by staining for intracellular IFNγ, CD8, CD45.1 and CD45.2 following Tag-V peptide stimulation ex vivo. Data are plotted as the ratio of IFNγ-producing CD8+ cells to Tag-V tetramer+ CD8+ cells. All points represent data acquired from individual animals. (C) Groups of 85 day-old SV11 mice (three mice/group) were administered combinatorial therapy and either the full or partial second round of treatment as in figure 6. At 140 days of age, splenocytes and brain lymphocytes were assessed for frequency of CD45.1+/CD45.2− (first transfer) Tag-V tetramer+ CD8+ lymphocytes. Error bars indicate standard error of the mean. No significant differences were observed between the groups using a 2-tailed t-test.
To address the possibility that the secondary treatment promoted expansion of the first donor TCR-V T cell population, we determined the frequency of CD45.1+/CD45.2− cells present in the spleen and lymph nodes of groups of SV11 mice that received two rounds of combinatorial immunotherapy versus only a single round. The results show that administration of the second treatment did not significantly change the frequency of TCR-V donor cells from the first donor pool (Fig. 7C). Although the second immunization produced a trend toward an increased frequency of CD45.1+/CD45.2− CD8+ cells in the spleen, this change was not statistically significant. These results suggest that the addition of new TCR-V T cells rather than expansion of TCR-V T cells from the first adoptive transfer is associated with the extended control of tumor progression observed in SV11 mice. Taken together, these results demonstrate that TCR-V cells from the initial adoptive transfer are maintained long-term in mice receiving a second round of combination immunotherapy, but that these cells fail to expand significantly in response to immunization and may be functionally compromised. Rather, extended control of tumor progression required the presence of a second bolus of TCR-V cells.
Discussion
Adoptive T cell transfer for the treatment of cancer requires strategies that subvert the development of tolerance and promote functional T cell accumulation at the tumor site. Examples of such modalities include modification of the immunologic environment with reagents such as granulocyte macrophage-colony stimulating factor or CpG, immunization, irradiation, and suppression of immune regulation (e.g. depletion of Tregs or administration of anti-CTLA-4) (26–28). The results presented here identify a combined strategy that targets separate stages of the T-CD8 response, priming versus secondary antigen encounter, to improve adoptive T cell immunotherapy against an otherwise weak tumor antigen in the context of established cancer.
We found that the timing of immunization following adoptive transfer is critical to achieve control of tumor progression by adoptively transferred T-CD8, with immunization administered at day 7 promoting optimal expansion of naïve TCR-V cells in SV11 mice. A possible explanation for this result may relate to differential kinetics of TCR-V cell activation in SV11 mice compared to non-transgenic B6 mice. In B6 mice, the first signs of TCR-V cell activation are detected between 24 and 48 hours post immunization (19). This result was attributed to the time required for antigen uptake, processing and presentation by APCs. In SV11 mice, however, endogenous Tag is immediately available for presentation to TCR-V cells, as evidenced by upregulation of CD69 on the responding T cells within 2 hours of adoptive transfer (19). We suggest that during the early phase of the response in SV11 mice, TCR-V cells triggered by the endogenous Tag are refractory to exogenous immunization delivered coincident with or soon after adoptive transfer. By day 7 post-adoptive transfer, following T cell contraction, the remaining TCR-V cells are responsive to immunization, as revealed by the robust expansion following immunization at day 7. This strong response could be explained by either an enhanced response to cross-presentation of exogenously-derived Tag or increased responsiveness to direct presentation of Tag-V on the Tag transformed cells. Indeed, we have shown previously that TCR-V T cell accumulation in B6 mice is poor when epitope V is only cross-presented, but dramatically increases when Tag-V additionally is directly presented by the cells used for immunization (21). Thus, a period of time may be required following priming against the endogenous Tag before TCR-V T cells are able to respond to antigen presentation by the Tag transformed cells. Together, these data indicate that a window of opportunity exists for expansion of the adoptively transferred T-CD8 between the period after initial recognition of the endogenous tumor antigen, when T cells are only weakly responsive to the vaccine, and before the onset of peripheral tolerance.
