

Abstract
Rhesus rhadinovirus (RRV), a primate γ-herpesvirus related to human Kaposi’s sarcoma-associated herpesvirus (KSHV), causes a similar pattern of pathogenesis. Previously, RRV was shown to express 7 pre-microRNAs (pre-miRNAs) in latently infected cells. Using deep sequencing, we analyzed the pattern of small RNA expression in vivo using latently RRV-infected B-cell lymphoma and retroperitoneal fibromatosis tissues. We identified 15 virally encoded pre-miRNAs in both tumors, including all previously reported RRV pre-miRNAs. Although all 15 RRV pre-miRNAs, like all 12 KSHV pre-miRNAs, are located 3′ to the conserved viral ORF71 gene and in the same transcriptional orientation, only one RRV miRNA is homologous to a KSHV miRNA. One previously identified RRV miRNA, miR-rR1-3, is actually a miRNA offset RNA (moRNA) derived from sequences located adjacent to pre-miR-rR1-3. Several other RRV-derived moRNAs were obtained, including one recovered >600 times. Together, this research provides a comprehensive list of the miRNAs and moRNAs encoded by RRV.
Keywords: Herpesviruses, microRNAs, rhesus rhadinovirus, Kaposi’s sarcoma-associated herpesvirus, post-transcriptional regulation, tumor virus
Introduction
MicroRNAs (miRNAs) are a family of ~22-nt non-coding RNAs capable of post-transcriptionally down-regulating the expression of target mRNAs (Bartel, 2004). They are typically transcribed as one arm of an ~80-nt RNA stem-loop found within a non-coding region of a capped and polyadenylated transcript referred to as a primary-miRNA (pri-miRNA) (Cai, Hagedorn, and Cullen, 2004; Lee et al., 2002; Lee et al., 2004; Zeng and Cullen, 2003). These stem-loops are bound by the RNase III enzyme Drosha and cleaved ~22 bp away from the loop to generate an ~60-nt RNA hairpin, bearing an ~2-nt 3′ overhang, referred to as the precursor-miRNA (pre-miRNA). The pre-miRNA is bound by Exportin5 (Lee et al., 2003; Yi et al., 2003) and exported to the cytoplasm where the pre-miRNA is recognized by Dicer, a second RNase III enzyme. Dicer cleaves ~22 bp from the base of the pre-miRNA to release the terminal loop and generate a second ~2-nt 3′ overhang (Hutvagner et al., 2001; Lee et al., 2002). One strand of the resultant miRNA duplex intermediate is then loaded into the RNA-induced silencing complex (RISC) (Hammond et al., 2000), while the remaining strand, referred to as the star or passenger strand, is degraded (Matranga et al., 2005). However, this discrimination, which is largely regulated by the stability of base pairing at the 5′ ends of the duplex, is rarely absolute. MiRNAs function as guides to direct RISC to mRNA transcripts bearing complementary target sequences, typically in the 3′UTR, leading to inhibition of protein expression (Martinez et al., 2002; Schwarz et al., 2002). While functional miRNA target sites on mammalian mRNAs are generally only partially complementary, full mRNA sequence complementarity to nucleotides 2 through 8 of the miRNA, referred to as the seed sequence, is commonly observed (Bartel, 2004).
MiRNA-offset RNAs (moRNAs) are a recently discovered class of small RNAs closely related to miRNAs (Shi et al., 2009). MoRNAs derive from sequences located immediately adjacent to the mature miRNA and miRNA star strands in the pri-miRNA precursor and have been recovered at low levels in several small RNA deep sequencing efforts (Babiarz et al., 2008; Jurak et al., 2010; Ruby et al., 2007; Shi et al., 2009; Umbach and Cullen, 2010). Although they appear to form “moRNA duplex intermediates” with their corresponding star strands, complete with short 3′ overhangs, the process by which they are generated remains unclear, and whether or not they are loaded into RISC and have functional activity is currently unknown. However, moRNA processing is clearly linked to the excision of the flanking miRNA duplex.
In addition to a relatively low level of moRNAs, several herpesviruses, of both human and non-human origin, have been found to express high levels of miRNAs, including the oncogenic human rhadinovirus Kaposi’s sarcoma-associated herpesvirus (KSHV). Sequencing analysis of small RNAs expressed in latently KSHV-infected human B cells has revealed that KSHV encodes 12 pre-miRNAs that are all located within a single cluster contained within the latency-associated region of the KSHV genome, 3′ of the ORF71 gene (Cai et al., 2005; Grundhoff, Sullivan, and Ganem, 2006; Pfeffer et al., 2005; Samols et al., 2005). As yet, only a small number of mRNA targets for the KSHV miRNAs have been identified. However, it appears that miRNA-induced down-regulation of both cellular and viral genes contributes to the creation of a cellular environment favorable for KSHV latency and/or replication (Umbach and Cullen, 2009).
Rhesus monkey rhadinovirus (RRV), a rhadinovirus that naturally infects rhesus macaques, is a close relative of KSHV that can induce a similar pattern of pathogenic effects, including abnormal B cell proliferation and cancer, in its simian host (Desrosiers et al., 1997; Orzechowska et al., 2008; Wong et al., 1999). RRV has been previously reported to encode seven pre-miRNAs, which were originally identified by conventional sequencing of cDNAs generated from small RNAs expressed in latently RRV-infected monkey vascular endothelial cells (MVECs) (Schäfer et al., 2007). Although transcripts targeted by RRV miRNAs have yet to be identified, RRV is genetically tractable, easily grown in cell culture and has a well-defined animal model, making it a potentially ideal system to study the contribution of virally encoded miRNAs to primate rhadinovirus pathogenesis both in vitro and in vivo (Bilello et al., 2006; DeWire et al., 2003; Estep et al., 2007).
