

Abstract
We have developed a multi-disciplinary approach combining molecular biology, delivery technology, combinatorial chemistry, and reversible masking to create improved systemic, targeted delivery of plasmid DNA while avoiding non-specific uptake in vivo. We initially used a well characterized model targeting the asialolglycoprotein receptor in the liver. Using our bilamellar invaginated vesicle (BIV) liposomal delivery system with reversible masking, we increased expression in the liver by 76-fold, nearly equaling expression in first-pass organs using non-targeted complexes, with no expression in other organs. The same technology was then applied to efficiently target delivery to a human tumor microenvironment model. We achieved efficient, targeted delivery by attachment of specific targeting ligands to the surface of our BIV complexes in conjunction with reversible masking to bypass non-specific tissues and organs. We identified ligands that target a human tumor microenvironment created in vitro by co-culturing primary human endothelial cells with human lung or pancreatic cancer cells. The model was confirmed by increased expression of tumor endothelial phenotypes including CD31 and VEGF-A, and prolonged survival of endothelial capillary-like structures. The co-cultures were used for high-throughput screening of a specialized small-molecule library to identify ligands specific for human tumor-associated endothelial cells in vitro. We identified small molecules that enhanced the transfection efficiency of tumor-associated endothelial cells, but not normal human endothelial cells or cancer cells. Intravenous injection of our targeted, reversibly masked complexes into mice, bearing human pancreatic tumor and endothelial cells, specifically increased transfection to this tumor microenvironment about 200-fold. Efficacy studies using our optimized targeted delivery of a plasmid encoding thrombospondin-1 eliminated tumors completely after five intravenous injections administered once every week.
Keywords: non-viral delivery, liposomes, targeted delivery, tumor vascular microenvironment, anti-cancer therapeutics, reversible masking
INTRODUCTION
Angiogenesis, the process of new blood vessel formation, is required for sustained cancer growth and metastasis.1, 2 Recent approval of antiangiogenic drugs, such as Bevacizumab, Sorafenib and Sunitinib, by the FDA supports the use of anti-angiogenesis as a strategy for the treatment of cancer.3, 4 However, these treatments are expensive and can have toxic side effects.5, 6 Therefore, investigators addressed the feasibility of using gene therapy due to its potential for prolonged production of therapeutic proteins and low cost. Pharmacokinetic studies showed that administration of therapeutic proteins might not be optimal when compared to delivery systems that potentially maintain elevated drug level continuously in the body.7–9 Delivery vehicles used for gene therapy include viral, non-viral, and bacterial vectors (Reviewed in: 10). Other delivery methods such as in vivo electroporation, ballistic and other needle-free delivery systems are also used (Reviewed in: 10). Much work has focused on the use of non-viral vectors due to diminished safety concerns and ease of manufacturing. Non-viral vectors have been used successfully in many pre-clinical and clinical studies (Reviewed in: 10).11–14
We previously reported the bilamellar invaginated vesicle (BIV) liposomal delivery system that has unique features and has proved effective for cancer gene therapy.11, 14–18 In our initial studies, we used a well characterized model targeting the asialolglycoprotein receptor in the liver in conjunction with our reversible masking technology to bypass uptake in non-target organs (Templeton, N.S. US Patent No. 7,037,520 B2 issued May 2, 2006). We then added targeted delivery using small molecules in conjunction with our reversible masking technology. A combinatorial library developed in lab of Burgess allows production of small molecules designed to bind proteins selectively.19–22 Members of the library resemble secondary structure motifs found at hot-spots in protein-ligand interactions, e.g. bivalent beta-turn mimics designed to have an affinity for cell surface receptors. Importantly, the bivalent small molecules can have selectivity for binding cell surface receptors. Here the strategy was adapted to produce bivalent molecules that have hydrocarbon tails, and preparation of functionalized BIV complexes from these is fast and routine in our lab. We believe that small molecules are needed for targeted delivery because larger ligands, including small peptides are immunogenic when multimerized on the surface of delivery vehicles including nanoparticles.23
We also developed a novel and robust in vitro screen to identify small molecule ligands from our combinatorial library to use for targeted delivery to human tumor vascular microenvironment. No endothelial culture model had been created that expresses most surface markers found in human tumor vasculature.24 Furthermore, recent publications have reported that tumor cells which are accessible to the circulation undergo a differentiation process called “vasculogenic mimicry” wherein they express vascular markers on their surface rather than tumor cell markers.25, 26 We generated co-cultures of tumor and endothelial cells to induce a human tumor vascular microenvironment phenotype in vitro and performed in vitro screening of our small molecule libraries.
Finally, our efficacy studies focused on the targeted delivery of plasmid DNA encoding the anti-angiogenic protein, human thrombospondin-1 (TSP1).27, 28 TSP1 is a secreted protein that can prevent angiogenesis, the formation of new blood vessels required to sustain tumor growth.29 The modified TSP1 mimetic ABT-510 has advanced to Phase II clinical trials to treat advanced cancer.29 Gene delivery of TSP1 significantly inhibits growth of various cancers and tumor microvessel density in animal models.12, 13, 30–32 Furthermore, TSP1 can also inhibit tumor growth by polarizing macrophages in the tumor microenvironment towards cytotoxic M1 differentiation.29 Here we show that targeted, reversibly masked delivery of a TSP1 expression plasmid significantly improves the efficacy of TSP1 gene therapy.
MATERIALS AND METHODS
Cell Culture
PANC-1, H1299, and MCF7 human cancer cell lines were purchased from the American Type Culture Collection (ATCC, Bethesda, MD). PANC-1 cells were cultured in high glucose DMEM. H1299 cells were cultured in RPMI-1640 medium, and MCF7 cells were cultured in Eagle’s Minimum Essential Medium. All the above media were supplemented with 10% fetal bovine serum (FBS). HUVEC was purchased from Lonza (Clonetics, Walkersville, MD) and grown in endothelial basal medium (Clonetics) supplemented with SingleQuots (Clonetics). HUVECs were cultivated at third to sixth passage. Co-culture of HUVECs and cancer cells was established after cell counting and plated at the ratio of approximately 10 – 30:1 HUVEC: cancer cells with the seeding density of 5,000 HUVECs/cm2. In some experiments, co-cultures were maintained in dual chamber Transwell systems which physically separated cancer cells from ECs while allowing free diffusion between the two cell populations through the 0.4 μm-sized microporous membrane (Corning).
Flow Cytometry
Cells were harvested and resuspended in 1xPBS at 10×106/ml. The cell suspension was incubated with anti-CD31:RPE (GeneTex, Irvine, CA) according to the manufacturer’s instructions. After washing, propidium iodide was added to the cell suspension to exclude dead cells in the analysis. Flow cytometry was performed on the BD LSRII (BD Biosciences, San Jose, CA) and analyzed by the CellQuest program with gates set on the forward scatter versus the side scatter.
Real-Time Quantitative RT-PCR
Human VEGF-A primers were synthesized containing the following sequences: forward 5′-TGGAATTGGATTCGCCATTT-3′ and reverse 5′-TGGGTGGGTGTGTCTACAGGA-3′. β-actin primer sequences were: forward 5′-CTGGAACGG TGAAGGTGACA-3′ and reverse 5′-AAGGGACTTCCTGTAACAATGCA-3′. Co-cultured cells were grown in transwell plates. Total RNA was extracted from the cells using Trizol (Invitrogen, Carlsbad, CA) following the manufacturer’s protocol and treated with DNase I (Invitrogen). One μg of total RNA was reverse-transcribed into cDNAs with an iScript cDNA synthesis kit (Bio-Rad, Hercules, CA) containing a mixture of oligo(dT) and random primers. Real-time PCR was performed on an ABI PRISM 7900HT Sequence Detection System (Applied Biosystems, Foster City, CA) using a DyNAmo HS SYBR Green qPCR kit (New England BioLabs, Finnzymes, Finland). Cycling conditions were the following: initial denaturation at 95°C for 10 min, followed by 40 cycles at 95°C for 15 s and 60°C for 1 min.
Western Blot
Eight days after co-culture in transwell dishes, 50 mg protein from EC lysates was loaded on 9% SDS-PAGE gels followed by Western transfer to nitrocellulose membranes (Hybond ECL; Amersham Pharmacia Biotech). The membranes were blocked with 5% nonfat milk in TBS (20 mM Tris-HCl, 150 mM NaCl [pH 7.4], and 0.05% Tween 20). After incubation with the primary human anti-VEGF antibody (R&D Systems, Minneapolis, MN) at 1 μg/mL for 2 h at room temperature (RT), the membranes were washed six times at 5-min intervals with TBS/0.05% Tween 20 and incubated with secondary anti-goat horseradish peroxidase-conjugated antibody (Transduction Laboratories, Lexington, KY).33
Tube Formation Assay
Eight days after co-culture in transwell plates, Matrigel (BD Biosciences, San Jose, CA) was added to the receiver chamber of a blank 6-well transwell plate at 4°C and incubated for 2 h at 37°C. After Matrigel had solidified, endothelial cells were trypsinized, counted and seeded on top at 5×105 cells/well. Cells were incubated for 16 h to allow the formation of capillary-like structures. To maintain the co-culture conditions, the cancer cells were cultivated in the upper chamber of the transwell plate. The tubular structure was observed daily to monitor morphology, integrity survival.
