

Abstract
In the present study, we investigated changes of cytosolic Ca2+ ([Ca2+]cyt), endoplasmic reticulum Ca2+ ([Ca2+]ER), and mitochondrial Ca2+(Ca2+m) in astrocytes following oxygen/glucose deprivation and reoxygenation (OGD/REOX). Two hours OGD did not cause changes in [Ca2+]cyt, but led to a significant increase in [Ca2+]ER. The elevation in [Ca2+]ER continued and reached a peak level (130 ± 2 μM) by 90 min REOX. An abrupt release of Ca2+ER occurred during 1.5–2.5 h REOX, which was accompanied with a delayed and sustained rise in [Ca2+]cyt. Moreover, Ca2+m content was increased significantly within 15 min REOX followed by a secondary rise (~ 4.5-fold) and a release of mitochondrial cytochrome c (Cyt c). Astrocytes exhibited translocation of Cyt c from mitochondria to ER and up regulation of ER stress protein p-eIF2α. Blocking Na+-K+-Cl− cotransporter isoform 1 (NKCC1) activity, either by its potent inhibitor bumetanide or genetic ablation, abolished release of ER Ca2+, delayed rise in [Ca2+]cyt and Ca2+m. Inhibition of the reverse mode operation of the Na+/Ca2+ exchanger (NCXrev) significantly attenuated OGD/REOX-mediated Cyt c release. In summary, our study illustrates that OGD/REOX triggers a time-dependent loss of Ca2+ homeostasis in cytosol and organelles (ER and mitochondria) in astrocytes. Collective stimulation of NKCC1 and NCXrev contributes to these changes.
Keywords: reactive astrocytes, mitochondrial Ca2+, ER Ca2+, ER stress, bumetanide
INTRODUCTION
Cytochrome c (Cyt c) has been suggested to function as a messenger in coordinating endoplasmatic reticulum (ER) and mitochondrial interactions and in accelerating apoptotic cell death (Boehning et al. 2003). Early in apoptosis, Cyt c translocates from mitochondria to ER where it selectively binds to inositol 1,4,5-triphosphate receptor (IP3R) and triggers sustained, oscillatory cytosolic Ca2+ increases, resulting in release of Cyt c from all mitochondria (Boehning et al. 2003). This phenomenon has been identified as a feed-forward mechanism that amplifies the apoptotic signals by a coordinated release of ER Ca2+ and Cyt c (Boehning et al. 2003; Boehning et al. 2004). Coimmunoprecipitation of Cyt c and IP3R type 1 (IP3R1) and/or ryanodine receptor type 2 (RyR2) was detected in gerbil hippocampus following transient brain ischemia (Beresewicz et al. 2006), suggesting that a coordinated release of ER Ca2+ and Cyt c may play a role in ischemic cell damage.
Release of Ca2+ from intracellular Ca2+ stores is a key component in astrocyte function under physiological conditions. This includes ATP-mediated Ca2+ release, which leads to a spatial expansion of astrocyte activation and plays an important role in coordination and synchronization of astrocyte responses to synaptic transmission (Smith et al. 2003; Takano et al. 2009). On the other hand, ER Ca2+ stores sequester Ca2+ to prevent intracellular Ca2+ overload in astrocytes in in vitro model of ischemia such as oxygen/glucose deprivation/reoxygenation (OGD/REOX) (Lenart et al. 2004). This event is accompanied with changes in mitochondrial function including increase of mitochondrial Ca2+ (Ca2+m) and depolarization of mitochondrial membrane potential (Ψm) (Kintner et al. 2007). However, the temporal changes in Ca2+ homeostasis of ER and mitochondria, as well as in mitochondrial Cyt c release are not well studied in astrocytes.
It has been demonstrated that non-NMDA mediated Ca2+ influx plays a significant role in astrocyte damage. For example, ischemia-induced astrocyte death depends on extracellular Ca2+ and is prevented by inhibition of the reverse mode of the Na+/Ca2+ exchanger (NCXrev) (Bondarenko et al. 2005). Pharmacological inhibition or genetic ablation of Na+-K+-Cl− cotransporter isoform 1 (NKCC1) attenuates Ca2+m overload and Ψm depolarization (Kintner et al. 2007). However, it is unknown whether the collective stimulation of NKCC1 and NCXrev plays a role in altering ER and mitochondrial Ca2+ signaling and Cyt c release in ischemic astrocytes.
In the present study, we detected changes in Ca2+ER, Ca2+m, Ca2+cyt as well as Cyt c release in cultured cortical astrocytes following 2 h OGD and 0–180 min REOX. We found that there was a concerted loss of Ca2+ER, Ca2+m, and Ca2+cyt homeostasis and release of Cyt c. Inhibition of NKCC1 and NCXrev activity significantly reduced mitochondrial and ER dysfunctions in astrocytes following OGD/REOX.
EXPERIMENTAL PROCEDURES
Materials
Eagle’s modified essential medium (EMEM) and Hanks balanced salt solution (HBSS) were from Mediatech Cellgro (Manassas, VA). Fetal bovine serum (FBS) was obtained from Valley Biomedical Inc. (Winchester, VA). Collagen-type I was from Collaborative Biomedical Products (Bedford, MA). Bumetanide, carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP), dibutyryl cyclic AMP (dBcAMP), sodium borohydride, and xestospongin C were purchased from Sigma Chemicals (St. Louis, MO). SEA0400 was a kind gift from Taisho Pharmaceutical Co. Ltd. (Omiya, Saitama, Japan). Fura-2 acetoxymethyl ester (Fura-2 AM), BR-A23187, MitoTracker green, mag-fura-2 AM, TO-PRO-3, and rhod 2-AM were from Invitrogen (Carlsbad, CA). 2-APB was from Tocris (Ellisville, MO). Mouse anti-Cyt c monoclonal antibodies (clone 6H2.B4 for immunofluorescence, clone 7H8.2C12 for western blotting) were purchased from BD Pharmingen (SanDiego, CA). Rabbit anti-MnSOD polyclonal antibody and rabbit anti-Calnexin polyclonal antibody were from Stressgen (Ann Arbor, MI). Rabbit anti-IP3R1 antiserum was from Millipore (Billerica, MA). Rabbit anti-phospho-eIF2α polyclonal antibody was from Cell Signaling Technology (Danvers, MA). Mouse anti-GFAP monoclonal antibody was from Sternberger Monoclonals (Lutherville, MD). Rabbit anti-Actin polyclonal antibody was from Santa Cruz Biotechnology (Santa Cruz, CA) Pluronic F-127 was from BASF Corp (Parsippany, NJ).
Animals and genotype analysis
NKCC1 homozygous mutant and wild-type mice (129/SvJ Black Swiss) were obtained by breeding gene-targeted NKCC1 heterozygous mutant mice (Flagella et al. 1999), and genotypeswere determined by polymerase chain reaction (PCR) analysis of DNA fromtail biopsies as described previously (Su et al. 2002)
Primary culture of mouse cortical astrocytes
Dissociated cortical astrocyte cultures were established as described before (Su et al. 2002). Cerebral cortices were removed from 1-day-old NKCC1+/+ or NKCC1−/− mice. The cortices were incubated in a trypsin solution (0.25 mg/ml of HBSS) for 25 min at 37°C. The dissociated cells were rinsed and resuspended in EMEM containing 10% FBS. Viable cells (1×104 cells/well) were plated in petri dishes (100 × 20 mm) or on glass coverslips (22 × 22 mm) in 6-well plates coated with collagen type-1. Cultures were maintained in a 5% CO2 atmosphere at 37°C and refed every 3 days throughout the study. To obtain morphologically differentiated astrocytes, confluent cultures (7 days in culture, DIV 7) were treated with EMEM containing 0.25 mM dBcAMP to induce differentiation. dBcAMP has been widely used to mimic neuronal influences on astrocyte differentiation (Swanson et al. 1997). Experiments were routinely performed in DIV 10–21 cultures.
