

Abstract
The papain/CLIK-148 coordinate system was employed as a model to study the interactions of a non-peptide thiocarbazate inhibitor of cathepsin L (1). This small molecule inhibitor, a thiol ester containing a diacyl hydrazine functionality and one stereogenic center, was most active as the S-enantiomer, with an IC50 of 56 nM; the R-enantiomer (2) displayed only weak activity (33 μM). Correspondingly, molecular docking studies with Extra Precision Glide revealed a correlation between score and biological activity for the two thiocarbazate enantiomers when a structural water was preserved. The molecular interactions between 1 and papain were very similar to the interactions observed for CLIK-148 (3a and 3b) with papain, especially with regard to the hydrogen bonding and lipophilic interactions of the ligands with conserved residues in the catalytic binding site. Subsequent docking of virtual compounds in the binding site led to the identification of a more potent inhibitor (5), with an IC50 of 7.0 nM. These docking studies revealed that favorable energy scores and correspondingly favorable biological activities could be realized when the virtual compound design included occupation of the S2, S3 and S1′ subsites by hydrophobic and aromatic functionalities of the ligand, and at least three hydrogen bonding contacts between the ligand and the conserved binding site residues of the protein.
Keywords: Extra Precision (XP) Glide docking, covalent binding, hydrogen bonding interactions, hydrophobic contributions, cysteine protease subsites
Introduction
Cathepsin L-like cysteine proteases have been involved in various disease states including malaria, leishmaniasis, Chagas' disease, African trypanosomiasis, toxoplasmosis, and amoebiasis, among others.1-7 These illnesses have generally been neglected by many pharmaceutical companies since projected drug development costs outweigh the purchasing power of the afflicted populations.8 With the formation of the NIH Molecular Libraries Screening Centers Network9 (MLSCN), academic centers in the network are providing the biological community with a wide range of small molecule biological probes, and in addition, have had the opportunity to focus on the discovery of new lead compounds for neglected disease targets (NDTs).8 The Penn Center for Molecular Discovery10 has recently developed a selective chemical probe for human cathepsin L,11 a cysteine protease implicated in a variety of NDTs. Since cathepsin L is highly homologous to the cysteine proteases expressed by parasites associated with NDTs,12, 13 the development of cathepsin L inhibitors could aid in the design of critically needed drugs for these diseases.
Many inhibitors of the parasitic cysteine proteases form covalent bonds with the active site Cys25 nucleophilic sulfur atom of their targeted enzymes. Such covalent binding is often apparently irreversible (or very slowly reversible), and may be required in order for inhibition to be sustained at the cellular level. More readily reversible covalent cysteine protease inhibitors have also been described,18 as have inhibitors that bind noncovalently.14 Understanding the molecular requirements for covalent attachment is of clear importance for designing new agents that will bind via this mechanism.
The X-ray coordinate system for papain/CLIK-148 (1cvz.pdb), solved at 1.7 Å resolution, is a representative example of the structure of a covalent ligand-bound cysteine protease complex.15 Since most of the amino acid residues that are involved in the binding of CLIK-148 to papain are conserved in cathepsin L, this publicly available high resolution structure has provided an excellent model for the successful design of highly active and specific cathepsin L inhibitors.15, 16 An alternative model would have been the structure of the inhibitor E-64 complexed to cathepsin L, but unfortunately, the coordinates for this structure are not publicly available.17 The coordinates for the structure of cathepsin L complexed to a noncovalently bound tripeptide are publicly available,14 but are less pertinent to our work because of the difference in the shape of the binding site in the tripeptide/cathepsin L complex compared with that of the covalently bound small molecule in papain (1cvz.pdb). We also explored the possibility of a homology model of cathepsin L based on the coordinates of 1cvz.pdb, however, considerable error was associated with subsequent dockings of this theoretical model, especially with regard to the positioning of critical hydrophobic groups in the non-prime subsites.18 We thus employed the papain/CLIK-148 structure for this study.
While molecular docking routines, including Schrodinger's XP Glide,19 adequately address hydrogen bonding, van der Waals, electrostatic, hydrophobic, and solvation contributions to binding, these algorithms do not routinely address covalent interactions between the ligand and the protein; covalent binding can only be addressed with quantum mechanical routines. Consequently, all XP Glide binding scores reported in the present study do not incorporate the contribution from the covalent interaction between the Cys25 sulfur and the electrophilic atom of the ligand. Instead, the distance between the nucleophilic sulfur of Cys25 and the electrophilic atom of the modeled ligand undergoing attack will be used to estimate the potential for covalent bond formation.
Although an activated electrophilic atom in the ligand is necessary for covalent binding to the protein, there are also non-covalent binding interactions that contribute significantly to biological activity. Such hydrogen bonding and hydrophobic interactions may be as important as the covalent bond formed between the ligand and the protein with regard to achieving high potency in vivo. Once the requisite hydrogen bonding atoms and hydrophobic functional groups on the ligand are correctly positioned within the binding site, the electrophilic center is placed in proximity to bind sulfur. This binding mechanism is illustrated by the succinyl epoxides represented by CLIK-14815 (3a and 3b) and E-64 (4).17, 20 These inhibitors have their origins in natural products, and have evolved to incorporate substituents for enzyme interactions beyond the electrophilic center that binds covalently to Cys25. The substituents (H-bond donors and acceptors as well as hydrophobic groups) may well play a pivotal role in positioning the ligand precisely for attack by the nucleophilic sulfur.
