

Introduction
HIV-1 protease (PR) cleaves Gag and Gag-Pol polyproteins during virus maturation. Gag mutations involving cleavage sites, in particular p7/p1, p1/p6, have been associated with drug resistance or recovery of replicative capacity (RC) both in the absence[1] and presence [2-4] of PR drug resistance associated mutations. Polymorphisms at these positions as well as the p24/p2 and p2/p7 cleavage site have also been documented in drug naïve patients [3, 5]. Furthermore, we and others have identified areas of the Gag protein from PI-treated patients that are associated with both drug resistance and recovery of RC (outside cleavage site regions) [6-8]. Polymorphisms throughout gag may therefore be relevant to response to PIs in naïve patients. Despite this body of data, phenotypic testing of clinical samples is still focused primarily on the protease gene. Phenotypic resistance test vectors have generally incorporated patient derived Protease and the 5′ end of gag (including p7/p1 and p1/p6 cleavage sites) into a gag containing vector, typically derived from an HIV-1 subtype B molecular clone. There exists therefore the possibility of misclassifying drug susceptibility based on such an approach.
Non-B subtypes of HIV-1 account for the vast majority of infections worldwide. As the WHO public health rollout of HAART progresses substantial numbers of patients will require second line therapy with ritonavir-boosted protease inhibitors (PI). Since little in vitro work has been undertaken on the basis of PI susceptibility in full length non B viruses we focused on PI drug susceptibility in wild type subtype A and C laboratory and clinical strains, not previously exposed to antiretrovirals, as these different subtypes provide a wide genetic diversity within protease and gag. We sought to explore the role of full-length gag as well as protease in determining PI susceptibility in such wild type viruses.
Materials and Methods
Construction of resistance test vectors
A replication defective, envelope deleted subtype B clone p8.91NSX, expressing gag-pol, tat and rev was used as previously described [6]. This vector was further modified by silencing an existing EcoRI site at the end of integrase and creating a new EcoRI site just after the end of protease using site directed mutagenesis to allow cloning of gag-protease sequences. The two vectors had indistinguishable replication capacities and drug susceptibility profiles (data not shown). We obtained the following molecular clones from the NIH AIDS Research and Reference Reagent program: (subtype) p97ZA012.1 (C)[9], p8CN006.29 (C)[9], pMJ4 (C)[10], p94CY017.41 (A)[11]. Additionally the gag-protease region was amplified from two treatment naïve patients, identified by phylogenetic analysis as having subtype A virus during routine genotypic testing at first HIV diagnosis. RNA was extracted from plasma virus (QIA amp® viral RNA extraction kit, Qiagen,) and reverse transcribed using Moloney-MLV-RT and random priming (Promega). Gag-pol coding sequence was amplified from cDNA by nested PCR using outer primers 5′gagout (GTG TGG AAA ATC TCT AGC AG) and RTrev1 (CTG GRA TAA CYT CTG CTT) and an inner primer set as described[6]. Both PCR steps were carried out using Expand® High Fidelity PCR kit (Boehringer Manheim). The subtype A gag-protease sequences were cloned into the vector backbone, and representative clones (12.2 and 32G) chosen that had the same pol sequence as determined from plasma virus population sequencing. We utilized the naturally occurring ApaI restriction site towards the end of gag to generate chimeras between the test samples and the subtype B reference virus as used by commercial phenotypic systems.
Generation of virus stocks and drug susceptibility testing
293T cells were cotransfected with a gag-pol expression vector containing cloned gag-protease sequences, and plasmids encoding VSV-G envelope and luciferase reporter genes, with PI drug susceptibility testing carried out as previously described [6]. Results, derived from at least two independent experiments (each in duplicate) were analyzed and IC50 determined using Graph Pad Prism 5 (GraphPad Software, Inc. La Jolla, CA, USA). Susceptibility testing for RT inhibitors differed in that supernatant containing resistance test vectors was harvested 48 hours post transfection, and used to infect fresh target 293T cells in the presence of serial dilutions of RT inhibitors in a 96 well plate format. Replicative capacity (RC) of these viruses was assessed by comparing the luciferase activity of recombinant virus to that of the wild type subtype B control virus in the absence of drug. Equal amounts of input plasmid DNA were used and we have previously shown that % infectivity correlates well with infectivity/ ng p24 in this system[6]. The drugs used in this study (ATV, DRV, LPV, SQV, AZT and NVP) were obtained from the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH.