OGD treatment
NKCC1+/+ or NKCC1−/− astrocytes on coverslips were rinsed twice with an isotonic OGD solution (pH 7.4) containing (in mM, at 37°C): 0 glucose, 26 NaHCO3, 120 NaCl, 5.36 KCl, 0.33 Na2HPO4, 0.44 KH2PO4, 1.27 CaCl2, 0.81 MgSO4. Cells were incubated in 1 ml of the OGD solution in a hypoxic incubator for 2 h (Forma Scientific Inc, model 3130, Marietta, OH), containing 94% N2, 1% O2 and 5% CO2. An orbital shaker (Thermolyne Inc, model M48215, Dubuque, IA) in the hypoxic chamber was used to facilitate equilibration of the hypoxic gases during the initial 30 min OGD. Normoxic control cells were incubated in 5% CO2 and atmospheric air for 2 h in a control HCO3−-MEM buffer containing 5.5 mM glucose. For REOX, OGD-treated cells were incubated with the HCO3−-MEM buffer under conditions identical to the normoxic controls. Cells grown in petri dishes were treated similarly as described above, except the solution volumes were 5 ml.
Cytosolic Ca2+ ([Ca2+]cyt) measurement
Astrocytes grown on coverslips were incubated with 5 μM fura-2 AM during 2 h OGD at 37°C (Lenart et al. 2004). For REOX, the coverslip was quickly (< 2 min) placed on an open-bath imaging chamber and superfused (1 ml/min) with HCO3−-MEM at 37° C. Using a Nikon TE 300 inverted epifluorescence microscope (40X oil immersion objective lens), astrocytes were excited every 5 min at 340 and 380 nm and the emission fluorescence at 510 nm recorded. Images were collected and analyzed with the MetaFluor (Molecular Devices, Sunnyvale, CA) image-processing software. At the end of each experiment, the cells were exposed to 1 mM MnCl2 and 5 μM BR-A23187 in Ca2+-free HEPES-MEM. The Ca2+ insensitive fluorescence was subtracted from each wavelength before calculations (Lenart et al. 2004). The MnCl2-corrected 340/380 emission ratios were converted to cytosolic Ca2+ concentration ([Ca2+]cyt) as described previously (Lenart et al. 2004).
ER Ca2+ ([Ca2+]ER) measurement
Astrocytes on coverslips were incubated with 4 μM mag-fura-2 AM and 0.02 % pluronic acid during 2 h OGD. In a previous study, we validated the selective determination of [Ca2+]ER by mag-fura-2 AM (Chen et al. 2008). The coverslip was placed on an open-bath imaging chamber in HEPES-MEM at 37°C. Cells were excited every 10 sec at 345 and 385 nm and the emission fluorescence images collected at 510 nm using Nikon TE 300 inverted epifluorescence microscope (40X objective lens). The Ca2+ER values were calculated using the equation . K was determined as 72 μM in astrocytes using solutions of known Ca2+ concentrations (Calcium Calibration Buffer Kit, Invitrogen, Carlsbad, CA). Rmin was obtained from a minimum F345/385 ratio in a Ca2+-free solution. Rmax was the maximum F345/385 ratio in a high Ca2+ solution (10 mM CaCl2).
Measurement of mitochondrial Ca2+ (Ca2+m)
Astrocytes on coverslips were incubated at 37°C for 60 min with 200 nM MitoTracker green and 9 μM rhod2-AM which was reduced in the presence of a minimum of sodium borohydride and 3 mM sodium succinate in HCO3− MEM (Marks et al. 2005; Kintner et al. 2007). Coverslips were placed in the perfusion chamber on the stage of the Leica DMIRE2 confocal microscope and superfused (1 ml/min) with HCO3−-MEM at 37° C. Cells (1–3 in the field) were visualized with a 60X oil-immersion objective and scanned sequentially for MitoTracker green (ex. 488 nm argon laser line, em. 500–545 nm) and rhod-2 (ex. 543 HeNe laser, em 544–677). The MitoTracker green signal was used to maintain focus prior to each sequential scan. Sequential scans were analyzed using the Leica confocal software. Average grayscale values were collected from regions of interest around mitochondrial clusters exhibiting colocalization of MitoTracker green and rhod-2. Ca2+m values were expressed as relative change of rhod-2 signals from the baseline values and summarized data represent the average of the calculated values from 7–10 cells/experiment as described before (Kintner et al. 2007).
Cyt c immunofluorescence staining
Cyt c release from mitochondria was determined by double staining of cells with specific antibody against Cyt c and specific antibody against mitochondrial protein manganese superoxide dismutase (MnSOD). Briefly, cells on coverslips were fixed in 4% paraformaldehyde in PBS for 15 min. After rinsing, cells were incubated with blocking buffer (10% normal goat serum, 0.4% Triton X-100 and 0.5% BSA in PBS) for 20 min followed by application of anti-Cyt c antibody (1:100 diluted in the blocking buffer) and anti-MnSOD antibody (1:50 diluted in the blocking buffer) for 1 h at 37°C. After rinsing in PBS, cells were incubated with goat anti-mouse secondary antibody IgG (H+L) conjugated to Alexa Fluor® 488 (1:200 dilution) and goat anti-rabbit secondary antibody IgG (H+L) conjugated to Alexa Fluor® 546 (1:200 dilution) for 1 h at 37°C. Nuclei were stained with TO-PRO-3 (1:1000 diluted in PBS) for 15 min at 37°C. Using the Leica DMIRE2 inverted confocal laser-scanning microscope, samples were excited at 488 nm (argon/krypton), 543 nm, and 633 nm and the emission fluorescence was recorded at 512–548 nm, 585–650 nm, or 650–750 nm, respectively. A single optical section of the cells was obtained with a 100X lens and Leica confocal software.
Quantitative analysis of the Cyt c release
Cyt c release from mitochondria was quantified by its co-localization with the mitochondrial protein MnSOD using Image J software (NIH, Bethesda, MD) with the Image J plug-in JACoP. An estimate of the colocalization events between both images was obtained by calculating the Pearson’s coefficient (Rp) (Bolte and Cordelieres 2006) and Manders’ coefficient (M2) (Manders et al. 1992). The Pearson’s coefficient is a global statistical approach that performs intensity correlation coefficient-based analysis with an Rp value ranging from 1 to −1 (1: complete positive correlation, −1: negative correlation, zero: no correlation). Manders’ coefficients (M1 and M2) are based on the Pearson’s correlation coefficient with average intensity values being taken out of the mathematical expression (Manders et al. 1992). Manders’ coefficients vary from 0 (non-overlapping) to 1 (100% co-localization). M2 is defined as the ratio of the “summed intensities of pixels from the MnSOD red image for which the intensity in the green channel is above zero” to the “total intensity in the red channel”(Manders et al. 1992). Therefore, M2 is a good indicator of the proportion of the red signal coincident with a signal in the green channel over its total intensity. For quantitative analysis, single optical sections were taken randomly from 5–7 different regions on each coverslip and 5 cells were analyzed from each optical section. The data were obtained from 3–6 independent cultures (n = 3–6).
