

Abstract
The paramyxovirus Simian Virus 5 (SV5) is a poor inducer of interferon (IFN) secretion in all cell types tested so far, including primary epithelial cells and primary human myeloid dendritic cells. SV5 is hypothesized to limit induction of antiviral responses through control of viral gene expression and production of the V protein antagonist. Plasmacytoid dendritic cells (pDCs) are known to uniquely express toll-like receptor (TLR)-7 and are a main producer of IFN-alpha among peripheral blood mononuclear cells in response to many viruses. Here, we tested whether SV5 would remain a poor inducer of IFN in primary human pDCs. The efficiency of SV5 infection of pDCs could be increased by an increasing multiplicity of infection. pDCs infected by both live and UV-inactivated SV5 induced large amounts of IFN-alpha secretion and resulted in upregulation of maturation markers CD80 and CD86. However, IL-6 secretion was not induced by SV5 infection. When TLR7 signaling was inhibited, SV5 induced less IFN secretion and CD80 expression, and there was a corresponding increase in number of infected cells. Similar effects were seen with inhibitors of cellular autophagy pathways, suggesting that the SV5 activation of pDC requires access to the cytoplasm and autophagic sampling of cytoplasmic contents. These results have implications for control of SV5 infections in vivo and for development of SV5 as a vaccine vector.
INTRODUCTION
The parainfluenza virus Simian Virus 5 (SV5) is a poor activator of antiviral responses in human cells (Choppin, 1964; Didcock et al 1999b; He et al., 2002; Wansley et al., 2005). SV5 encodes the V protein as an inhibitor of host cell antiviral responses, and this contrasts with many other paramyxoviruses such as Sendai virus (SeV) and measles virus (MeV) which encode both a V protein and a family of C proteins which counteract innate responses (Lamb and Parks, 2007). The poor activation of host cell responses by SV5 infection is thought to be largely due to two main factors: the actions of the viral V protein and control of viral RNA synthesis. A major function of the SV5 V protein is the inhibition of IFN signaling, which occurs through V-mediated targeting of signal transducer and activator of transcription 1 (STAT1) for ubiquitylation and degradation (Didcock et al. 1999a). The SV5 V protein also blocks activation of the IFN-beta promoter during virus infection or following transfection of dsRNA (Andrejeva et al, 2004; He et al., 2002). The paramyxovirus V protein inhibits IFN-beta induction by targeting the IFN-inducible RNA helicase encoded by the melanoma differentiation-associated gene 5 (mda-5; Andrejeva et al., 2004). By contrast, the alternative RNA helicase retinoic acid-inducible gene I (RIG-I) does not appear to be inhibited by V protein (Childs et al. 2007). V protein is thought to also act as a decoy substrate for kinases that activate IFN regulatory factor 3 (Lu et al., 2008). In support of these V functions, a recombinant SV5 that encodes a truncated V protein lacking the highly conserved cys-rich domain is a potent activator of IFN-beta and proinflammatory cyotokine synthesis (He et al., 2002).
A second major factor contributing to limited activation of cellular responses is the ability of SV5 to control levels and types of viral RNA generated during replication. This is evident from our finding that an SV5 mutant with substitutions in the genomic promoter overexpresses viral RNA and also induces IFN and cytokines through RIG-I pathways, even though this mutant expresses a functional V protein (Manuse and Parks, 2009). Similarly, potent antiviral responses are induced by an SV5 P/V mutant which overexpresses viral RNA (Wansley and Parks, 2002), but importantly, expression of the WT P subunit of the viral polymerase restores normal levels of viral gene expression and reduces host cell responses (Dillon and Parks, 2007). Together, these data support a model whereby SV5 infection fails to activate antiviral responses during a robust replication cycle, except under conditions where V protein is defective or when synthesis of viral RNA components is elevated over a threshold. This model raises the question of whether SV5 would still be a poor inducer of antiviral responses in cells that sense virus by mechanisms that are independent of virus replication.
Dendritic cells (DCs) play a critical role in sensing viral infections to activate both innate and adaptive immune responses. Two major DC subsets exist within human peripheral blood (Shortman and Liu, 2002): myeloid dendritic cells (mDCs) and plasmacytoid dendritic cells (pDCs). mDCs are very efficient at phagocytosis and antigen presentation that can result in strong activation of T cells (Banchereau et al., 2000; Reis e Sousa, 2001). SV5 can establish highly productive infections of primary human mDC, and similar to epithelial cells, these infected mDC do not secrete high levels of IFN or cytokines (Arimilli et al., 2006; 2007). Consistent with this finding, SV5 infected mDC are poor activators of T cell function in vitro (Arimilli et al, 2006).
By contrast to mDC, pDCs are much less efficient at phagocytosis, antigen presentation, and activation of T cells. Instead, this DC subset responds to virus exposure by rapidly producing abundant amounts of IFN-alpha (Barchet et al, 2005; Liu, 2005). pDC differ from mDC in their profile of Toll-like Receptor (TLR) expression (Blasius and Beutler, 2010). While mDCs express TLR1, 2, 4, 5, 6, and 8, pDCs have high expression of TLR7 and TLR9 (Shortman and Liu, 2002) which recognize ssRNA and CpG DNA, respectively (Bauer et al. 2001; Diebold et al., 2004). pDCs have been demonstrated to produce IFN-alpha in response to virus by mechanisms that are either dependent or independent of virus replication. For example, pDCs produce IFN-alpha in response to herpes simplex virus-2, West Nile virus, and Hepatitis C virus independent of viral replication (Liang et al., 2009; Lund et al., 2003; Silva et al., 2007). On the other hand, IFN-alpha secretion from pDCs infected with negative strand viruses such as vesicular stomatitis virus (VSV), respiratory syncytial virus (RSV) and Sendai virus has been shown to depend on replication (Hornung et al., 2004; Lee et al., 2007; Waibler et al., 2008). Consistent with virus replication in the cytoplasm, the cellular autophagy machinery which directs cytoplasmic contents to the endosome is required for Sendai virus and VSV replication-dependent activation of pDC (Lee et al., 2007).
Given that WT SV5 is a poor inducer of host cell cytokine synthesis during replication in both epithelial and mDC cultures, we have addressed the question of whether SV5 will also be a poor inducer of IFN from pDC which can detect virus infections by a replication–independent mechanism. Using primary cultures of human pDC, our results support a model whereby IFN-alpha induction from infected pDC is independent of virus replication, but is dependent on signaling through TLR7 and autophagy pathways. Thus, SV5 activation of human pDC differs from other negative strand viruses since it is not dependent on virus replication but involves signaling through TLR7 and autophagy pathways.
MATERIALS AND METHODS
Cells, viruses and reagents
Plasmacytoid dendritic cells (pDC) were prepared from human peripheral blood mononuclear cells harvested as described previously (Arimilli et al., 2006). Briefly, whole blood from healthy donors was subjected to Ficoll-Hypaque (Isolymph, CTL, Deer Park, NY) density gradient centrifugation. pDCs were isolated by magnetic activated cell sorting (MACS) using microbeads containing anti-CD304 (Miltenyi Biotech). Cells were cultured in RPMI (Lonza) with 10% FBS (heat inactivated), 1% Penicillin/Streptomycin, 1% non-essential amino acids, 1 mM Sodium Pyruvate, 2 mM L-glutamine, 10 mM HEPES, with 50 ng/ml Interleukin(IL)-3 (Miltenyi Biotech, Auburn, CA) to promote cell viability.
