

Abstract
Flavodiiron proteins (FDPs) catalyze reductive scavenging of dioxygen and nitric oxide in air sensitive microorganisms. FDPs contain a distinctive non-heme diiron/flavin mononucleotide (FMN) active site. Alternative mechanisms for the nitric oxide reductase (NOR) activity have been proposed consisting of either protonation of a diiron-bridging hyponitrite or “super-reduction” of a diferrous-dinitrosyl by the proximal FMNH2 in the rate-determining step. In order to test these alternative mechanisms, we examined a deflavinated FDP (deflavo-FDP) from Thermotoga maritima. The deflavo-FDP retains an intact diiron site but does not show multi-turnover NOR or O2 reductase (O2R) activity. Reactions of the reduced (diferrous) deflavo-FDP with nitric oxide were examined by UV-vis absorption, EPR, resonance Raman, and FTIR spectroscopies. Anaerobic addition of nitric oxide up to 1 NO:diferrous deflavo-FDP results in formation of a diiron-mononitrosyl complex characterized by a broad S = 1/2 EPR signal arising from antiferromagnetic coupling of an S = 3/2 {FeNO}7 with an S = 2 Fe(II). Further addition of NO results in two reaction pathways, one of which produces N2O and the diferric site and the other of which produces a stable diiron-dinitrosyl complex. Both NO-treated and as-isolated deflavo-FDPs regain full NOR and O2R activities upon simple addition of FMN. The production of N2O upon addition of NO to the mononitrosyl deflavo-FDP supports the hyponitrite mechanism, but the concomitant formation of a stable diiron-dinitrosyl complex in the deflavo-FDP is consistent with a super-reduction pathway in the flavinated enzyme. We conclude that a diiron-mononitrosyl complex is an intermediate in the NOR catalytic cycle of FDPs.
Flavodiiron proteins (FDPs)1 are widespread among bacteria, archaea, and some protozoan pathogens (1–6). FDPs play important roles in the responses to oxidative and nitrosative stresses in microaerobic and anoxic environments by reductively scavenging dioxygen and nitric oxide according to Scheme 1.
Scheme 1.

The relative levels of NADH:dioxygen reductase (O2R) vs NADH:nitric oxide oxidoreductase (NOR) activities (at saturating NROR/Rd) vary significantly among FDPs, but these variations have yet to be correlated with specific structural features. FDPs are soluble cytoplasmic enzymes that are unrelated to the membrane-bound denitrifying NORs (7–9). The minimum functional unit of all structurally characterized FDPs is a “head-to-tail” homodimer (Figure 1) (6, 10–11). The N-terminal domain of each subunit contains a non-heme diiron site (Fe1–Fe2 distance 3.2–3.6 Å), while the C-terminal domain binds a flavin mononucleotide (FMN) cofactor ~4 Å away from the diiron site in the opposite subunit (Figure 1). In almost all structurally characterized FDPs, each iron of the diiron site contains two histidine ligands and a terminal monodentate carboxylate ligand from either aspartate or glutamate. Bridging aspartate carboxylate and oxo/hydroxo ligands complete the diiron coordination sphere resulting in two 5-coordinate irons. Solvent or other exogenous small molecules occupy a buried pocket above the diiron site in these FDP structures. An obvious inference is that O2 or NO diffuses into this pocket and binds to the sixth iron coordination sites during enzymatic turnover. Minimal O2R and NOR mechanisms can, thus, be envisioned involving precursor diferrous-substrate complexes leading to reductive formation of either water from O2 or N2O from NO. According to the reaction stoichiometry (Scheme 1), a single O2R turnover requires four reducing equivalents (presumably provided endogenously by the diferrous site and proximal FMNH2), whereas a single NOR turnover apparently requires only two reducing equivalents.
Figure 1.