Our observation that α-CD40 conditioning of SV11 mice increases the magnitude of the T-CD8 response toward the endogenous tumor antigen and accumulation at the tumor site is consistent with previous studies showing α-CD40-enhanced T-CD8 responses (25, 29). Initial studies investigating the effect of α-CD40 conditioning on treatment of solid tumors reported that CD40 expression was required on host APCs, but not necessarily by the tumor, (30) and that α-CD40-mediated immunotherapy was most efficient in mice with high tumor burden (31). This suggests that sufficient tumor antigen must be available for APC-mediated priming of T-CD8 responses. This effect has been attributed to an increase in the functional maturation of APCs that have acquired the endogenous tumor antigens by cross-presentation, resulting in more efficient stimulation of T-CD8 (32). Thus, in the setting of adoptive immunotherapy, α-CD40 conditioning enhances the otherwise weak initial stimulation of donor T cells by host APCs. The alternate possibility that α-CD40 directly enhances T-CD8, as reported by Bourgeois and colleagues (33), is not ruled out by our studies since TCR-V cells upregulate CD40 following transfer into SV11 mice (data not shown).
The success of combinatorial therapy lies in the potential for the separate modalities to complement one another, thereby augmenting the anti-tumor response. The present study indicates that α-CD40 conditioning and properly-timed immunization synergistically enhance the TCR-V cell response to SV11 tumors. One explanation for this observation is that the two modalities target different sources of tumor antigen – whereby α-CD40 enhances the response towards the endogenous Tag and immunization provides a potent source of exogenous antigen. While α-CD40 conditioning is likely to target APCs presenting tumor-derived Tag-V, cellular immunization could impact APCs that acquire the antigen for cross-presentation and also provide a form of directly-presented Tag-V. Given the requirements for both cross- and direct-presentation in the maximal expansion of TCR-V cells in B6 mice (21), it may be surmised that α-CD40 conditioning optimizes the initial response to endogenous Tag following adoptive transfer, generating a potently activated TCR-V cell population that responds avidly to “secondary” encounter with antigen derived from immunization delivered on day 7. Indeed, direct presentation of Tag-V by the Tag transformed cells may play a significant role in the observed expansion, as immunization at day 7 with transporter associated with antigen processing (TAP) knockout cells expressing the V-only Tag leads to only limited expansion of TCR-V cells in SV11 mice (data not shown).
Prolonged T cell persistence in SV11 mice following combinatorial therapy might be explained by enhanced survival due to up-regulation of apoptosis-resistance genes such as bcl-2 or the Ca++- independent protein kinase Cθ T cell activation-involved enzyme, important for T cell survival and complete differentiation into cytokine-producing T-CD8 (34). Additionally, increased expression of receptors for survival cytokines such as interleukin (IL)-7 or IL-15 may prolong T cell survival (35, 36). The elicitation of third signal pro-inflammatory cytokines, such as IL-12 (37), tumor necrosis factor-α (38), or IL-23 (39) may result from distinct contributions by either α-CD40 mAb or immunization. Recent studies investigating the role of CD40 ligation in facilitating dendritic cell activation and IL-12 production demonstrate both in vitro and in vivo that CD40 cross-linking alone does not sufficiently induce IL-12 production, but requires another signal such as a toll-like receptor ligand (40, 41). This necessary, yet absent signal may be fulfilled by vaccination. In a renal cell adenocarcinoma mouse model, synergy was observed in the T-CD8-mediated anti-tumor response elicited following anti-CD40 mAb and IL-2 administration (42). It was postulated that α-CD40 may function to bypass the need for CD4+ T cell help by direct APC maturation, while CD4+ T cell-derived IL-2 may promote enhanced survival of expanded T cells (43). Thus in the SV11 model, immunization may induce IL-2 and other cytokines necessary for optimizing survival of TCR-V cells in an otherwise tolerizing environment. Alternatively, the combination treatment may simply amplify the overall cytokine/chemokine milieu. We note that a similar effect could not be achieved by repeated booster immunizations with B6/Tag V-only cells (data not shown), suggesting that each treatment indeed makes a unique contribution.
The enhanced effect of combinatorial therapy on tumor progression was associated with both earlier kinetics of high level T cell accumulation and extended T cell persistence at the tumor site. Administration of α-CD40 mAb induces high-levels of TCR-V cells in the periphery that can be detected 7 days following adoptive transfer, while the immunization approach required an additional week to achieve this same level. When given in combination, the earlier and more prolonged persistence of TCR-V cells in the brain provides both a more rapid and enduring effect on the tumor. Alternatively, the combinatorial therapy could instill unique functional properties to the responding T-CD8 such that these cells are more effective on a per cell basis than those resulting from either treatment in isolation, although we did not detect any major functional differences in our analysis.