Recombinant SV5 encoding the enhanced green fluorescent protein (SV5-EGFP) as an additional gene between HN and L was generated from cDNA as described previously (Arimilli et at., 2007). Viruses were grown in MDBK cells and purified by centrifugation through a 25% glycerol cushion (Capraro et al., 2008).
Infection, inhibitors, IFN ELISA and cell surface labeling
Cells were mock infected or infected in suspension at the mois indicated in the figure legends. As positive control for TLR7 activation, cells were treated with gardiquimod (InvivoGen, San Diego, CA) at a concentration of 1 ug/ml at the time of infection. The TLR7 inhibitor IRS661 is an oligonucleotide with the sequence 5’-TGCTTGCAAGCTTGCAAGCA-3’ (Panwar et al, 2007), and was added 30 min prior to infection at a concentration of 2.8 uM. Chloroquine (Sigma) was used at a concentration of 2.5 ug/ml. Wortmanin and 3-methyladenine (3-MA) were added 10 min prior to infection at 10 nM and 1 mM, respectively.
IFN-alpha and Interleukin-6 in cell culture supernatant were measured by an ELISA (PBL Biosciences, Piscataway, NJ) and values were normalized to 2 × 105 cells in order to compare between experiments. To detect cell surface markers and EGFP expression, cells were stained with the following conjugated monoclonal antibodies: CD11c, CD123, CD80, and CD86 (BD Pharmigen, San Diego, CA). Samples were analyzed on a FACScan instrument using Cell Quest software (Benton Dickinson, San Diego, CA). Statistical analysis of data was by the Student’s t-test type 2 tailed method.
Microscopy
Microscopy was carried out as described previously using a Nikon Eclipse microscope and a 20x lens (Manuse and Parks, 2009). Phase and fluorescent images were captured using a QImaging digital camera and processed using QCapture software (QImaging, Surry, BC, Canada). Exposure times were set manually to be constant between samples.
RESULTS
SV5 can productively infect human pDCs
Since human pDCs represent only ~0.5% of PBMCs (Jing et al., 2009), pDCs were isolated from whole blood PBMCs by positive selection using anti-CD304 antibody-conjugated microbeads. It is known that pDCs express cell surface markers that are distinct from that seen with myeloid DCs (Shortman and Liu, 2002). As shown in Fig. 1A, our pDC preparations displayed the previously defined profile of high CD123 (IL-3 receptor) expression and lack of CD11c expression. This contrasted with the profile of myeloid DCs, which express high levels of CD11c and very low levels of CD123 (Fig. 1A).
Figure 1. SV5 productively infects pDC in vitro.
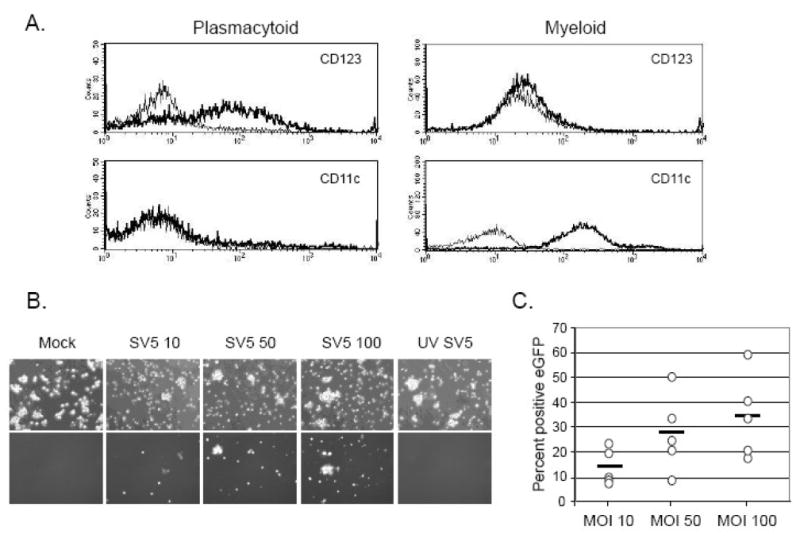
A) Markers on plasmacytoid DCs. pDCs were isolated by positive selection from PBMCs from whole human blood using CD304 microbeads. After overnight culturing, cells were stained for surface expression of CD123 and CD11c (bold lines) or with an isotype control (thin lines) before analysis by flow cytometry. For comparison, monocyte precursors were isolated from PBMCs from whole human blood by positive selection using CD14 microbeads, and myeloid DC were generated as described previously (Arimilli et al, 2007) before staining for surface markers in parallel. B) pDCs were mock infected, infected at an moi of 10, 50 or 100 with SV5-EGFP or infected with UV inactivated SV5-EGFP at an moi of 100. Phase (top panel) or flourescence (bottom panel) micrographs were taken at 24 h pi. C) pDCs from multiple donors (each donor is represented by a symbol) were mock infected or infected at an moi of 10, 50 or 100 with SV5-EGFP. At 24 h pi the number of EGFP-positive cells was quantified by flow cytometry. Bar represents the average of three donors for each moi.
To determine the extent to which SV5 could infect pDCs, cells were mock-infected or infected at an moi of 10, 50 or 100 PFU/cell with SV5 that was engineered to encode EGFP as an additional gene between HN and L (Arimilli et al. 2007). As shown in Fig. 1B, a low number of pDCs expressed EGFP when infected at an moi of 10, but this was substantially increased by an increasing moi. The percent of pDCs that were positive for SV5-derived EGFP expression was quantified for five different donors by flow cytometry (Fig. 1C). At an moi of 10, on average ~15% of the pDC population was EGFP-positive, but an moi of 100 resulted in an average of 35% of the pDCs being positive for EGFP expression. In addition, de novo viral NP and M protein expression was detected by metabolic labeling and immunoprecipitation of extracts from cells infected with live SV5-EGFP but not with UV-inactivated SV5-EGFP (data not shown). These data demonstrate that SV5 can productively infect pDCs and that a greater number of cells can be infected with increasing moi.
Live and UV-treated SV5 induce similar IFN-alpha secretion and maturation marker expression from pDC
Previous results have shown that some paramyxoviruses such as measles virus and respiratory syncytial virus block activation of human pDC responses (Schlender et al., 2005). To determine the response of human pDCs to SV5 infection, cells were mock infected, infected with live SV5-EGFP or UV-inactivated SV5-EGFP at an moi of 100, or treated with the TLR7 agonist Imiquimod as a positive control. Twenty four h later, media were analyzed by ELISA for levels of IFN-alpha. As shown in Fig 2A, pDC isolated from three different donors responded to the positive control TLR-7 agonist imiquimod by secreting large amounts of IFN-alpha. Similarly, infection with live SV5-EGFP induced very high levels of IFN-alpha from pDCs, but this same level of IFN was also seen using UV inactivated virus. The kinetics of IFN-alpha secretion was nearly identical for pDC treated with live and with UV-inactivated SV5-EGFP (Fig. 2B), and this lagged substantially from the rapid induction by imiquimod. The level of IFN-alpha secretion was not altered by increasing moi above 10, since very similar levels were detected using live virus at mois of 10, 50 and 100 but also by UV inactivated virus. Thus, while increasing moi leads to increased numbers of infected cells (Fig. 1), it has little effect on levels of IFN-alpha production (Fig. 2).
Figure 2. IFN-alpha production from SV5 infected pDCs.