Preparation of BIV DNA:Liposome Complexes and Reversible Masking Agents
Plasmids p4241 and p4119 were generous gifts from Robert Debs (California Pacific Medical Center Research Institute, San Francisco, CA). They encode the luciferase and CAT genes, respectively. pCMV-THBS-1 was a kind gift from David Roberts (National Institutes of Health, Bethesda, MD) and encodes the TSP1 gene.27, 28 All plasmids were grown under ampicillin selection in DH5α Escherichia coli. Plasmid DNA was purified by anion exchange chromatography using the Qiagen Endo-Free Plasmid Giga Kit (Qiagen, Hilden Germany). DOTAP and DOTAP:Chol BIV liposomes, BIV DNA:liposome complexes (BIV complexes) were prepared as previously described 15, except that synthetic cholesterol (Sigma-Aldrich, St. Louis, MO) was used at a ratio of 50:45 DOTAP:cholesterol. Succinylated asialofetuin was prepared and coated onto BIVs as previously described.15 Reversible Masking Agent #1 (RM #1) is a pendant-modified PEG NHS ester having 5 pendant carboxyl groups added to a PEG polymer and is 5000 MW. Reversible Masking Agent #2 (RM #2) is n-dodecyl-β-D-maltopyranoside, approximately 511 MW. Reversible Masking Agents were added to complexes just prior to injections in mice. Zeta potential analyses were performed using a Delsa 440SX zeta-potential analyzer (Beckman-Coulter).
Bivalent Small Molecule Production
Briefly, through selective coupling β-turn monovalent small molecules were mixed in solution to produce bivalent small molecules: homodimers, KB991 – KB1005, and heterodimers, KB1006 – KB 1140. During the process, only potassium carbonate was required to affect the coupling. Boc-protected monomeric compounds were treated with 30% TFA in CH2Cl2 for 4 h at 25°C. The solvent was removed and residue was re-dissolved in DMSO to make a solution of 0.03 M. The dichlorotriazine linker scaffold and K2CO3 were sequentially added. The resulting suspension was sonicated for 15 min and rocked for 7 days. DMSO was lyophilized, and aqueous HCl solution (5%, about 0.5 mL) was added to the above solid residue and sonicated for 3 min. Most of the compounds were precipitated in acidic solutions. After centrifugation, the pellets were dried and saved. In order to coat the bivalent small molecules onto the surface of BIV complexes, a hydrocarbon tail was included in the molecules for insertion into the surface lipid bilayer. Compounds (about 10.0 mg) were initially dissolved in 1.0 mL THF/H2O (v:v = 1:1). CuSO4 solution (1.0 M, 10 μL) was added and followed by Cu powder (1.0 mg). After that procedure, azidooctadecane in THF solution (0.1 mmol, 0.2 mL) was added, and the resulting suspension was stirred at 25°C for 24 h. The suspension was filtered through a glass pipette filled with silica gel using 30% methanol in CH2Cl2 as eluents. The solution was dried and concentrated to the final products. After synthesis, the solid compounds were dissolved in 1:1 chloroform:methanol in glass test tubes. Thin films were produced at the bottom of the tubes under a steady stream of argon gas under the tissue culture hood. The films were dissolved in sterile water to produce a 5 mg/mL stock and subjected to sonication (Lab-Line Trans-sonic 820/H) at 50°C. Aliquots of the reconstituted compounds were stored at −80°C.
In Vitro Delivery and High Throughput Luciferase Assay
We used our high throughput assay to identify bivalent compounds attached to the surface of BIV complexes that internalize into endothelial cells in a human tumor microenvironment more efficiently than non-targeted BIV complexes. Our assay features a luciferase reporter gene and a dedicated plate reader luminometer, the Luminoskan Ascent, certified for ultra-sensitive detection of luciferase expression (Thermo Electron Corp., Waltham, MA) that has 3 injectors/robotic dispensers. The Luminoskan allows many different sample formats from single 10 cm tissue culture dishes to 384-well plates, and has a high degree of sensitivity (<1 fmol ATP/well) for observing small differences in addition to a high dynamic range for samples (>9 decades over whole gain setting area). If the plasmid DNA encoding luciferase is internalized and efficiently transported to the nucleus, then bioluminescence is detected in cells grown in the wells of the plates. The read out is fast, enabling rapid testing of functionalized BIV complexes in a one-bivalent compound-per-well format. Normal HUVECs were used for controls, and delivery to the tumor cells alone or to the co-cultures was compared. Luminoskan data was used to identify the bivalent compounds that produce the highest levels of luciferase gene expression in HUVECs that are co-cultured with human tumor cells and not in normal HUVEC cells or in the tumor cells. Approximately 150 members of the small molecule library were tested at various concentrations on the surface of BIV-luciferase complexes. Optimal transfection time, amount of complexes used for transfection, the optimal integration and lag time were also determined.
Briefly, 7 days after co-culture, cells were harvested and 50 μL cell suspension was seeded to 96-well dishes at 2×104 cells/well. Complexes were prepared as previously described.15 The compounds were diluted to concentrations including 0.5, 10, 200, 500 pg compound/μg DNA encapsulated in the complexes. 1 μL of compound was pipeted slowly into the center of 10 μL of BIV-luciferase DNA complexes that were pre-loaded in 96-well plates and followed by incubation at RT overnight for maximal coating. The following day, cells were transfected with 0.52 μL compound-coated BIV complexes which was diluted to 5 μL and placed into 45 μL serum free medium. Cells were grown in cell culture medium post-transfection. For co-cultures of HUVEC with H1299 cells, DOTAP BIV liposomes were used, and cells were transfected for 4 h. For co-cultures of HUVEC and PANC-1 cells, DOTAP:Chol BIV liposomes were used, and cells were transfected for 2 h. At 24 h post-transfection, cells were lysed using 1% Triton X-100 (Sigma-Aldrich, St. Louis, MO) followed by high throughput luciferase assay using the Luminoskan Ascent to detect gene expression. One sec of integration time and 14 sec of lag time were applied during the assay. Transfection efficiencies of the compound coated BIV liposomal complexes were compared to that of uncoated complexes. Triplicates were measured for each condition. All the dilutions were made in 5% dextrose in water (D5W).
Human Tumor Microenvironment-Pancreatic Cancer Mouse Model
HUVEC and PANC-1 co-cultured cells were harvested and resuspended in 1xPBS after 8 days in co-culture. A 500 μL cell suspension containing 2×106 co-cultured cells (about 1×106 PANC-1 cells) was IP injected into each 8~10 week-old severe combined immunodeficient (SCID) mouse. All animal procedures were performed in accordance with the Baylor College of Medicine (Houston, TX) institutional guidelines using an approved animal protocol.
In Vivo Targeted Delivery and CAT Assay
At 8 weeks post-IP injections of co-cultures detailed above, BIV-CAT DNA complexes were prepared and coated with the small molecule KB1023 at 500 pg compound/μg DNA as discussed above. The complexes were mixed with various concentrations of reversible masking reagent, RM #2, n-dodecyl-β-D-maltopyranoside (Anatrace, Maumee, OH), just prior to intravenous (IV) injections into mice. Each mouse was injected with a total volume of 110 μL complexes containing 35 μg of p4119 CAT DNA. At 14 h post-IV injection, mice were sacrificed, tissues were harvested, and total protein was extracted as previously described.15 CAT protein production was measured using the CAT ELISA kit (Roche, Indianapolis, IN) following the manufacturer’s instructions. Protein concentration was determined using the Micro BCA kit (Pierce) following the manufacturer’s instructions.
Studies targeting the asialoglycoprotein receptor in Balb/c mice were performed as previously described 15 except that reversible masking using RM #1 was added to these studies. Ten mice were IV injected per group. In these studies 50 ug of CAT plasmid, p4119, were encapsulated in BIVs and organs were harvested 24 h post-injection.
Anti-Cancer Therapy
At 2 weeks post-IP injections of the co-cultures detailed above, in vivo delivery was performed using the protocol described above except that 35 μg TSP1 plasmid DNA was encapsulated in the BIV-KB1023 coated complexes and 11mM reversible masking reagent #2 was used prior to IV injections. Injections were performed every two weeks for a total of three injections. In a different experimental group, injections were performed weekly for a total of five injections. Two weeks after the final injection (8 weeks post-IP injection of the co-cultures to establish the human tumor microenvironment-human tumor model), the mice were sacrificed and tumor size was measured. Intra-abdominal tumors and other organs (liver, lungs, spleen, pancreas and colon) were dissected followed by fixation in 10% neutral buffered formalin.
Statistical Analysis
Data were expressed as means ± SEM. Experimental and control groups were compared using ANOVA and the unpaired student t test. P<0.05 was considered significant.
RESULTS
Human tumor microenvironment in vitro model
Tumor cells secrete growth factors and cytokines to initiate and stimulate angiogenesis to generate a microenvironment optimal for tumor growth (Reviewed in: 7). We established an in vitro human tumor microenvironment model by co-culturing human umbilical vein endothelial cells (HUVEC) with human H1299 non-small cell lung carcinoma cells (H1299 co-cultures) or human PANC-1 ductal pancreatic adenocarcinoma (PANC-1 co-cultures). We first looked for changes in the endothelial markers over time to indicate the transition of normal endothelium to tumor vasculature endothelium. An increase in CD31 on the endothelial cells has been reported to occur at this transition as detected by flow cytometry.34 Our flow cytometry data (Fig. 1b,c) shows that this transition occurs between days 8 and 9 in co-culture with H1299 cells. A majority of the HUVECs expressed low levels of CD31 (Fig. 1a, gated by R4). Only few HUVECs expressed high levels of CD31 (gated by R2). After 8 days in the H1299 co-culture, the percentage of high level, CD31 expressing endothelial cells increased by 113% compared to that of the HUVEC control (Fig. 1b; 16.86% versus 7.9%). The increased CD31 expression was significantly higher at 264% after 9 days in co-culture compared to the HUVEC controls (Fig. 1c; 29.96% versus 8.23%). This increased CD31 expression persisted beyond day 9.
Figure 1. Increase in endothelial CD31+ expression after co-culture.