The dramatic but transient effect of α-CD40 conditioning on the accumulation of TCR-V cells observed here in SV11 mice is reminiscent of some previous findings. In particular, we previously found that the T-CD8 response targeting Tag epitope I was dramatically enhanced by α-CD40 conditioning in mice that develop Tag-induced pancreatic tumors, but these T cells were completely eliminated from the host within three weeks (44). A similar transient effect was observed by Kedl et al. (45) using an ovalbumin-expressing transplantable mouse melanoma model. These authors demonstrated that long-term survival and function of ovalbumin-specific T cells could be achieved when viral immunization was combined with α-CD40 administration. We show here that cell-based immunization can rescue adoptively transferred T-CD8 from α-CD40-induced rapid attrition in an autochthonous tumor model. To be most effective, a-CD40 therapy may need to be provided at the time when transferred T cells first encounter the tumor antigen as opposed to the time of immunization. Bronte et al. (46) demonstrated that systemic administration of agonistic α-CD40 mAb was most effective when delivered at the time of tumor challenge rather than the time of immunization, implicating antigen derived from the growing tumor as contributory to enhanced elicitation of α-CD40-driven, T-CD8-mediated tumor immunity.
An important finding of this study is that multiple rounds of combinatorial therapy, but not the individual components alone, led to a highly significant increase in control of established tumors. A fresh dose of TCR-V T cells was required to achieve further control of tumor progression despite the persistence of T cells from the first dose at the tumor site. This result indicates that tumor progression was not due to selection of epitope V escape variants, but rather suggests that T cells from the initial transfer were no longer able to inhibit tumor growth even following a booster immunization. This conclusion is supported by observations that TCR-V T cells from the first transfer failed to respond significantly to the second immunization, suggesting that each transfer of TCR-V T cells may be effective for only a fixed amount of time. Whether this window of effectiveness varies for each round of combination therapy remains unknown.
Given the myriad avenues of immune escape from tumor-targeted immunotherapy, the need for protocols that include multiple anti-tumor modalities has become increasingly evident. Recent clinical trials have demonstrated the safety and potential efficacy of α-CD40 mAb administration in cancer patients (47, 48), paving the way for investigations that incorporate this reagent in combinatorial protocols for immunotherapy of cancer. As shown in other solid tumor models, effective α-CD40 mAb-mediated therapy is likely a race between having sufficient tumor antigen present for amplification of T cells and becoming overwhelmed by disease. Together with the dramatic enhancement of the anti-tumor response observed in the current studies, this information warrants new inquiry into the use of α-CD40 mAb with adoptive T cell transfer in human trials, and the potential to significantly expand T cell responses with well-timed immunization.
Acknowledgments
We thank Jeremy Haley and Melanie Epler for excellent technical assistance. This work was supported by Research Scholar Grant 04-059-1-LIB from the American Cancer Society. KSOC was supported by KO8 award #416-67HY from the National Institutes of Health.
Footnotes
Financial Disclosure: All authors have declared there are no financial conflicts of interest in regards to this work.