However, an accurate functional analysis of RRV miRNAs cannot be accomplished without knowledge of the full complement of RRV miRNAs. Although many protein-coding genes encoded by KSHV and RRV show obvious homology, there appears to be little conservation of miRNAs at the sequence level (Schäfer et al., 2007), making it difficult to determine if all RRV miRNAs have been identified. To comprehensively identify all miRNAs encoded by RRV, cDNA libraries generated from small RNAs expressed in two distinct RRV-induced tumors, a retroperitoneal fibromatosis (RF) and a B-cell lymphoma, were subjected to deep sequencing. In addition to recovering all previously reported RRV miRNAs (Schäfer et al., 2007), deep sequencing also identified 8 novel viral pre-miRNAs. It was also determined that the RRV miRNA previously identified as miR-rR1-3 is actually a moRNA that has silencing activity similar to conventional miRNAs. These data not only identify the full complement of miRNAs encoded by RRV but also shed light on the origin and function of the as yet poorly characterized moRNAs.
Results
Deep sequencing of small RNAs from RRV-infected tumor samples
Tissue from a latently RRV-infected RF (18483) and B-cell lymphoma (19286) were obtained post-mortem from RRV-infected rhesus macaques. Small RNAs 18 to 24 nts in length were isolated from both samples and used to prepare cDNA libraries for Solexa/Illumina sequencing (Umbach and Cullen, 2010). Deep sequencing returned 4,512,216 usable reads from the RF sample and 2,440,978 reads from the B cell lymphoma sample. Of these, 571,791 and 303,871 sequences, respectively, were identified as cellular miRNAs by comparison to the miRBase 13.0 database. As the miRNA transcriptome of rhesus macaques remains incompletely defined, this likely represents an underestimate. Of the remaining reads derived from the RF and B-cell lymphoma samples, 242,095 and 127,407, respectively, mapped to the RRV genome (AF083501). The vast majority of these viral sequences (>99%) were identified as RRV miRNAs and in both tumors this represented ~30% of the total miRNA reads obtained. A comprehensive list of the major mature RRV miRNA isoforms recovered during this analysis is presented in supplementary tables 1 and 2.
In addition to recovering all the previously reported RRV miRNAs, miR-rR1-1 through miR-rR-7 (Schäfer et al., 2007), 8 novel pre-miRNAs, designated here as miR-rR1-8 through miR-rR1-15, were also identified in both the RF and B-cell lymphoma sample (Table 1). Secondary structure predictions demonstrated that the loci encompassing all novel RRV miRNA candidates could be readily folded into the hairpin structures typical of canonical pri-miRNAs (Fig. 1) (Bartel, 2004). Additionally, both the 5p and 3p arms of the miRNA duplex intermediate were recovered for six of the eight novel RRV pre-miRNAs and these displayed the predicted ~2-nt 3′ overhangs (Bartel, 2004), thus further confirming that these are indeed genuine miRNAs.
Table 1.
Viral miRNAs and moRNAs recovered from RRV-induced retroperitoneal fibromatosis tissue and B-cell lyphomas.
| Retroperitoneal Fibromatosis (18483) | B-cell lymphoma (19286) | |||||||
|---|---|---|---|---|---|---|---|---|
| miRNA | moRNA | miRNA | moRNA | |||||
| 5p | 3p | 5p | 3p | 5p | 3p | 5p | 3p | |
| miR-rR1-1 | 1,456 | 1 | 1,984 | 1 | 1 | |||
| miR-rR1-2 | 1,582 | 1,159 | ||||||
| miR-rR1-3 | 1,921 | 4,400 | 12 | 807 | 1,930 | 19 | 2 | |
| miR-rR1-4 | 37 | 1,363* | 20 | 479* | ||||
| miR-rR1-5 | 11,124 | 531 | 4,492 | 223 | ||||
| miR-rR1-6 | 9,727 | 61 | 177 | 7,633 | 56 | 434 | ||
| miR-rR1-7 | 1,600 | 555 | 590 | 274 | ||||
| miR-rR1-8 | 7 | 770 | 20 | 1,029 | ||||
| miR-rR1-9 | 288 | 14 | 4 | 501 | 20 | 12 | ||
| miR-rR1-10 | 111 | 1 | 102 | 6 | ||||
| miR-rR1-11 | 291 | 212 | 195 | 329 | ||||
| miR-rR1-12 | 330 | 19 | 298 | 17 | ||||
| miR-rR1-13 | 9 | 191,963 | 11 | 95,377 | ||||
| miR-rR1-14 | 75 | 1 | 40 | 2 | ||||
| miR-rR1-15 | 58 | 13,273 | 23 | 7,938 | ||||
The recovery frequency of each miRNA or moRNA is shown, with bolded values indicating small RNAs that have not been previously reported. Both miRNAs and moRNAs are presented as 5p reads, i.e. derived from the 5′ arm of the pri-miRNA stem-loop, or as 3p reads. An asterisk indicates a strain-specific RRV miRNA sequence isoform that differs from the isoform previously reported (Schäfer et al., 2007).
Figure 1.

Predicted secondary structures of the novel primary miRNA hairpins. Predicted pri-miRNA stem-loops formed by the novel RRV miRNAs identified during deep sequencing of small RNAs derived from RRV-infected tumor samples. Red sequences indicate the dominant form of the mature miRNA, while blue sequences indicate the less-abundant star strand, if recovered. It should be noted that the pri-miRNA stem is generally ~11 bp longer than the mature pre-miRNA hairpin (Zeng, Yi, and Cullen, 2005).
All novel RRV miRNAs identified mapped to the viral latency-associated region (Fig. 2). Moreover, these novel RRV miRNAs were found interspersed among, and in the same transcriptional orientation as, the previously identified RRV miRNAs. This suggests that expression of the entire RRV miRNA locus is coordinated and that all RRV miRNAs likely derive from a single pri-miRNA precursor, as previously observed for the KSHV miRNAs (Cai and Cullen, 2006).
Figure 2.

Genomic location of the RRV pre-miRNAs. These are shown relative to viral protein coding genes (black arrows) located adjacent to the RRV latency-associated region. White arrows indicate previously identified RRV miRNAs, while gray arrows indicate novel RRV miRNAs identified in this study. Transcriptional orientations of viral genes and miRNAs are as indicated.