Following standard nomenclature,21 oligopeptides binding to a peptidase are cleaved between the S1 and S1′ subsites of the protein, with subsite S numbers increasing in both directions the farther the subsite is from the cleavage site. Occupation of the prime and non-prime subsites in the protein also affects selectivity within the cysteine protease family. For example, a derivative of the naturally occurring inhibitor E-64 (E-64c), binds covalently via the Cys25 sulfur, but is non-selective, inactivating most cysteine proteases. On the other hand, in the complex of CLIK-148 bound to papain (1cvz.pdb), CLIK-148, a selective cathepsin L inhibitor, is precisely positioned in the active site. Here, in addition to the formation of the Cys25 covalent bond, several papain residues participate both in hydrogen bonding and hydrophobic interactions with the succinyl epoxide inhibitor (CLIK-148), including Gln19, Cys25, Gly66, Asp158, Trp177, and Ser205 (Table 2). In addition, the inhibitor lies deeply buried in both the S2 and S1′ subsites. Disruptions of the interactions with the binding site residues can lead to significant loss of biological activity. Other diastereomers of the epoxy succinyl ligands, containing up to 4 stereogenic centers (16 possible stereoisomers) have been isolated and tested in selected cysteine proteases; not surprisingly, some are inactive when the stereogenicities of the ligands are changed. However, since the succinyl epoxide natural products contain at least three stereogenic centers, understanding the effect of stereo inversion on activity is not straightforward in these systems. In the present study, we have explored the effect of stereoinversion directly, through a multi-disciplinary approach employing chemical, computational, and biological techniques.
Table 2.
Amino acid residues in papain and corresponding residues in cathepsin L and the interactions they make with CLIK-148 and the 3 inhibitors in the acyl hydrazine series: 1, 2, and 5.
| Residue in papain | Corresponding Residue in cathepsin L | Types of binding interactionsa for compounds 1, 2, 5 and CLIK-148 |
|---|---|---|
| Gln19 | Gln19 | H-bondingb (CLIK-148, 1) |
| Cys25 | Cys25 | Covalent bond (CLIK-148, 1, 2, 5) H-bonding (1, 5) |
| Gly66 | Gly68 | H-bonding (CLIK-148, 1, 2, 5) |
| Asp158 | Asp162 | H-bonding (CLIK-148, 1, 5) |
| His159 | His163 | H-bonding (2, 5) |
| Trp177 | Trp189 | Hydrophobic/aromatic (CLIK-148, 1, 5) H-bonding (1) |
| Ser205 | Ala214 | Hydrophobic (CLIK-148, 1, 5) |
Interactions were observed between the ligand and papain for the highest ranking poses obtained from ExtraPrecision Glide docking. Each compound was docked to papain without a covalent attachment to the Cys25 sulfur. A structural water was present in the catalytic site, near Asp158.
hydrogen bonding.
Materials and Methods
Molecular Docking Studies
Molecular docking studies were performed using Maestro version 8.0 and Extra Precision Glide (XP Glide), version 4.5.19 All ligands were docked flexibly to papain, which served as a model for cathepsin L. The coordinates for the papain/CLIK-148 complex (1cvz.pdb) were downloaded from the RCSB Protein Data Bank.22 In this structure, CLIK-148 is covalently bound to papain. To prepare the system for docking, the covalent bond between the Cys25 sulfur and the epoxide carbon was manually deleted. The protein was then prepared for subsequent grid generation and docking using the Protein Preparation Wizard tool supplied with Glide. Using this tool, all hydrogen atoms were added to papain, the protonation states for histidine residues were optimized, crystallographic waters not deemed to be important for ligand binding were deleted, and the entire protein was minimized. The structural water near Asp158 was preserved; it was re-oriented and adjusted using the commands within the Protein Preparation Wizard tool available within Glide, so that hydrogen bonding between the ligand and the protein could be established. In order to validate the XP Glide algorithm for subsequent docking studies of the cathepsin L inhibitors, the non-covalently bound CLIK-148 was extracted from the complex and prepared for single ligand docking using the LigPrep application with the OPLS_2005 force field. Next, a grid was prepared for re-docking CLIK-148 into papain using the Receptor Grid Generation tool in Glide. With non-covalently bound CLIK-148 in place, the centroid of the workspace ligand was chosen to define the grid box. The option to dock ligands similar in size to the workspace ligand was selected for determining the grid sizing. For this single ligand docking, the extra precision mode was selected. The default settings for scaling the van der Waals radii were selected: a scaling factor of 0.8 and a partial charge cutoff of 0.25. No constraints were defined for the docking runs. The highest scoring docking pose (orientation plus conformation) returned for CLIK-148 was overlaid with the starting protein complex. For subsequent molecular docking of compounds 1, 2 and 5 in the binding site of papain, LigPrep was used for energy minimizations of these small molecules with the OPLS_2005 force field. Using the initial grid generated for papain, the extra precision docking was repeated for each molecule (1, 2, and 5), as described above. The orientation of the structural water near the Asp 158 was maintained for these docking studies.
Kinetic Analysis
Reaction progress curves for cathepsin L-catalyzed hydrolysis of Z-Phe-Arg-AMC at various inhibitor concentrations23 were used to estimate the rate constants for inhibition by compound 1. Baseline fluorescence readings (reaction mixture with no substrate) were fit to a cubic polynomial using the MATLAB polyfit function, and the resulting curve was subtracted from time course measurements for each inhibitor concentration. Raw fluorescence readings were linear with respect to AMC concentration. The time-zero reading for baseline fluorescence and the maximum average fluorescence reading were used as the minimum (0 μM) and maximum (1 μM) values for the linear scale, respectively.
Progress curves for each inhibitor concentration were fit to a 5-parameter inhibition kinetic model:
The best-fitting parameters were determined using APPSPACK optimization software with a linear least squares objective function.23 It was also necessary to estimate an additional parameter, representing the time-delay until the first experimental reading was taken (ca. 150 sec).