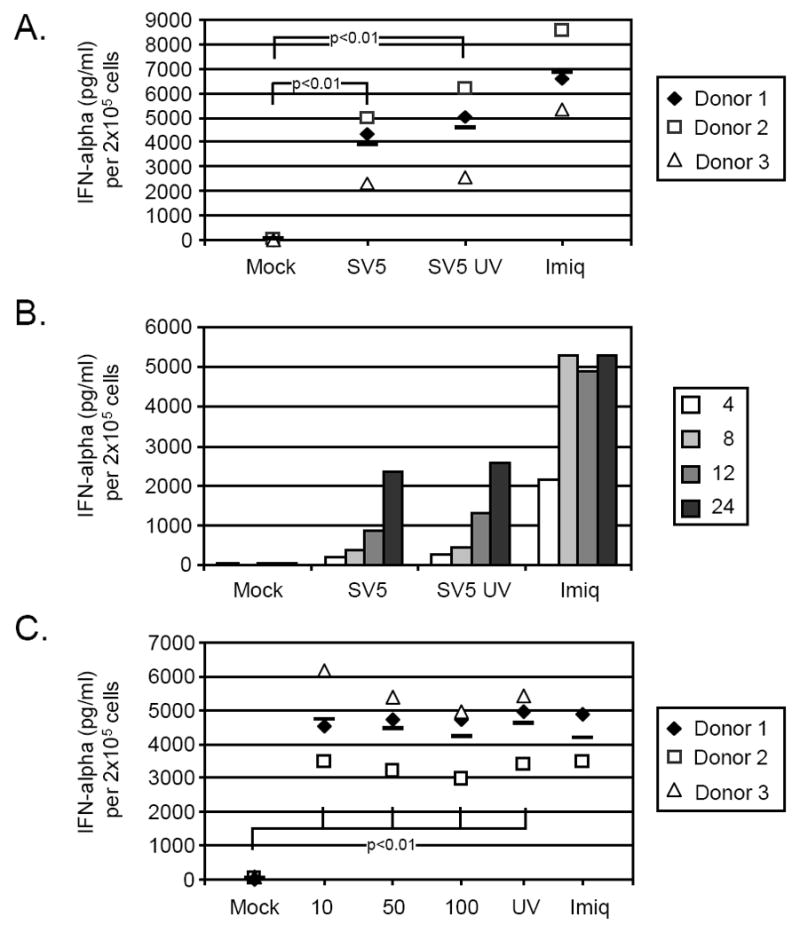
A) IFN induction. pDCs from three donors (each is represented by a different symbol) were mock infected, infected with live SV5-EGFP or UV-treated SV5-EGFP at an moi of 100, or treated with 1 ug/ml Imiquimod as a positive control. At 24 h pi supernatant was harvested and analyzed by ELISA for level of IFN-alpha. p values for SV5 and UV treated SV5 are shown. B) Timecourse of IFN induction. pDCs from one representative donor were treated as described for panel A. At the indicated hrs pi, supernatant was harvested and analyzed by ELISA for levels of IFN-alpha. C) Effect of moi on IFN induction. pDC were infected at the indicated mois and treated as described in panel A. Levels of IFN-alpha secretion were determined at 24 h pi. In panels A and C, bars represent mean levels for the three donors. For panel C, data for all conditions had a significance of p<0.004 when compared to mock sample.
In Fig. 2, each symbol represents data from cells obtained from an individual donor. It is noteworthy that in Fig. 2, for a given treatment there was some variability among donors in the level of IFN-alpha induction. However, for each donor the relative IFN-alpha induction from pDC treated with live virus, UV treated virus and imiquimod was remarkably consistent.
While myeloid DC are thought to be the most potent antigen presenting cells, pDC can also increase expression of cell surface costimulatory markers CD80 and CD86 (Banchereau et al. 2000; Reis e Sousa, 2001). As shown in Fig 3 for three donors, infection of pDCs with SV5-EGFP resulted in upregulation of CD80 (panel A) and CD86 (panel B) above that seen for mock infected cells (indicated by the dotted line). Increases in CD80 expression were consistently seen, but were not statistically significant compared to mock samples. By contrast, changes in CD86 were highly significant (p<0.005). Similar to IFN-alpha induction, maturation marker expression did not differ between live or UV-inactivated virus.
Figure 3. Increased maturation marker expression on SV5 infected pDCs.
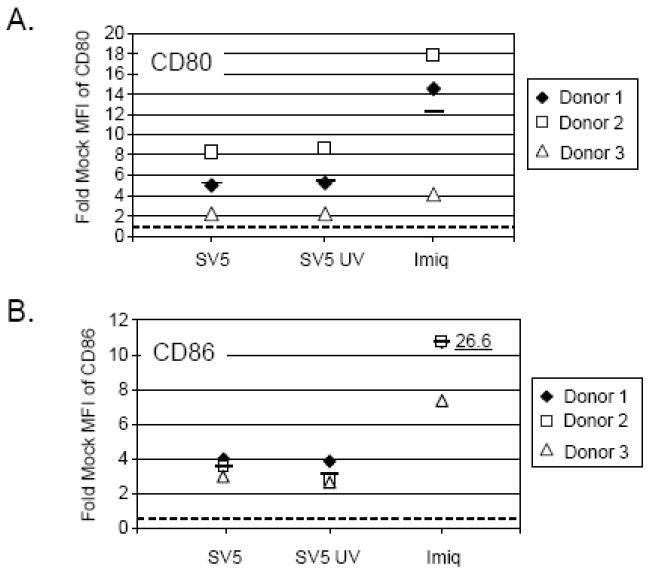
Human pDCs from three donors (each represented by a different symbol) were mock infected, infected with live SV5 or UV-inactivated SV5 at an moi of 100, or treated with 1 ug/ml Imiquimod as a positive control. At 24 h pi cells were harvested and surface labeled for CD80 (panel A) and CD86 expression (panel B). pDCs were analyzed by flow cytometry and results are expressed as fold mock mean fluorescent intensity (MFI) of maturation marker expression. The dashed line indicates the level of expression for mock infected cells set at one. The average of three donors for each condition is represented by a bar. Mean level of maturation marker upregulation for imiquimod treated pDCs is shown as 26.6. For panel B, data from SV5 and UV-treated SV5 samples had p values of 0.0008 and 0.005, respectively.
Interestingly, although exposure of human pDC to SV5 induced both IFN-alpha secretion and upregulation of maturation markers (Figs 2 and 3), there was no significant increase in the secretion of IL-6 (Fig. 4). This result is similar to the IFN and cytokine responses found for influenza virus infection of mouse pDC which differed from that seen with CpG treatment (Iparraguirre et al., 2008).
Figure 4. Induction of IL-6 from human pDC by a TLR7 agonist but not by SV5.
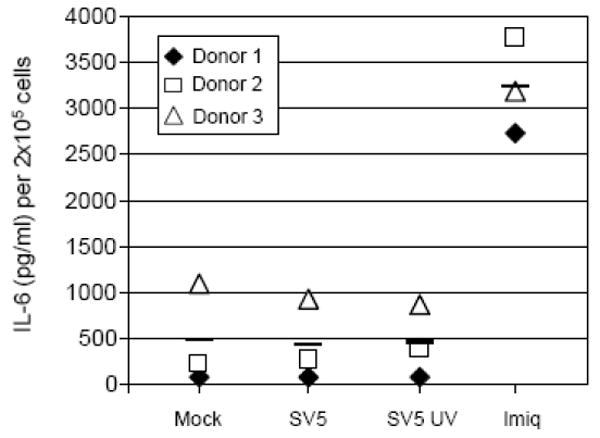
Human pDC were treated as described in the legend to Fig. 3, and at 24 h post infection levels of secreted IL-6 were determined by ELISA. The average of three donors for each condition is represented by a bar.