Identification of RRV moRNAs in vivo
The in-depth sequence coverage afforded by deep sequencing permitted the recovery of several previously unidentified small RRV RNAs, including the pre-miRNA terminal loop of miR-rR1-2 and several RRV moRNAs (Table 1 and data not shown). Importantly, it is apparent that the small RNA previously annotated as miR-rR1-3 is actually a moRNA derived from the pri-miR-rR1-3 precursor, now that the 5p and 3p arms of the miRNA duplex intermediate have both been recovered at high levels (Table 1 and Fig. 4A). These sequences will be designated miR-rR1-3-5p and miR-rR1-3-3p, while the previously designated “miR-rR1-3” and its corresponding star strand will be referred to as moR-rR1-3-5p and moR-rR1-3-3p, respectively. Of note, the previous effort to identify RRV miRNAs failed to recover either miR-rR1-3-5p or -3p and recovered the moR-rR1-3-5p sequence only once (Schäfer et al., 2007).
Figure 4.

Activity of miR-rR1-3-3p and moR-rR1-3-5p. (A) Predicted secondary structure of the pri-miR-rR1-3 hairpin. The sequence of mature miR-rR1-3-3p is indicated in red and the miR-rR1-3-5p star strand shown in blue. The dominant arm of moR-rR1-3 is indicated in teal and the corresponding star strand in green. (B) Schematic of the full-length and 5′ truncated miR-rR1-3 expression constructs expanded to show relative sizes and locations of mature miRNAs, moRNAs and cleavage intermediates. MiRNAs and moRNAs indicated in bold designate the dominant arms recovered from deep sequencing. The arrow indicates the 5′ border of the 5′ truncated ΔmiR-rR1-3 expression construct. Hatched yellow boxes represent basal stem sequences of pri-miR-rR1-12. (C) Inhibitory activity of miR-rR1-3-3p (red) and moR-rR1-3-5p (teal) revealed in transfected 293T cells using luciferase-based indicator plasmids bearing two perfectly complementary target sites. Values are displayed relative to a control indicator bearing an unrelated sequence (gray), which was set at 100%. Average of three independent experiments with standard deviation indicated.
Expression levels of novel RRV miRNAs in vivo and in latently infected MVECs
Expression of the novel RRV miRNAs identified in Table 1 was further confirmed by stem-loop qRT-PCR using custom primers designed to assay the most abundant isoform of each miRNA recovered from deep sequencing (Supp. Tables 1 and 2). Assay of the original RF and B-cell lymphoma samples used for deep sequencing confirmed the expression of miR-rR1-3, miR-rR1-8, miR-rR1-9, miR-rR1-11, miR-rR1-13, miR-rR1-14 and miR-rR1-15 in both samples (Fig. 3A). Of all the miRNAs assayed, miR-rR1-14 was consistently expressed at the lowest level, while the remaining miRNAs were expressed at fairly comparable levels.
Figure 3.
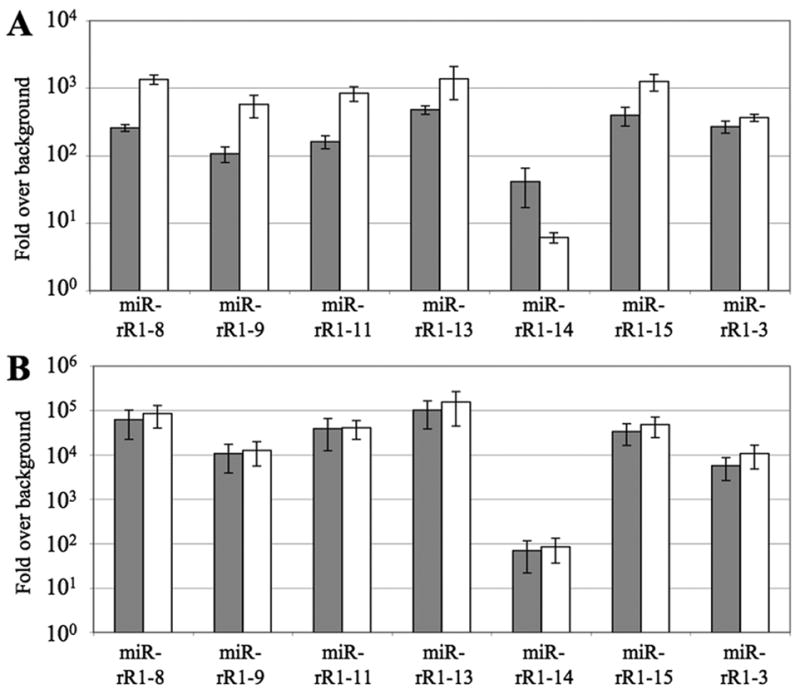
RRV miRNA expression levels determined by qRT-PCR. All values are given as fold-expression relative to an uninfected control sample and are normalized to levels of a cellular miRNA, miR-16. (A) Expression levels of RRV miRNAs in the RF tumor sample (18483; gray) and the B cell lymphoma (19286; white). RNA recovered from RRV-uninfected tissue derived from the same animals served as the negative controls. (B) Expression levels of RRV miRNAs in latently infected MVECs harvested 23 (gray) or 44 (white) dpi. RNA from uninfected MVECs served as the negative control.
To determine if the previous lack of recovery of the novel RRV miRNAs listed in Table 1 was due to their low expression in the RRV infected MVECs originally used as a source of infected cell RNA (Schäfer et al., 2007), we also used qRT-PCR to analyze latently RRV-infected MVECs harvested at 23 or 44 days post-infection (dpi). This yielded results similar to those seen with the tumor samples, with miR-rR1-14 again being the least expressed miRNA and the remaining miRNAs detected at fairly comparable levels (Fig. 3B). Interestingly, the level of RRV miRNAs did not appear to change between 23 and 44 dpi, suggesting that viral miRNAs may reach a steady-state level during latent MVEC infection.
Technical difficulties (such as biased G:C or A:U content, or the potential for the sequence to homodimerize) precluded the design of RT-PCR primers for, and thus the verification of, miR-rR1-10 and miR-rR1-12 expression. However, given the predicted secondary structures of the loci surrounding both miRNAs (Fig. 1) and the recovery of the miR-rR1-12 star strand from deep sequencing (Table 1), we believe that these are also authentic RRV miRNAs.