Subcellular fractionation
Cells were harvested by gently scraping plates with a cell scraper and washing once with ice-cold PBS. The cell pellets were resuspended in 1 ml ice cold buffer A [250 mM sucrose, 10 mM Tris-HCl at pH 7.5, 1 mM EGTA and protease inhibitors, as described previously (Sun et al. 1995)]. Cells were then homogenized on ice using a 1-ml glass Dounce homogenizer with a tight-fitting pestle (95% of cells were disrupted as detected by Trypan blue staining). Crude lysates were centrifuged at 1,000g for 15 min at 4°C to remove nuclei and unbroken cells in the resulting pellet (P1). The low-speed supernatant was collected and then subjected to a 10,000g centrifugation for 15 min, yielding the 10,000g pellet (P2, containing mitochondria and heavy ER). The supernatant from the P2 pellet was centrifuged at 15,000g in order to remove any remaining mitochondria. Finally, the 15,000g supernatant was separated into cytosol (S3) and light membrane (P3, light ER) fractions by centrifugation at 100,000g for 1 h. The 100,000g supernatant was collected as the S3 fraction and the pellet was resuspended in 60 μl of buffer A. The P2 fraction was washed twice by resuspending cells in 100 μl buffer A and pelleting (10,000g for 15 min). After the final wash, the P2 fraction was resuspended in 60 μl buffer A. The protein content in each fraction (P2, S3, P3) was measured with the bicinchoninic acid method.
Gel Electrophoresis and Western Blotting
Protein samples and pre-stained molecular mass markers (Bio-Rad, Hercules, CA) were denatured in SDS reducing 5X sample buffer and boiled at 90°C for 5 min before gel electrophoresis. The protein samples (40–100 μg/lane) were loaded and separated by SDS-PAGE (4–15% gradient gel or 10% gel). After transferring to a nitrocellulose membrane, the blots were blocked in 7.5% nonfat dry milk in Tris-buffered saline (TBS) for 2 h at room temperature and then incubated with a primary antibody at 4°C overnight. After rinsing, the blots were incubated with horseradish peroxidase-conjugated secondary IgG for 1.5 h at room temperature. Bound antibody was visualized using the enhanced chemiluminescence assay (Amersham Corp, Piscataway, NJ). Relative changes in protein expression were estimated from the integrated pixel intensity of each protein band using Image J software. Mouse anti-Cyt c monoclonal antibody (clone 7H8.2C12) (1:800), rabbit anti-IP3R1 antiserum (1:1000), rabbit anti-MnSOD polyclonal antibody (mitochondria marker, 1:2000), rabbit anti-Calnexin (CNX) polyclonal antibody (microsomal constituent marker, 1:1000), rabbit anti- the alpha subunit of eukaryotic initiation factor 2 (p-eIF2α) polyclonal antibody (1:500), and mouse anti-actin monoclonal antibody (1:3000) were used.
Statistics
Throughout the study, n values represented the number of cultures used in each experiment. Standard error of mean values for each experimental group was reported as a gauge of the accuracy of the calculated mean. For western blotting assay, standard deviation was reported in the summary data. Statistical significance was determined by either Student’s t-test or an ANOVA (Bonferroni post-hoc test) for multiple comparisons at a confidence of 95% (p < 0.05).
RESULTS
Release of Cyt c from mitochondria in astrocytes following in vitro ischemia
Normoxic control astrocytes had a punctate perinuclear pattern of Cyt c expression (Figure 1A). Cyt c signals were colocalized with immunostaining of mitochondrial protein Mn-superoxide dismutase (MnSOD) (thin arrow, Figure 1A). At 60 min REOX following 2 h OGD, mitochondria were swollen (*, Figure 1B) with punctate Cyt c distribution. By 160 min REOX, in some astrocytes, Cyt c immunostaining was absent and only MnSOD immunoreactive signals were detected in mitochondria (arrowhead, Figure 1C). Moreover, Cyt c immunoreactivity was enhanced in other astrocytes in which MnSOD immunostaining signals were clearly not colocalized with Cyt c (Figure 1C, thick arrow). In contrast, in astrocytes treated with 10 μM bumetanide, a potent inhibitor of NKCC1, the Cyt c release at 160 min REOX was absent (Figure 1D, thin arrow) and accompanied with less mitochondrial swelling. Similar to the bumetanide-treated astrocytes, NKCC1−/− astrocytes exhibited more resistance to the OGD/REOX-mediated mitochondrial damage. No detectable Cyt c release at 160 min REOX was observed in NKCC1−/− astrocytes (Figure 1E, thin arrow). In a positive control study for Cyt c release, 10 μM FCCP exposure (4 min) triggered mitochondrial swelling and non-colocalization of Cyt c and MnSOD (Figure 1F, arrowhead, thick arrow), which is similar to the ischemic astrocytes.
Figure 1. Release of Cyt c from mitochondria in astrocytes following OGD/REOX.
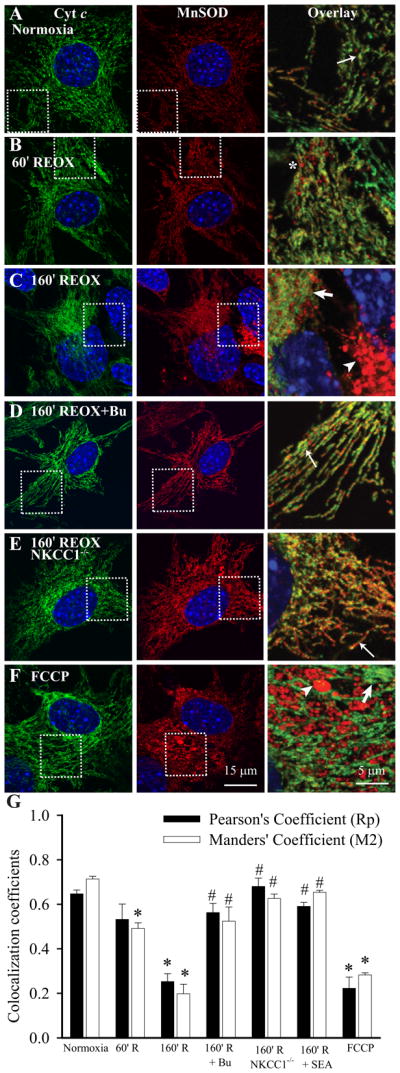
Localization of Cyt c was visualized by staining of astrocytes with an anti-Cyt c antibody (green) and with an antibody against the mitochondrial marker protein MnSOD (red). Nuclei were stained with TO-PRO-3 dye (blue). Overlay: corresponding Cyt c and MnSOD signals in the boxed region (3X magnification). A. Normoxic control conditions. B. 60 min REOX following 2 h OGD. C. 160 min REOX. D. 160 min REOX, treated with bumetanide (Bu, 10 μM) during 0–160 min REOX. E. Astrocytes from NKCC1−/− mice at 160 min REOX. F. Astrocytes treated with 10 μM FCCP for 4 min. Thin arrow: colocalized Cyt c and MnSOD signals. Thick arrow: non-colocalized Cyt c and MnSOD signals. Arrowhead: cells with MnSOD signals only. *: swollen mitochondria. G. Summary data of Rp and M2. The Cyt c release in astrocytes was analyzed with Image J software and the JACop plug-in. In the SEA0400 study, SEA0400 (1 μM) was present during 0–160 min REOX. Data are means ± SEM; n = 3–13. * p < 0.05 vs. normoxia; # p < 0.05 vs. 160 min REOX.
Quantitative analysis of Cyt c release in astrocytes
As shown in Figure 1G, the Rp was 0.65 ± 0.02 and the M2 was 0.72 ± 0.02 in the normoxic control astrocytes, reflecting the relative co-localization of Cyt c and MnSOD under this condition. At 60 min REOX when there was moderate Cyt c release, Rp was reduced to 0.53 ± 0.07 and M2 dropped to 0.49 ± 0.03 (p < 0.05). Moreover, when Cyt c and MnSOD became non-colocalized at 160 min REOX, Rp decreased further to 0.25 ± 0.04 (p < 0.05), accompanied with a deep drop in the M2 value (0.20 ± 0.05, p < 0.05). Interestingly, cells treated with 10 μM bumetanide or cells from NKCC1−/− mice exhibited significantly less reduction in Rp and M2 values at 160 min REOX (p < 0.05). On the other hand, in cells where Cyt c release was triggered with 10 μM FCCP, Rp and M2 values were significantly reduced (p < 0.05), similar to those at 160 min REOX.