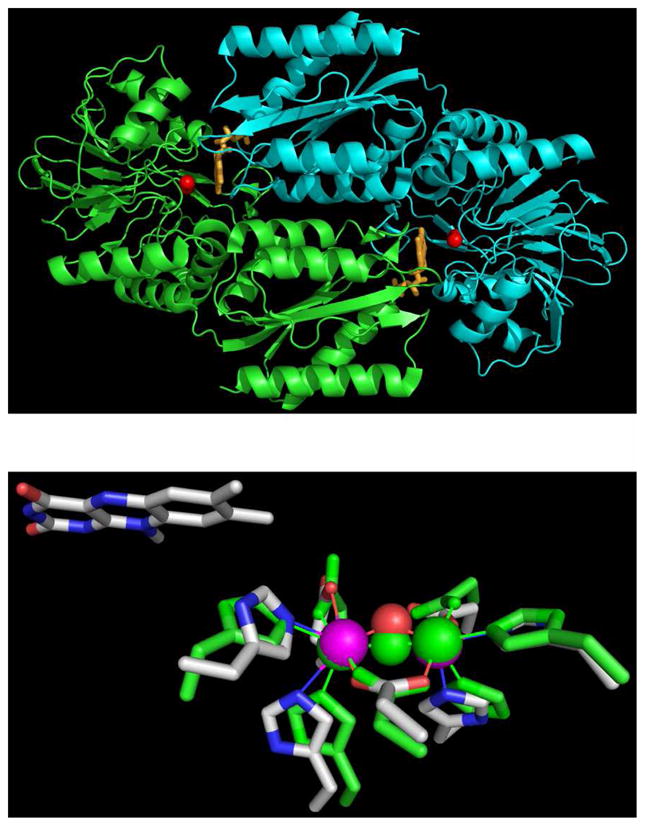
Top Panel: homodimeric backbone structure of M. thermoacetica FDP (PDB ID 1YCF) showing iron atoms as red spheres and FMN as orange sticks. Bottom Panel: superposition of the diiron sites in Mt FDP (PDB ID 1YCF) and Tm FDP (PDB ID 1VME). For the Mt FDP diiron site, iron ligand side chains are depicted as CPK-colored sticks, iron as purple spheres, and bridging solvent as a red sphere. All corresponding Tm FDP atoms are colored green. Proximal isoalloxazine ring of FMN in Mt FDP is also shown. Images and superposition were generated in PyMOL (DeLano Scientific LLC).
The His/carboxylate/solvent-bridged diiron sites of FDPs are reminiscent of those in subunit R2 of ribonucleotide reductase (12–15), the hydroxylase component of soluble methane monooxygenase (MMOH) (16–18), and the Δ9-stearoyl-acyl carrier protein desaturase (Δ9D) (19–20), although there is no detectable amino acid sequence homology between any of these latter enzymes and FDPs. While the diferrous sites of both MMOH and R2 form NO adducts, neither of these proteins exhibits significant NOR activity (21–23). The NO adduct of reduced R2 was characterized as a symmetric, magnetically coupled diferrous-dinitrosyl (21, 23), ([{FeNO}7]2 in Enemark and Feltham’s notation (24)). The reasons for the striking difference in reactivity towards NO of seemingly similar diiron sites in FDPs vs other non-heme diiron proteins are unclear. One possible explanation is that the proximal FMN cofactor in FDPs plays a more integral role in catalytic turnover than simply re-reducing the diiron site back to diferrous after its oxidation to the diferric state by two NO molecules. Super-reduction of a diferrous-dinitrosyl precursor to a ferrous-nitroxyl a diferrous-dinitroxyl, i.e. [{FeN(H)O}8•{FeNO}7] or [{FeN(H)O}8]2, by the proximal FMNH2 has been proposed to provide an energetically favorable route for proton delivery and N-N bond formation leading to the release of N2O and water (Scheme 2) (5, 11). A computational study, however, suggested an alternative mechanism in which the binding of NO to one iron forms a diiron-mononitrosyl complex before the {FeNO}7 unit reacts with a second NO to produce a diferric-hyponitrite intermediate (Scheme 2) (25). An analogous hyponitrite pathway is also presumed to occur in the denitrifying NORs (8, 26–28).
Scheme 2.