Activity and expression of moR-rR1-3
MoRNAs were first identified in lower eukaryotes (Shi et al., 2009) and, more recently, viral moRNAs have also been identified in cells infected by KSHV, HSV-1 and HSV-2 (Jurak et al., 2010; Umbach and Cullen, 2010). The defining characteristic of moRNAs is that they derive from sequences located either immediately 5′ or 3′ to a pre-miRNA, and it is therefore apparent that one end of a moRNA must be generated by Drosha cleavage of the pri-miRNA. However, it has remained unclear whether moRNAs are loaded into RISC and even how moRNAs are processed. It is possible that the other end of the moRNA is generated either by a second cleavage by Drosha, in the nucleus, or by Dicer cleavage, in the cytoplasm. It has also been suggested that moRNAs might simply be breakdown products generated at low levels during miRNA processing (Babiarz et al., 2008), analogous, for instance, to the pre-miRNA loop sequences that are sometimes recovered (Umbach and Cullen, 2010).
To determine if moR-rR1-3-5p has the ability to down-regulate complementary transcripts in a manner similar to traditional miRNAs, and is therefore loaded into RISC, an expression construct containing the pri-miR-rR1-3 region (Fig. 4B) was co-transfected into 293T cells along with luciferase based indicator constructs bearing an artificial, perfect target site for either miR-rR1-3-3p or moR-rR1-3-5p. When compared to expression of a luciferase indicator bearing an unrelated target site, miR-rR1-3-3p specifically down-regulated expression of its cognate indicator by ~10-fold (Fig. 4C). In contrast, moR-rR1-3-5p induced a more moderate but still significant ~3-fold down-regulation of its indicator. These data therefore strongly suggest that moR-rR1-3-5p is able to program RISC.
Examination of the miR-rR-3 locus revealed that miR-rR1-3-5p is encoded only 51 nt 3′ of the 3p arm of the pre-miR-rR-12 hairpin (Fig. 4B). This is perhaps surprising given previous reports demonstrating that Drosha prefers to cleave pri-miRNA stem-loops flanked by unstructured RNA sequence (Zeng and Cullen, 2005). (It should be noted that the pri-miRNA stem-loop is ~11 bp longer than the pre-miRNA stem, as shown in Fig. 1). Although we were not able to assay miR-rR1-12 levels by RT-PCR for technical reasons, both strands of the predicted miR-rR1-12 miRNA duplex intermediate were recovered by deep sequencing, as were both strands of the miR-rR1-3-3p duplex intermediate (Table 1). Moreover, miR-rR1-3-3p expression was also readily detected by RT-PCR (Fig. 3). Therefore, despite their close physical proximity, miR-rR1-12 and miR-rR1-3 are clearly both expressed in latently RRV-infected cells.
In an attempt to directly examine miR-rR1-3-3p and moR-rR1-3-5p expression levels, RNA was harvested from 293T cells transfected with the same full-length miR-rR1-3 expression construct (Fig. 4). Hybridization with a miR-rR1-3-3p-specific probe resulted in detection of the mature miRNA at the expected size of ~20 nt (Fig. 5). The larger band of between 40 and 50 nts is presumably the pre-miR-rR1-3 precursor, although this migrated somewhat faster than the expected ~56-nt size (Fig. 4B).
Figure 5.
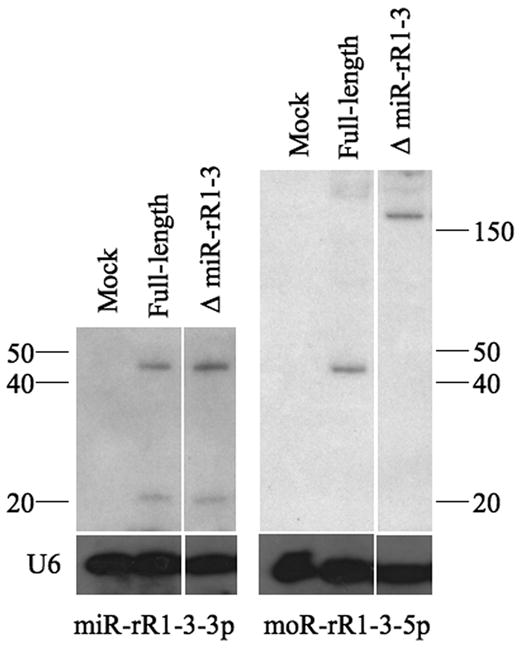
Detection of RRV small RNAs. Northern blots for miR-rR1-3-3p (left panel) and moR-rR1-3-5p (right panel) using RNA samples derived from 293T cells transfected with the indicated expression plasmids. Expression constructs used and expected fragment sizes are diagramed in Fig. 4B. Endogenous U6 RNA served as a loading control.
When the same samples were hybridized with a moR-rR1-3-5p-specific probe, no RNA with the expected ~18-nt size of moR-rR1-3-5p was detected. This was not unexpected, given the low frequency of recovery of this moRNA by deep sequencing (Table 1). However, a larger band of between 40 and 50 nts was detected by the moR-rR1-3-5p specific probe. This is presumably a moRNA processing intermediate that includes the moRNA sequence as well as 5′ flanking sequences up to the 3′ end of miR-rR1-12-3p (Fig. 4B). However, similar to the pre-miR-rR1-3 band, this band also appeared slightly smaller than the expected 51-nt size. To confirm the identity of this band, we generated a second moR-rR1-3 expression plasmid by deleting sequences located 5′ to the loop of the adjacent pri-miR-rR1-12 stem-loop (Fig. 4), which should block miR-rR1-12 processing without affecting miR-rR1-3 expression. The 51-nt moR-rR1-3-5p-specific band resulting from Drosha cleavage of both the miR-rR1-12 and miR-rR1-3 pri-miRNA stem-loops should therefore be lost and replaced by a predicted 157-nt band extending from the 5′ miR-rR1-3 Drosha cleavage site to the transcription start site. Indeed, the “ΔmiR-rR1-R3” expression vector was found to generate the predicted ~160-nt miR-rRi-3-5p specific band, but no ~45-nt specific band, in transfected 293T cells (Fig. 5), yet did not affect the pattern of miR-rR1-3 specific bands.