Biological Testing
Analog activity analyses were conducted with the following assay buffer: 20 mM sodium acetate, 1 mM EDTA, and 5 mM DTT, pH 5.5. Confirmatory results were obtained utilizing the following assay conditions, replacing DTT with cysteine in the assay buffer. Compounds were serially diluted in DMSO and transferred into a 96-well Corning 3686 assay microplate to give 16 dilutions ranging from 50 μM to 1.5 nM. Human liver cathepsin L (Calbiochem 219402) was activated by incubating with assay buffer for 30 min. Assay buffer consisted of 20 mM sodium acetate, 1 mM EDTA, and 5 mM cysteine, pH 5.5. Upon activation, cathepsin L (300 pM) was incubated with 1 μM Z-Phe-Arg-AMC substrate and test compound in 100 μL of assay buffer for 1 h at room temperature. Fluorescence of AMC released by enzyme-catalyzed hydrolysis of Z-Phe-Arg-AMC was read on a PerkinElmer Envision microplate reader (excitation 355 nm, emission 460 nm). Data was scaled using internal controls and fit to a four parameter logistic model (IDBS XLfit equation 205) to obtain IC50 values in triplicate.
Results and Discussion
We recently described the identification of a potent, non-peptide inhibitor of cathepsin L (1, Figure 1), with an IC50 of 56 nM under defined conditions.11 This thiocarbazate contains a diacyl hydrazine functionality and a single stereogenic center. The most active congener proved to be the S-enantiomer, with an IC50 of 56 nM. The R-enantiomer (2, Figure 1), described here, was only modestly active against cathepsin L (IC50 = 33 μM). Molecular docking studies were initiated on both enantiomers, in order to probe the importance of the elements that contribute to binding. As a validation study, we first examined the non-covalent docking of the ring-opened epoxide form of CLIK-148 (3b, Figure 2) in papain. Initially, the orientation of the water molecule near Asp 148 was not suitable for hydrogen bonding with the ligand or with Asp 148. Using the routines available in XP Glide, this water molecule was re-orientated so that adequate hydrogen bonding could be established (Figure 5). Then, after breaking the covalent bond to Cys25 and re-docking CLIK-148 into the binding site, the experimentally derived binding mode was reproduced (Figure 5). In this docking pose (orientation plus conformation), with a score of -9.27 kcal/mol, CLIK-148 hydrogen bonds to the protein through side chain and backbone atoms of Gly66, Cys25, Gln19, and Asp158. The pyridine group occupies a hydrophobic aromatic pocket in papain in the S1′ subsite near Trp177 and the phenylalanine of the ligand occupies the S2 subsite. Taken together, these features play a major role in positioning the ligand appropriately for covalent attachment to the protein. In the highest scoring pose, the distance between the Cys25 sulfur and the electrophilic carbon of the ligand (epoxide ring carbon) is about 3 Å.
Figure 1.

Cathepsin L inhibitors 1 (S-enantiomer) and 2 (R-enantiomer).
Figure 2.

CLIK-148 in epoxide form (3a) and epoxide ring-opened form (3b), and E-64 (4).
Figure 5.
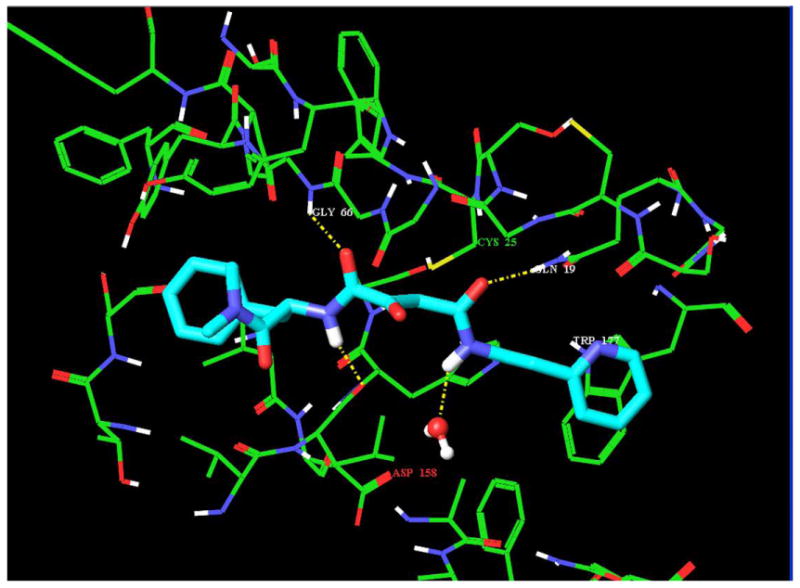
CLIK-148 docked to papain after breaking the covalent bond to Cys25 sulfur. The hydrogen bonding interactions (yellow dashed lines) between ligand and protein involve Gly66, Cys25, Gln19 and the backbone CO of Asp158; the aromatic/hydrophobic interactions in the S1′ subsite involve Trp177.