Multiple sequence alignment
was carried out using the program Muscle (www.drive5.com/muscle/) with manual alignment in Se-Al (tree.bio.ed.ac.uk).
Results
Discrepancy between gag-protease and protease alone vectors in PI susceptibility
We tested a panel of six viruses against the following PIs: Atazanavir (ATV), Darunavir (DRV), Lopinavir (LPV) and Saquinavir (SQV). Fold changes relative to subtype B are shown in Table 1. For full-length gag-pro constructs we observed a range of PI susceptibilities, with 5 out of 6 showing reduced susceptibility for at least one drug (using a generous biological cut-off of FC>2, which is greater than the assay variability as determined by the standard deviations of repeated IC50 values for the reference virus). Overall, there was a 5-fold reduction in susceptibility to at least one drug in half the samples tested, with the greatest reductions seen for lopinavir, with more variable changes in susceptibility for atazanavir and saquinavir.
Table 1.
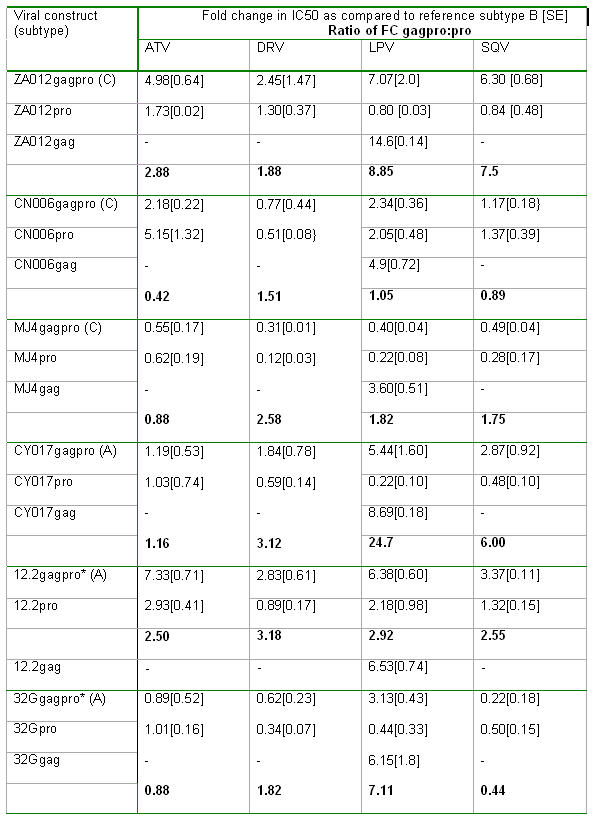
Importantly however, when specific proteases from these viruses were tested in the context of the subtype B gag (table 1), as in most commercially available phenotypic drug resistance assays, a reversal of this reduction in susceptibility was generally observed. This use of the protease alone vector resulted in overestimating the LPV susceptibility of CY017 by nearly 25 times, of ZA012 by nearly 9 times and for 32G by a factor of 7, compared to the gag-protease vector (table 1).
Gag alone confers reduced PI susceptibility
In order to determine if the determinants of reduced susceptibility in some of the viruses were located in gag, we examined the profile of our panel of gag sequences fused not with their cognate protease, but with the reference subtype B protease (Fig 1A) and compared these to the full-length cognate gag-protease samples with regard to LPV susceptibility (Table 1). We confirmed that gag alone determined reduced susceptibility to PI. Notably, ZA012 gag with B protease showed 14-fold reduced susceptibility as compared to subtype B gag-pro. This experiment also demonstrates that the cognate protease restores susceptibility to PI and in the extreme case of MJ4, can completely reverse a partially resistant phenotype to a hyper-susceptible one, in relation to subtype B.