We have previously described two SV5 mutants that are potent inducers of IFN secretion through production of dsRNA and activation of RIG-I signaling (Manuse and Parks, 2009; 2010; Wansley and Parks, 2002). As shown in Fig. 5, pDC infected with these mutants also induced high levels of IFN-alpha. For some donors (e.g. donors 1 and 2), the leader mutant and P/V mutant consistently induced slightly lower and slightly higher IFN levels compared to WT SV5-EFGP, respectively, but these levels were not substantially different. These data indicate that in pDCs, WT SV5 induces approximately the same level of IFN secretion as SV5 mutants which potently induce IFN through replication dependent pathways (Manuse and Parks, 2009). Taken together, these results indicate that WT SV5 is a potent inducer of IFN-alpha and surface maturation marker expression in pDCs and that activation of these cells does not require virus replication.
Figure 5. Activation of IFN-alpha secretion by SV5 RNA synthesis mutants.
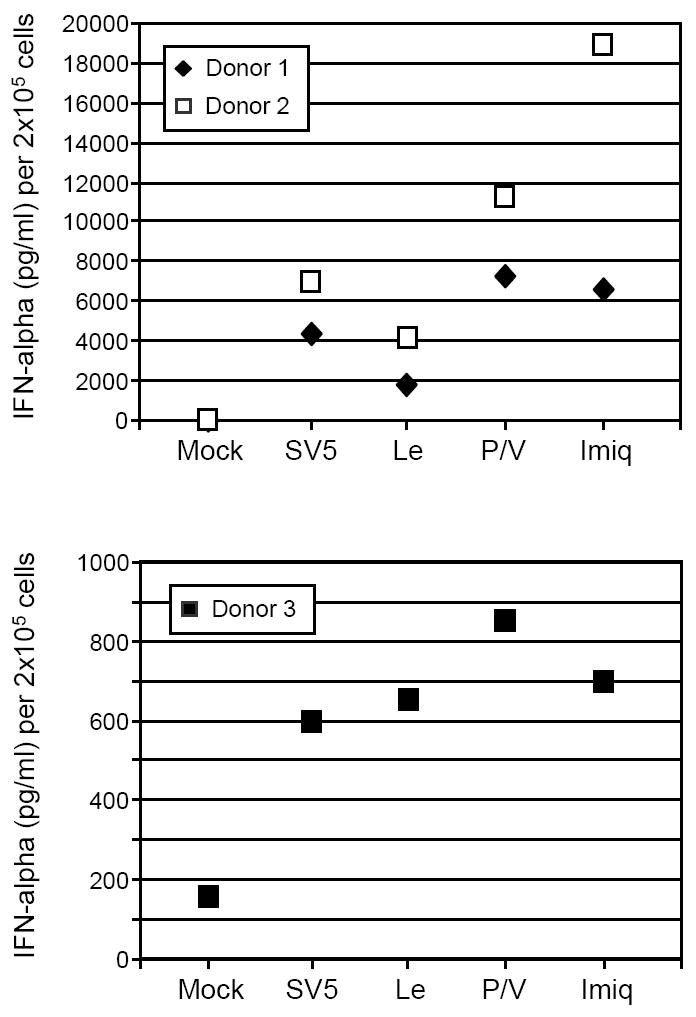
pDC were infected with SV5-EGFP or with the SV5 mutants containing substitutions in the leader promoter (Le) or the P/V gene (P/V). Alternatively, cells were treated with the TLR7 ligand imiquimod. At 24 h pi, levels of secreted IFN-alpha were determined by ELISA. Results are from two separate experiments with two donors. Data for two donors with similar levels of IFN are shown in the top panel. Data from a donor with low levels of IFN is graphed in the lower panel.
Effect of endosomal and TLR7 inhibitors on SV5 activation of pDC responses
pDC can sense viral infections through TLR7 or TLR9, both of which exist within pDC endosomal compartments (Lee et al., 2003; Mark et al. 2004). Since SV5 is an RNA virus, we hypothesized that the RNA-sensing TLR7 pathway was involved in pDC responses to infection, and that inhibitors of endosomal acidification would block responses to SV5. To test this hypothesis, pDCs were pre-treated with the endosomal inhibitor chloroquine and then mock infected or infected with the live or UV-treated SV5-EGFP at an moi of 100. Twenty four h later, media were analyzed by ELISA for IFN-alpha. As shown in Fig 6A for three independent donors, live and UV-inactivated SV5-EGFP induced ~6,000 pg/ml of IFN-alpha (open symbols), and this activation was reduced to ~1000-2000 pg/ml by chloroquine treatment (closed symbols).
Figure 6. Inhibitors of TLR7 decrease IFN-alpha secretion and CD80 expression in SV5-infected pDC.
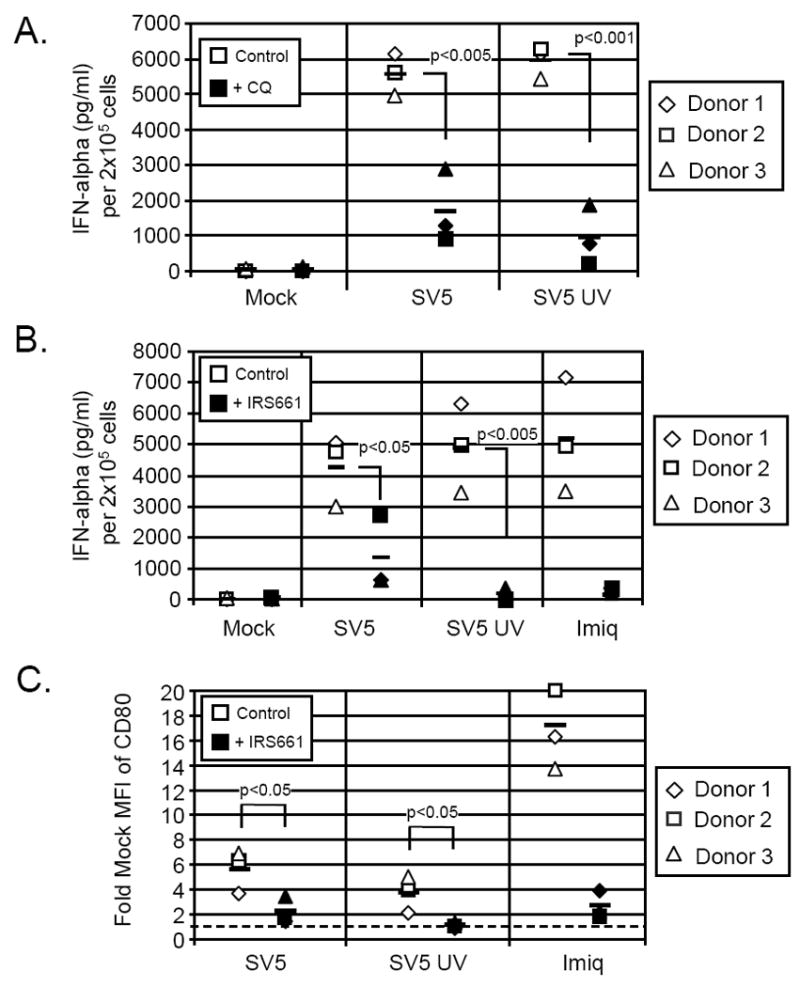
A) Effect of chloroquine. pDCs from three donors were mock treated (control samples) or pre-treated with 2.5 ug/ml of chloroquine (filled symbols). Cells were then mock infected or infected with live or UV-inactivated SV5 at an moi of 100. Twenty-four h pi supernatant was harvested and analyzed for levels of IFN-alpha by ELISA. B) Effect of TLR7 inhibitor. pDCs from three donors were pre-treated with 2 uM IRS661 (closed symbols) or were left untreated (open symbols) for 30 minutes. Cells were then mock infected or infected as described for panel A. Twenty-four h pi supernatant was analyzed for IFN-alpha by ELISA. C) Effect of TLR7 inhibitor on maturation markers. Cells were treated as described in panel B and at 24 h pi cells were stained for CD80 surface expression and analyzed by flow cytometry. Results are expressed as fold mock mean fluorescent intensity (MFI) of CD80 expression. The dashed line indicates the level of expression for mock infected cells set at one. The average of three donors for each condition is represented by the bar.