References
- 1.Rosenberg SA. Progress in human tumour immunology and immunotherapy. Nature. 2001;411:380–4. doi: 10.1038/35077246. [DOI] [PubMed] [Google Scholar]
- 2.Boon T, Coulie PG, Van den Eynde BJ, van der Bruggen P. Human T cell responses against melanoma. Ann Rev Immunol. 2006;24:175–208. doi: 10.1146/annurev.immunol.24.021605.090733. [DOI] [PubMed] [Google Scholar]
- 3.Renner C, Kubuschok B, Trumper L, Pfreundschuh M. Clinical approaches to vaccination in oncology. Ann Hematol. 2001;80:255–66. doi: 10.1007/s002770100324. [DOI] [PubMed] [Google Scholar]
- 4.Gattinoni L, Powell DJ, Jr, Rosenberg SA, Restifo NP. Adoptive immunotherapy for cancer: building on success. Nat Rev Immunol. 2006;6:383–93. doi: 10.1038/nri1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ashton-Rickardt PG, Bandeira A, Delaney JR, et al. Evidence for a differential avidity model of T cell selection in the thymus. Cell. 1994;76:651–63. doi: 10.1016/0092-8674(94)90505-3. [DOI] [PubMed] [Google Scholar]
- 6.Lotz C, Ferreira EA, Drexler I, et al. Partial tyrosinase-specific self tolerance by HLA-A*0201-restricted cytotoxic T lymphocytes in mice and man. Int J Cancer. 2004;108:571–9. doi: 10.1002/ijc.11602. [DOI] [PubMed] [Google Scholar]
- 7.Bakker AB, van der Burg SH, Huijbens RJ, et al. Analogues of CTL epitopes with improved MHC class-I binding capacity elicit anti-melanoma CTL recognizing the wild-type epitope. Int J Cancer. 1997;70:302–9. doi: 10.1002/(sici)1097-0215(19970127)70:3<302::aid-ijc10>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 8.Deeths MJ, Kedl RM, Mescher MF. CD8+ T cells become nonresponsive (anergic) following activation in the presence of costimulation. J Immunol. 1999;163:102–10. [PubMed] [Google Scholar]
- 9.Herndon JM, Stuart PM, Ferguson TA. Peripheral deletion of antigen-specific T cells leads to long-term tolerance mediated by CD8+ cytotoxic cells. J Immunol. 2005;174:4098–104. doi: 10.4049/jimmunol.174.7.4098. [DOI] [PubMed] [Google Scholar]
- 10.Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol. 2006;6:295–307. doi: 10.1038/nri1806. [DOI] [PubMed] [Google Scholar]
- 11.Melief CJ, Toes RE, Medema JP, van der Burg SH, Ossendorp F, Offringa R. Strategies for immunotherapy of cancer. Adv Immunol. 2000;75:235–82. doi: 10.1016/s0065-2776(00)75006-1. [DOI] [PubMed] [Google Scholar]
- 12.Waldmann TA. Effective cancer therapy through immunomodulation. Annual Rev Med. 2006;57:65–81. doi: 10.1146/annurev.med.56.082103.104549. [DOI] [PubMed] [Google Scholar]
- 13.Palmiter RD, Chen HY, Messing A, Brinster RL. SV40 enhancer and large-T antigen are instrumental in development of choroid plexus tumors in transgenic mice. Nature. 1985;316:457–60. doi: 10.1038/316457a0. [DOI] [PubMed] [Google Scholar]
- 14.Van Dyke TA, Finlay C, Miller D, Marks J, Lozano G, Levine AJ. Relationship between simian virus 40 large tumor antigen expression and tumor formation in transgenic mice. J Virol. 1987;61:2029–32. doi: 10.1128/jvi.61.6.2029-2032.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deckhut AM, Lippolis JD, Tevethia SS. Comparative analysis of core amino acid residues of H-2Db-restricted cytotoxic T-lymphocyte recognition epitopes in simian virus 40 T antigen. J Virol. 1992;66:440–7. doi: 10.1128/jvi.66.1.440-447.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lippolis JD, Mylin LM, Simmons DT, Tevethia SS. Functional analysis of amino acid residues encompassing and surrounding two neighboring H-2Db-restricted cytotoxic T lymphocyte epitopes in simian virus 40 tumor antigen. J Virol. 1995;69:3134–46. doi: 10.1128/jvi.69.5.3134-3146.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mylin LM, Deckhut AM, Bonneau RH, et al. Cytotoxic T lymphocyte escape variants, induced mutations, and synthetic peptides define a dominant H-2Kb-restricted determinant in simian virus 40 tumor antigen. Virology. 1995;208:159–72. doi: 10.1006/viro.1995.1139. [DOI] [PubMed] [Google Scholar]