Figure 1.

(A) Schematic diagram of chimeras used Key: X (test sequence), B (reference subtype B sequence), MA (matrix, or p17), CA (capsid, or p24), p2 (spacer peptide 1), p7 (nucleocapsid), p1 (spacer peptide 2), p6, pro (protease), RT (reverse transcriptase). (B) Mean replicative capacity of study viruses expressed as % of wild type subtype B reference (log10) (C) Mean fold change in IC50 for the panel of viruses against AZT. (D) Comparison of mean fold change in LPV and DRV IC50 using cognate gag-pro-RT versus gag-pro for two viruses (12.2 and MJ4).
There was variation in replicative capacity between constructs (Figure 1B). Interestingly the cognate gag-protease combination had a lower RC than gag with a subtype B protease in both ZA012 and 32G. There was no direct correlation between PI susceptibility and RC; for example there was an 8.9 fold difference in LPV FC for gag-protease versus protease alone in ZA012, but no difference in RC (Figure 1B, compare white and grey bars). As a control for any potential impact of replication capacity on drug susceptibility, we performed phenotypic susceptibility testing for the panel of viral constructs against the RT inhibitors zidovudine (Figure 1C) and nevirapine (data not shown). For both drugs, the fold changes relative to the subtype B reference were less than 1.5 fold across all constructs tested. We also confirmed that use of cognate gag-pro or cognate gag-pro-RT did not affect PI susceptibility results (Figure 1D).
Finally, we analysed gag and protease sequences from the seven viruses tested (Figures S1 and S2). Comparison of gag cleavage sites (in boxes) shows that most are highly conserved, with the exception of p7/p2 (Figure S1, orange box). Protease sequence analysis showed no major resistance mutations and typical non-B polymorphisms in subtypes A and C, some of which are classified as minor resistance mutations (Figure S2).
Conclusions
Overall these data demonstrate that virus specific protease confers variable susceptibility to PI, and more importantly suggest that consideration of protease alone may overestimate PI susceptibility in viruses not previously exposed to this class of drugs. This could lead to misclassification of susceptibility for some isolates, and potentially also limits assessment of the genotype-phenotype relationships in drug experienced patients.
Given the expanding body of evidence showing that gag mutations are known to contribute to PI resistance in treated patients, and in light of our data we recommend that full gag sequences should be included in recombinant virus assays to determine PI susceptibility in clinical isolates. We would also encourage a rapid expansion of full-length gag sequencing in patients failing PI-based therapy, to populate the relevant relational databases increasingly used to assess the clinical impact of HIV drug resistance. Gag genotypic determination to guide patient care can be then be evaluated once specific Gag determinants of PI susceptibility have been identified.
Lower clinical cut-offs of 5.2 and 10 fold have been proposed for ATV and LPV response respectively [12, 13]. Although fold changes in our system may not be directly comparable, we were concerned to observe that some viruses approached and even exceeded these values. In the ACTG 5142 study 23% of patients starting boosted lopinavir had a viral load >50 copies/ml after 2 years, as compared to 11% for NNRTI based regimens (p=0.003)[14]. Patients with viral failure after boosted PI (bPI) in this and other studies infrequently have major protease resistance mutations[15, 16], although there is a lack of data on evolution of Gag mutations. A better understanding of determinants in Gag reducing susceptibility to PI might explain these observations.
Finally, our findings may have important implications for the public health approach to ART. Increasing numbers of patients are moving to PI-based second line therapy following the rollout of HARRT, and combined LPV/ritonavir is the most widely available PI in resource poor settings. Some of the subtype A and C constructs studied demonstrated particularly high FCs for this agent, and as resistance testing is not routinely performed in these patients, further research is clearly warranted.