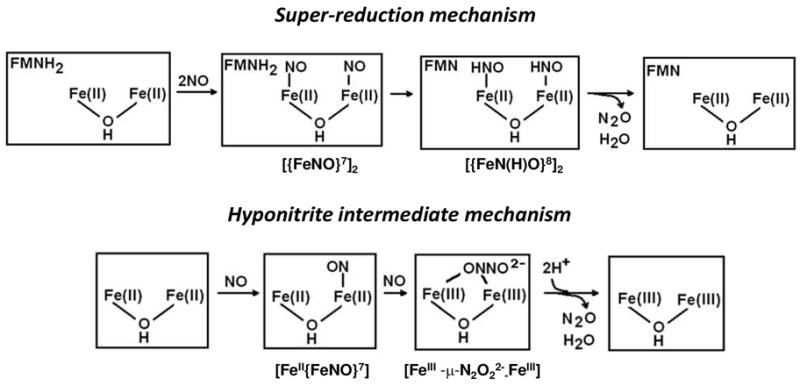
Thus, in the super-reduction mechanism FMNH2 is essential for turnover of the diferrous-dinitrosyl, whereas in the hyponitrite intermediate mechanism, protonation of the spontaneously formed diferric-hyponitrite intermediate leads directly to release of N2O without participation of the FMN. In principle these mechanisms could be distinguished by examining the reactivity of the diiron site with NO in the absence of the FMN cofactor. In 2004, a crystal structure of an FDP from the thermophilic bacteria, Thermotoga maritima (Tm) was deposited into the Protein Data Bank (PDB code: 1VME) by the Joint Center for Structural Genomics. The diiron site structure in Tm FDP is very similar to that in other FDPs (Figure 1), but the proximal FMN cofactor is absent in the deposited Tm FDP structure. This latter structure, thus, suggests the possibility of characterizing the reactivity of the diiron site in the flavin-free Tm FDP.
Tm is classified as an anaerobic bacteria. Elevated Tm FDP levels are observed in Tm cultures exposed to low levels of dioxygen, suggesting a role for this FDP in oxidative stress protection (29). We have found no reports of the response of Tm to nitric oxide exposure or of reactions of Tm FDP with nitric oxide. In this study, we show that flavin-containing Tm FDP has NOR activity and characterize the reactions of the FMN-free Tm FDP (deflavo-FDP) with nitric oxide.
MATERIALS AND METHODS
Protein preparations
All protein concentrations are expressed either in monomers or, where indicated, on the basis of FMN content. The expression and purification of recombinant Tm FDP (GeneID TM0755), Tm rubredoxin (Rd), and Tm NADH:rubredoxin oxidoreductase (NROR) (31), iron, protein and flavin analyses (3, 30) were carried out as described elsewhere.
FDP deflavination and reflavination
Approximately 20 mg of as-isolated Tm FDP was applied to a 50-mL Phenyl Sepharose column (GE Healthcare) with buffer A (50 mM MOPS, 100 mM Na2HPO4, 100 mM KC2H3O2 and 1.2 M NH4SO4 at pH 7.4) to immobilize the FDP. The column pH was then lowered using a 20-column-volume gradient from buffer A to buffer B (50 mM MOPS, 100mM Na2HPO4, 100 mM KC2H3O2 and 1.2 M NH4SO4 at pH 2.5). FMN (yellow band) was eluted from the column at pH 2.5 with buffer B. The column pH was returned to pH 7.3 using a 20-column-volume gradient from buffer B to buffer A. The deflavinated FDP (deflavo-FDP) was eluted with 50 mM MOPS, pH 7.3. Excess salt was removed by washing the concentrated deflavo-FDP with 50 mM MOPS, pH 7.3 in an Amicon 30,000 NMWL centrifugal filter unit (Millipore) and the protein was stored in 50 mM MOPS at pH 7.4. Deflavo-FDP was quantitatively reflavinated (reflavo-FDP) by incubating a 5 mole FMN:1 mole (in monomer) deflavo-FDP mixture ([approximate FDP] = 2 mM) at 37 °C for ~15 minutes. Excess FMN was removed by centrifugal filtration washing with 50 mM MOPS pH 7.3 until the flow-through was colorless. In situ reflavination was accomplished as described below.