To test the role of TLR7 in pDC activation by SV5, cells were treated prior to infection with IRS661, an oligonucleotide inhibitor that has been shown to be specific for TLR7 signaling (Wang et al, 2006). In cells treated for 30 minute with the inhibitor (closed symbols, Fig. 6B), levels of IFN-alpha induced by live SV5-EGFP (open symbols) were substantially reduced, while only background levels were seen with UV-inactivated virus or with stimulation by imiquimod (Fig 6B). Similar results were seen for maturation markers, where addition of the TLR7 inhibitor gave a significant reduction in levels of cell surface CD80 surface expression after exposure to SV5 (Fig. 6C). Together these results with two different inhibitors support the hypothesis that SV5 activates pDC through TLR7 signaling.
The number of pDCs that were positive for SV5-derived EGFP expression increased upon inhibition of TLR signaling by either IRS 661 (Fig. 7A) or chloroquine (Fig. 7B). With IRS661 treatment, the effect of TLR7 inhibition on number of infected cells was dramatic, with the number of infected cells increasing from 25 to 50 percent EGFP-positive, while chloroquine treatment had a lesser effect. Additionally, the level of viral gene expression as calculated by the mean fluorescent intensity of the EGFP positive population was also enhanced when TLR7 signaling was inhibited (data not shown). These data indicate that the SV5 activates pDCs through TLR7 signaling, and that inhibition of TLR7 decreases IFN production and increases both the percentage of infected cells as well as the level of viral gene expression.
Figure 7. TLR7 inhibition increases the number of SV5 infected pDCs.
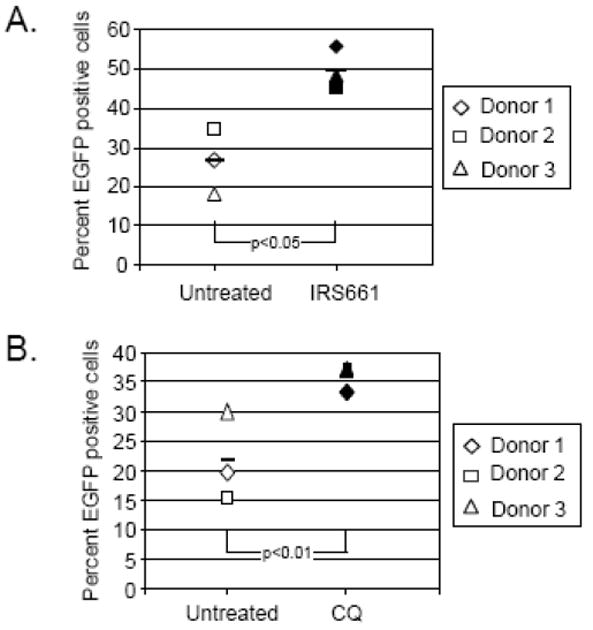
pDCs from three different donors (each represented by a different symbol) were left untreated (open symbols) or pre-treated (closed symbols) with 2 uM IRS661 (panel A) or 2.5 ug/ml chloroquine (panel B). Cells were then infected at an moi of 100 with SV5-EGFP and at 24 h pi were harvested and analyzed for EGFP expression by flow cytometry. The average of three donors for each condition is represented by the bar.
Inhibitors of autophagy reduce SV5 activation of pDC responses
Activation of SV5 infected pDCs could result from direct exposure of virus to endosomal TLR7 prior to fusion with the plasma membrane. Alternatively, previous results have shown that VSV and Sendai virus activate pDC in a replication-dependent manner after access to the cytoplasm, and the viral RNAs are thought to be targeted to the endosome by autophagy since activation is blocked by autophagy inhibitors (Lee et al., 2007). To determine the role of autophagy in SV5-mediated pDC activation, cells were mock treated or treated with the autophagy inhibitors wortmanin (WM) and 3-methyladenine (3MA) at concentrations used in previous studies (Lee et al. 2007), and levels of SV5-induced IFN-alpha were determined. As shown in Fig. 8, IFN induction by VSV in control samples was lower than that seen with SV5-EGFP, but most importantly VSV-mediated IFN secretion was blocked by prior treatment with both WM and 3-MA as reported previously (Lee et al. 2007). Similarly, IFN induced by live SV5-EGFP or UV inactivated virus was dramatically reduced by treatment with WM or 3-MA. These results are important as they demonstrate that a virus (e.g. SV5) which activates pDC IFN responses by a replication-independent mechanism requires the same cellular autophagy machinery as a virus (e.g. VSV) that activates through replication-dependent pathways.
Figure 8. Autophagy inhibitors block IFN-alpha secretion from SV5-infected pDCs.
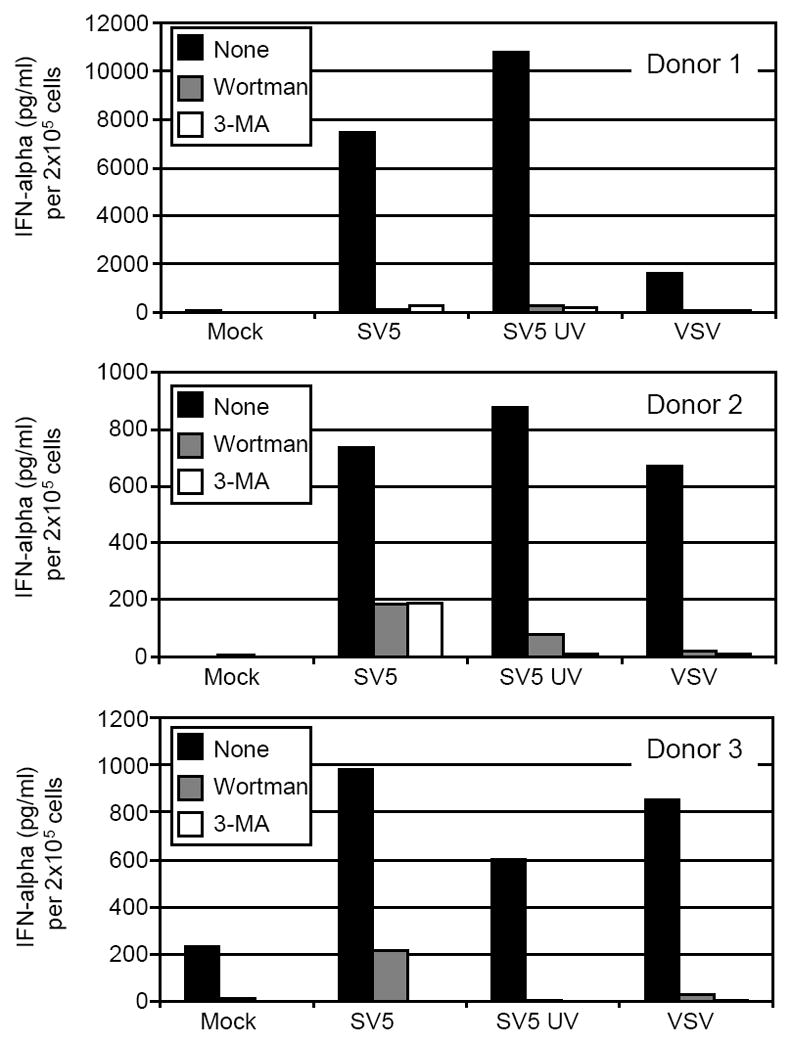
Human pDCs from three different donors were left untreated (black bars) or pre-treated with Wortmanin (gray bars) or 3-MA (white bars). Cells were then mock infected or infected with live or UV-inactivated SV5 at an moi of 100. Twenty-four h pi supernatant was harvested and analyzed for levels of IFN-alpha by ELISA.