We established co-cultures at a plating ratio of 30:1 HUVEC:H1299 after counting the cells. Endothelial cells were stained by r-phycoerythin-conjugated anti-CD31 Ab and the endothelial cell population (gated by R3) was measured using flow cytometry post-seeding on a daily basis. The vast majority of the endothelial cells expressed low levels of CD31 (gated by R4). A few endothelial cells expressed a high level of CD31 (gated by R2). At 8 days after co-culture, the endothelial cell population of the co-culture significantly increased in CD31 expression compared to that of the HUVEC control (c). The enhancement in CD31 expression was far greater after 9 days in co-culture (b, c) compared to the HUVEC control (a, c). *P=0.026.
A VEGF-A autocrine loop is activated in tumor vasculature, and expression of the VEGF receptors and VEGF-A are increased at both the mRNA and protein levels.35 VEGF-A is also a key pro-angiogenic factor and stimulates endothelial cell proliferation and migration, prolongs endothelial cell survival, and sustains capillary-like tubular structures that are formed by endothelial cells.36, 37 Figure 2 shows increased expression of VEGF-A as detected by quantitative RT-PCR (Fig. 2a; 225%) and by Western blotting (Fig. 2b; 160%) in the endothelial cell chamber of PANC-1 co-cultures at day 8 in co-culture in two-chamber transwell plates with 0.4 μm-sized microporous membranes.
Figure 2. Enhanced endothelial VEGF-A expression after co-culture.

Co-cultures were established at a plating ratio of 20:1 or 10:1 HUVEC:PANC-1 and cultivated in two-chamber transwell dishes for 8 days. Endothelial cells were harvested and VEGF-A expression was measured using real-time RT-PCR and Western blotting. Endothelial VEGF-A expression increased at transcriptional (a) and translational (b) levels after co-culture compared to that of the HUVEC control. *P<0.01, N=6.
When plated on Matrigel, endothelial cells transiently form capillary-like tubular networks in vitro. At 16 hours (h) after plating on Matrigel, our assays showed no significant difference in tube formation between HUVECs (Fig. 3a) and endothelial cells of the PANC-1 co-cultures (Fig. 3b; co-cultured for 8 days in transwell plates). The tubular structure of the HUVEC control started to degrade at 48 h and by 72 h was almost completely degraded (Fig. 3c). In contrast, a significant amount of tubular structure survived at 72 h in endothelial cells from the PANC-1 co-culture (Fig. 3d) and continued to survive for 11 more days (Fig. 3e–f). When the PANC-1 cell inserts were removed from the transwell plates, no difference in tube survival between the endothelial cells separated from the PANC-1 co-culture and the HUVECs was detected. Our data suggest that factors produced by co-culture with the cancer cells prolong the survival of the endothelial tubular structure of the co-culture, perhaps due to increased VEGF-A expression.
Figure 3. Prolonged tube survival after co-culture.

At 8 days after co-culture in transwell dishes at a plating ratio of 10:1 HUVEC:PANC-1, endothelial cells were harvested and seeded on Matrigel. At 16 h, both the HUVEC control (a) and endothelial cells of the co-culture (b) form capillary-like tubular structures. These structures started to degrade 48 h later. By 72 h, the tubular structure of the HUVEC control was almost completely degraded (c). However, a significant amount of tubular structures survived in the co-culture (d). These structures maintained an excellent tubular network and survived for 11 more days (e, f).
High-throughput in vitro screening of small molecule libraries for targeted delivery
The Burgess group prepared libraries of 15 homodimeric and 135 heterodimeric bivalent compounds that are “semi-peptidic” β-turn analogs. Their general structure is shown in Figure 4a. Significantly, these compounds can incorporate any amino acid side chains, so they can be designed to mimic turns at any hot spot that involves that motif. These compounds are different than other compound libraries that have been prepared before insofar as they have polar “warhead” functionalities (mimics 1 and 2)38 and hydrophobic tails. The small molecule peptidomimetics used in prior studies are active against some protein-protein interaction targets which have β-turn hot-spots.20, 21 One compound bound to TrkA receptors on neurons and has applications for stroke recovery and neurodegenerative disorders including dementia.22, 39 For the custom libraries used in our work, two monovalent mimics were combined through chemical steps requiring only potassium carbonate for coupling to form bivalent homodimers or heterodimers (see Materials and Methods for further details). This modification greatly enhances the affinity of the compounds for cell surface receptors. Unlike most combinatorial syntheses, no protecting groups are involved in the last steps of this approach, so the final product does not have to be purified from protecting group residues and added scavenger materials. A hydrocarbon tail was structurally incorporated for coating of the compounds to the surface of liposomal complexes. The libraries used for our work were newly synthesized and are not those used for screening compounds that bound to TrkA receptors.
Figure 4. Bivalent small molecule structure and library screening.
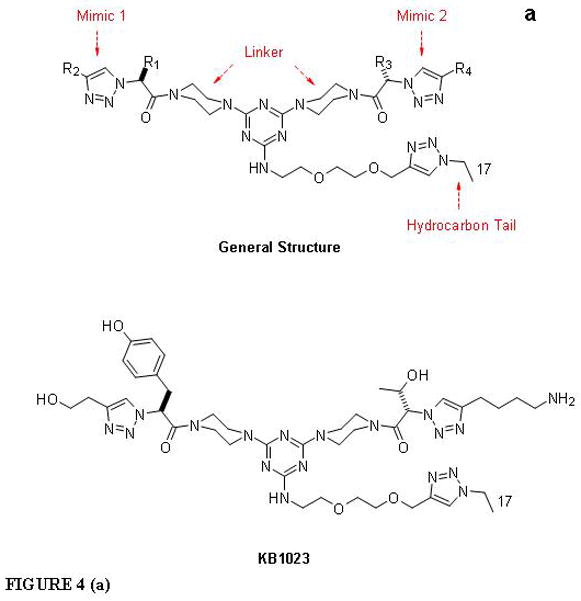

The general structure of the bivalent small molecule (a) includes two β-turn mimics for interaction with cell surface receptors, a hydrocarbon tail for insertion into BIV liposomal complexes, and a linker. The structure of our “hit” molecule, KB1023, is also shown. Our high-throughput luciferase assay (b) was used to screen for tumor endothelial cell-specific targeting ligands. At 7 days after co-culture, cells were harvested and seeded to 96-well plates at 2×104 cells/well. On the same day, BIV-luciferase DNA:liposome complexes were prepared followed by coating of compounds at various compound:DNA ratios. The coated complexes were incubated at RT overnight. The following day, cells were transfected with 50 μL of serum free medium that contained 0.52 μL coated complexes. Transfection was ended by replacing the transfection medium with cell culture medium containing serum. At 24 h post-transfection, cells were lysed and the cell lysate was loaded to 96-well plates at 20 uL/well for luciferase assay using the Luminoskan plate reader.
A novel, high-throughput assay was developed to screen the small molecule libraries for ligands that target human endothelial cells in a tumor microenvironment (Fig. 4b). Highly sensitive and accurate detection systems are required for successful high throughput screens. Furthermore, delivery into the cell nucleus for the detection of potential ligand binding and internalization across the cell membrane is most direct and ultimately reliable. Luciferase expression produced by plasmid DNA delivered to the nucleus meets these criteria; it is an experimentally straight-forward, and well-established technology. We used a Luminoskan Ascent plate luminometer (Thermo Labsystems) to achieve highly sensitive high-throughput quantitation of transfection efficiency.
The co-cultures versus cancer cells versus HUVECs were screened in vitro against the homodimer or heterodimer bivalent small molecule libraries using our high-throughput screen. Hits were defined as those compounds that increased transfection of the luciferase plasmid by at least 100% (+2 on the Y axis) over transfections using BIV complexes alone. BIVs are highly efficient in transfecting cells in culture40 and in vivo.15 Therefore, in order to find ligands to enhance transfection in vitro, we used half the amount of complexes required for optimal transfection of the cells in culture. In screening the libraries, we identified a compound KB1023 that specifically enhanced the transfection efficiency by 100% in the PANC-1 co-culture (Fig. 5a), but not in PANC-1 cells (Fig. 5b) or HUVEC (Fig. 5c) alone. The structure of KB1023 is shown in Figure 4a.
Figure 5. Human pancreatic and lung tumor vascular microenvironment targeting ligands.



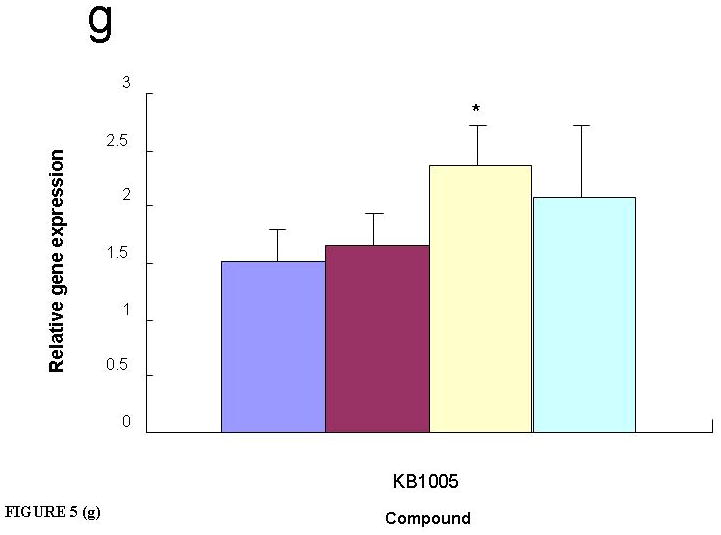
Compound KB1023 increased the transfection efficiency of the PANC-1+HUVEC co-culture (a), but not PANC-1 cells (b) or HUVECs (c). KB1061 enhanced the transfection efficiency in the co-culture of H1299 and HUVEC (d), but not in H1299 cells (e) or HUVECs (f). Compound KB1005 is required for targeted delivery to PANC-1 cells (g). Luciferase gene expression was compared to that of uncoated liposomal complexes. *P<0.05.