To further investigate the role of ionic dysregulation in OGD/REOX-induced Cyt c release from mitochondria, we used SEA0400, a selective inhibitor of NCXrev activity. In astrocytes treated with the NCXrev inhibitor SEA0400 (1 μM) during REOX, the Cyt c release at 160 min REOX was absent (data not shown). The summary data in Figure 1G illustrate that cells treated with SEA0400 did not exhibit significant reduction in Rp (0.59 ± 0.02) and in M2 values (0.65 ± 0.01) at 160 min REOX. This was in contrast to the low Rp (0.25 ± 0.04) and M2 (0.20 ± 0.05) values in the 160 min REOX groups. These data further demonstrates a role for NCXrev in mitochondrial damage in ischemic astrocytes.
Colocalization of Cyt c and MnSOD in astrocytes was further analyzed by plotting the distribution of Rp and M2 values. The majority of normoxic control astrocytes exhibited high Rp or M2 values with a normal distribution (Kolmogorov-Smirnov normality test, p > 0.2, Suppl. Figure 1 A, C). In contrast, following 2 h OGD/160 min REOX, the number of astrocytes with low Rp or M2 values were increased and the cell distribution drastically shifted to the left part of the x-axis (Suppl. Figure 1 A, C). Moreover, the OGD/REOX-treated cells were not normally distributed (Kolmogorov-Smirnov normality test, p < 0.05). On the other hand, either pharmacological inhibition of NKCC1 with bumetanide or genetic ablation prevented the Cyt c release. Cells maintained a normal distribution pattern with high values of Rp or M2 (Suppl. Figure 1 B, D). Taken together, these analyses clearly demonstrate a release of Cyt c from mitochondria in astrocytes following OGD/REOX, and the protective effects of NKCC1 inhibition with bumetanide or by genetic ablation.
Delayed elevation in [Ca2+]cyt following OGD/REOX
We investigated the time course of changes in [Ca2+]cyt following OGD/REOX. As shown in Figure 2A, there were no changes in [Ca2+]cyt after 2 h OGD. Interestingly, [Ca2+]cyt started to rise at ~ 120 min REOX at a rate of ~ 8 nM/min and reached a plateau (358 ± 25 nM) between 160–180 min REOX. Moreover, the timing of the Ca2+cyt increase was close to the release of significant Cyt c at 160 min REOX. In contrast, inhibition of NKCC1 activity with bumetanide abolished the delayed rise in [Ca2+]cyt (Figure 2B). To test the role of NCXrev in delayed Ca2+ dysregulation, we examined whether SEA0400 (1μM), a potent inhibitor of NCXrev, can block Ca2+ rise during OGD/REOX. Inhibition of NCXrev prevented the delayed rise in [Ca2+]cyt (Figure 2B), which is similar to the effects of NKCC1 inhibition.
Figure 2. Delayed elevation in [Ca2+]cyt following OGD/REOX.
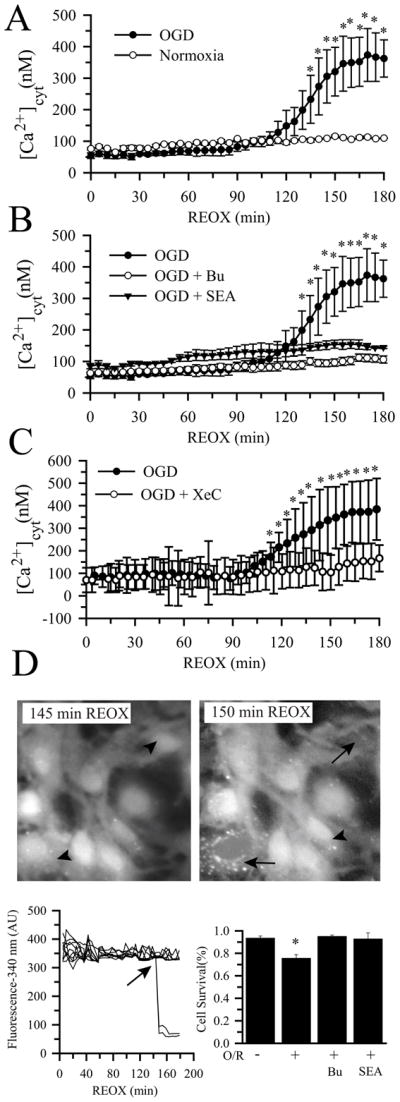
A. [Ca2+]cyt was monitored in single NKCC1+/+ astrocytes during 0–180 min REOX following 2 h OGD. Normoxic control were sister NKCC1+/+ astrocytes subjected to 3 h normoxic superfusion. B. Effects of bumetanide and SEA0400 on [Ca2+]cyt. Bumetanide (5 μM) or SEA0400 (1 μM) was present during REOX. Data are means ± SEM; n = 3–4, * p < 0.05 vs. normoxia. C. Effects of xestospongin C (XeC). Xestospongin C (20μM) was present during REOX only. Data are means ± SD (n = 20 cells). * p < 0.05 vs. OGD/REOX + xestospongin C. D. Summary data of astrocyte death. Upper panel: fura-2 fluorescence in live astrocytes (arrowhead) and dying astrocytes (loss of fura-2 signal, arrow). Lower panels: sudden loss of fura-2 fluorescence (340 nm wavelength) in dying astrocytes during OGD/REOX (O/R). Data are means ± SEM; n = 3–4, * p < 0.05 vs. normoxia.
We speculated that the delayed elevation of [Ca2+]cyt may result from IP3R-mediated Ca2+ release from ER. As shown in Figure 2C, inhibition of IP3R with its specific inhibitor xestospongin C (20 μM) largely blocked the delayed rise in [Ca2+]cyt, suggesting ER Ca2+ stores as the source for the post-OGD Ca2+ rise in the cytosol. Moreover, we observed a sudden loss of fura-2 fluorescence signal in some astrocytes during 120–180 min REOX (Figure 2 D-left panel, arrow). These dying cells with damaged membrane integrity failed to retain the dye. As shown in the lower panel of Figure 2 D-right panel, normoxic astrocytes exhibited a basal level of cell death (6% ± 2 %). In contrast, 24 ± 3 % astrocytes died during 180 min REOX following 2 h OGD. Most importantly, a time-dependent cell death analysis revealed that the majority of cell death occurred during 125–180 min REOX, when the cytosolic Ca2+ dysregulation was triggered (Suppl. Figure 2). Inhibition of NKCC1 or NCXrev activity with Bu or SEA0400 prevented the OGD/REOX-induced astrocyte cell death (Figure 2D).
Biphasic changes in [Ca2+]ER and Ca2+m following OGD/REOX
The delayed rise in [Ca2+]cyt may result from the release of Ca2+ER. To test this hypothesis, changes of [Ca2+]ER were determined. Figure 3A shows that [Ca2+]ER was increased from a resting level of 27 ± 3 μM to 65 ± 3 μM at 2 h OGD (p < 0.05). During REOX, [Ca2+]ER was further augmented and reached a peak value of 130 ± 2 μM by 90 min REOX. However, after ~ 90 min REOX, Ca2+ER was abruptly released, occurring at the time when [Ca2+]cyt was increasing. Ca2+ER appeared to start refilling at 3 h REOX. These findings demonstrate that OGD/REOX causes Ca2+ER biphasic dysregulation in astrocytes. The release of Ca2+ER may in part contribute to the detrimental delayed increase in Ca2+cyt.
Figure 3. Biphasic changes in [Ca2+]ER and Ca2+m following OGD/REOX.