Discussion
In this manuscript, we report the use of deep sequencing technology to detect the expression of 15 distinct RRV pre-miRNA species in a latently RRV-infected B-cell lymphoma and RF tumor harvested from two rhesus macaques infected with the 17577 strain of RRV. Previously, conventional sequencing was used to identify seven RRV pre-miRNAs expressed in MVECs latently infected with RRV strain 26-95 (Schäfer et al., 2007), and all of these were also detected in vivo. Interestingly, one RRV miRNA, miR-rR1-4-3p, does differ by a single nucleotide between these two RRV strains (Fig. 6A), but as this single nucleotide difference lies outside the miRNA seed sequence, it may have little or no effect on the pattern of mRNAs regulated by this viral miRNA.
Figure 6. Sequence conservation of RRV miRNAs.
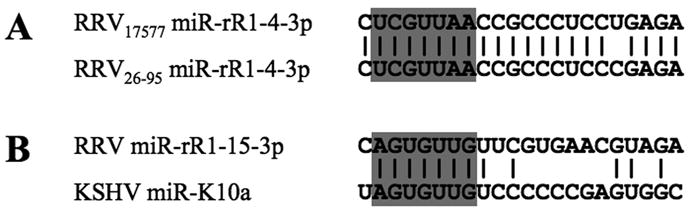
(A) This alignment contrasts the sequences of the RRV miRNA miR-rR1-4-3p in the RRV strains 17577, used in this report, and 26-95, used in a previous publication (Schäfer et al., 2007). (B) Sequence alignment of miR-rR1-15 with the KSHV miRNA miR-K10a. In both cases, the conserved miRNA seed regions are indicated, as are conserved bases.
RRV is a close relative of KSHV and both viruses cause a similar pattern of disease in immunodeficient hosts. However, a comparison of the sequences of the 15 RRV miRNAs described in this manuscript and earlier (Schäfer et al., 2007) with the known KSHV miRNAs (Cai et al., 2005; Grundhoff, Sullivan, and Ganem, 2006; Pfeffer et al., 2005; Samols et al., 2005) identifies only one example of significant sequence homology. Specifically, the newly described miR-rR1-15 miRNA bears an identical seed sequence to the previously described KSHV miRNA miR-K10a (Fig. 6B). However, as the mRNA species targeted by miR-K10a remain to be defined, the significance of this conservation is currently unclear. Nevertheless, the observed homology does imply that these two viral miRNAs are likely performing similar functions in RRV- and KSHV-infected cells that, uniquely, have been conserved during primate rhadinovirus miRNA evolution.
An interesting feature of KSHV miR-K10a is that a significant proportion of this miRNA is edited by an adenosine deaminase, resulting in the modification of the adenosine located at position 2 of miR-K10a to inosine (Fig. 6B). This modification changes the miR-K10a seed and generates an alternative miRNA isoform called miR-K10b (Pfeffer et al., 2005). However, we did not detect a significant level of editing of miR-rR1-15 or, indeed, of any RRV-encoded miRNA (data not shown).
Although the miRNAs encoded by RRV and KSHV are therefore largely distinct, all 12 viral pri-miRNAs encoded by KSHV and all 15 pri-miRNAs encoded by RRV are found in the same genomic location in both viruses, within the latency-associated region and 3′ to the shared ORF71 gene (Fig. 2) (Cai et al., 2005; Pfeffer et al., 2005; Samols et al., 2005). This implies that RRV and KSHV evolved from a common ancestor that first acquired one or more miRNA stem-loops in the latency region of the genome and then underwent evolutionary divergence, leading to the current pattern of virus-specific miRNA sequences.
As noted in previous deep sequencing surveys and RT-PCR verification studies (Git et al., 2010; Umbach and Cullen, 2010), we observed only a modest correlation between miRNA expression levels as assessed by deep sequencing or qRT-PCR. For example, RRV miR-rR1-14 was the least frequently recovered miRNA by deep sequencing (Table 1), which correlates well with the qRT-PCR data, where it was also the least expressed RRV miRNA of those assayed (Fig. 3). In contrast, miR-rR1-8 and miR-rR1-11, which were both recovered at low to intermediate levels by deep sequencing, were both detected at relatively high levels by qRT-PCR. In fact, levels of miR-rR1-8 and miR-rR1-11 detected by qRT-PCR were comparable to the levels of miR-rR1-13 and miR-rR1-15, even though these latter two miRNAs were recovered at 10 to 100 greater frequencies by deep sequencing. These discrepancies likely reflect biases inherent in both the deep sequencing and qRT-PCR methodologies. Notably, deep sequencing results can be influenced by linker ligation and PCR amplification biases, whereas stem-loop qRT-PCR is influenced by PCR primer annealing efficiency as well as by the inability of each primer set to efficiently detect more than a single miRNA 3′ sequence isoform, which may underestimate total miRNA expression levels.
In spite of these caveats, it is worth comparing the level of recovery of the previously reported RRV miRNAs (Schäfer et al., 2007) with the levels of the novel viral miRNAs identified in this study to determine if the novel miRNAs were not previously recovered simply due to the fact that they are expressed at low levels in latently infected MVECs. While deep sequencing frequencies of individual RRV miRNAs varied greatly, this does not appear to be the case; two novel miRNAs, miR-rR1-13 and miR-rR1-15, were the two most abundant miRNAs recovered from deep sequencing in both the RF and B-cell lymphoma samples and in the latently infected MVECs. This may imply that the different RRV strains used in this study (RRV17577) or the earlier study (RRV26-95) (Schäfer et al., 2007) express individual miRNAs at different levels. However, sequence analysis indicates that all the newly reported RRV miRNAs are in fact present and intact in the RRV26-95 strain.
Overall, the RRV miRNA profiles derived from the RF and B-cell lymphoma samples are remarkably similar; deep sequencing recovered a similar pattern of mature miRNAs and star strands from both samples. Specifically, the more abundant arm of each pre-miRNA hairpin was identical in both tumor samples except for miR-rR1-11, where numbers for both the 5p and 3p arms were similar in both samples. Together, these data suggests that the in vivo RRV miRNA expression profiles are relatively consistent regardless of tumor type and that we have likely identified a comprehensive list of viral pre-miRNAs expressed during latent RRV infection.