We also screened the libraries for small molecule hits to human endothelium in a H1299 non-small cell lung carcinoma microenvironment. We identified a different compound, KB1061, which increased transfection efficiency in H1299 co-cultures (Fig. 5d), but not H1299 cells (Fig. 5e) or HUVEC (Fig. 5f) alone. For targeted delivery to PANC-1 cells, compound KB1005 is required (Fig. 5g), and is therefore different than the compound required to target delivery to human pancreatic tumor microenvironment. The hits from the initial screen of these co-cultures were validated in transwell cultures in which the tumor and endothelial cells were transfected separately. These data were identical to those obtained from the differential screening results of the co-cultures versus HUVEC alone, and showed that KB1023 or KB1061 only increased the transfection efficiency of the endothelial cell compartment grown on the bottom chambers of the transwell cultures containing PANC-1 or H1299 tumor cells in the upper chambers, respectively. Ideally, we had planned to identify one ligand that could universally target delivery to endothelial cells in the human tumor microenvironment. However, consistent with the known heterogeneity of the tumor microenvironment associated with different cancers, our data show that multiple ligands are required to achieve enhanced delivery to the different human tumor microenvironment phenotypes. Several markers that are specifically expressed on the surface of endothelial cells undergoing angiogenic responses have been identified and used for targeted delivery24, 41–48 of phage particles, drugs, therapeutic antibodies, and other reagents. Interestingly, gene expression pattern analyses24, 42 and subtractive proteomic mapping48 have shown many differences and some similarities in the markers found on the surface of tumor vasculature endothelial cells from different tumor types. In tumor microenvironments, endothelial cells interact with tumor cells, immune cells, pericytes, fibroblasts, pericytes and the extracellular matrix (ECM). Tumor cells can alter the gene expression and phenotype of endothelial cells directly via a paracrine mechanism or indirectly, such as by altering the ECM (Reviewed In: 49).
Reversible Masking and targeted delivery to the asialoglycoprotein receptor
Reversible masking initially provides temporary shielding of the positive charge of BIV complexes during delivery in order to bypass non-target organs and then provides re-exposed charge at the target cell surface to allow fusogenic entry. Therefore, the mask that provides shielding of charge dissociates from the BIV complexes and is, therefore, reversible. Cells are negatively charged on the surface, and specific cell types vary in their density of negative charge. These differences in charge density can influence the ability of cells to be transfected. In fact certain viruses also have a partial positive charge around key subunits of viral surface proteins responsible for binding to and internalization through target cell surface receptors.50–55 We believe that maintenance of adequate positive charge on the surface of complexes is essential to drive cell entry.
Overall charge of complexes was measured on a zeta potential analyzer (Delsa 440SX, Beckman-Coulter). BIV complexes 45.5 mV in surface charge transfect cells at the highest levels (Fig. 6a,b). Whereas, BIV complexes coated with the reversible masking agent that are 4.8 mV in charge do not significantly transfect cells, tissues or organs (Templeton, N.S. US Patent No. 7,037,520 B2 issued May 2, 2006). Therefore, the overall charge of complexes must be shielded briefly post-injection and then re-exposed when transfecting the target cell. For these studies we used a large reversible mask, RM #1, a pendant-modified PEG NHS ester having 5 pendant carboxyl groups added to a PEG polymer that is 5000 MW.
Figure 6. Targeted delivery to the asialoglycoprotein receptor using RM #1.
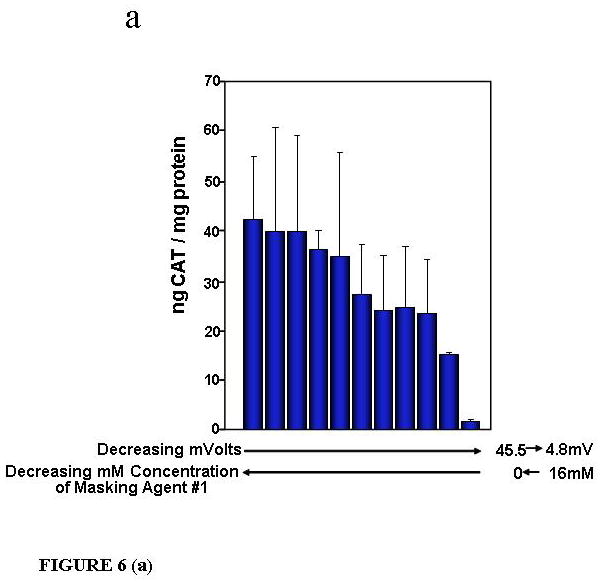
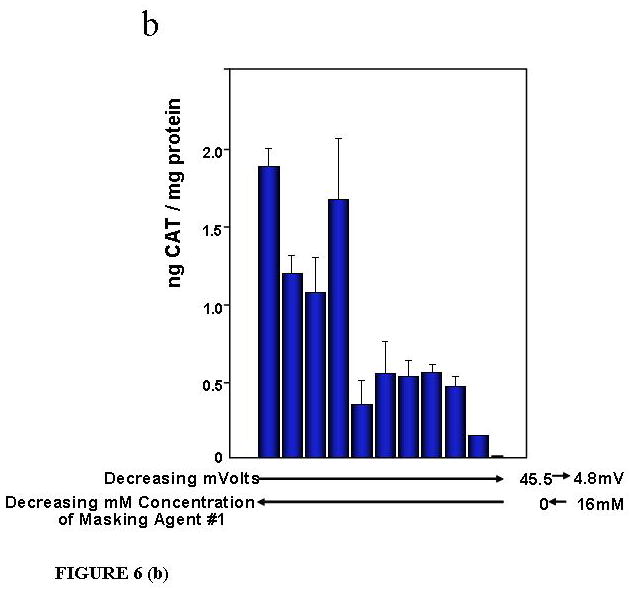
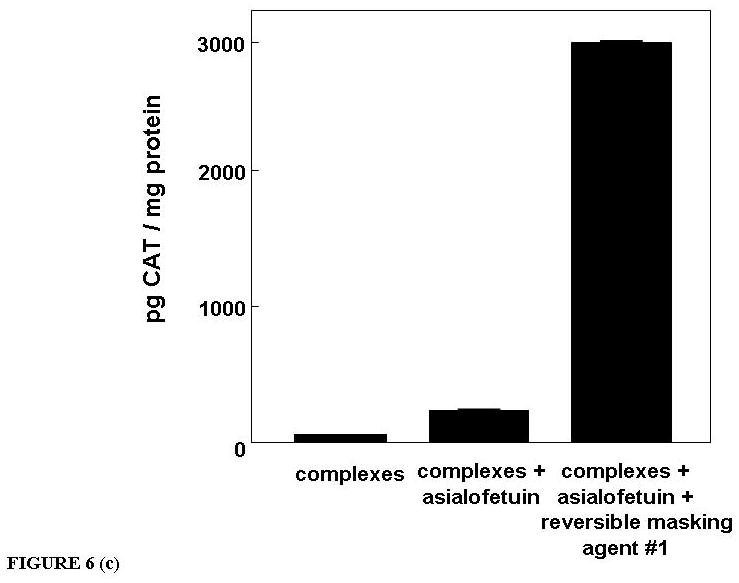
CAT production was measured 24 h post-IV injections of BIV-CAT complexes with different mM concentrations of RM #1 ranging between 0 mM to 16 mM (a,b). Each mouse was IV injected with 50 ug of p4119 CAT DNA encapsulated in BIVs, and 5 Balb/c mice were injected per group. Increasing concentrations of RM #1 decreased the overall charge of complexes to 4.8 mV that nearly eliminated CAT production in the lungs (a) and heart (b). CAT production was also measured in the liver for Balb/c mice injected with BIV-CAT complexes, complexes + succinylated asialofetuin, or complexes + succinylated asialofetuin + 16 mM RM #1 (c). Each mouse was IV injected with 50 ug of p4119 CAT DNA encapsulated in BIVs, and 10 Balb/c mice were injected per group. The presence of succinylated asialofetuin increased expression approximately 7-fold from 40 to 275 pg CAT/mg protein. Adding RM #1 just prior to IV injections increased expression approximately 76-fold from 40 to 3025 pg CAT/mg protein.
Targeted delivery for treatment of cancer is further complicated by the lack of known cell surface receptors to use for efficient targeting. Even the well-studied HER2/neu receptor is slightly elevated only on a small percent (about 10%) of breast cancer cells and does not internalize ligands, including antibodies and antibody fragments, as efficiently as other known ligand-receptor interactions. For example, the affinity and internalization of the asialoglycoprotein for the asialoglycoprotein receptor is the most robust and efficient ligand-receptor interaction identified to date and, therefore, is often used for proof-of-principle studies, including those presented here (Templeton, N.S. US Patent No. 7,037,520 B2 issued May 2, 2006).15 However, this system falls short of perfection because the asialoglycoprotein receptor is found both on the surface of liver hepatocytes and Kupffer cells, thereby allowing some delivery to be cleared rapidly from the circulation. In addition, as discussed above, the asialoglycoprotein is too large. Optimal ligands should be small (about 500 MW or less, e.g. drugs and small molecules), should have high affinity and internalization into unique receptors found exclusively on the target cells, and should be non-immunogenic/non-toxic particularly when multimerized on the surface of delivery vehicles including liposomes.
However, we performed proof-of-principle studies targeting the asialoglycoprotein receptor with BIV-CAT DNA complexes coated with or without succinylated asialofetuin and with or without reversible masking, RM #1 (Fig. 6c). Succinylated asialofetuin is also a large ligand and required the use of a large reversible mask. CAT production was measured in the liver for Balb/c mice injected with BIV-CAT complexes, complexes + succinylated asialofetuin, or complexes + succinylated asialofetuin + 16 mM RM #1 (Fig. 6c). Each mouse was IV injected with 50 ug of p4119 CAT DNA encapsulated in BIVs, and 10 Balb/c mice were injected per group. Tissues were harvested 24 h post-IV injection. The presence of succinylated asialofetuin increased expression approximately 7-fold from 40 to 275 pg CAT/mg protein. Adding RM #1 just prior to IV injections increased expression approximately 76-fold from 40 to 3025 pg CAT/mg protein.