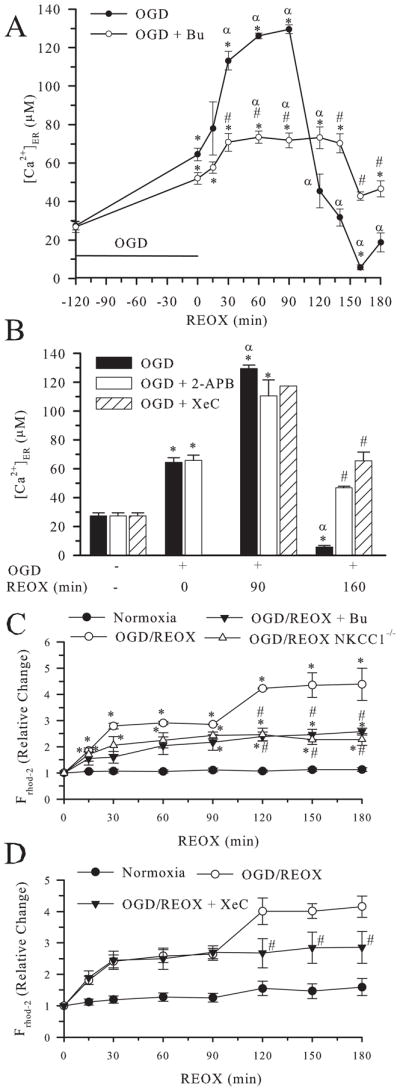
A. [Ca2+]ER was determined in NKCC1+/+ astrocytes under normoxic control conditions and at 0–180 min REOX following 2 h OGD. In the bumetanide study, bumetanide (5 μM) was present during REOX. B. [Ca2+]ER was determined in NKCC1+/+ astrocytes treated with 2-APB, a nonspecific IP3R inhibitor (100 μM) during 0 min, 90 min, or 160 min REOX, or a specific IP3R inhibitor xestospongin C (20 μM, XeC). Data are means ± SEM; n = 3–9 (for XeC at 90 min REOX, n = 1). * p < 0.05 vs. normoxia, # p < 0.05 vs. OGD/REOX untreated; α p < 0.05 vs. 0 min REOX. C. Relative change in Ca2+m was determined in NKCC1+/+ and NKCC1−/− astrocytes under normoxic conditions or at 0–180 min of REOX. Data are means ± SEM; n = 3–5. * p < 0.05 vs. normoxia, # p < 0.05 vs. OGD/REOX. D. Relative change in Ca2+m was determined in NKCC1+/+ astrocytes under normoxic conditions or at 0–180 min of REOX. Xestospongin C (XeC, 20 μM) was present during REOX only. Data are means ± SEM; n = 8–35 cells. # p < 0.05 vs. OGD/REOX.
Consistent with its effect on [Ca2+]cyt, inhibition of NKCC1 activity with bumetanide during REOX not only significantly attenuated REOX-mediated augmentation of Ca2+ER loading, but also blocked REOX-triggered Ca2+ER release (Figure 3A). To investigate whether IP3R plays a role in Ca2+ER release, inhibition of IP3R with a non-selective inhibitor 2-APB (100 μM) was tested. As shown in Figure 3B, 2-APB did not significantly affect accumulation of Ca2+ER during 0–90 min REOX, but it reduced the release of Ca2+ER during 2–3 h REOX. To further confirm the role of the IP3R in the OGD/REOX-mediated release of Ca2+ER, we also used the specific IP3R blocker xestospongin C (20 μM). The presence of xestospongin C during OGD/REOX did not affect Ca2+ER accumulation at 90 min REOX, but it did prevent depletion of Ca2+ER stores at 160 min REOX (Figure 3B). Taken together, these data suggest that REOX-triggered Ca2+ER release is in part due to IP3R activation.
To further investigate whether REOX-triggered Ca2+ER release would affect mitochondrial Ca2+ homeostasis and mitochondrial function, we next monitored the relative change in the rhod-2 florescence in mitochondria following 2 h OGD. Three hours of normoxic perfusion did not result in significant changes in Ca2+m (Figure 3C). However, within 15 min of REOX following 2 h OGD, there was a significant increase in Ca2+m that reached a plateau by 30 min REOX (~ 3-fold, Figure 3C). Interestingly, there was a second increase in Ca2+m between 90–120 min of REOX, when Ca2+ER release and increases in [Ca2+]cyt occurred. Inhibition of NKCC1 either with bumetanide or by genetic ablation did not prevent the initial post-OGD loading of Ca2+m, but completely blocked the secondary rise in Ca2+m (Figure 3C). Interestingly, inhibition of IP3R with xestospongin C also abolished the second phase increase in Ca2+m, implying that the source for this second Ca2+m rise is ER (Figure 3D). Taken together, the data suggest that the OGD/REOX-induced released of Ca2+ER is partially buffered by mitochondria and this may facilitate mitochondrial Cyt c release.
Translocation of Cyt c from mitochondria to ER at 160 min REOX
Translocation of Cyt c into ER may play a role in IP3R activation and Ca2+ER release. We measured relative changes of Cyt c content in different subcellular fractionations (mitochondria and heavy endoplasmic reticulum; cytosol; and endoplasmic reticulum) following OGD/REOX. Figure 4A shows that the cytosol fraction lacked mitochondrial MnSOD and ER marker protein calnexin (CNX) expression. No MnSOD was detected in ER fraction (P3). Thus, relatively pure cytosol and ER fractions were obtained in this study. However, the P2 fraction (mitochondria and heavy endoplasmic reticulum) contained both MnSOD and CNX. The impurity of mitochondrial fraction is less relevant to this study, because our focus is on Cyt c translocation to the ER fraction (P3).
Figure 4. Translocation of Cyt c to ER following OGD/REOX.

A. Cyt c content in mitochondria (P2), cytosol (S3), and ER (P3) fractions was determined. The mitochondrial protein MnSOD and the ER protein calnexin (CNX) were used to determine purity of the fractions and to ensure equal protein loading. 10 μM bumetanide was present during REOX in the drug treated group. B. Representative immunoblot of Cyt c in ER fraction. CNX on the same blot was probed for sample loading control. Summary data are the ratio of Cyt c/CNX intensities. Data are means ± SEM, n = 7. * p < 0.05 vs. 160 min REOX.
Under normoxic control conditions, Cyt c was largely located in mitochondria and only a trace level of Cyt c was detected in ER fraction (Figure 4A). At 120 min REOX after 2 h OGD, the Cyt c level in ER was elevated and further increased by 160 min REOX. The Cyt c/CNX ratio was increased by ~ 2-fold at 160 min REOX (Figure 4B). Moreover, inhibition of NKCC1 with 10 μM bumetanide significantly reduced the amount of Cyt c translocated to ER fraction at 160 min REOX (p < 0.05).
Changes of ER stress protein following OGD/REOX
Ca2+ER dysregulation can lead to the unfolded protein response and ER stress development (Groenendyk and Michalak 2005). We examined whether expression of ER stress proteins such as the phosphorylated form of eIF2α (p-eIF2α) was altered. As shown in Figure 5A, expression of p-eIF2α protein was increased at 2 h OGD and reached a peak level (~3.5-fold) at 120 min REOX (p < 0.05), when ER Ca2+ depletion occurred. p-eIF2α remained elevated at 3 h REOX. Inhibition of NKCC1 activity with bumetanide during 0–3 h REOX attenuated the up-regulation of p-eIF2α protein (Figure 5B). These results imply that Ca2+ER dysregulation was accompanied by transient ER unfolded protein responses during 2 h OGD and 0–3 h REOX.
Figure 5. ER stress protein expression.

A. OGD/REOX-mediated up-regulation of p-eIF2α. Upper panel: Representative blot showing expression of p-eIF2α protein in normoxic control, 2 h OGD, or 1–3 h REOX. The same blot was probed with anti-actin antibody as a loading control. Lower panel: Summary graph of the relative expression of p-eIF2α protein/actin ratio. Data are means ± SEM. n = 3–4. * p < 0.05 vs. Con. B. Representative blot showing that the expression of p-eIF2α protein was reduced in astrocytes treated with 10 μM bumetanide during 0–3 h REOX (Upper panel). Lower panel: Summary graph of p-eIF2α protein/actin ratio. Data are means ± SEM n = 3–4. * p < 0.05 vs. OGD/REOX.