DISCUSSION
Host cell responses to viral infections can be initiated by receptors that detect and respond to viral replication products. For example, the cytoplasmic helicases RIG-I and MDA-5 can detect viral RNA replication products and initiate synthesis of IFN and other antiviral cytokines (Childs et al., 2007; Yoneyama et al. 2005). In all of the human cell types which we have examined so far including epithelial, fibroblast and mDC, SV5 establishes highly productive infections with only minimal activation of antiviral responses (Wansley and Parks, 2002; Arimilli et al, 2006). The only examples of SV5 strongly inducing cytokine secretion from human cells is when the virus encodes an altered V protein (Dillon et al. 2005; He et al., 2002; Wansley and Parks, 2002) or when viral RNA synthesis is elevated by mutations in viral proteins other than V protein (Manuse and Parks, 2009; Young et al. 2006). Thus, for the SV5 P/V and Leader mutants described previously, antiviral responses require virus replication and are mediated in large part by RIG-I signaling pathways (Manuse and Parks, 2010). Viral infections can also be detected by mechanisms that are not dependent on virus replication, including interactions with TLRs (Boehme and Compton, 2004). The overall goal of this work was to address the question of whether SV5 would also be a poor inducer of IFN from pDC which utilize TLR7 to respond to viruses through mechanisms that are not dependent on virus replication.
In primary human pDCs, WT SV5 activated very potent IFN-alpha secretion, but importantly this was not dependent on virus replication. This was evident by our findings that IFN-alpha secretion: 1) was similar between pDC exposure to UV-inactivated and live infectious SV5, 2) did not increase with increasing moi above 10, and 3) was very similar between WT SV5 and SV5 mutants that overexpress viral gene products. This latter finding is important support that SV5 activation of antiviral responses in human pDC is not dependent on virus replication, as was shown previously for responses to these mutants in epithelial cell types (Manuse and Parks, 2009; 2010). To our knowledge, this is the first human cell type in which WT SV5 infection induces a strong IFN response. This work highlights the correlation between the type of pattern recognition receptor expressed in a cell (e.g. TLR7) and the extent to which host cell responses are activated during viral infection.
Inhibitor studies indicate that TLR7 played an important role in responding to both live and UV-inactivated SV5. Since TLR7 senses ssRNA, it is likely that this pattern recognition receptor is directly detecting the viral genome. The current model for SV5 entry depends on fusion at the cell surface resulting in the deposition of the viral genome into the cytoplasm of the cell (Lamb, 1993). This raises the question of how the nucleocapsid-bound viral genome is detected by TLR7 which exists within endosomes. One possibility is that pDCs have mechanisms for rapid endocytosis from the plasma membrane such that some virus particles are endocytosed as an alternative outcome to fusion with the plasma membrane to deposit the nucleocapid in the cytoplasm. At least some live virus particles are able to enter the cytoplasm and begin viral gene transcription as evidenced by GFP expression, especially at higher multiplicities of infection. Interestingly, very similar levels of IFN-alpha secretion are seen with low and high moi infections, which indicate that there may be a low threshold for number of genomes needed for TLR7 activation.
Alternatively, viral genomes that are deposited into the cytoplasm by fusion may gain access to endosomal TLR7 by means of autophagy, a cellular process that delivers cytoplasmic components to intracellular vesicles including lysosomes and endosomes (Mizushima, 2009). Autophagy has been implemented during virus infection by regulating programmed cell death and in controlling antiviral responses to infection (Orvedahl, and Levine, 2009). Although we have not carried out parallel experiments with VSV in this study, previous work has demonstrated that VSV requires replication to induce IFN production from pDCs (Iwasaki, 2007; Lee et al., 2007). Furthermore, VSV induction of IFN-alpha is through autophagy-dependent mechanisms (Lee et al., 2007). Our results differ from previous reports with Sendai virus and VSV by demonstrating that SV5 which activates pDC independent of replication also requires autophagy pathways.
While only a few studies of paramyxovirus infection of human pDCs have been previously reported, the published data revealed differences between viruses in the degree and mechanism of pDC activation. In one report, respiratory syncytial virus (RSV) strain A2 and human metapneumovirus induced IFN-alpha secretion from infected pDCs, but also blocked a further increase in IFN production upon challenge with a TLR9 agonist (Guerrero-Plata, et al., 2006). Other investigators reported that pDCs are not activated for IFN secretion by exposure to either the A2 strain or clinical isolates of RSV or by measles virus, whereas the RSV strain Long induced large amounts of type I IFN (Schlender et al., 2005). Similarly, SeV induces IFN-alpha from pDC in a replication-dependent mechanism (Lee et al., 2007) which contrasts with our results here for SV5. Together, these studies illustrate different mechanisms for IFN induction employed by members of this diverse family of viruses. Future work will focus on whether this is a reflection of the different mechanisms paramyxoviruses employ to counteract host cell responses.
When TLR7 pathways were inhibited, SV5 infected pDC secreted less IFN-alpha as expected, but there was also a higher percentage of cells that were positive for GFP expression. This may have resulted from decreased IFN-alpha signaling, which is known to upregulate many genes that inhibit virus replication. However, IFN signaling should not be a factor in SV5-infected cells, due to actions of the V protein in targeting STAT1 for degradation (Didcock et al., 1999b). V-mediated STAT1 degradation is rapid and can even be catalyzed by V that is contained within the incoming virion particle (Precious et al. 2007). Thus, increased SV5 replication in the presence of a TLR7 inhibitor is not likely to be due to changes in IFN signaling. As an alternative, TLR7 inhibition may increase virus replication due to inhibition of intracellular pathways that are directly activated by TLR7 signaling. This proposal is consistent with the finding that TLR7-mediated induction of antiviral genes does not require IFN signaling, but rather TLR7 can directly induce antiviral gene products (Domizio et al., 2009). Thus, while the SV5 V protein can block the effect of IFN-alpha production by infected cells, TLR7 may still inhibit SV5 replication through a direct intracellular activation of antiviral pathways that is independent of signaling.