As discussed above, we believe that use of small molecules for targeted delivery is ideal and smaller ligands should be able to use smaller reversible masks. Therefore we accomplished decreasing the overall charge of BIV complexes by adding increasing amounts of the small neutral lipid, n-dodecyl-β-D-maltopyranoside (RM #2), approximately 511 MW (Fig. 7; Templeton, N.S. US Patent No. 7,037,520 B2 issued May 2, 2006). CAT production was measured in human MCF7 breast cancer cells 24 h post-transfection with BIV-CAT complexes with different mM concentrations of RM #2 ranging between 0 mM to 12 mM. Concentrations of RM #2 from 8 mM to 12 mM eliminated CAT production.
Figure 7. Measuring the effect of RM #2 in vitro.
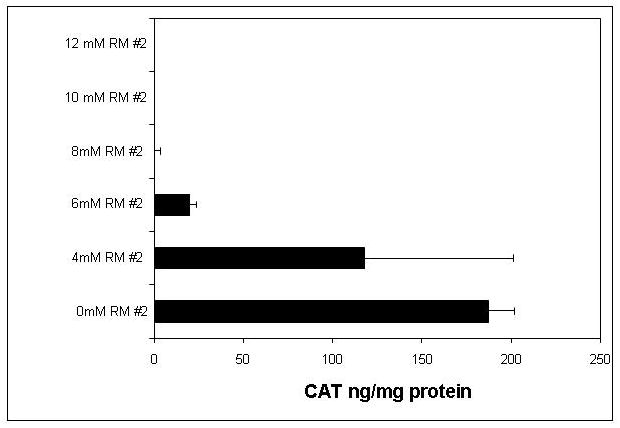
CAT production was measured in human MCF7 breast cancer cells 24 h post-transfection with BIV-CAT complexes with different mM concentrations of RM #2 ranging between 0 mM to 12 mM. Concentrations of RM #2 from 8 mM to 12 mM eliminated CAT production.
Targeting human tumor microenvironment in vivo
Previous studies have demonstrated that human endothelial cells can incorporate into functional vasculature when grafted into immune-deficient mice.56 Thus, we implanted co-cultures of PANC-1 cells with HUVEC to generate tumors that contain a human vascular component. We confirmed the targeting of KB1023 in vivo and optimized delivery using reversible masking to bypass non-specific uptake post-intravenous (IV) injection. At 9 days in co-culture, the PANC-1 co-cultures were injected intraperitoneally (IP) into SCID mice to establish a human pancreatic tumor microenvironment+PANC-1 tumor model. We assessed targeted delivery 8 weeks post-IP injections when pancreatic tumors were about 400 mm3. When KB1023 coated BIV-CAT DNA complexes were IV injected into our PANC-1 co-culture model in SCID mice, the vast majority was delivered to the lungs and hearts non-specifically (Fig. 8a). Only a small portion was delivered to the tumor tissue (Fig. 8b). This result is consistent with other reports that showed the majority of the DNA:liposome complexes delivered to the lung post-IV injections.15, 57 Therefore, we created a novel “reversible masking” approach that is far more efficient than PEGylation for minimizing non-specific delivery while maintaining far higher levels of target cell transfection. We avoid uptake in the lungs and other non-specific target organs using “shielding/deshielding compounds” that can be added to the complexes used for targeted delivery just prior to injection or administration in vivo (Templeton, N.S. US Patent No. 7,037,520 B2 issued May 2, 2006). Our strategy uses neutral, small molecular weight lipids (about 500 MW and lower), e.g. n-dodecyl-β-D-maltopyranoside (RM #2). Because these lipids are small and not charged, they are loosely associated with the surface of BIV complexes and are removed in the bloodstream by the time they reach the target cell.
Figure 8. In vivo targeting and optimization using RM #2.
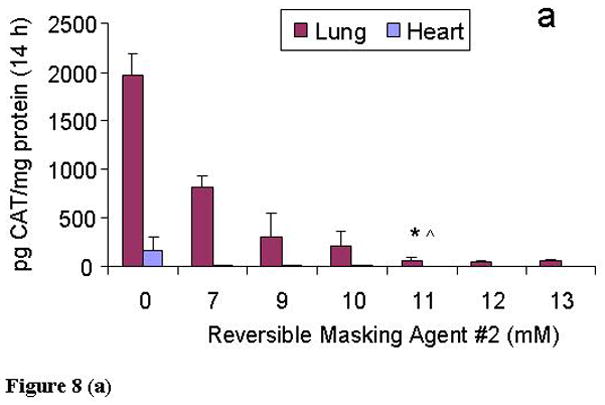
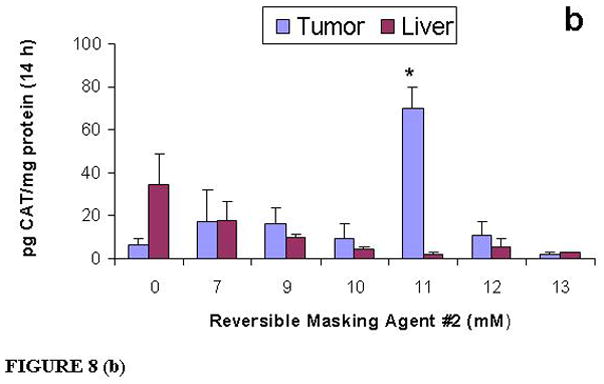
At 14 h post-IV injections, the majority of KB1023 coated liposomal complexes was transfected non-specifically in the lungs and hearts (a). When injecting using reversible masking (RM #2), and with increasing RM agent concentration, non-specific uptake by lungs and hearts decreased significantly. At 11 mM RM #2, the lungs and hearts showed little to no non-specific uptake (a), while delivery and subsequent gene expression in the tumor tissue increased about 10-fold (c). No increased delivery was found in other non-specific tissues, such as liver (b). To dissociate the human tumor vasculature from the tumor tissue in order to do the CAT assays and protein assays was prohibitive; therefore, the 10-fold increased delivery to tumor vascular microenvironment is a low estimate. Because the tumor endothelium is approximately 5% of the entire tumor volume, the increased targeted delivery to the tumor vascular microenvironment is most likely about 200-fold greater than delivery using uncoated BIV complexes alone. Further increasing RM #2 beyond 11 mM did not increase the delivery to tumor tissue and instead diminished delivery and subsequent gene expression. Therefore, for targeted delivery to the tumor microenvironment, using 11 mM RM #2 is optimal. #CAT expression was measured 14 h post-IV injection and compared to the control (0 mM) using targeted delivery without RM. *P<0.01. ^P<0.05. N=4~5 per group.
The reversible mask can be optimized for delivery to a given target organ while bypassing delivery to non-target organs and tissues. Figure 8b shows that 11 mM RM #2 in a 110 μL injection volume was required to bypass delivery of BIV-CAT DNA liposome complexes to lungs and heart post-IV injection (expression reduced by greater than 97%). Correspondingly, delivery of KB1023 coated BIV-CAT DNA complexes + 11 mM RM #2 showed approximately 10-fold increased delivery to tumor tissue (Fig. 8b) that included the human tumor microenvironment of the PANC-1 co-culture model in SCID mice compared to delivery of uncoated BIV complexes alone (0 mM). To dissociate the human tumor vasculature from the tumor tissue in order to perform the CAT assays and protein assays was prohibitive; therefore, the 10-fold increased delivery to the human tumor microenvironment is a low estimate. Several other investigators also measure only the increased delivery into the tumor due to low amounts of endothelial cells that cannot be dissociated to produce enough cell mass for measurements of gene expression and total protein.58 Because the tumor endothelium is approximately 5% of the entire tumor volume, the increased targeted delivery to the tumor vasculature is most likely about 200-fold greater than delivery using uncoated BIV complexes alone. Therefore, if the endothelial cells within the tumor could be measured for CAT production and normalized to protein, there would be approximately 1400 pg CAT/mg protein. This level is in the range of CAT production produced 14 h post-IV injections of uncoated BIV-CAT DNA complexes in the lungs (Fig 8a). Our in vivo results, combined with our in vitro data further suggest that KB1023 targeted the tumor microenvironment cells and not the cancer cells of the PANC-1 co-culture in SCID mice; however, we cannot exclude the possibility that a target expressed by both cells mediates the observed targeting of the coated BIV complexes. Although our in vitro assessment suggested that use of 8 mM RM #2 might be sufficient to bypass non-specific uptake in vivo, in fact 11 mM RM #2 was required. However, this in vitro prediction was more useful than the in vivo assessment using a different reversible masking agent, RM #1, requiring the use of 16 mM in vivo. Therefore, when testing new reversible masks, in vitro studies can be useful for narrowing the range of RM to test in vivo.
The increased CAT expression obtained with reversible masking was also specific for tumor versus liver. Figure 8b shows that KB1023 coated BIV-CAT DNA complexes + reversible mask (RM #2) did not increase the CAT expression in liver, and at 11 mM RM #2 expression in the liver was negligible. Therefore, targeting was specific and did not increase uptake and clearance of the complexes by the Kupffer cells in the liver. Further increasing the amount of RM #2 above 11 mM did not result in further increase of CAT expression in the tumors. Instead, the expression decreased showing that 11 mM RM #2 was the optimal concentration to use in the PANC-1 co-culture model in SCID mice.