Since the validation study revealed that XP Glide could accurately reproduce the experimentally derived binding mode of CLIK-148, thiocarbazate 1 was docked into the binding site of papain (Figures 6 and 9a); the di-imide functionality in 1 was maintained in the anti orientation with respect to the geometry of the NH bonds (Figure 6b). The highest scoring pose obtained had a binding energy of -9.03 kcal/mol, very close to the energy value observed in our validation study with CLIK-148/papain (-9.27 kcal/mol). Furthermore, the orientations of the ligands (CLIK-148 and 1) bound to papain were strikingly similar (Figure 6b). For the 1/papain complex, Gln19, Cys25, Gly66, Asp158, and Trp177 all participated in hydrogen bonding interactions with the ligand (Figure 6b), as observed for CLIK-148/papain. In addition, both the pyridine group of CLIK-148 and the 2-ethylphenyl anilide group of 1 occupied the S1′ subsite containing Trp177. The indole group of 1 occupied the S2 subsite, near Ser 205 (papain residue), in a fashion similar to that of the phenylalanine group of CLIK-148. The Ser 205 residue in papain corresponds to the Ala 214 residue in cathepsin L; this is the only residue in the binding site of cathepsin L that is not identical to the aligned residue in papain. However, both Ala and Ser side chains are small enough to accommodate the bulky hydrophobic groups present in both inhibitors (phenylalanine in CLIK-148 and 2-ethylphenyl anilide in 1),15 so this difference would appear to be negligible. Both CLIK-148 and 1 fully occupy the S1′ and S2 subsites. The tert-butoxy group of the NHBoc in compound 1 sits in the S3 subsite, occupying a large hydrophobic cleft. In addition, the amino acid-derived NH of inhibitor 1 hydrogen bonds to the backbone carbonyl of Asp 148, a conserved binding site residue. Inhibitors that span from the S to the S1′ subsites have high potency and selectivity toward cysteine proteases.15, 24 Compound 1 demonstrated selectivity towards cathepsin L versus other cysteine proteases, including cathepsins V, S, B, and K, with the greatest selectivity index observed for cathepsin K (150).23 The details of a structure activity relationship study for cathepsin L inhibition in the carbazate series are being published separately25, but reveal that removal of the NHBoc group causes a dramatic loss of cathepsin L inhibitory activity. This single synthetic change to the carbazate scaffold of 1 removes both the potential for hydrogen bonding with Asp 148 and the hydrophobic contact of the tert-butoxy group in the S3 subsite, resulting in a 400-fold reduction in cathepsin L inhibition.
Figure 6.
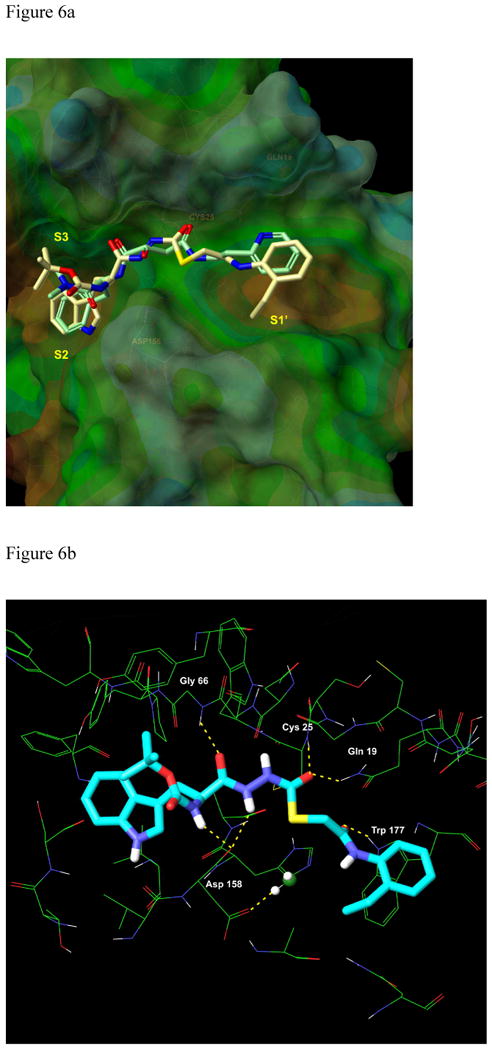
a. Overlay of CLIK-148(light green carbons with heteroatom coloring) with compound 1 (tan carbons with heteroatom coloring) in the binding site of papain. b. Binding of 1 (S-enantiomer) to papain. Six critical hydrogen bonding interactions between the ligand and the protein are established. The papain residues that participate in hydrogen bonding, Gln19, Cys25, Gly 66, Asp158, and Trp177, are identical to the corresponding residues in cathepsin L.
Figure 9.
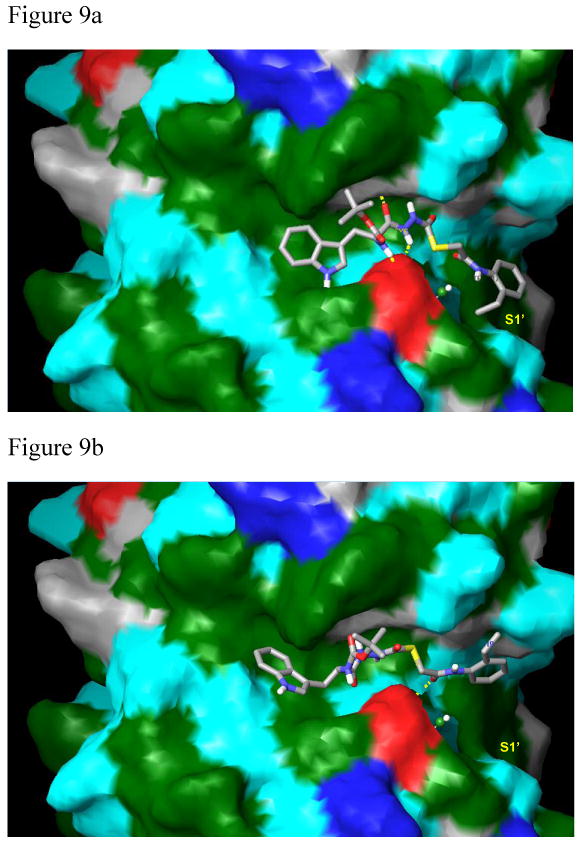
Compounds 1 (9a) and 2 (9b) bound to papain. The protein surface was calculated in Maestro (Schrodinger, Inc.) with molecular properties color-coded as follows: green: strongly hydrophobic; red: negatively charged (Asp158 residue); dark blue: positively charged; cyan and grey: hydrophilic. 9a. Compound 1 binds via a strong hydrophobic interaction within the S1′ subsite consisting of an aromatic/aromatic interaction between Trp177 and the 2-ethylphenyl anilide group of the ligand. In addition, there are strong hydrophobic interactions between the indole of the ligand within the S2 subsite and between the tert-butoxy group in the S3 subsite (XP Glide score: -9.04 kcal/mol). 9b. The phenethyl group of 2 lies completely outside of the S1′ hydrophobic binding pocket and the indole of 2 looses significant hydrophobic contacts in the S2 subsite. The tert-butoxy group of the ligand also looses hydrophobic contacts within the S3 subsite. Critical hydrogen bonds are absent in this complex, resulting in the lower XP Glide score of -7.03 kcal/mol, and the weakened biological activity (33 μM).