Our previous studies with animal models of SV5 infection indicate that despite being a poor inducer of IFN in tissue culture cells, virus growth in vivo is limited and virus is cleared from the respiratory tract by ~7 days post infection (Capraro et al., 2008). Thus, at least in the ferret model SV5 does not establish long term infections of the respiratory tract contrary to what is predicted by its behavior in tissue culture cells. Our finding that pDCs produce high levels of IFN in response to SV5 by a replication-independent mechanism provides some insight into possible limitations on growth in vivo. We propose that pDC recruited to the site of SV5 infection respond to progeny virions by producing high levels of IFN-alpha, which in turn limits SV5 growth and aids in virus clearance by immune mechanisms.
Acknowledgments
We thank members of the Parks lab and Dr. Doug Lyles for helpful comments on the project. This work was supported by NIH grants AI42023, DC009619 and AI060642 (GDP, SBM). M.J.M. was supported by NIH Training Award Grant T32-AI007401.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andrejeva J, Childs KS, Young DF, Carlos TS, Stock N, Goodbourn S, Randall RE. The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN promoter. Proc Natl Acad Sci U S A. 2004;101:17264–17269. doi: 10.1073/pnas.0407639101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arimilli S, Alexander-Miller MA, Parks GD. A Simian Virus 5 (SV5) P/V Mutant Is Less Cytopathic than Wild-Type SV5 in Human Dendritic Cells and Is a More Effective Activator of Dendritic Cell Maturation and Function. J Virol. 2006;80:3416–3427. doi: 10.1128/JVI.80.7.3416-3427.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arimilli S, Johnson JB, Alexander-Miller MA, Parks GD. TLR-4 and -6 agonists reverse apoptosis and promote maturation of simian virus 5-infected human dendritic cells through NFkB-dependent pathways. Virology. 2007;365:144–156. doi: 10.1016/j.virol.2007.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu Y-J, Pulendran B, Palucka K. Immunobiology of Dendritic Cells. Annu Rev Immunol. 2000;18:767–811. doi: 10.1146/annurev.immunol.18.1.767. [DOI] [PubMed] [Google Scholar]
- Barchet W, Cella M, Colonna M. Plasmacytoid dendritic cells--virus experts of innate immunity. Semin Immunol. 2005;17:253–261. doi: 10.1016/j.smim.2005.05.008. [DOI] [PubMed] [Google Scholar]
- Bauer S, Kirschning CJ, Hacker H, Redecke V, Hausmann S, Akira S, Wagner H, Lipford GB. human TLR9 confers responsiveness to bacterial DNA via species-specific CpG motif recognition. Proc Natl Acad Sci U S A. 2001;98:9237–9242. doi: 10.1073/pnas.161293498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasius AL, Beutler B. Intracellular Toll-like Receptors. Immunity. 2010;32:305–315. doi: 10.1016/j.immuni.2010.03.012. [DOI] [PubMed] [Google Scholar]
- Boehme KW, Compton T. Innate sensing of viruses by toll-like receptors. J Virol. 2004;78:7867–7873. doi: 10.1128/JVI.78.15.7867-7873.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capraro GA, Johnson JB, Kock ND, Parks GD. Virus growth and antibody responses following respiratory tract infection of ferrets and mice with WT and P/V mutants of the paramyxovirus Simian Virus 5. Virology. 2008;376:416–428. doi: 10.1016/j.virol.2008.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childs KS, Stock N, Ross C, Andrejeva J, Hilton L, Skinner M, Randall R, Goodbourn S. mda-5, but not RIG-I, is a common target for paramyxovirus V proteins. Virology. 2007;359:190–200. doi: 10.1016/j.virol.2006.09.023. [DOI] [PubMed] [Google Scholar]
- Choppin PW. Multiplication of a myxovirus (SV5) with minimal cytopathic effects and without interference. Virology. 1964;23:224–233. doi: 10.1016/0042-6822(64)90286-7. [DOI] [PubMed] [Google Scholar]
- Didcock L, Young DF, Goodbourn S, Randall RE. The V Protein of Simian Virus 5 Inhibits Interferon Signalling by Targeting STAT1 for Proteasome-Mediated Degradation. J Virol. 1999a;73:9928–9933. doi: 10.1128/jvi.73.12.9928-9933.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didcock L, Young DF, Goodbourn S, Randall RE. Sendai virus and SV5 block activation of IFN-responsive genes: importance of virus pathogenesis. J Virol. 1999b;73:3125–3133. doi: 10.1128/jvi.73.4.3125-3133.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diebold S, Kaisho S, Hemmi H, Akira S, Reis e Sousa C. Innate antiviral responses by means of TLR-7-mediated recognition of single-stranded RNA. Science. 2004;303:1529–15431. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- Dillon PJ, Parks GD. A Role for the Phosphoprotein P Subunit of the Paramyxovirus Polymerase in Limiting Induction of Host Cell Antiviral Responses. J Virol. 2007;81:11116–11127. doi: 10.1128/JVI.01360-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domizio D, Blum A, Gallagher-Gambarelli M, Molens JP, Chaperot L, Pumas J. TLR7 stimulation in human plasmacytoid dendritic cells leads to the induction of early IFN-inducible gene in the absence of type I IFN. Blood. 2009;114:1794–1802. doi: 10.1182/blood-2009-04-216770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero-Plata A, Casola A, Suarez G, Yu X, Spetch L, Peeples ME, Garofalo RP. Differential Response of Dendritic Cells to Human Metapneumovirus and Respiratory Syncytial Virus. Am J Respir Cell Mol Biol. 2006;34:320–329. doi: 10.1165/rcmb.2005-0287OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B, Paterson RG, Stock N, Durbin JE, Durbin RK, Goodbourn S, Randall RE, Lamb RA. Recovery of Paramyxovirus Simian Virus 5 with a V Protein Lacking the Conserved Cysteine-rich Domain: The Multifunctional V Protein Blocks both Interferon-[beta] Induction and Interferon Signaling. Virology. 2002;303:15–32. doi: 10.1006/viro.2002.1738. [DOI] [PubMed] [Google Scholar]
- Hornung V, Schlender J, Guenthner-Biller M, Rothenfusser S, Endres S, Conzelmann K-K, Hartmann G. Replication-dependent potent IFN-alpha induction in human plasmacytoid DC by a single stranded RNA virus. J Immuol. 2004;173:5935–5943. doi: 10.4049/jimmunol.173.10.5935. [DOI] [PubMed] [Google Scholar]
- Iparraguirre A, Tobias JW, Hensley SE, Masek KS, Cavanagh LL, Rendl M, Hunter CA, Ertl HC, von Andrian UH, Weninger W. Two distinct activation states of plasmacytoid dendritic cells induced by influenza virus and CpG 1826 oligonucleotide. J Leuko Biol. 2008;83:610–20. doi: 10.1189/jlb.0807511. [DOI] [PubMed] [Google Scholar]
- Iwasaki A. Role of autophagy in innate viral recognition. Autophagy. 2007;3:354–356. doi: 10.4161/auto.4114. [DOI] [PubMed] [Google Scholar]