Tumor growth inhibition
After optimizing in vivo targeting using the CAT reporter gene, we tested the efficacy of our targeted delivery in tumor growth prevention using anti-angiogenic TSP1 as the therapeutic gene. Figure 9 shows the in vivo efficacy data generated after IV injections of BIV-TSP1 DNA complexes with or without the small molecule (ligand), KB1023, and with or without reversible masking (RM #2) into our human tumor microenvironment+PANC-1 tumor bearing mice. As shown in Figure 9, mice treated with KB1023 coated BIV-TSP1 DNA complexes demonstrated significant suppression of pancreatic cancer growth. Tumor growth was inhibited by 87.99% compared to BIV liposome injection controls (average tumor volume was 47.27mm3 versus 393.54 mm3). When targeted delivery was combined with reversible masking, tumor growth was inhibited by 98.67% with the average tumor volume of 5.25mm3 compared to the liposome injected controls at 393.54 mm3. All of these results were produced after a total of three IV injections administered about once every two weeks. Furthermore, when we increased the overall TSP1 therapeutic dosage by increasing the total number of injections to a total of five that were administered once per week, tumor growth was further suppressed. The tumors were completely eliminated for five of seven mice, and the average tumor volume was 0.7mm3 for the other two mice. Mice treated with untargeted delivery of TSP1 demonstrated tumor growth retardation by 62.69% with the mean volume of 146.82mm3. Interestingly, we exceeded the efficacy data reported for TSP1 anti-angiogenic gene therapy approaches using viral vectors, adenovirus 32 or adeno-associated virus 12, cationic polymers 31, or bacterial vectors, Salmonella choleraesuis 30. Some viral 59–63 and non-viral vectors 64, 65 have been used that demonstrate good delivery to tumor endothelium; however, these delivery vehicles have not been used to deliver the TSP1 gene. Presumably, our increased efficacy using TSP1 gene therapy is attributed to high levels of specific delivery and gene expression exclusively in the human tumor microenvironment.
Figure 9. Tumor growth inhibition using targeted delivery of the TSP1 gene.
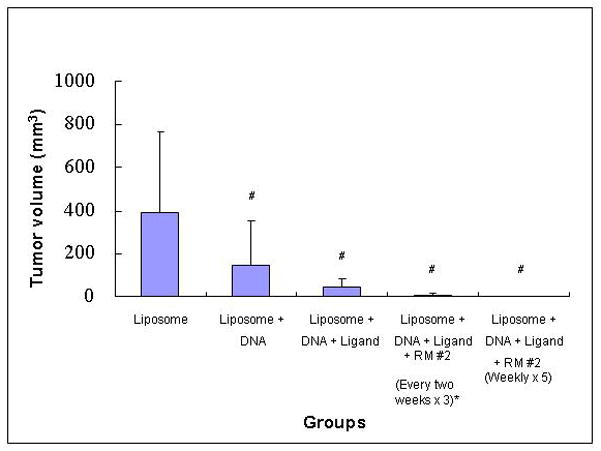
At 2 weeks post-IP injection of the co-cultures, BIV liposomal complexes that encapsulated 35 μg TSP1 DNA were coated with the ligand KB1023 and co-injected with 11 mM reversible masking using RM #2 and IV injected into each mouse. The injections were performed every two weeks for a total of three IV injections. At 2 weeks after the final injection (8 weeks post-IP injections of co-cultures), mice were sacrificed to compare intra-abdominal tumor size measured with calipers. Mice treated with human tumor microenvironment targeted delivery of TSP1 demonstrated significant cancer growth delay compared to control mice with only liposomes injected. When targeted delivery was combined with optimal reversible masking using RM #2, tumor growth was suppressed to a greater extent nearly eradicating the tumors. Tumor growth was further suppressed when the treatment was enhanced to weekly injections for a total of five IV injections. Five out of 7 mice had no detectable tumors. *N=20. Other groups have 5~7 mice per group. #P<0.05.
DISCUSSION
Tumor vasculature is morphologically abnormal, and the vascular endothelial cells differ from normal endothelial cells at molecular and functional levels.66 Cross-talk between tumor endothelium and tumor cells creates a microenvironment optimal for tumor growth. Establishing robust and appropriate animal models to understand the biology of the tumor microenvironment and to identify agents that target this environment by high-throughput screening is essential for developing the most effective anti-cancer therapies. Isolation of tumor endothelial cells from tumor tissue using magnetic beads is a powerful approach to discover tumor endothelial markers24, however the cost and low yield of this approach precludes routine study of this tumor endothelium. Another concern is that the tumor endothelial phenotype is lost soon after isolation from the tumor microenvironment.66 To maintain this important crosstalk between tumor and endothelial cells, we created an in vitro human tumor microenvironment model by co-culturing lung or pancreatic cancer cells with HUVECs. Under optimal conditions, we confirmed the stable expression of tumor endothelial phenotypes. In particular, the up-regulation of VEGF-A was meaningful because investigators have shown that tumor angiogenesis is driven primarily by VEGF-A, and targeting the VEGF-A pathway has been effective in cancer treatment.67 Our model offers a robust platform for the discovery of novel anti-angiogenic compounds such as VEGF-A blockers. The model also permits the study of VEGF-A withdrawal and normalization of vasculature because the stimuli from cancer cells can be conveniently removed from the system. In summary, our approach is simple and feasible, and incorporates the dynamic communication between tumor and endothelial cells. Therefore, it can serve as a good model for the study of the tumor microenvironment. The fact that we used this model to successfully identify several tumor microenvironment-targeting ligands further supports this concept. In vitro, we demonstrated that these ligands selectively transfect endothelial cells exposed to soluble factors produced by tumor cells. Thus, they may increase vector delivery by targeting tumor endothelium in vivo, but we can not exclude the possibility that they also inhibit tumor growth by delivering the TSP1 plasmid to human tumor cells or other stromal cells derived from the murine host. However, we believe that our human tumor microenvironment model provides an advance beyond the typically used model in which delivery of therapeutic agents is tested on human tumor xenografts that incorporate mouse vascular endothelium. These models can be misleading when translating these therapies into human tumors where the targeting ligands must recognize human tumor vasculature.
In this work, we greatly improved the specific delivery of BIV liposomes by introducing small ligands that target delivery to the tumor vascular microenvironment and using reversible masking that provides for bypass of non-specific organs and tissues. Targeted delivery might be possible using small peptides that are multimerized on the surface of liposomes, but these can generate immune responses after repeated injections, particularly systemically, and peptides can preclude penetration and delivery across the interstitial pressure gradient of tumors. Other larger ligands including antibodies, antibody fragments, proteins, partial proteins, etc. are far more refractory than using small peptides for targeted delivery on the surface of liposomes. Our optimized targeted delivery was highly efficient in cancer growth prevention in mouse xenograft models, and the fact that our targeting ligands are small molecules should allow for repeated injections indefinitely without generating immune responses. Additionally, our delivery systems including the ligand (<500 MW) and reversible masking reagent are non-immunogenic and non-toxic, and safe for clinical usage. Moreover, our IV administration once every two weeks or once per week is convenient and could be widely used in medical practice. Therefore, our targeted, reversibly masked delivery system has great potential for effective anti-cancer therapy.
Although we have initiated work to identify the receptor to which KB1023 binds, we have not yet identified this receptor. One possibility is a cell surface receptor that is exclusively expressed on some pancreatic tumor vascular cells or other cells in the tumor microenviroment. Studies have discovered tumor endothelium marker (TEM) that is uniquely expressed on specific types of tumors as well as several pan-TEMs.24, 42, 68 Secondly, the potential receptor might be a molecule that is expressed at relatively low levels on normal endothelial cells and up-regulated on some pancreatic tumor endothelial cells or other cells in the tumor microenviroment. The search for better cell surface receptors to use for targeted delivery is critical and achievable using our approach reported here. Significantly, knowing the function and identity of the best receptors is not required for this targeting strategy. A method developed in the Burgess lab allows production of small molecules designed to bind proteins selectively. Importantly, the bivalent small molecules have both selectivity for binding cell surface receptors, and will resemble secondary structure motifs found at hot-spots in protein-ligand interactions. Bivalent beta-turn mimics were designed that have an affinity for cell surface receptors. Although we did not identify the ligand’s receptor to date, we can still use our targeted delivery system in the clinic for anti-angiogenic cancer therapy. In fact, many drugs have been approved by FDA before fully understanding their mechanism. We have reported an extremely effective anti-cancer therapeutic approach. Furthermore, our targeted, reversibly masked BIV delivery system using small molecules that target delivery to other diseased target cells could also be applied to the treatment of diseases and disorders other than cancer and metastases.
Acknowledgments
We thank GeneExcel, Inc. (now part of Gradalis, Inc.), the National Institutes of Health (MH070040, GM076261), the Intramural Research Program of the NIH, NCI, Center for Cancer Research, and the Robert A. Welch Foundation for the financial support of this project.