- 18.Schell TD. In vivo expansion of the residual tumor antigen-specific CD8+ T lymphocytes that survive negative selection in simian virus 40 T-antigen-transgenic mice. J Virol. 2004;78:1751–62. doi: 10.1128/JVI.78.4.1751-1762.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ryan CM, Schell TD. Accumulation of CD8+ T cells in advanced-stage tumors and delay of disease progression following secondary immunization against an immunorecessive epitope. J Immunol. 2006;177:255–67. doi: 10.4049/jimmunol.177.1.255. [DOI] [PubMed] [Google Scholar]
- 20.Schell TD, Mylin LM, Georgoff I, Teresky AK, Levine AJ, Tevethia SS. Cytotoxic T-lymphocyte epitope immunodominance in the control of choroid plexus tumors in simian virus 40 large T antigen transgenic mice. J Virol. 1999;73:5981–93. doi: 10.1128/jvi.73.7.5981-5993.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Otahal P, Hutchinson SC, Mylin LM, Tevethia MJ, Tevethia SS, Schell TD. Inefficient cross-presentation limits the CD8+ T cell response to a subdominant tumor antigen epitope. J Immunol. 2005;175:700–12. doi: 10.4049/jimmunol.175.2.700. [DOI] [PubMed] [Google Scholar]
- 22.Mylin LM, Schell TD, Roberts D, et al. Quantitation of CD8+ T-lymphocyte responses to multiple epitopes from simian virus 40 (SV40) large T antigen in C57BL/6 mice immunized with SV40, SV40 T-antigen-transformed cells, or vaccinia virus recombinants expressing full-length T antigen or epitope minigenes. J Virol. 2000;74:6922–34. doi: 10.1128/jvi.74.15.6922-6934.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Falk K, Rotzschke O, Stevanovic S, Jung G, Rammensee H-G. Allele-specific motifs revealed by sequencing of self-peptides eluted from MHC molecules. Nature. 1991;351:290–6. doi: 10.1038/351290a0. [DOI] [PubMed] [Google Scholar]
- 24.Kusmartsev S, Nagaraj S, Gabrilovich DI. Tumor-associated CD8+ T cell tolerance induced by bone marrow-derived immature myeloid cells. J Immunol. 2005;175:4583–92. doi: 10.4049/jimmunol.175.7.4583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Diehl L, den Boer AT, Schoenberger SP, et al. CD40 activation in vivo overcomes peptide-induced peripheral cytotoxic T-lymphocyte tolerance and augments anti-tumor vaccine efficacy. Nat Med. 1999;5:774–9. doi: 10.1038/10495. [DOI] [PubMed] [Google Scholar]
- 26.Dalyot-Herman N, Bathe OF, Malek TR. Reversal of CD8+ T cell ignorance and induction of anti-tumor immunity by peptide-pulsed APC. J Immunol. 2000;165:6731–7. doi: 10.4049/jimmunol.165.12.6731. [DOI] [PubMed] [Google Scholar]
- 27.van Elsas A, Hurwitz AA, Allison JP. Combination immunotherapy of B16 melanoma using anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and granulocyte/macrophage colony-stimulating factor (GM-CSF)-producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation. J Exp Med. 1999;190:355–66. doi: 10.1084/jem.190.3.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang LX, Shu S, Plautz GE. Host lymphodepletion augments T cell adoptive immunotherapy through enhanced intratumoral proliferation of effector cells. Cancer Res. 2005;65:9547–54. doi: 10.1158/0008-5472.CAN-05-1175. [DOI] [PubMed] [Google Scholar]
- 29.French RR, Chan HT, Tutt AL, Glennie MJ. CD40 antibody evokes a cytotoxic T-cell response that eradicates lymphoma and bypasses T-cell help. Nat Med. 1999;5:548–53. doi: 10.1038/8426. [DOI] [PubMed] [Google Scholar]
- 30.van Mierlo GJD, den Boer AT, Medema JP, et al. CD40 stimulation leads to effective therapy of CD40- tumors through induction of strong systemic cytotoxic T lymphocyte immunity. Proc Natl Acad Sci U S A. 2002;99:5561–6. doi: 10.1073/pnas.082107699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nguyen LT, Elford AR, Murakami K, et al. Tumor growth enhances cross-presentation leading to limited T cell activation without tolerance. J Exp Med. 2002;195:423–35. doi: 10.1084/jem.20010032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schoenberger SP, Toes RE, van der Voort EI, Offringa R, Melief CJ. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature. 1998;393:480–3. doi: 10.1038/31002. [DOI] [PubMed] [Google Scholar]