Supplementary Material
Acknowledgements
We would like to thank Didier Trono, EPFL, Lausanne, Switzerland for the p8.9 and pMDG plasmids, and Nigel Temperton for the pCSFLW plasmid encoding luciferase. The following reagents were obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: ATV, DRV, LPV, SQV, AZT, NVP; pMJ4 from Drs. Thumbi Ndung'u, Boris Renjifo and Max Essex; HIV-1 p98CN006 from Dr. Robert Bollinger and the UNAIDS Network for HIV Isolation and Characterization; p97ZA012.1 from Drs. Cynthia M. Rodenburg, Beatrice H. Hanhn, Feng Gao, and the UNAIDS Network for HIV Isolation and Characterisation; p94CY017.41 from Drs. Stanley A. Trask, Feng Gao, Beatrice H. Hahn, and the Aaron Diamond AIDS Research Centre.
Funding source: RG is funded by the Wellcome Trust (WT081772MA). We acknowledge funding through the NIHR UCLH/UCL Comprehensive Biomedical Research Centre. The research leading to these results has also received funding from the European Community's Seventh Framework Programme (FP7/2007-2013) under the project “Collaborative HIV and Anti-HIV Drug Resistance Network (CHAIN)” – grant agreement n° 223131.
RKG is supported by a Wellcome Trust Clincal Research Training Fellowship
Abbreviations
- ATV
atazanavir
- DRV
darunavir
- LPV
lopinavir
- SQV
saquinavir
- AZT
zidovudine
- NVP
nevirapine
Footnotes
GenBank Accession number for 12.2: EU927369 and 32G: bankit1299707
Conflicts of interest
RG has received educational travel grants from Gilead Sciences, Bristol Myers Squibb and Boehringer-Ingelheim.
DP has acted as a consultant for Bristol Myers Squibb, Johnson&Johnson, Boehringer Ingelheim, Roche, and Gilead Sciences.
AK, ALM, GJT and CMP have no conflicts of interest
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Ravindra K GUPTA, University College London Medical School, Windeyer Institute 46 Cleveland Street London W1T 4JF Tel: 0207 679 9226 rebmrag@ucl.ac.uk.
Arinder KOHLI, University College London Medical School, Windeyer Institute 46 Cleveland Street London W1T 4JF Tel: 0207 679 9226 arinder.kohli@kcl.ac.uk.
Adele L. McCORMICK, University College London Medical School, Windeyer Institute 46 Cleveland Street London W1T 4JF Tel: 0207 679 9226 Adele.McCormick@ucl.ac.uk
Greg J. TOWERS, University College London Medical School, Windeyer Institute 46 Cleveland Street London W1T 4JF g.towers@ucl.ac.uk
Deenan PILLAY, University College London Medical School, Windeyer Institute 46 Cleveland Street London W1T 4JF and Centre for Infections Health Protection Agency 61 Colindale Avenue London NW9.
Chris M PARRY, University College London Medical School, Windeyer Institute 46 Cleveland Street London W1T 4JF and Centre for Infections Health Protection Agency 61 Colindale Avenue London NW9.
References
- 1.Nijhuis M, van Maarseveen NM, Lastere S, Schipper P, Coakley E, Glass B, et al. A novel substrate-based HIV-1 protease inhibitor drug resistance mechanism. PLoS Med. 2007;4:e36. doi: 10.1371/journal.pmed.0040036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bally F, Martinez R, Peters S, Sudre P, Telenti A. Polymorphism of HIV type 1 gag p7/p1 and p1/p6 cleavage sites: clinical significance and implications for resistance to protease inhibitors. AIDS Res Hum Retroviruses. 2000;16:1209–1213. doi: 10.1089/08892220050116970. [DOI] [PubMed] [Google Scholar]