DISCUSSION
OGD/REOX disrupts ER Ca2+ store homeostasis
In this study, we found that 2 h of OGD led to an ~2.4 fold Ca2+ accumulation in ER stores. During REOX, Ca2+ER continued to rise until an abrupt release of ER Ca2+ occurred between 90–160 min REOX. Interestingly, the release of Ca2+ from ER stores was accompanied by a delayed rise in Ca2+cyt. These findings demonstrate that astrocyte ER Ca2+ stores buffer cytosolic Ca2+ in an attempt to maintain cytosolic Ca2+ homeostasis following OGD/REOX. An increase in IP3R-sensitive ER Ca2+ stores has been revealed in astrocytes under conditions of Na+/K+ ATPase inhibition or chronic hypoxia (Golovina and Blaustein 2000; Smith et al. 2003). The absence of detectable increases in Ca2+cyt during early REOX suggests that ER is located near the sites of Ca2+ influx across the plasma membrane and facilitates Ca2+ uptake into ER stores. Indeed, it has been reported that NCX-1 and Na+/K+ ATPase are co-distributed in the plasma membrane regions of smooth muscle and are also in register with the sarcoplasmic reticulum (Moore et al. 1993). Furthermore, Goldman et al. showed that when NCXrev is stimulated by application of ouabain and reduction of extracellular Na+, NCXrev-induced increases in Ca2+cyt can be buffered by ER (Goldman et al. 1994). Our findings further indicate that excessive Ca2+ influx via stimulation of NCXrev in ischemic astrocytes can be rapidly taken up by ER.
Here we report first-line evidence that astrocytes deplete ER Ca2+ stores in an in vitro model of ischemia. The release of Ca2+ from ER stores between 90 –160 min of REOX was associated with the loss of Ca2+cyt homeostasis as well as with a secondary increase in Ca2+m and the release of Cyt c from mitochondria. Interestingly, depletion of ER Ca2+ stores was muted by both non-specific (2-APB) and specific (xestospongin C) inhibitors of IP3R, suggesting a primary role for IP3R in the ER Ca2+ depletion in astrocytes. Both global or focal cerebral ischemia cause hydrolysis of poly-PI and formation of IP3 through activation of PLC (Lin et al. 1991; Yoshida et al. 1986). Group I metabotropic glutamate receptor (mGluR) stimulates poly-PI turnover and therefore can influence IP3 levels (Conn and Pin 1997). Activation of mGluR5 has been shown to trigger a sustained astrocyte Ca2+ oscillation in the ischemic core and penumbra following photothrombosis ischemia, in part due to IP3-induced Ca2+ release (Ding et al. 2009; Bruno et al. 2001). Our findings further suggest that IP3R-mediated ER Ca2+ depletion in astrocytes may occur in ischemic brain particularly during the reperfusion period and exacerbate astrocyte damage.
Effects of ER Ca2+depletion on astrocyte function
Loss of Ca2+ER homeostasis can affect many functions of the ER, including changes in protein folding and in ER stress (Orrenius et al. 2003). Depletion of Ca2+ER stores with thapsigargin can lead to ER stress (Paschen and Doutheil 1999) which induces suppression of protein synthesis, polyribosomal disaggregation, and activation of ER stress genes (Doutheil et al. 1997; Mengesdorf et al. 2001). This ER stress response is almost identical to the responses found in transient cerebral ischemia (Althausen et al. 2001; Kumar et al. 2003; DeGracia et al. 1999). Glial degeneration after hypoxia-ischemia or focal ischemia has been detected in immature and mature brains with both apoptotic or necrosis cell death features (Gelot et al. 2009; Giffard and Swanson 2005). The underlying cell damage mechanisms involve activation of caspase-dependent signaling pathways and oxidative stress (Ouyang et al. 2007). In our current study, we observed that OGD/REOX caused a sharp, sustained elevation of ER stress protein p-eIF2α, which was accompanied with dysregulation of ER Ca2+ and ~ 25% cell death. This suggests that ER Ca2+ dysregulation and ER stress may also contribute to astrocyte demise in ischemic brains. Future experiments should include investigating whether these changes occur in reactive astrocytes in in vivo ischemic brains.
It is well established that Ca2+ transients and/or synchronized Ca2+ transients (Ca2+ oscillations) represent a form of astrocyte excitability and play an important role in astrocyte-astrocyte as well as in astrocyte-neuron intercellular communication (Koizumi 2010). These regulated increases in astrocyte [Ca2+]cyt can result from release of Ca2+ from ER stores and trigger gliotransmitter release (glutamate, purines). Astrocytes respond to hypoxia/ischemia insults by developing reactive astrogliosis, which is reflected by up-regulation of GFAP expression, astrocyte hypertrophy, and proliferation (Xiong et al. 2009; Hwang et al. 2008; Ouyang et al. 2007). Reactive astrocytes change in their ability to release, take up, and metabolize gliotransmitters. Thus, they may cause unusual excitation of adjacent local neuronal networks and participate in pathophysiology of epilepsy after brain injury (Koizumi 2010). In our current study,~ 75% of the astrocytes did not die, but exhibited depletion of Ca2+ER and sustained elevation in [Ca2+]cyt following OGD/REOX. If these changes also occur in reactive astrocytes following in vivo cerebral ischemia, they may represent the dysfunction of reactive astrocytes and contribute to ischemic brain damage. This speculation requires confirmation in a future study.
Mitochondrial dysfunction following OGD/REOX
In our study, longer REOX periods (90–160 min REOX) resulted in secondary increases in Ca2+m. Interestingly, the secondary increase in Ca2+m occurred in the same time period when Ca2+ER release and [Ca2+]cyt were elevated. Increases in Ca2+m can directly induce formation of the mitochondrial permeability transition pore (PTP) in the inner mitochondrial membrane (Ichas et al. 1997) and cause the depolarization of the mitochondrial membrane potential. Reichert and colleagues reported that inhibition of PTP by cyclosporin A could prevent loss of mitochondrial membrane potential in astrocytes and block the subsequent Cyt c release triggered by 45–60 min of OGD (Reichert et al. 2001).
In our current study, we found that 120 min REOX following 2 h OGD triggered Cyt c translocation from mitochondrial fraction to ER fraction. The amount of Cyt c translocated to ER increased further at 160 min REOX. One possible explanation for the translocation of Cyt c to ER is the close physical association or “privileged communication” between ER and mitochondria (Rizzuto et al. 1998). The lack of significant Cyt c accumulation in cytosol has been reported in astrocytes after OGD/REOX (Reichert et al. 2001), which has been attributed to rapid modification and/or degradation of Cyt c after its release from mitochondria. It should be noted that in the current study, we consistently observed abundant Cyt c signals in the mitochondrial fraction (regardless of experimental conditions) as assayed by the immunoblotting, compared to the immunostaining data. This discrepancy may in part be attributed to the epitope of Cyt c being readily exposed under the denatured conditions of immunoblotting, thereby enhancing the antibody binding.
We hypothesized that the released Cyt c binds to IP3R in ER and facilitate Ca2+ER release. Cyt c binding to the C terminus of IP3R1 has been confirmed in yeast 2-hybrid analysis in vitro and in HeLa and PC12 cell lines (Boehning et al. 2003). Furthermore, the binding of Cyt c to IP3R1, as well as to ryanodine receptor type 2, was detected in gerbil hippocampus following transient brain ischemia (Beresewicz et al. 2006). However, recent studies suggest that IP3R2 plays an important role in ER Ca2+ release in cortical and hippocampal astrocytes (Sheppard et al. 1997; Petravicz et al. 2008). Future studies are required to determine whether the released Cyt c binds to IP3R1 or IP3R2 in astrocytes following OGD/REOX and whether it enhances Ca2+ER release.