O2R/NOR activity assays and N2O production measurements
NADH-dependent O2R and NOR activities were measured spectrophotometrically by monitoring NADH consumption rates (ε340 = 6220 M−1cm−1), as reported earlier (31). Assays were typically carried out in 50 mM MOPS pH 7.4 with 0.1 mM EDTA, and the sequential addition of NADH (100–200 μM), Rd (2–10 μM), NROR (0.1 μM for the O2-reduction and 0.2 μM for the NO-reduction), and FDP (50 nM−1 μM for the O2-reduction and 9 to 17 μM for the NO-reduction (concentrations on an FMN or diiron basis). Small volumes of aqueous O2- or NO-saturated stock solutions were injected into the initially anaerobic assay solutions to initiate the reactions. Alternatively, gaseous air or 0.1 atm NO was introduced into the headspace of the initially anaerobic UV-vis cuvette containing the assay solutions, which were stirred in order to maximize equilibration with the headspace gas.
The production of N2O was monitored by FTIR spectroscopy with the ν(N-N) mode of N2O at 2230 cm−1, as previously reported (28). Briefly, protein solutions ranging from 1 to 2 mM were made anaerobic by prolonged purging with argon on a Schlenk line and reduced with 8 mM dithionite followed by removal of excess reduction agents with desalting spin columns (Zebra, Pierce) in a glove box containing less than 1 ppm O2 (Vacuum Atmospheres Company). A diethylamine NONOate (Cayman Chemical, Ann Arbor, MI) stock solution was prepared using a ε250 = 6,500 M−1cm−1 extinction coefficient in 0.01 M NaOH, and aliquot additions to deoxymyoglobin were used to confirm the concentration of the NO produced. Quickly after the addition of the NONOate stock aliquot the reduced protein, a 30-μL droplet of sample was deposited on a CaF2 window and sealed by dropping a second CaF2 window to complete the FTIR liquid cell. The optical pathlength was controlled by a 100-μm Teflon spacer. FTIR spectra were obtained at room temperature on a Bruker Tensor 27 equipped with a liquid-N2-cooled MCT detector and purged with compressed air, dried, depleted of CO2 (Purge gas generator, puregas LLC). Sets of 100-scan accumulations were acquired at 4-cm−1 resolution, until no further growth of the 2230 cm−1 band was observed. The FTIR spectrum of a buffer blank was also collected in the same conditions for background subtraction.
Preparation of NO adducts
Deflavo-FDP was made anaerobic by prolonged purging with either argon or nitrogen gas on a Schlenk line before being transferred to an anaerobic glove box. Fully reduced enzyme was obtained either by titration to achieve a slight stoichiometric excess of sodium dithionite or by addition of excess dithionite followed by removal of the reducing agent with desalting spin columns. Stoichiometric addition of NO to fully reduced protein was achieved by adding small aliquots of NO-saturated solutions after determining the stock concentration by titration against deoxymyoglobin. Alternatively, NONOate was added into reduced protein solutions in Eppendorf tubes. In both cases, these additions were made just prior to transfer into EPR tubes, Raman capillaries or FTIR cells. To prepare the NO adducts observed at high NO concentrations, the headspaces over reduced protein solutions was replaced with NO (14NO purchased from Airgas and treated with 1 M KOH solution; 15NO and 15N18O from Aldrich) directly in UV-vis cuvettes, EPR tubes, or Raman capillaries. For FTIR measurements, the NO adducts were prepared in Eppendorf tubes before immediate transfer to CaF2 cells.