Figures 9a and 9b. Compounds 1 and 2 shown in Connolly surface models of the protein.
As presented above, the R-enantiomer 2 had a cathepsin L inhibitory activity of only 33 μM. In an attempt to understand why this enantiomer was virtually inactive, we docked 2 into the binding site of papain. The highest scoring pose for 2 obtained from this XP Glide docking study (depicted in Figure 7) had a docking score of -7.0, two kcal/mol lower than the score for the S-enantioner 1. In addition, key hydrogen bonding and hydrophobic contacts that are established in the complexes of the active S-enantiomer (1) with papain, and in CLIK-148 with papain are completely disrupted for the R-enantiomer (2) in this binding site (Figures 8, 9a, and 9b). While the S-enantiomer 1 makes at least six hydrogen bonding contacts to the active site residues and large hydrophobic contacts within the S1′, S2 and S3 subsites (Figures 6a and 6b), the R-enantiomer forms only two hydrogen bonds to papain and does not occupy the S1′ subsite at all (Figures 8 and 9b). This change in stereogenicity from S (1) to R (2) reveals a dramatic shifting of the 2-ethylphenyl anilide group out of this critical subsite in these docking studies (cf. Figures 9a and 9b). The key hydrogen bond made between the NH of the Trp residue in the S-enantiomer 1 with Asp 148 is also absent in the binding of the R-enantiomer 2 (Table 2). In addition, the indole of 2 makes fewer significant hydrophobic contacts in the S2 subsite (near Ser 205) than the indole of 1. These observed differences in binding interactions between 1 and 2 and the corresponding difference in docking scores provide a cogent rationale for the observed decrease in the cathepsin L inhibitory activity of 2 vs. 1 of almost three orders of magnitude (Table 1). Also noteworthy is the increase in the distance of the electrophilic carbonyl carbon in 2 to the Cys25 to 4.4 Å, suggesting that covalent bond formation might be less favorable.
Figure 7.
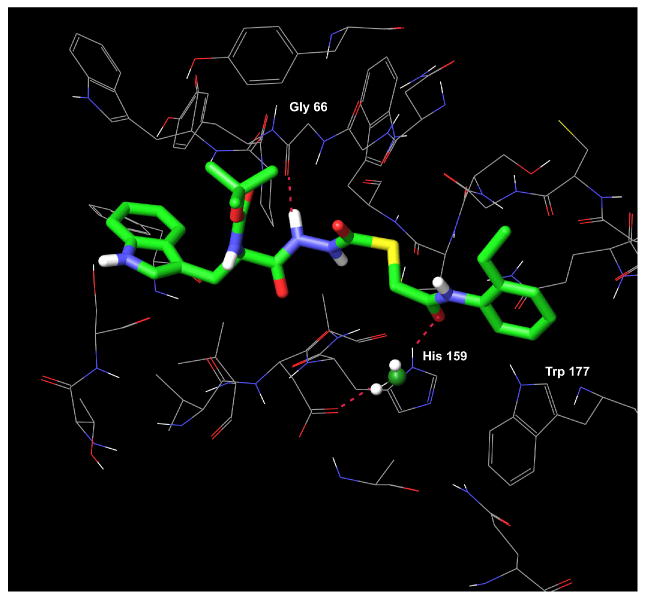
Compound 2 (R-enantiomer) in the binding site of papain. Only two hydrogen bonds are made between 2 and the binding site residues.
Figure 8.
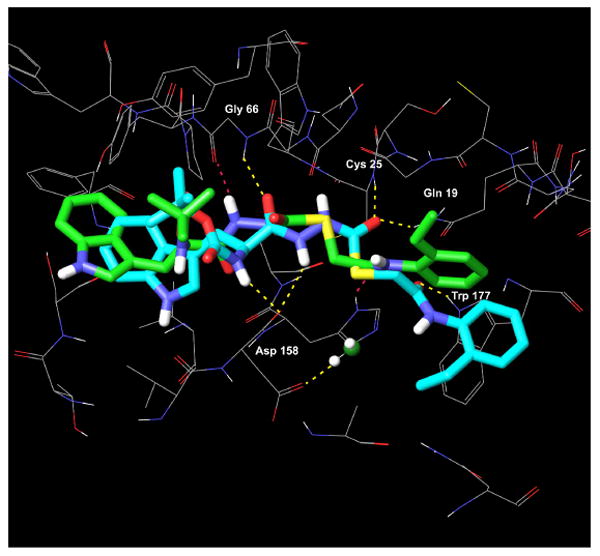
Overlay of 1 (S-enantiomer, green carbons with heteroatom coloring) with the significantly less active R-enantiomer (2, cyan carbons with heteroatom coloring) in the binding site of papain. The hydrogen bonds made between 1 and papain (yellow dashed lines) are lost when the R-enantiomer (2) binds to papain; the R-enantiomer makes only 2 hydrogen bonds to papain (red dashed lines). The hydrophobic interaction between the 2-ethylphenyl anilide group of 1 and Trp177 in the S1′ subsite is absent in the binding of 2 to papain.
Table 1.