In the current study, we also observed that the NKCC1 inhibitor bumetanide significantly blocked the secondary increase in Ca2+m, release of Ca2+ER and Cyt c. NKCC1 activity following OGD leads to intracellular Na+ overload that stimulates Ca2+ entry via NCX rev (Kintner 2007). Inhibition of NCXrev activity by SEA0400 abolished the Cyt c release in the current study. These results imply thatNKCC1 coupled with NCX rev play a role in promoting influx of Ca2+ following OGD/REOX, which may eventually overwhelm the buffering capacity of ER and mitochondria and triggers Cyt c release and the collapse of mitochondrial function.
In summary, we report here that 2 h OGD/0–3 h REOX triggered biphasic changes of Ca2+ER and a delayed increase of Ca2+cyt and Ca2+m in astrocytes. The abrupt Ca2+ER release during 90–160 min was accompanied with a second rise of Ca2+m and a profound Cyt c release from mitochondria. The translocation of Cyt c to ER may enhance IP3R-dependent Ca2+ER release. Interestingly, inhibition of ion transporters such as NKCC1 and NCXrev prevented many of these changes. Taken together, the coupling of Ca2+ ER signaling and mitochondrial dysfunction may play an important role in astrocyte damage following ischemia.
Supplementary Material
Acknowledgments
This work was supported in part by an NIH grant R01NS38118 and AHA EIA 0540154 (D. Sun), NIH grant P30 HD03352 (Waisman Center), an NIH grant R01HL61974 (G. E. Shull), and China Scholarship Council Postgraduate Scholarship Program (Y. Liu).
ABBREVIATIONS
- Bu
bumetanide
- dBcAMP
dibutyryl cyclic AMP
- [Ca2+]cyt
calcium concentration cytoplasmic
- [Ca2+]ER
calcium concentration endoplasmic reticulum
- Ca2+m
mitochondrial calcium
- CNX
calnexin
- Cyt c
cytochrome c
- DIV
days in culture
- ER
endoplasmatic reticulum
- FBS
fetal bovine serum
- FCCP
carbonyl cyanide-p-trifluoromethoxyphenylhydrazone
- Fura-2 AM
Fura-2 acetoxymethyl ester
- HBSS
Hanks balanced salt solution
- IP3R
inositol 1,4,5-triphosphate receptor
- M2
Manders’ coefficient
- Ψm
mitochondrial membrane potential
- mGluR
Group I metabotropic glutamate receptor
- MnSOD
manganese superoxide dismutase
- NCXrev
reverse mode function of Na+/Ca2+ exchanger
- NKCC1
Na+-K+-Cl− cotransporter isoform 1
- OGD/REOX
oxygen glucose deprivation and reoxygenation
- p-eIF2α
phosphorylated eukaryotic initiation factor 2 alpha
- PTP
mitochondrial permeability transition pore
- Rp
Pearson’s coefficient
- RyR2
ryanodine receptor type 2
- XeC
xestospongin C
References
- Althausen S, Mengesdorf T, Mies G, Olah L, Nairn AC, Proud CG, Paschen W. Changes in the phosphorylation of initiation factor eIF-2 alpha, elongation factor eEF-2 and p70 S6 kinase after transient focal cerebral ischaemia in mice. J Neurochem. 2001;78:779–787. doi: 10.1046/j.1471-4159.2001.00462.x. [DOI] [PubMed] [Google Scholar]
- Beresewicz M, Kowalczyk JE, Zablocka B. Cytochrome c binds to inositol (1,4,5) trisphosphate and ryanodine receptors in vivo after transient brain ischemia in gerbils. Neurochem Int. 2006;48:568–571. doi: 10.1016/j.neuint.2005.11.020. [DOI] [PubMed] [Google Scholar]
- Boehning D, Patterson RL, Sedaghat L, Glebova NO, Kurosaki T, Snyder SH. Cytochrome c binds to inositol (1,4,5) trisphosphate receptors, amplifying calcium-dependent apoptosis. Nat Cell Biol. 2003;5:1051–1061. doi: 10.1038/ncb1063. [DOI] [PubMed] [Google Scholar]
- Boehning D, Patterson RL, Snyder SH. Apoptosis and calcium: new roles for cytochrome c and inositol 1,4,5-trisphosphate. Cell Cycle. 2004;3:252–254. [PubMed] [Google Scholar]
- Bolte S, Cordelieres FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- Bondarenko A, Svichar N, Chesler M. Role of Na+-H+ and Na+-Ca2+ exchange in hypoxia-related acute astrocyte death. Glia. 2005;49:143–152. doi: 10.1002/glia.20107. [DOI] [PubMed] [Google Scholar]
- Bruno V, Battaglia G, Copani A, D’Onofrio M, Di Iorio P, De Blasi A, Melchiorri D, Flor PJ, Nicoletti F. Metabotropic glutamate receptor subtypes as targets for neuroprotective drugs. J Cereb Blood Flow Metab. 2001;21:1013–1033. doi: 10.1097/00004647-200109000-00001. [DOI] [PubMed] [Google Scholar]
- Chen X, Kintner DB, Luo J, Baba A, Matsuda T, Sun D. Endoplasmic reticulum Ca2+ dysregulation and endoplasmic reticulum stress following in vitro neuronal ischemia: role of Na+-K+-Cl− cotransporter. J Neurochem. 2008;106:1563–1576. doi: 10.1111/j.1471-4159.2008.05501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- DeGracia DJ, Adamczyk S, Folbe AJ, et al. Eukaryotic initiation factor 2 alpha kinase and phosphatase activity during postischemic brain reperfusion. Exp Neurol. 1999;155:221–227. doi: 10.1006/exnr.1998.6986. [DOI] [PubMed] [Google Scholar]
- Ding S, Wang T, Cui W, Haydon PG. Photothrombosis ischemia stimulates a sustained astrocytic Ca2+ signaling in vivo. Glia. 2009;57:767–776. doi: 10.1002/glia.20804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doutheil J, Gissel C, Oschlies U, Hossmann KA, Paschen W. Relation of neuronal endoplasmic reticulum calcium homeostasis to ribosomal aggregation and protein synthesis: implications for stress-induced suppression of protein synthesis. Brain Res. 1997;775:43–51. doi: 10.1016/s0006-8993(97)00899-8. [DOI] [PubMed] [Google Scholar]
- Flagella M, Clarke LL, Miller ML, et al. Mice lacking the basolateral Na-K-2Cl cotransporter have impaired epithelial chloride secretion and are profoundly deaf. J Biol Chem. 1999;274:26946–26955. doi: 10.1074/jbc.274.38.26946. [DOI] [PubMed] [Google Scholar]
- Gelot A, Villapol S, Billette dV, Renolleau S, Charriaut-Marlangue C. Astrocytic demise in the developing rat and human brain after hypoxic-ischemic damage. Dev Neurosci. 2009;31:459–470. doi: 10.1159/000232564. [DOI] [PubMed] [Google Scholar]
- Giffard RG, Swanson RA. Ischemia-induced programmed cell death in astrocytes. Glia. 2005;50:299–306. doi: 10.1002/glia.20167. [DOI] [PubMed] [Google Scholar]
- Goldman WF, Yarowsky PJ, Juhaszova M, Krueger BK, Blaustein MP. Sodium/calcium exchange in rat cortical astrocytes. J Neurosci. 1994;14:5834–5843. doi: 10.1523/JNEUROSCI.14-10-05834.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golovina VA, Blaustein MP. Unloading and refilling of two classes of spatially resolved endoplasmic reticulum Ca2+ stores in astrocytes. Glia. 2000;31:15–28. doi: 10.1002/(sici)1098-1136(200007)31:1<15::aid-glia20>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- Groenendyk J, Michalak M. Endoplasmic reticulum quality control and apoptosis. Acta Biochim Pol. 2005;52:381–395. [PubMed] [Google Scholar]