- 3.Cote HC, Brumme ZL, Harrigan PR. Human immunodeficiency virus type 1 protease cleavage site mutations associated with protease inhibitor cross-resistance selected by indinavir, ritonavir, and/or saquinavir. J Virol. 2001;75:589–594. doi: 10.1128/JVI.75.2.589-594.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Doyon L, Croteau G, Thibeault D, Poulin F, Pilote L, Lamarre D. Second locus involved in human immunodeficiency virus type 1 resistance to protease inhibitors. J Virol. 1996;70:3763–3769. doi: 10.1128/jvi.70.6.3763-3769.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Malet I, Roquebert B, Dalban C, Wirden M, Amellal B, Agher R, et al. Association of Gag cleavage sites to protease mutations and to virological response in HIV-1 treated patients. J Infect. 2007;54:367–374. doi: 10.1016/j.jinf.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 6.Parry CM, Kohli A, Boinett CJ, Towers GJ, McCormick AL, Pillay D. Gag determinants of fitness and drug susceptibility in protease inhibitor-resistant human immunodeficiency virus type 1. J Virol. 2009;83:9094–9101. doi: 10.1128/JVI.02356-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gatanaga H, Suzuki Y, Tsang H, Yoshimura K, Kavlick MF, Nagashima K, et al. Amino acid substitutions in Gag protein at non-cleavage sites are indispensable for the development of a high multitude of HIV-1 resistance against protease inhibitors. J Biol Chem. 2002;277:5952–5961. doi: 10.1074/jbc.M108005200. [DOI] [PubMed] [Google Scholar]
- 8.Myint L, Matsuda M, Matsuda Z, Yokomaku Y, Chiba T, Okano A, et al. Gag non-cleavage site mutations contribute to full recovery of viral fitness in protease inhibitor-resistant human immunodeficiency virus type 1. Antimicrob Agents Chemother. 2004;48:444–452. doi: 10.1128/AAC.48.2.444-452.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rodenburg CM, Li Y, Trask SA, Chen Y, Decker J, Robertson DL, et al. Near full-length clones and reference sequences for subtype C isolates of HIV type 1 from three different continents. AIDS Res Hum Retroviruses. 2001;17:161–168. doi: 10.1089/08892220150217247. [DOI] [PubMed] [Google Scholar]
- 10.Ndung'u T, Renjifo B, Essex M. Construction and analysis of an infectious human Immunodeficiency virus type 1 subtype C molecular clone. J Virol. 2001;75:4964–4972. doi: 10.1128/JVI.75.11.4964-4972.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao F, Vidal N, Li Y, Trask SA, Chen Y, Kostrikis LG, et al. Evidence of two distinct subsubtypes within the HIV-1 subtype A radiation. AIDS Res Hum Retroviruses. 2001;17:675–688. doi: 10.1089/088922201750236951. [DOI] [PubMed] [Google Scholar]
- 12.Kempf DJ, Isaacson JD, King MS, Brun SC, Sylte J, Richards B, et al. Analysis of the virological response with respect to baseline viral phenotype and genotype in protease inhibitor-experienced HIV-1-infected patients receiving lopinavir/ritonavir therapy. Antivir Ther. 2002;7:165–174. [PubMed] [Google Scholar]
- 13.Naeger LK, Struble KA. Effect of baseline protease genotype and phenotype on HIV response to atazanavir/ritonavir in treatment-experienced patients. Aids. 2006;20:847–853. doi: 10.1097/01.aids.0000218548.77457.76. [DOI] [PubMed] [Google Scholar]
- 14.Riddler SA, Haubrich R, DiRienzo AG, Peeples L, Powderly WG, Klingman KL, et al. Class-sparing regimens for initial treatment of HIV-1 infection. N Engl J Med. 2008;358:2095–2106. doi: 10.1056/NEJMoa074609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kempf DJ, King MS, Bernstein B, Cernohous P, Bauer E, Moseley J, et al. Incidence of resistance in a double-blind study comparing lopinavir/ritonavir plus stavudine and lamivudine to nelfinavir plus stavudine and lamivudine. J Infect Dis. 2004;189:51–60. doi: 10.1086/380509. [DOI] [PubMed] [Google Scholar]
- 16.Delaugerre C, Flandre P, Chaix ML, Ghosn J, Raffi F, Dellamonica P, et al. Protease inhibitor resistance analysis in the MONARK trial comparing first-line lopinavir-ritonavir monotherapy to lopinavir-ritonavir plus zidovudine and lamivudine triple therapy. Antimicrob Agents Chemother. 2009;53:2934–2939. doi: 10.1128/AAC.01643-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.