References
- 1.Folkman J, Kalluri R. Cancer without disease. Nature. 2004 Feb 26;427(6977):787. doi: 10.1038/427787a. [DOI] [PubMed] [Google Scholar]
- 2.Tandle A, Blazer DG, 3rd, Libutti SK. Antiangiogenic gene therapy of cancer: recent developments. J Transl Med. 2004 Jun 25;2(1):22. doi: 10.1186/1479-5876-2-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ferrara N, Hillan KJ, Gerber HP, Novotny W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov. 2004 May;3(5):391–400. doi: 10.1038/nrd1381. [DOI] [PubMed] [Google Scholar]
- 4.Faivre S, Demetri G, Sargent W, Raymond E. Molecular basis for sunitinib efficacy and future clinical development. Nat Rev Drug Discov. 2007 Sep;6(9):734–45. doi: 10.1038/nrd2380. [DOI] [PubMed] [Google Scholar]
- 5.Eskens FA, Verweij J. The clinical toxicity profile of vascular endothelial growth factor (VEGF) and vascular endothelial growth factor receptor (VEGFR) targeting angiogenesis inhibitors; a review. Eur J Cancer. 2006 Dec;42(18):3127–39. doi: 10.1016/j.ejca.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 6.Verheul HM, Pinedo HM. Possible molecular mechanisms involved in the toxicity of angiogenesis inhibition. Nat Rev Cancer. 2007 Jun;7(6):475–85. doi: 10.1038/nrc2152. [DOI] [PubMed] [Google Scholar]
- 7.Kerbel RS. Tumor angiogenesis. N Engl J Med. 2008 May 8;358(19):2039–49. doi: 10.1056/NEJMra0706596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Siemann DW, Chaplin DJ, Horsman MR. Vascular-targeting therapies for treatment of malignant disease. Cancer. 2004 Jun 15;100(12):2491–9. doi: 10.1002/cncr.20299. [DOI] [PubMed] [Google Scholar]
- 9.Nolan DJ, Ciarrocchi A, Mellick AS, Jaggi JS, Bambino K, Gupta S, et al. Bone marrow-derived endothelial progenitor cells are a major determinant of nascent tumor neovascularization. Genes Dev. 2007 Jun 15;21(12):1546–58. doi: 10.1101/gad.436307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Templeton NS, editor. Gene and Cell Therapy: Therapeutic Mechanisms and Strategies. 3. Boca Raton, FL: CRC Press, Taylor & Francis Group; 2008. [Google Scholar]
- 11.Ramesh R, Saeki T, Templeton NS, Ji L, Stephens LC, Ito I, et al. Successful treatment of primary and disseminated human lung cancers by systemic delivery of tumor suppressor genes using an improved liposome vector. Mol Ther. 2001 Mar;3(3):337–50. doi: 10.1006/mthe.2001.0266. [DOI] [PubMed] [Google Scholar]
- 12.Zhang X, Xu J, Lawler J, Terwilliger E, Parangi S. Adeno-associated virus-mediated antiangiogenic gene therapy with thrombospondin-1 type 1 repeats and endostatin. Clin Cancer Res. 2007 Jul 1;13(13):3968–76. doi: 10.1158/1078-0432.CCR-07-0245. [DOI] [PubMed] [Google Scholar]
- 13.Xu M, Kumar D, Stass SA, Mixson AJ. Gene therapy with p53 and a fragment of thrombospondin I inhibits human breast cancer in vivo. Mol Genet Metab. 1998 Feb;63(2):103–9. doi: 10.1006/mgme.1997.2654. [DOI] [PubMed] [Google Scholar]
- 14.Liu S, Ballian N, Belaguli NS, Patel S, Li M, Templeton NS, et al. PDX-1 acts as a potential molecular target for treatment of human pancreatic cancer. Pancreas. 2008 Aug;37(2):210–20. doi: 10.1097/MPA.0b013e31816a4a33. [DOI] [PubMed] [Google Scholar]
- 15.Templeton NS, Lasic DD, Frederik PM, Strey HH, Roberts DD, Pavlakis GN. Improved DNA: liposome complexes for increased systemic delivery and gene expression. Nat Biotechnol. 1997 Jul;15(7):647–52. doi: 10.1038/nbt0797-647. [DOI] [PubMed] [Google Scholar]
- 16.Tirone TA, Fagan SP, Templeton NS, Wang X, Brunicardi FC. Insulinoma-induced hypoglycemic death in mice is prevented with beta cell-specific gene therapy. Ann Surg. 2001 May;233(5):603–11. doi: 10.1097/00000658-200105000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shi HY, Liang R, Templeton NS, Zhang M. Inhibition of breast tumor progression by systemic delivery of the maspin gene in a syngeneic tumor model. Mol Ther. 2002 Jun;5(6):755–61. doi: 10.1006/mthe.2002.0602. [DOI] [PubMed] [Google Scholar]
- 18.Yotnda P, Davis AR, Hicks MJ, Templeton NS, Brenner MK. Liposomal enhancement of the antitumor activity of conditionally replication-competent adenoviral plasmids. Mol Ther. 2004 Apr;9(4):489–95. doi: 10.1016/j.ymthe.2004.01.018. [DOI] [PubMed] [Google Scholar]
- 19.Park C, Burgess K. Facile macrocyclizations to beta-turn mimics with diverse structural, physical, and conformational properties. J Comb Chem. 2001 May-Jun;3(3):257–66. doi: 10.1021/cc010003n. [DOI] [PubMed] [Google Scholar]
- 20.Reyes S, Pattarawarapan M, Roy S, Burgess K. Preferred secondary structures as a possible driving force for macrocyclization. Tetrahedron. 2000;56:9809–18. [Google Scholar]
- 21.Burgess K. Solid-phase syntheses of beta-turn analogues to mimic or disrupt protein-protein interactions. Acc Chem Res. 2001 Oct;34(10):826–35. doi: 10.1021/ar9901523. [DOI] [PubMed] [Google Scholar]
- 22.Bruno MA, Clarke PB, Seltzer A, Quirion R, Burgess K, Cuello AC, et al. Long-lasting rescue of age-associated deficits in cognition and the CNS cholinergic phenotype by a partial agonist peptidomimetic ligand of TrkA. J Neurosci. 2004 Sep 15;24(37):8009–18. doi: 10.1523/JNEUROSCI.1508-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schroeder U, Graff A, Buchmeier S, Rigler P, Silvan U, Tropel D, et al. Peptide nanoparticles serve as a powerful platform for the immunogenic display of poorly antigenic actin determinants. J Mol Biol. 2009 Mar 13;386(5):1368–81. doi: 10.1016/j.jmb.2008.11.023. [DOI] [PubMed] [Google Scholar]
- 24.St Croix B, Rago C, Velculescu V, Traverso G, Romans KE, Montgomery E, et al. Genes expressed in human tumor endothelium. Science. 2000 Aug 18;289(5482):1197–202. doi: 10.1126/science.289.5482.1197. [DOI] [PubMed] [Google Scholar]
- 25.Folberg R, Maniotis AJ. Vasculogenic mimicry. APMIS. 2004 Jul-Aug;112(7–8):508–25. doi: 10.1111/j.1600-0463.2004.apm11207-0810.x. [DOI] [PubMed] [Google Scholar]
- 26.Hendrix MJ, Seftor EA, Kirschmann DA, Quaranta V, Seftor RE. Remodeling of the microenvironment by aggressive melanoma tumor cells. Ann N Y Acad Sci. 2003 May;995:151–61. doi: 10.1111/j.1749-6632.2003.tb03218.x. [DOI] [PubMed] [Google Scholar]
- 27.Weinstat-Saslow DL, Zabrenetzky VS, VanHoutte K, Frazier WA, Roberts DD, Steeg PS. Transfection of thrombospondin 1 complementary DNA into a human breast carcinoma cell line reduces primary tumor growth, metastatic potential, and angiogenesis. Cancer Res. 1994 Dec 15;54(24):6504–11. [PubMed] [Google Scholar]
- 28.Isenberg JS, Hyodo F, Ridnour LA, Shannon CS, Wink DA, Krishna MC, et al. Thrombospondin 1 and vasoactive agents indirectly alter tumor blood flow. Neoplasia. 2008 Aug;10(8):886–96. doi: 10.1593/neo.08264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Isenberg JS, Martin-Manso G, Maxhimer JB, Roberts DD. Regulation of nitric oxide signalling by thrombospondin 1: implications for anti-angiogenic therapies. Nat Rev Cancer. 2009 Mar;9(3):182–94. doi: 10.1038/nrc2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee CH, Wu CL, Shiau AL. Systemic administration of attenuated Salmonella choleraesuis carrying thrombospondin-1 gene leads to tumor-specific transgene expression, delayed tumor growth and prolonged survival in the murine melanoma model. Cancer Gene Ther. 2005 Feb;12(2):175–84. doi: 10.1038/sj.cgt.7700777. [DOI] [PubMed] [Google Scholar]
- 31.Xu M, Chen QR, Kumar D, Stass SA, Mixson AJ. In vivo gene therapy with a cationic polymer markedly enhances the antitumor activity of antiangiogenic genes. Mol Genet Metab. 1998 Jul;64(3):193–7. doi: 10.1006/mgme.1998.2705. [DOI] [PubMed] [Google Scholar]
- 32.Liu P, Wang Y, Li YH, Yang C, Zhou YL, Li B, et al. Adenovirus-mediated gene therapy with an antiangiogenic fragment of thrombospondin-1 inhibits human leukemia xenograft growth in nude mice. Leuk Res. 2003 Aug;27(8):701–8. doi: 10.1016/s0145-2126(02)00346-6. [DOI] [PubMed] [Google Scholar]
- 33.Ma X, Ottino P, Bazan HE, Bazan NG. Platelet-activating factor (PAF) induces corneal neovascularization and upregulates VEGF expression in endothelial cells. Invest Ophthalmol Vis Sci. 2004 Sep;45(9):2915–21. doi: 10.1167/iovs.04-0128. [DOI] [PubMed] [Google Scholar]
- 34.Allport JR, Weissleder R. Murine Lewis lung carcinoma-derived endothelium expresses markers of endothelial activation and requires tumor-specific extracellular matrix in vitro. Neoplasia. 2003 May-Jun;5(3):205–17. doi: 10.1016/S1476-5586(03)80053-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ria R, Vacca A, Russo F, Cirulli T, Massaia M, Tosi P, et al. A VEGF-dependent autocrine loop mediates proliferation and capillarogenesis in bone marrow endothelial cells of patients with multiple myeloma. Thromb Haemost. 2004 Dec;92(6):1438–45. doi: 10.1160/TH04-06-0334. [DOI] [PubMed] [Google Scholar]