- Jing Y, Shaheen E, Drake RR, Chen N, Gravenstein S, Deng Y. Aging is associated with a numerical and functional decline in plasmacytoid dendritic cells, whereas myeloid dendritic cells are relatively unaltered in human peripheral blood. Human Immunol. 2009;70:777–784. doi: 10.1016/j.humimm.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb RA. Paramyxovirus Fusion: A Hypothesis for Changes. Virology. 1993;197:1–11. doi: 10.1006/viro.1993.1561. [DOI] [PubMed] [Google Scholar]
- Lamb RA, Parks GD. Paramyxoviridae: the viruses and their replication. In: Fields B, Knipe D, Howley P, editors. Fields Virology. 5. Lippincott Williams and Wilkins Publishers; Philadelphia, Pa: 2007. pp. 1449–1496. [Google Scholar]
- Lee HK, Lund JM, Ramanathan B, Mizushima N, Iwasaki A. Autophagy-Dependent Viral Recognition by Plasmacytoid Dendritic Cells. Science. 2007;315:1398–1401. doi: 10.1126/science.1136880. [DOI] [PubMed] [Google Scholar]
- Lee J, Chuang T-H, Redecke V, She L, Pitha PM, Carson DA, Raz E, Cottam HB. Molecular basis for the immunostimulatory activity of guanine nucleoside analogs: Activation of Toll-like receptor 7. Proc Natl Acad Sci U S A. 2003;100:6646–6651. doi: 10.1073/pnas.0631696100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang H, Russell RS, Yonkers NL, McDonald D, Rodriguez B, Harding CV, Anthony DD. Differential Effects of Hepatitis C Virus JFH1 on Human Myeloid and Plasmacytoid Dendritic Cells. J Virol. 2009;83:5693–5707. doi: 10.1128/JVI.02671-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y-J. IPC: Professional Type 1 Interferon-Producing Cells and Plasmacytoid Dendritic Cell Precursors. Annu Rev Immunol. 2005;23:275–306. doi: 10.1146/annurev.immunol.23.021704.115633. [DOI] [PubMed] [Google Scholar]
- Lu LL, Puri M, Horvath CM, Sen GC. Select paramyxoviral V proteins inhibit IRF3 activation by acting as alternative substrates for inhibitor of kB kinase epsilon (IKKe)/TBK1. J Biol Chem. 2008;283:14269–14276. doi: 10.1074/jbc.M710089200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund J, Sato A, Akira S, Medzhitov R, Iwasaki A. Toll-like Receptor 9-mediated Recognition of Herpes Simplex Virus-2 by Plasmacytoid Dendritic Cells. J Exp Med. 2003;198:513–520. doi: 10.1084/jem.20030162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manuse MJ, Parks GD. Role for the Paramyxovirus Genomic Promoter in Limiting Host Cell Antiviral Responses and Cell Killing. J Virol. 2009;83:9057–9067. doi: 10.1128/JVI.01055-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manuse MJ, Parks GD. TLR3-dependent Upregulation of RIG-I Leads to Enhanced Cytokine Production From Cells Infected with the Parainfluenza Virus SV5. Virology. 2010;397:231–241. doi: 10.1016/j.virol.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark R, Jochen M, Tanja G, Peter L, Grayson BL, Hermann W, Stefan B. Toll-like receptor 9 binds single-stranded CpG-DNA in a sequence- and pH-dependent manner. Eur J Immunol. 2004;34:2541–2550. doi: 10.1002/eji.200425218. [DOI] [PubMed] [Google Scholar]
- Mizushima N. Physiological function of autophagy. Curr Top Microbiol Immunol. 2009;335:71–84. doi: 10.1007/978-3-642-00302-8_3. [DOI] [PubMed] [Google Scholar]
- Orvedahl A, Levine B. Autophagy in mammalian antiviral immunity. Curr Top Microbiol Immunol. 2009;335:267–285. doi: 10.1007/978-3-642-00302-8_13. [DOI] [PubMed] [Google Scholar]
- Pawar RD, Ramanjaneyulu A, Kulkarni OP, Lech M, Segerer S, Anders H-J. Inhibition of Toll-Like Receptor-7 (TLR7) or TLR7 plus TLR-9 Attenuates Glomerulonephritis and Lung Injury in Experimental Lupus. J Am Soc Nephrol. 2007;18:1721–1731. doi: 10.1681/ASN.2006101162. [DOI] [PubMed] [Google Scholar]
- Precious BL, Carlos TS, Goodbourne S, Randall RE. Catalytic turnover of STAT1 allows PIV5 to dismantle the interferon-induced antiviral state of cells. Virology. 2007;368:114–121. doi: 10.1016/j.virol.2007.06.024. [DOI] [PubMed] [Google Scholar]
- Reis e Sousa C. Dendritic cells as sensors of infection. Immunity. 2001;14:495. doi: 10.1016/s1074-7613(01)00136-4. [DOI] [PubMed] [Google Scholar]
- Schlender J, Hornung V, Finke S, Gunthner-Biller M, Marozin S, Brzozka K, Moghim S, Endres S, Hartmann G, Conzelmann K-K. Inhibition of Toll-Like Receptor 7- and 9-Mediated Alpha/Beta Interferon Production in Human Plasmacytoid Dendritic Cells by Respiratory Syncytial Virus and Measles Virus. J Virol. 2005;79:5507–5515. doi: 10.1128/JVI.79.9.5507-5515.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shortman K, Liu Y-J. Mouse and human dendritic cell subtypes. Nat Rev Immunol. 2002;2:151–161. doi: 10.1038/nri746. [DOI] [PubMed] [Google Scholar]
- Silva MC, Guerrero-Plata A, Gilfoy FD, Garofalo RP, Mason PW. Differential Activation of Human Monocyte-Derived and Plasmacytoid Dendritic Cells by West Nile Virus Generated in Different Host Cells. J Virol. 2007;81:13640–13648. doi: 10.1128/JVI.00857-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waibler Z, Detje CN, Bell JC, Kalinke U. Matrix protein mediated shutdown of host cell metabolism limits vesicular stomatitis virus-induced interferon-alpha responses to plasmacytoid dendritic cells. Immunobiology. 2008;212:887–894. doi: 10.1016/j.imbio.2007.09.003. [DOI] [PubMed] [Google Scholar]
- Wang JP, Liu P, Latz E, Golenbock DT, Finberg RW, Libraty DH. Flavivirus Activation of Plasmacytoid Dendritic Cells Delineates Key Elements of TLR7 Signaling beyond Endosomal Recognition. J Immunol. 2006;177:7114–7121. doi: 10.4049/jimmunol.177.10.7114. [DOI] [PubMed] [Google Scholar]
- Wansley EK, Parks GD. Naturally occurring substitutions in the P/V gene convert the noncytopathic paramyxovirus SV5 into a virus that induces type I interferon and cell death. J Virology. 2002;76:10109–10121. doi: 10.1128/JVI.76.20.10109-10121.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wansley EK, Dillon PJ, Gainey MD, Tam J, Cramer SD, Parks GD. Growth sensitivity of a recombinant simian virus 5 P/V mutant to type I interferon differs between tumor cell lines and normal primary cells. Virology. 2005;335:131–144. doi: 10.1016/j.virol.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Yoneyama M, Kikuchi M, Matsumoto K, Imaizumi T, Miyagishi M, Taira K, Foy E, Loo YM, Gale M, Jr, Akira S, Yonehara S, Kato A, Fujita T. Shared and unique functions of the DexD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J Immunol. 2005;175(5):2851–8. doi: 10.4049/jimmunol.175.5.2851. [DOI] [PubMed] [Google Scholar]
- Young VA, Dillon PJ, Parks GD. Variants of the paramyxovirus SV5 with accelerated or delayed gene expression activate proinflammatory cytokine synthesis. Virology. 2006;350:90–102. doi: 10.1016/j.virol.2006.01.006. [DOI] [PubMed] [Google Scholar]