As discussed above, the small RNA previously annotated as miR-rR1-3 is more accurately designated moR-rR1-3-5p. Unfortunately, assay of moR-rR1-3-5p expression levels by RT-PCR was not possible due to the inability of the stem-loop primers to distinguish the mature moRNA from the highly abundant 5′ flanking region generated by Drosha cleavage of pri-miR-rR1-3 (Fig. 5). On the other hand, we were able to demonstrate that moR-rR1-3-5p has at least moderate inhibitory activity using a luciferase-based indicator assay (Fig. 4), thus strongly suggesting that this moRNA is loaded into RISC and can target complementary transcripts for inhibition in a manner identical to miRNAs. This argues that moRNA biogenesis occurs in the cytoplasm and likely requires cleavage by Dicer.
We speculate that sufficient base-pairing at the base of the pri-miRNA stem (Fig. 4A) can transiently hold the sequences flanking the pri-miRNA together after pre-miRNA excision by Drosha, thus allowing the 2-nt 3′ overhang on the dsRNA “pre-moR” precursor to be bound, and possibly stabilized, by Exportin5 leading to nuclear export. We hypothesize that this moRNA precursor can also remain at least transiently base-paired in the cytoplasm, thus allowing Dicer to bind the 3′ overhang resulting from Drosha cleavage. Dicer cleavage of the stem could then generate a “moRNA duplex intermediate”, which would then be available for RISC loading and subsequent transcript targeting. The existence of such moRNA duplex intermediates, closely analogous in structure to miRNA duplex intermediates, is strongly suggested by the recovery of both moR-5p and moR-3p RNAs from several viral pri-miRNAs, including miR-rR1-3 in this manuscript, miR-K4 and miR-K10 in KSHV (Umbach and Cullen, 2010) and miR-H4 and miR-H6 in HSV-1 (Jurak et al., 2010).
In support of this hypothesis, we note that viral moRNAs tend to arise from pri-miRNA stem-loops that are closely flanked, 5′ and/or 3′, by other pri-miRNAs stem-loops. Coordinated Drosha cleavage of this cluster of three pri-miRNAs would generate short RNAs flanking the central stem that may transiently maintain the short stem derived from the base of the pri-miRNA stem-loop (Figs. 1 and 4A). Of note, the most abundant species of RRV moRNA recovered by deep sequencing was derived from sequences adjacent to pre-miR-rR1-6. The pri-miR-rR1-6 hairpin is closely flanked upstream by pri-miR-rR1-15 and downstream by pri-miR-rR1-7. Drosha cleavage of pri-miR-rR1-6 and these two flanking pri-miRNAs is predicted to generate 5′ and 3′ flanking fragments 59 and 87 nts in length, respectively. These sequences may be short enough so that the pri-miRNA basal stem is sufficient to keep the two fragments base-paired for export by Exportin 5 and cleavage by Dicer to generate the final moRNA, moR-rR1-6-5p (Table 1). Similarly, the moR-rR1-3 analyzed in Figs. 4 and 5, the second most abundant moRNA recovered from deep sequencing, is also closely flanked 5′ by pre-miR-rR1-12 (Fig. 4) and 3′ by pre-miR-rR1-4. Although Drosha cleavage is predicted to result in a 3′ flanking fragment of 373 nt, the 5′ flanking fragment is expected to be only 51 nts long (Figs. 4B and 5). Again, the short length of the 5′ flanking sequence may allow this RNA fragment to remain base-paired to the 3′ flanking sequence (Fig. 4A) during nuclear export and subsequent Dicer cleavage. Similarly, moR-K4-5p, detected in KSHV-infected cells, is predicted to derive from the 73 nt sequence that separates miR-K4 from miR-K3.
If the close proximity of pri-miRNA stems indeed facilitates moRNA biogenesis, then this would explain why moRNAs have been reported more frequently in viruses, with their tightly packed genomes, than in cells. Nevertheless, moRNA biogenesis is clearly still very inefficient in comparison to miRNA biogenesis, as evidenced by the far lower moRNA frequencies recovered by deep sequencing (Table 1) and the lower inhibitory activity observed (Fig. 4C). Whether moRNAs are expressed at levels sufficient to exert a significant phenotypic effect in any setting, including herpesvirus-infected cells, currently remains unclear.
Materials and Methods
Animal tissues
RF tumor and B-cell lymphoma tissues were derived from RRV17577-infected animals 18483 and 19286, respectively, and have been characterized and described (Orzechowska et al., 2008). Approximately 100 mg of frozen tumor tissue or control tissue (muscle) from each animal was placed into 500 μl of Tri-reagent (Applied Biosystems, Foster City, CA) and homogenized with a tissue tearor for RNA extraction according to manufacturer’s directions.
Solexa/Illumina sequencing and data analysis
cDNA libraries for deep sequencing were prepared as outlined previously (Umbach and Cullen, 2010). Thirty micrograms of total RNA from RRV-infected tumor samples were used to isolate small RNAs (18–24 nt) that were then copied into cDNA using linkers described in the Solexa Digital Gene Expression Kit but chemically modified as described in Lau et al. (2001). Importantly, the 3′ linker (IDT, Coralville, IA) was 5′ adenylated and 3′ blocked to minimize ligation artifacts. Raw sequence data were returned in FASTA format and 3′ linker sequences removed. The sequence list was then indexed, collapsed and size filtered for sequences 18- to 24-nts in length to generate a final list for MegaBLAST analysis. Sequences were analyzed against the RRV genome (AF083501) and the mature human miRNA dataset from miRBase (release 13.0) using the formatdb, megablast, blastoutparse and filter alignment scripts from the miRDeep software package (Friedlander et al., 2008).
Quantitation of miRNA expression by qRT-PCR
RRV miRNA expression levels were analyzed using the TaqMan MicroRNA Assay System (Applied Biosystems, Foster City, CA) according to the manufacturer’s directions. Briefly, 10 ng of total RNA was used as a template for reverse transcription. The sample was diluted to 60 μl, of which 8 μl was used for stem-loop qRT-PCR. Custom primers for detecting the most abundant isoform of each viral miRNA were designed by Applied Biosystems (Foster City, CA). Each reverse transcription reaction was performed twice independently, and each reaction was used for triplicate PCR. All values were normalized to the ubiquitously expressed cellular miRNA, miR-16, and all viral miRNA levels are displayed as fold-expression relative to RNA derived from uninfected tissue samples harvested from the same animal, or from uninfected MVECs, which were calculated as outlined by (Livak and Schmittgen, 2001).