Cathepsin L inhibitory activities and Extra Precision (XP) Glide docking scores for compounds 1, 2, 5, and CLIK-148.
| Compound | IC50 (μM)c | XP Glide docking score (kcal/mol)b |
|---|---|---|
| 1 | 0.056 ± 0.001 | -9.03 |
| CLIK-148 | < 0.100a | -9.27 |
| 2 | 33 ± 1.75 | -7.11 |
| 5 | 0.007 ± 0.001 | -10.00 |
Tsuge et al.15 have reported the catL inhibitory activity of CLIK-148 as 63% inhibition at 10-7 M, which could be exrapolated to an IC50 of < 100 nM.
The scores represent the highest ranking poses obtained from ExtraPrecision Glide docking of each compound bound to papain, without the covalent attachment to the Cys25 sulfur. In each case, a structural water was present in the catalytic site, near Asp158 (papain numbering).
IC50 determinations were carried out in triplicate.
These mechanistic insights into the binding site interactions of 1 suggested additional room within the S1′ subsite, which led to the design of compound 5. This oxocarbazate analog of 1 was designed to contain the requisite S stereogenicity, an oxygen in place of the thiol ester sulfur, and a large hydrophobic/aromatic group (quinoline) to occupy further the S1′ subsite. When this virtual compound (5, Figure 10) was docked into the binding site of papain, the best pose obtained had a score of -10.00, a score improved by 1 kcal/mol compared to that of our initial lead 1 (-9.03 kcal/mol). This compound was synthesized and found to be more potent than 1, with an enzyme inhibitory activity of 7 nM against cathepsin L. In the highest scoring docking pose for this compound, three hydrogen bonds are formed between 5 and the protein; moreover, the tetrahydroquinoline group on the ligand occupies the large hydrophobic pocket with Trp177 in the S1′ subsite (Figure 10). Changing the sulfur in 1 to an oxygen in 5 leads to a change in orientation of the ester bond, making a new interaction with His159 possible. This hydrogen bond is also observed in the binding of CLIK-148 to papain (Table 2). In both inhibitors (1 and 5), the carbazate carbonyl carbons are oriented for nucleophilic attack by Cys25, with the distances from the Cys sulfur to the carbonyl carbon in both ligands in the three angstrom range.
Figure 10.
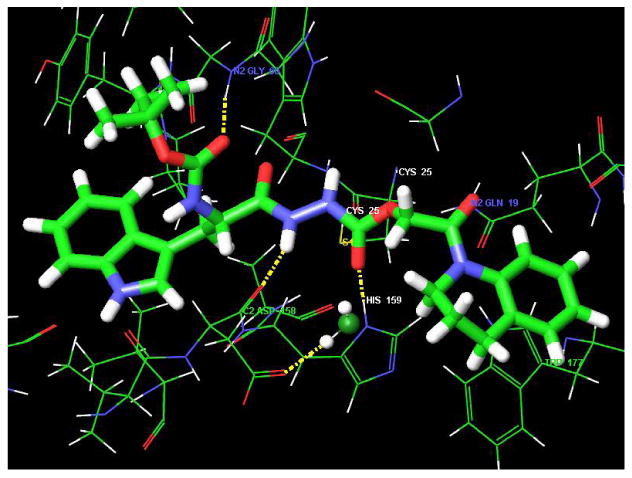
Compound 5 bound to papain with the tetrahydro-isoquinoline group fully occupying the S1′ subsite. The IC50 for cathepsin L inhibition is 7 nM.
The structure of pro-cathepsin L (1mhw.pdb) was also explored in molecular docking studies with ligand 1. However, only very poor XP Glide scores could be obtained from these studies. The two highest scoring docking poses for 1 in the binding site of the pro-cathepsin L structure had scores of 1.15 and 6.74 kcal/mol. When the interactions between 1 and the pro-cathepsin L structure were examined, severe steric clashes between the indole of the ligand and the Leu 69 side chain were observed (a distance of 0.61 angstroms between the ligand and the Leu side chain). This residue corresponds to Tyr 67 in papain. However, in 1mhw.pdb, the Leu 69 side chain is pointing into the binding site cavity, whereas in papain, the Tyr 67 side chain hydroxyl is 6.11 angstroms removed from any atom in 1, and no unfavorable contacts are observed. Further unfavorable contacts were also observed between ligand 1 and the backbone atoms surrounding the Cys 25 residue in the pro-cathepsin L structure, and only one hydrogen bond was observed between the ligand and the conserved binding site residues. A homology model of cathepsin L based on the coordinates of CLIK-148 bound to papain was also generated (MOE software, CCG, Inc.). Docking scores for 1 in the binding site of the resulting theoretical model were somewhat better than those obtained for the pro-cathepsin L structure (-3.82 and -2.40 kcal/mol for the two highest scoring poses of 1 bound to the model structure), but these scores were still unfavorable. Since significantly better scores were realized for ligand dockings of 1 with the papain structure than with either the pro-cathepsin L structure or the theoretical model, this experimentally-derived system (1cvz.pdb) was used directly for all docking studies of the carbazate ligands.
To compare our docking analysis with the kinetic behavior of compound 1, we constructed a 5-parameter ODE model of reversible inhibitor binding and fit the model to reaction progress curves measured at various inhibitor concentrations (see Materials and Methods section). The best-fitting parameters were k1 = 2.3 μM-1s-1, k-1 = 0.30 s-1, kcat = 4.0 s-1, kon = 0.024 μM-1s-1, and koff = 2.2 × 10-5 s-1. Most notably, the rate of inhibitor dissociation (koff) from cathepsin L was extremely slow, leading to a Ki = koff / kon = 0.890 nM.23 Alternative reaction schemes for steady-state and irreversible inhibitor binding were also tested, but did not fit the data as well as the 5-parameter model. Taken together, the results from the docking and kinetic analyses suggest a covalent but slowly reversible mechanism of inhibition that is aided by strong non-covalent interactions.