- 33.Bourgeois C, Rocha B, Tanchot C. A role for CD40 expression on CD8+ T cells in the generation of CD8+ T cell memory. Science. 2002;297:2060–3. doi: 10.1126/science.1072615. [DOI] [PubMed] [Google Scholar]
- 34.Barouch-Bentov R, Lemmens EE, Hu J, et al. Protein kinase C-theta is an early survival factor required for differentiation of effector CD8+ T cells. J Immunol. 2005;175:5126–34. doi: 10.4049/jimmunol.175.8.5126. [DOI] [PubMed] [Google Scholar]
- 35.Schluns KS, Kieper WC, Jameson SC, Lefrancois L. Interleukin-7 mediates the homeostasis of naive and memory CD8 T cells in vivo. Nat Immunol. 2000;1:426–32. doi: 10.1038/80868. [DOI] [PubMed] [Google Scholar]
- 36.Li XC, Demirci G, Ferrari-Lacraz S, et al. IL-15 and IL-2: a matter of life and death for T cells in vivo. Nat Med. 2001;7:114–8. doi: 10.1038/83253. [DOI] [PubMed] [Google Scholar]
- 37.Schmidt CS, Mescher MF. Adjuvant effect of IL-12: conversion of peptide antigen administration from tolerizing to immunizing for CD8+ T cells in vivo. J Immunol. 1999;163:2561–7. [PubMed] [Google Scholar]
- 38.Wu TH, Pabin CN, Qin Z, et al. Long-term suppression of tumor growth by TNF requires a Stat1- and IFN regulatory factor 1-dependent IFN-gamma pathway but not IL-12 or IL-18. J Immunol. 2004;172:3243–51. doi: 10.4049/jimmunol.172.5.3243. [DOI] [PubMed] [Google Scholar]
- 39.Oppmann B, Lesley R, Blom B, et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000;13:715–25. doi: 10.1016/s1074-7613(00)00070-4. [DOI] [PubMed] [Google Scholar]
- 40.Schulz O, Edwards AD, Schito M, et al. CD40 triggering of heterodimeric IL-12 p70 production by dendritic cells in vivo requires a microbial priming signal. Immunity. 2000;13:453–62. doi: 10.1016/s1074-7613(00)00045-5. [DOI] [PubMed] [Google Scholar]
- 41.Krug A, Towarowski A, Britsch S, et al. Toll-like receptor expression reveals CpG DNA as a unique microbial stimulus for plasmacytoid dendritic cells which synergizes with CD40 ligand to induce high amounts of IL-12. Eur J Immunol. 2001;31:3026–37. doi: 10.1002/1521-4141(2001010)31:10<3026::aid-immu3026>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 42.Murphy WJ, Welniak L, Back T, et al. Synergistic anti-tumor responses after administration of agonistic antibodies to CD40 and IL-2: coordination of dendritic and CD8+ cell responses. J Immunol. 2003;170:2727–33. doi: 10.4049/jimmunol.170.5.2727. [DOI] [PubMed] [Google Scholar]
- 43.Granucci F, Andrews DM, Degli-Esposti MA, Ricciardi-Castagnoli P. IL-2 mediates adjuvant effect of dendritic cells. Trends Immunol. 2002;23:169–71. doi: 10.1016/s1471-4906(02)02187-7. [DOI] [PubMed] [Google Scholar]
- 44.Otahal P, Knowles BB, Tevethia SS, Schell TD. Anti-CD40 conditioning enhances the T(CD8) response to a highly tolerogenic epitope and subsequent immunotherapy of simian virus 40 T antigen-induced pancreatic tumors. J Immunol. 2007;179:6686–95. doi: 10.4049/jimmunol.179.10.6686. [DOI] [PubMed] [Google Scholar]
- 45.Kedl RM, Jordan M, Potter T, Kappler J, Marrack P, Dow S. CD40 stimulation accelerates deletion of tumor-specific CD8(+) T cells in the absence of tumor-antigen vaccination. Proc Natl Acad Sci U S A. 2001;98:10811–6. doi: 10.1073/pnas.191371898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bronte V, Cingarlini S, Apolloni E, et al. Effective genetic vaccination with a widely shared endogenous retroviral tumor antigen requires CD40 stimulation during tumor rejection phase. J Immunol. 2003;171:6396–405. doi: 10.4049/jimmunol.171.12.6396. [DOI] [PubMed] [Google Scholar]
- 47.Vonderheide RH. Prospect of targeting the CD40 pathway for cancer therapy. Clin Cancer Res. 2007;13:1083–8. doi: 10.1158/1078-0432.CCR-06-1893. [DOI] [PubMed] [Google Scholar]
- 48.Vonderheide RH, Flaherty KT, Khalil M, et al. Clinical activity and immune modulation in cancer patients treated with CP-870,893, a novel CD40 agonist monoclonal antibody. J Clin Oncol. 2007;25:876–83. doi: 10.1200/JCO.2006.08.3311. [DOI] [PubMed] [Google Scholar]