Cell culture and RNA
293T cells were maintained in Dulbecco’s modified Eagle medium (Sigma-Aldrich, St. Louis, MO) supplemented with 10% fetal bovine serum. Primary rhesus MVEC cultures (MVEC21150 CGB-5, kindly provided by Dr. Jay Nelson) were maintained in Endothelial Cell medium 2 (EBM-2; Cambrex BioScience, Walkersville, MD) supplemented with Single Quots (Cambrex). MVEC cultures were infected with approximately 2 × 105 infectious units of RRV17577-GFP (S. Wong, unpublished results), passed when confluent and maintained for the indicated length of time. Total RNA was isolated using Tri-reagent (Applied Biosystems, Foster City, CA) according to the manufacturer’s directions.
Expression vectors, indicator plasmids and luciferase assays
Renilla luciferase indicators for miR-rR1-3-3p and moR-rR1-3-5p were cloned by annealing synthetic oligonucleotides perfectly complementary to either miR-rR1-3-3p or moR-rR1-3-5p into the XbaI and ClaI sites downstream of the Renilla gene in the pNL-SIN-CMV-RLuc vector, as previously described (Gottwein, Cai, and Cullen, 2006). The full-length miR-rR1-3 expression construct was generated using primers centered around the pre-miR-rR1- 3 hairpin to PCR a 262-bp fragment from 293T cells infected with a RRV-GFP virus (Schäfer et al., 2007), which was then cloned into the BamHI and EcoRI sites of pcDNA3 (Invitrogen, Carlsbad, CA). The ΔmiR-rR1-3 expression construct is similar to the full-length construct, except that the 5′ end is truncated to the miR-rR1-12 terminal loop. 293T cells were transfected with 25 ng of the indicator plasmid together with 1 μg of each expression construct. Samples were harvested in lysis buffer 24 hours post-transfection and luciferase expression assayed using the Renilla Luciferase Assay System (Promega, Madison, WI). Expression levels are displayed relative to values obtained with a control indicator construct bearing an irrelevant target site. Final values and standard deviations represent the average of three independent experiments. Transfections were performed in duplicate so that the parallel sample could be harvested for Northern blot analysis.
Northern blots
MiRNA Northern blots were carried out as previous described (Cai, Hagedorn, and Cullen, 2004). Briefly, 30 μg of total RNA was separated on 15% TBE-urea gels (Bio-Rad, Hercules, CA) and then transferred onto nitrocellulose membranes. Following UV cross-linking, membranes were probed with radioactively end-labeled DNA oligonucleotides complementary to each target miRNA or moRNA. Blots were then probed for endogenous U6 RNA, which served as a loading control.
Supplementary Material
Acknowledgments
This research was supported by National Institutes of Health grants R01-AI067968 (B.R.C.), R01-CA075922 (S.W.W.) and RR000163 (S.W.W.). J.L.U. was supported by T32-CA009111. We thank Dr. Jay Nelson (Vaccine and Gene Therapy Institute, Oregon Health & Science University, Beaverton, OR) for the MVECs used in this research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Babiarz JE, Ruby JG, Wang Y, Bartel DP, Blelloch R. Mouse ES cells express endogenous shRNAs, siRNAs, and other Microprocessor-independent, Dicer-dependent small RNAs. Genes Dev. 2008;22(20):2773–85. doi: 10.1101/gad.1705308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–97. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Bilello JP, Morgan JS, Damania B, Lang SM, Desrosiers RC. A genetic system for rhesus monkey rhadinovirus: use of recombinant virus to quantitate antibody-mediated neutralization. J Virol. 2006;80(3):1549–62. doi: 10.1128/JVI.80.3.1549-1562.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Cullen BR. Transcriptional origin of Kaposi’s sarcoma-associated herpesvirus microRNAs. J Virol. 2006;80(5):2234–42. doi: 10.1128/JVI.80.5.2234-2242.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Hagedorn CH, Cullen BR. Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA. 2004;10(12):1957–66. doi: 10.1261/rna.7135204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Lu S, Zhang Z, Gonzalez CM, Damania B, Cullen BR. Kaposi’s sarcoma-associated herpesvirus expresses an array of viral microRNAs in latently infected cells. Proc Natl Acad Sci U S A. 2005;102(15):5570–5. doi: 10.1073/pnas.0408192102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desrosiers RC, Sasseville VG, Czajak SC, Zhang X, Mansfield KG, Kaur A, Johnson RP, Lackner AA, Jung JU. A herpesvirus of rhesus monkeys related to the human Kaposi’s sarcoma-associated herpesvirus. J Virol. 1997;71(12):9764–9. doi: 10.1128/jvi.71.12.9764-9769.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWire SM, Money ES, Krall SP, Damania B. Rhesus monkey rhadinovirus (RRV): construction of a RRV-GFP recombinant virus and development of assays to assess viral replication. Virology. 2003;312(1):122–34. doi: 10.1016/s0042-6822(03)00195-8. [DOI] [PubMed] [Google Scholar]
- Estep RD, Powers MF, Yen BK, Li H, Wong SW. Construction of an infectious rhesus rhadinovirus bacterial artificial chromosome for the analysis of Kaposi’s sarcoma-associated herpesvirus-related disease development. J Virol. 2007;81(6):2957–69. doi: 10.1128/JVI.01997-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedlander MR, Chen W, Adamidi C, Maaskola J, Einspanier R, Knespel S, Rajewsky N. Discovering microRNAs from deep sequencing data using miRDeep. Nat Biotechnol. 2008;26(4):407–15. doi: 10.1038/nbt1394. [DOI] [PubMed] [Google Scholar]