Conclusions
The molecular-level determinants of cathepsin L inhibitory activity have been identified via molecular docking experiments of cathepsin L inhibitors in the binding site of papain. Utilizing the papain system as a model for cathepsin L, we found that the docking scores paralleled the experimental bioactivities for cathepsin L inhibition, and could be employed as a guide in selecting new molecules for synthesis. To this end, compound 5, designed to incorporate the key hydrogen bonding, hydrophobic and aromatic elements necessary for cathepsin L activity, proved to be the most active compound in the series of ligands studied thus far.
The docking studies reported here provide an understanding of the importance of the non-covalent determinants of bioactivity in the cathepsin L enzyme, and as a result, we have correlated the binding energy scores of selected cathepsin L inhibitors with biological activity. We conclude that the potential for covalent attachment of the Cys25 sulfur to the electrophilic center in the ligand is insufficient for generating high inhibitory potency against cathepsin L using our standard assay conditions. Our study supports this claim, given the extremely low potency of the R-enantiomer 2 (IC50 = 33 μM), and the observed loss of critical binding elements in the binding site model of 2 docked to papain. Even though the R-enantiomer contains the same activated thiocarbonyl group as found in the S-enantiomer, stereo inversion at a distant center in the molecule completely disrupts the other binding components that are critical for high potency. Notably, the 2-ethylphenyl anilide group in 2 does not make the necessary hydrophobic contacts in the S1′ aromatic pocket that are required for cathepsin L inhibition. Other key hydrogen bonding and hydrophobic contacts are also lost in the binding of 2 to papain. Additional derivatives in both the thiol ester and ester series that probe the binding features within this chemical series have been designed and synthesized, and as such are the subject of a companion chemistry publication describing the structure activity relationships.25
Supplementary Material
Figure 3.

Cathepsin L inhibitor 5 (S stereocenter); IC50 7 nM against cathepsin L.
Figure 4.
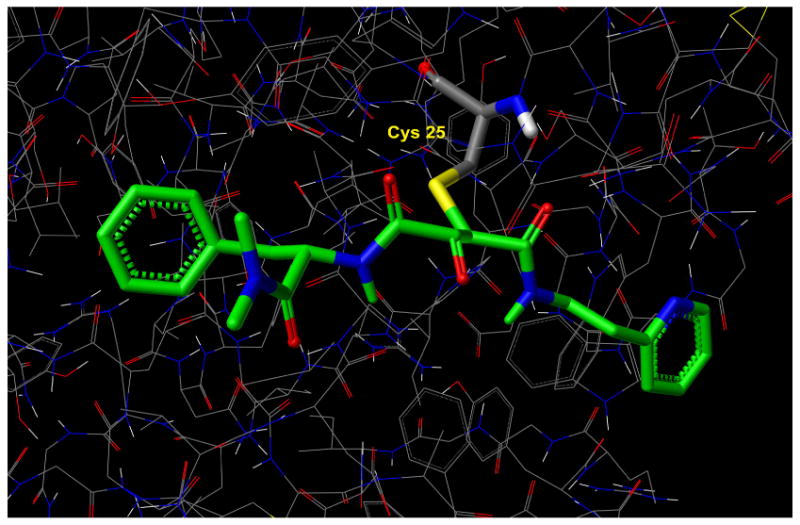
X-ray structure of papain/CLIK-148 (1cvz.pdb) depicting covalent bond between the Cys25 sulfur of papain and the epoxide carbon of CLIK-148. The epoxide is illustrated in its ring-opened form.
Footnotes
Supporting Information Available The coordinate files (pdb format) for the papain coordinate system derived from 1cvz.pdb, with the Cys 25 sulfur to ligand bond manually deleted (papain_mpb.pdb), plus the coordinates for compounds 1 (mpb_compound1.pdb), 2 (mpb_compound2.pdb), and 5 (mpb_compound5.pdb) in the same coordinate system as papain. This material is available free of charge via the internet at pubs.acs.org.
References
- 1.McKerrow JH, Engel JC, Caffrey CR. Cysteine protease inhibitors as chemotherapy for parasitic infections. Bioorg Med Chem. 1999;7:639–644. doi: 10.1016/s0968-0896(99)00008-5. [DOI] [PubMed] [Google Scholar]
- 2.Que X, Reed SL. Cysteine proteinases and the pathogenesis of amebiasis. Clin Microbiol Rev. 2000;13(2):196–206. doi: 10.1128/cmr.13.2.196-206.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rafati S, Salmanian AH, Hashemi K, Schaff C, Belli S, Fasel N. Identification of Leishmania major cysteine proteinases as targets of the immune response in humans. Mol Biochem Parasitol. 2001;113(1):35–43. doi: 10.1016/s0166-6851(00)00377-7. [DOI] [PubMed] [Google Scholar]
- 4.Salas F, Fichmann J, Lee GK, Scott MD, Rosenthal PJ. Functional expression of falcipain, a Plasmodium falciparum cysteine proteinase, supports its role as a malarial hemoglobinase. Infect Immun. 1995;63(6):2120–2125. doi: 10.1128/iai.63.6.2120-2125.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Teo CF, Zhou XW, Bogyo M, Carruthers VB. Cysteine protease inhibitors block Toxoplasma gondii microneme secretion and cell invasion. Antimicrob Agents Chemother. 2007;51:679–688. doi: 10.1128/AAC.01059-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tomas AM, Miles MA, Kelly JM. Overexpression of cruzipain, the major cysteine proteinase of Trypanosoma cruzi, is associated with enhanced metacyclogenesis. Eur J Biochem. 1997;244(2):596–603. doi: 10.1111/j.1432-1033.1997.t01-1-00596.x. [DOI] [PubMed] [Google Scholar]
- 7.Werbovetz KA. Target-based drug discovery for malaria, leishmaniasis, and trypanosomiasis. Curr Med Chem. 2000;7(8):835–860. doi: 10.2174/0929867003374615. [DOI] [PubMed] [Google Scholar]
- 8.Pecoul B. New Drugs for Neglected Diseases: From Pipeline to Patients. PLoS Medicine. 2004;1:19–22. doi: 10.1371/journal.pmed.0010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Molecular Libraries Screening Center Network. http://nihroadmap.nih.gov/molecularlibraries/
- 10.Penn Center for Molecular Discovery. http://www.seas.upenn.edu/∼pcmd.