- 36.Neufeld G, Cohen T, Gengrinovitch S, Poltorak Z. Vascular endothelial growth factor (VEGF) and its receptors. FASEB J. 1999 Jan;13(1):9–22. [PubMed] [Google Scholar]
- 37.Cai J, Ahmad S, Jiang WG, Huang J, Kontos CD, Boulton M, et al. Activation of vascular endothelial growth factor receptor-1 sustains angiogenesis and Bcl-2 expression via the phosphatidylinositol 3-kinase pathway in endothelial cells. Diabetes. 2003 Dec;52(12):2959–68. doi: 10.2337/diabetes.52.12.2959. [DOI] [PubMed] [Google Scholar]
- 38.Angell Y, Chen D, Brahimi F, Saragovi HU, Burgess K. A combinatorial method for solution-phase synthesis of labeled bivalent beta-turn mimics. J Am Chem Soc. 2008 Jan 16;130(2):556–65. doi: 10.1021/ja074717z. [DOI] [PubMed] [Google Scholar]
- 39.Maliartchouk S, Feng Y, Ivanisevic L, Debeir T, Cuello AC, Burgess K, et al. A designed peptidomimetic agonistic ligand of TrkA nerve growth factor receptors. Mol Pharmacol. 2000 Feb;57(2):385–91. [PubMed] [Google Scholar]
- 40.Yotnda P, Chen DH, Chiu W, Piedra PA, Davis A, Templeton NS, et al. Bilamellar cationic liposomes protect adenovectors from preexisting humoral immune responses. Mol Ther. 2002 Mar;5(3):233–41. doi: 10.1006/mthe.2002.0545. [DOI] [PubMed] [Google Scholar]
- 41.Nanda A, St Croix B. Tumor endothelial markers: new targets for cancer therapy. Curr Opin Oncol. 2004 Jan;16(1):44–9. doi: 10.1097/00001622-200401000-00009. [DOI] [PubMed] [Google Scholar]
- 42.Madden SL, Cook BP, Nacht M, Weber WD, Callahan MR, Jiang Y, et al. Vascular gene expression in nonneoplastic and malignant brain. Am J Pathol. 2004 Aug;165(2):601–8. doi: 10.1016/s0002-9440(10)63324-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carson-Walter EB, Watkins DN, Nanda A, Vogelstein B, Kinzler KW, St Croix B. Cell surface tumor endothelial markers are conserved in mice and humans. Cancer Res. 2001 Sep 15;61(18):6649–55. [PubMed] [Google Scholar]
- 44.Neri D, Bicknell R. Tumour vascular targeting. Nat Rev Cancer. 2005 Jun;5(6):436–46. doi: 10.1038/nrc1627. [DOI] [PubMed] [Google Scholar]
- 45.Oh P, Li Y, Yu J, Durr E, Krasinska KM, Carver LA, et al. Subtractive proteomic mapping of the endothelial surface in lung and solid tumours for tissue-specific therapy. Nature. 2004 Jun 10;429(6992):629–35. doi: 10.1038/nature02580. [DOI] [PubMed] [Google Scholar]
- 46.Thorpe PE. Vascular targeting agents as cancer therapeutics. Clin Cancer Res. 2004 Jan 15;10(2):415–27. doi: 10.1158/1078-0432.ccr-0642-03. [DOI] [PubMed] [Google Scholar]
- 47.Arap W, Pasqualini R, Ruoslahti E. Cancer treatment by targeted drug delivery to tumor vasculature in a mouse model. Science. 1998 Jan 16;279(5349):377–80. doi: 10.1126/science.279.5349.377. [DOI] [PubMed] [Google Scholar]
- 48.Pasqualini R, Koivunen E, Kain R, Lahdenranta J, Sakamoto M, Stryhn A, et al. Aminopeptidase N is a receptor for tumor-homing peptides and a target for inhibiting angiogenesis. Cancer Res. 2000 Feb 1;60(3):722–7. [PMC free article] [PubMed] [Google Scholar]
- 49.Jung YD, Ahmad SA, Liu W, Reinmuth N, Parikh A, Stoeltzing O, et al. The role of the microenvironment and intercellular cross-talk in tumor angiogenesis. Semin Cancer Biol. 2002 Apr;12(2):105–12. doi: 10.1006/scbi.2001.0418. [DOI] [PubMed] [Google Scholar]
- 50.Repits J, Sterjovski J, Badia-Martinez D, Mild M, Gray L, Churchill MJ, et al. Primary HIV-1 R5 isolates from end-stage disease display enhanced viral fitness in parallel with increased gp120 net charge. Virology. 2008 Sep 15;379(1):125–34. doi: 10.1016/j.virol.2008.06.014. [DOI] [PubMed] [Google Scholar]
- 51.Cilliers T, Nhlapo J, Coetzer M, Orlovic D, Ketas T, Olson WC, et al. The CCR5 and CXCR4 coreceptors are both used by human immunodeficiency virus type 1 primary isolates from subtype C. J Virol. 2003 Apr;77(7):4449–56. doi: 10.1128/JVI.77.7.4449-4456.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee E, Hall RA, Lobigs M. Common E protein determinants for attenuation of glycosaminoglycan-binding variants of Japanese encephalitis and West Nile viruses. J Virol. 2004 Aug;78(15):8271–80. doi: 10.1128/JVI.78.15.8271-8280.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Markoff L, Falgout B, Chang A. A conserved internal hydrophobic domain mediates the stable membrane integration of the dengue virus capsid protein. Virology. 1997 Jun 23;233(1):105–17. doi: 10.1006/viro.1997.8608. [DOI] [PubMed] [Google Scholar]
- 54.Reeves JD, Schulz TF. The CD4-independent tropism of human immunodeficiency virus type 2 involves several regions of the envelope protein and correlates with a reduced activation threshold for envelope-mediated fusion. J Virol. 1997 Feb;71(2):1453–65. doi: 10.1128/jvi.71.2.1453-1465.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Andeweg AC, Boers PH, Osterhaus AD, Bosch ML. Impact of natural sequence variation in the V2 region of the envelope protein of human immunodeficiency virus type 1 on syncytium induction: a mutational analysis. J Gen Virol. 1995 Aug;76( Pt 8):1901–7. doi: 10.1099/0022-1317-76-8-1901. [DOI] [PubMed] [Google Scholar]
- 56.Alajati A, Laib AM, Weber H, Boos AM, Bartol A, Ikenberg K, et al. Spheroid-based engineering of a human vasculature in mice. Nat Methods. 2008 May;5(5):439–45. doi: 10.1038/nmeth.1198. [DOI] [PubMed] [Google Scholar]
- 57.Thurston G, McLean JW, Rizen M, Baluk P, Haskell A, Murphy TJ, et al. Cationic liposomes target angiogenic endothelial cells in tumors and chronic inflammation in mice. J Clin Invest. 1998 Apr 1;101(7):1401–13. doi: 10.1172/JCI965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hood JD, Bednarski M, Frausto R, Guccione S, Reisfeld RA, Xiang R, et al. Tumor regression by targeted gene delivery to the neovasculature. Science. 2002 Jun 28;296(5577):2404–7. doi: 10.1126/science.1070200. [DOI] [PubMed] [Google Scholar]
- 59.Tseng JC, Granot T, Digiacomo V, Levin B, Meruelo D. Enhanced specific delivery and targeting of oncolytic Sindbis viral vectors by modulating vascular leakiness in tumor. Cancer Gene Ther. 2009 Oct 2; doi: 10.1038/cgt.2009.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hofmann A, Wenzel D, Becher UM, Freitag DF, Klein AM, Eberbeck D, et al. Combined targeting of lentiviral vectors and positioning of transduced cells by magnetic nanoparticles. Proc Natl Acad Sci U S A. 2009 Jan 6;106(1):44–9. doi: 10.1073/pnas.0803746106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nicol CG, Denby L, Lopez-Franco O, Masson R, Halliday CA, Nicklin SA, et al. Use of in vivo phage display to engineer novel adenoviruses for targeted delivery to the cardiac vasculature. FEBS Lett. 2009 Jun 18;583(12):2100–7. doi: 10.1016/j.febslet.2009.05.037. [DOI] [PubMed] [Google Scholar]
- 62.Popkov M, Jendreyko N, McGavern DB, Rader C, Barbas CF., 3rd Targeting tumor angiogenesis with adenovirus-delivered anti-Tie-2 intrabody. Cancer Res. 2005 Feb 1;65(3):972–81. [PubMed] [Google Scholar]
- 63.Hesse A, Kosmides D, Kontermann RE, Nettelbeck DM. Tropism modification of adenovirus vectors by peptide ligand insertion into various positions of the adenovirus serotype 41 short-fiber knob domain. J Virol. 2007 Mar;81(6):2688–99. doi: 10.1128/JVI.02722-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tan PH, Manunta M, Ardjomand N, Xue SA, Larkin DF, Haskard DO, et al. Antibody targeted gene transfer to endothelium. J Gene Med. 2003 Apr;5(4):311–23. doi: 10.1002/jgm.358. [DOI] [PubMed] [Google Scholar]
- 65.Driessen WH, Fujii N, Tamamura H, Sullivan SM. Development of peptide-targeted lipoplexes to CXCR4-expressing rat glioma cells and rat proliferating endothelial cells. Mol Ther. 2008 Mar;16(3):516–24. doi: 10.1038/sj.mt.6300388. [DOI] [PubMed] [Google Scholar]
- 66.Hida K, Hida Y, Shindoh M. Understanding tumor endothelial cell abnormalities to develop ideal anti-angiogenic therapies. Cancer Sci. 2008 Mar;99(3):459–66. doi: 10.1111/j.1349-7006.2007.00704.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004 Jun 3;350(23):2335–42. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- 68.Hida K, Hida Y, Amin DN, Flint AF, Panigrahy D, Morton CC, et al. Tumor-associated endothelial cells with cytogenetic abnormalities. Cancer Res. 2004 Nov 15;64(22):8249–55. doi: 10.1158/0008-5472.CAN-04-1567. [DOI] [PubMed] [Google Scholar]