- Git A, Dvinge H, Salmon-Divon M, Osborne M, Kutter C, Hadfield J, Bertone P, Caldas C. Systematic comparison of microarray profiling, real-time PCR, and next-generation sequencing technologies for measuring differential microRNA expression. RNA. 2010;16(5):991–1006. doi: 10.1261/rna.1947110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottwein E, Cai X, Cullen BR. A novel assay for viral microRNA function identifies a single nucleotide polymorphism that affects Drosha processing. J Virol. 2006;80(11):5321–6. doi: 10.1128/JVI.02734-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundhoff A, Sullivan CS, Ganem D. A combined computational and microarray-based approach identifies novel microRNAs encoded by human gamma-herpesviruses. RNA. 2006;12(5):733–50. doi: 10.1261/rna.2326106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond SM, Bernstein E, Beach D, Hannon GJ. An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature. 2000;404(6775):293–6. doi: 10.1038/35005107. [DOI] [PubMed] [Google Scholar]
- Hutvagner G, McLachlan J, Pasquinelli AE, Balint E, Tuschl T, Zamore PD. A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science. 2001;293(5531):834–8. doi: 10.1126/science.1062961. [DOI] [PubMed] [Google Scholar]
- Jurak I, Kramer MF, Mellor JC, van Lint AL, Roth FP, Knipe DM, Coen DM. Numerous conserved and divergent microRNAs expressed by herpes simplex viruses 1 and 2. J Virol. 2010;84(9):4659–72. doi: 10.1128/JVI.02725-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau NC, Lim LP, Weinstein EG, Bartel DP. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science. 2001;294(5543):858–62. doi: 10.1126/science.1065062. [DOI] [PubMed] [Google Scholar]
- Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, Lee J, Provost P, Radmark O, Kim S, Kim VN. The nuclear RNase III Drosha initiates microRNA processing. Nature. 2003;425(6956):415–9. doi: 10.1038/nature01957. [DOI] [PubMed] [Google Scholar]
- Lee Y, Jeon K, Lee JT, Kim S, Kim VN. MicroRNA maturation: stepwise processing and subcellular localization. EMBO J. 2002;21(17):4663–70. doi: 10.1093/emboj/cdf476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, Kim VN. MicroRNA genes are transcribed by RNA polymerase II. Embo J. 2004;23(20):4051–60. doi: 10.1038/sj.emboj.7600385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Martinez J, Patkaniowska A, Urlaub H, Luhrmann R, Tuschl T. Single-stranded antisense siRNAs guide target RNA cleavage in RNAi. Cell. 2002;110(5):563–74. doi: 10.1016/s0092-8674(02)00908-x. [DOI] [PubMed] [Google Scholar]
- Matranga C, Tomari Y, Shin C, Bartel DP, Zamore PD. Passenger-strand cleavage facilitates assembly of siRNA into Ago2-containing RNAi enzyme complexes. Cell. 2005;123(4):607–20. doi: 10.1016/j.cell.2005.08.044. [DOI] [PubMed] [Google Scholar]
- Orzechowska BU, Powers MF, Sprague J, Li H, Yen B, Searles RP, Axthelm MK, Wong SW. Rhesus macaque rhadinovirus-associated non-Hodgkin lymphoma: animal model for KSHV-associated malignancies. Blood. 2008;112(10):4227–34. doi: 10.1182/blood-2008-04-151498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer S, Sewer A, Lagos-Quintana M, Sheridan R, Sander C, Grasser FA, van Dyk LF, Ho CK, Shuman S, Chien M, Russo JJ, Ju J, Randall G, Lindenbach BD, Rice CM, Simon V, Ho DD, Zavolan M, Tuschl T. Identification of microRNAs of the herpesvirus family. Nat Methods. 2005;2(4):269–76. doi: 10.1038/nmeth746. [DOI] [PubMed] [Google Scholar]
- Ruby JG, Stark A, Johnston WK, Kellis M, Bartel DP, Lai EC. Evolution, biogenesis, expression, and target predictions of a substantially expanded set of Drosophila microRNAs. Genome Res. 2007;17(12):1850–64. doi: 10.1101/gr.6597907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samols MA, Hu J, Skalsky RL, Renne R. Cloning and identification of a microRNA cluster within the latency-associated region of Kaposi’s sarcoma-associated herpesvirus. J Virol. 2005;79(14):9301–5. doi: 10.1128/JVI.79.14.9301-9305.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäfer A, Cai X, Bilello JP, Desrosiers RC, Cullen BR. Cloning and analysis of microRNAs encoded by the primate gamma-herpesvirus rhesus monkey rhadinovirus. Virology. 2007;364(1):21–7. doi: 10.1016/j.virol.2007.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz DS, Hutvagner G, Haley B, Zamore PD. Evidence that siRNAs function as guides, not primers, in the Drosophila and human RNAi pathways. Mol Cell. 2002;10(3):537–48. doi: 10.1016/s1097-2765(02)00651-2. [DOI] [PubMed] [Google Scholar]
- Shi W, Hendrix D, Levine M, Haley B. A distinct class of small RNAs arises from pre-miRNA-proximal regions in a simple chordate. Nat Struct Mol Biol. 2009;16(2):183–9. doi: 10.1038/nsmb.1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umbach JL, Cullen BR. The role of RNAi and microRNAs in animal virus replication and antiviral immunity. Genes Dev. 2009;23(10):1151–64. doi: 10.1101/gad.1793309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umbach JL, Cullen BR. In-depth analysis of Kaposi’s sarcoma-associated herpesvirus microRNA expression provides insights into the mammalian microRNA-processing machinery. J Virol. 2010;84(2):695–703. doi: 10.1128/JVI.02013-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong SW, Bergquam EP, Swanson RM, Lee FW, Shiigi SM, Avery NA, Fanton JW, Axthelm MK. Induction of B cell hyperplasia in simian immunodeficiency virus-infected rhesus macaques with the simian homologue of Kaposi’s sarcoma-associated herpesvirus. J Exp Med. 1999;190(6):827–40. doi: 10.1084/jem.190.6.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi R, Qin Y, Macara IG, Cullen BR. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev. 2003;17(24):3011–6. doi: 10.1101/gad.1158803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y, Cullen BR. Sequence requirements for micro RNA processing and function in human cells. RNA. 2003;9(1):112–23. doi: 10.1261/rna.2780503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y, Cullen BR. Efficient processing of primary microRNA hairpins by Drosha requires flanking nonstructured RNA sequences. J Biol Chem. 2005;280(30):27595–603. doi: 10.1074/jbc.M504714200. [DOI] [PubMed] [Google Scholar]
- Zeng Y, Yi R, Cullen BR. Recognition and cleavage of primary microRNA precursors by the nuclear processing enzyme Drosha. EMBO J. 2005;24(1):138–48. doi: 10.1038/sj.emboj.7600491. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.