- 11.Myers MC, Shah PP, Diamond SL, Huryn DM, Smith AB., 3rd Identification and Synthesis of a Unique Thiocarbazate Cathepsin L Inhibitor. Bioorg Med Chem Lett. 2008;18:210–214. doi: 10.1016/j.bmcl.2007.10.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vicik R, Busemann M, Gelhaus C, Stiefl N, Scheiber J, Schmitz W, Schulz F, Mladenovic M, Engels B, Leippe M, Baumann K, Schirmeister T. Aziridide-based inhibitors of cathepsin L: synthesis, inhibition activity, and docking studies. Chem Med Chem. 2006;1(10):1126–1141. doi: 10.1002/cmdc.200600106. [DOI] [PubMed] [Google Scholar]
- 13.Vicik R, Hoerr V, Glaser M, Schultheis M, Hansell E, McKerrow J, Holzgrabe U, Caffrey C, Ponte-Sucre A, Moll H, Stich G, Schirmeister T. Aziridine-2,3-dicarboxylate inhibitors targeting the major cysteine protease of Trypanosoma brucei as lead trypanocidal agents. Bioorg Med Chem Lett. 2006;16:2753–2757. doi: 10.1016/j.bmcl.2006.02.026. [DOI] [PubMed] [Google Scholar]
- 14.Chowdhury SF, Sivaraman J, Wang J, Devanathan G, Lachance P, Qi H, Menard R, Lefebvre J, Konishi Y, Cygler M, Sulea T, Purisima EO. Design of Noncovalent Inhibitors of Human Cathepsin L. From the 96-Residue Proregion to Optimized Tripeptides. J Med Chem. 2002;45(24):5321–5329. doi: 10.1021/jm020238t. [DOI] [PubMed] [Google Scholar]
- 15.Tsuge H, Nishimura T, Tada Y, Asao T, Turk D, Turk V, Katunuma N. Inhibition mechanism of cathepsin L-specific inhibitors based on the crystal structure of papain-CLIK148 complex. Biochem Biophys Res Commun. 1999;266:411–6. doi: 10.1006/bbrc.1999.1830. [DOI] [PubMed] [Google Scholar]
- 16.Katunuma N, Murata E, Kakegawa H, Matsui A, Tsuzuki H, Tsuge H, Turk D, Turk V, Fukushima M, Tada Y, Asao T. Structure based development of novel specific inhibitors for cathepsin L and cathepsin S in vitro and in vivo. FEBS Lett. 1999;458(1):6–10. doi: 10.1016/s0014-5793(99)01107-2. [DOI] [PubMed] [Google Scholar]
- 17.Fujishima A, Imai Y, Nomura T, Fujisawa Y, Yamamoto Y, Sugawara T. The crystal structure of human cathepsin L complexed with E-64. FEBS Lett. 1997;407(1):47–50. doi: 10.1016/s0014-5793(97)00216-0. [DOI] [PubMed] [Google Scholar]
- 18.A homology model of cathepsin L based on the coordinates of papain/CLIK 148 (1cvz.pdb) was constructed using the MOE software available from the Chemical Computing Group, Inc. Subsequent dockings of thiocarbazate 1 against the resulting theoretical model were conducted using the XP Glide docking algorithm (Schrodinger, Inc.). Poor docking scores for thiocarbazate 1 were obtained due to the lack of contacts made in the S2 subsite, since key residues in this site had shifted considerably from their original positions in papain during homology model construction.
- 19.Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein-Ligand Complexes. J Med Chem. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 20.Kim MJ, Yamamoto D, Matsumoto K, Inoue M, Ishida T, Mizuno H, Sumiya S, Kitamura K. Crystal structure of papain-E64-c complex. Binding diversity of E64-c to papain S2 and S3 subsites. Biochemical Journal. 1992;287(3):797–803. doi: 10.1042/bj2870797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schechter I, Berger A. On the size of the active site in proteases. I. Papain. Biochem Biophys Res Commun. 1967;27(2):157–62. doi: 10.1016/s0006-291x(67)80055-x. [DOI] [PubMed] [Google Scholar]
- 22.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Research. 2000;28:235–242. doi: 10.1093/nar/28.1.235. http://www.rcsb.org. [DOI] [PMC free article] [PubMed]
- 23.Shah PP, Myers MC, Beavers MP, Purvis JE, Jing H, Grieser HJ, Sharlow ER, Napper AD, Huryn DM, Cooperman BS, Smith AB, 3rd, Diamond SL. Kinetic Characterization and Molecular Docking of a Novel, Potent, and Selective Slow-Binding Inhibitor of Human Cathepsin L. doi: 10.1124/mol.108.046219. Manuscript submitted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.LaLonde JM, Zhao B, Smith WW, Janson CA, DesJarlais RL, Tomaszek TA, Carr TJ, Thompson SK, Oh HJ, Yamashita DS, Veber DF, Abdel-Meguid SS. Use of Papain as a Model for the Structure-Based Design of Cathepsin K Inhibitors: Crystal Structures of Two Papain-Inhibitor Complexes Demonstrate Binding to S′-Subsites. J Med Chem. 1998;41(23):4567–4576. doi: 10.1021/jm980249f. [DOI] [PubMed] [Google Scholar]
- 25.Myers MC, Shah PP, Beavers MP, Diamond SL, Smith AB, 3rd, Huryn DM. Thiocarbazate inhibitors of cathepsin L: A structure activity study defining a novel chemotype. Bioorg Med Chem Lett. Manuscript submitted for publication. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.