- Hwang IK, Yoo KY, An SJ, et al. Late expression of Na+/H+ exchanger 1 (NHE1) and neuroprotective effects of NHE inhibitor in the gerbil hippocampal CA1 region induced by transient ischemia. Exp Neurol. 2008;212:314–323. doi: 10.1016/j.expneurol.2008.04.007. [DOI] [PubMed] [Google Scholar]
- Ichas F, Jouaville LS, Mazat JP. Mitochondria are excitable organelles capable of generating and conveying electrical and calcium signals. Cell. 1997;89:1145–1153. doi: 10.1016/s0092-8674(00)80301-3. [DOI] [PubMed] [Google Scholar]
- Kintner DB, Luo J, Gerdts J, Ballard AJ, Shull GE, Sun D. Role of Na+-K+-Cl− cotransport and Na+/Ca2+ exchange in mitochondrial dysfunction in astrocytes following in vitro ischemia. Am J Physiol Cell Physiol. 2007;292:C1113–C1122. doi: 10.1152/ajpcell.00412.2006. [DOI] [PubMed] [Google Scholar]
- Koizumi S. Synchronization of Ca2+ oscillations: involvement of ATP release in astrocytes. FEBS J. 2010;277:286–292. doi: 10.1111/j.1742-4658.2009.07438.x. [DOI] [PubMed] [Google Scholar]
- Kumar R, Krause GS, Yoshida H, Mori K, DeGracia DJ. Dysfunction of the unfolded protein response during global brain ischemia and reperfusion. J Cereb Blood Flow Metab. 2003;23:462–471. doi: 10.1097/01.WCB.0000056064.25434.CA. [DOI] [PubMed] [Google Scholar]
- Lenart B, Kintner DB, Shull GE, Sun D. Na-K-Cl cotransporter-mediated intracellular Na+ accumulation affects Ca2+ signaling in astrocytes in an in vitro ischemic model. J Neurosci. 2004;24:9585–9597. doi: 10.1523/JNEUROSCI.2569-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin TN, Liu TH, Xu J, Hsu CY, Sun GY. Brain polyphosphoinositide metabolism during focal ischemia in rat cortex. Stroke. 1991;22:495–498. doi: 10.1161/01.str.22.4.495. [DOI] [PubMed] [Google Scholar]
- Manders EM, Stap J, Brakenhoff GJ, van Driel R, Aten JA. Dynamics of three-dimensional replication patterns during the S-phase, analysed by double labelling of DNA and confocal microscopy. J Cell Sci. 1992;103:857–862. doi: 10.1242/jcs.103.3.857. [DOI] [PubMed] [Google Scholar]
- Marks JD, Boriboun C, Wang J. Mitochondrial nitric oxide mediates decreased vulnerability of hippocampal neurons from immature animals to NMDA. J Neurosci. 2005;25:6561–6575. doi: 10.1523/JNEUROSCI.1450-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mengesdorf T, Althausen S, Oberndorfer I, Paschen W. Response of neurons to an irreversible inhibition of endoplasmic reticulum Ca2+-ATPase: relationship between global protein synthesis and expression and translation of individual genes. Biochem J. 2001;356:805–812. doi: 10.1042/0264-6021:3560805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore ED, Etter EF, Philipson KD, Carrington WA, Fogarty KE, Lifshitz LM, Fay FS. Coupling of the Na+/Ca2+ exchanger, Na+/K+ pump and sarcoplasmic reticulum in smooth muscle. Nature. 1993;365:657–660. doi: 10.1038/365657a0. [DOI] [PubMed] [Google Scholar]
- Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol. 2003;4:552–565. doi: 10.1038/nrm1150. [DOI] [PubMed] [Google Scholar]
- Ouyang YB, Voloboueva LA, Xu LJ, Giffard RG. Selective dysfunction of hippocampal CA1 astrocytes contributes to delayed neuronal damage after transient forebrain ischemia. J Neurosci. 2007;27:4253–4260. doi: 10.1523/JNEUROSCI.0211-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschen W, Doutheil J. Disturbances of the functioning of endoplasmic reticulum: a key mechanism underlying neuronal cell injury? J Cereb Blood Flow Metab. 1999;19:1–18. doi: 10.1097/00004647-199901000-00001. [DOI] [PubMed] [Google Scholar]
- Petravicz J, Fiacco TA, McCarthy KD. Loss of IP3 receptor-dependent Ca2+ increases in hippocampal astrocytes does not affect baseline CA1 pyramidal neuron synaptic activity. J Neurosci. 2008;28:4967–4973. doi: 10.1523/JNEUROSCI.5572-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichert SA, Kim-Han JS, Dugan LL. The mitochondrial permeability transition pore and nitric oxide synthase mediate early mitochondrial depolarization in astrocytes during oxygen-glucose deprivation. J Neurosci. 2001;21:6608–6616. doi: 10.1523/JNEUROSCI.21-17-06608.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, Pozzan T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science. 1998;280:1763–1766. doi: 10.1126/science.280.5370.1763. [DOI] [PubMed] [Google Scholar]
- Sheppard CA, Simpson PB, Sharp AH, Nucifora FC, Ross CA, Lange GD, Russell JT. Comparison of type 2 inositol 1,4,5-trisphosphate receptor distribution and subcellular Ca2+ release sites that support Ca2+ waves in cultured astrocytes. J Neurochem. 1997;68:2317–2327. doi: 10.1046/j.1471-4159.1997.68062317.x. [DOI] [PubMed] [Google Scholar]
- Smith IF, Boyle JP, Plant LD, Pearson HA, Peers C. Hypoxic remodeling of Ca2+ stores in type I cortical astrocytes. J Biol Chem. 2003;278:4875–4881. doi: 10.1074/jbc.M209206200. [DOI] [PubMed] [Google Scholar]
- Su G, Kintner DB, Flagella M, Shull GE, Sun D. Astrocytes from Na(+)-K(+)-Cl(−) cotransporter-null mice exhibit absence of swelling and decrease in EAA release. Am J Physiol Cell Physiol. 2002;282:C1147–C1160. doi: 10.1152/ajpcell.00538.2001. [DOI] [PubMed] [Google Scholar]
- Sun D, Lytle C, O’Donnell ME. Astroglial cell-induced expression of Na-K-Cl cotransporter in brain microvascular endothelial cells. Am J Physiol. 1995;269:C1506–C1512. doi: 10.1152/ajpcell.1995.269.6.C1506. [DOI] [PubMed] [Google Scholar]
- Swanson RA, Liu J, Miller JW, Rothstein JD, Farrell K, Stein BA, Longuemare MC. Neuronal regulation of glutamate transporter subtype expression in astrocytes. J Neurosci. 1997;17:932–940. doi: 10.1523/JNEUROSCI.17-03-00932.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano T, Oberheim N, Cotrina ML, Nedergaard M. Astrocytes and ischemic injury. Stroke. 2009;40:S8–12. doi: 10.1161/STROKEAHA.108.533166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong M, Yang Y, Chen GQ, Zhou WH. Post-ischemic hypothermia for 24h in P7 rats rescues hippocampal neuron: association with decreased astrocyte activation and inflammatory cytokine expression. Brain Res Bull. 2009;79:351–357. doi: 10.1016/j.brainresbull.2009.03.011. [DOI] [PubMed] [Google Scholar]
- Yoshida S, Ikeda M, Busto R, Santiso M, Martinez E, Ginsberg MD. Cerebral phosphoinositide, triacylglycerol, and energy metabolism in reversible ischemia: origin and fate of free fatty acids. J Neurochem. 1986;47:744–757. doi: 10.1111/j.1471-4159.1986.tb00675.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.