Molecular spectroscopies
UV-vis absorption spectra were recorded on either Varian Cary 50 or Ocean Optics USB 2000 spectrophotometers in anaerobic UV-vis cuvettes, EPR tubes, Raman capillaries or FTIR sandwiched cells. EPR spectra were obtained with a Bruker E500 X-band EPR spectrometer equipped with a superX microwave bridge and a dual-mode cavity with a helium flow cryostat (ESR900, Oxford Instrument, Inc.). Quantitation of the EPR signals was performed under nonsaturating conditions by double integration and comparison with series of concentrations of Cu(II)EDTA and Fe(II)EDTA(NO) standards. RR spectra were obtained using a custom McPherson 2061/207 spectrograph (set at 0.67 m with variable gratings) equipped with a liquid-N2-cooled CCD detector (LN-1100PB, Princeton Instruments). The 458-nm excitation laser was derived from an Ar laser (Innova 90, Coherent). A long-pass filter (RazorEdge, Semrock) was used to attenuate Rayleigh scattering. RR spectra were collected at room temperature in a 90° scattering geometry on samples mounted on a reciprocating translation stage. To assess the photosensitivity of the NO adduct, rapid acquisitions with minimal laser power and continuous sample translation were compared with longer data acquisitions on static samples. Frequencies were calibrated relative to indene and aspirin standards and are accurate to ±1 cm−1. Polarization conditions were optimized using CCl4 and indene. The integrity of the RR samples was confirmed by direct monitoring of their UV-vis absorption spectra in Raman capillaries before and after laser exposure. Typical enzyme concentrations for RR experiments were 1 mM. FTIR photolysis experiments were carried out as described previously (27–28). Approximately 15 μL of 1.2 mM protein solutions were loaded in FTIR cells with a 15-μm pathlength. After confirming the presence of the NO adduct by UV-vis absorption spectroscopy, the FTIR cell was mounted to a closed-cycle cryogenic systems (Displex and Omniplex, Advanced Research System) and placed inside the sample compartment of the FTIR instrument. The sample was kept in the dark during cooling below 30 K. FTIR spectra were obtained on a Perkin-Elmer System 2000 and a Bruker Tensor 27, both equipped with a liquid-N2-cooled MCT detector. Sets of 1000-scan accumulations were acquired at 4-cm−1 resolution. Photolysis of the nitrosyl complexes were performed by continuous illumination of the sample directly in the FTIR sample chamber using a 50-W tungsten lamp after filtering out heat and NIR emissions. The complete reversibility of the photolysis processes described below was confirmed by reproducing the same FTIR difference spectra after annealing the samples above 40 K.
RESULTS
Characterization of Tm FDP and deflavo-FDP
Heterologous expression in E. coli and purification of Tm FDP produces an enzyme only partially (20–30%) loaded with FMN (assuming 1 FMN per protein monomer constitutes 100% loading). The FMN loading can be increased to ~50% if the growth temperature of the E. coli culture is increased from 37 to 42 °C at the time of induction of FDP overexpression (31). Multiple preparations of Tm FDP contain 2.0 ± 0.2 Fe per protein monomer irrespective of the FMN content. These observations are similar to those described in a previous report on heterologously expressed Tm FDP (29). O2R and NOR activities of Tm FDP were measured via NADH consumption using the Tm redox protein components, NROR (the gene for which is adjacent to that encoding the FDP) and Rd (Scheme 1). The steady-state activity parameters kcat and kcat/KM, determined from fitting to Michaelis-Menten plots (Figure S1), are 4 s−1 and 2 × 106 M−1s−1, respectively, for O2R activity, and 0.05 s−1 and 700 M−1 s−1 for NOR activity. Neither activity is observed if any one of the protein components (FDP, NROR or Rd) is omitted from the assay solutions. The relatively low NOR activity of Tm FDP prevents a reliable assessment of cooperativity, which, for some FDPs, is evident from the sigmoidal forms of the Michaelis-Menten plots (31).
Essentially complete removal of FMN from as-isolated Tm FDP is achieved by an acid-wash (pH 2.5) of the protein bound to phenyl sepharose prior to elution of deflavo-FDP at pH 7.3. The deflavo-FDP consistently retains 2 Fe per protein monomer. As expected, the UV-vis absorption features of FMN are absent from the spectrum of the Tm deflavo-FDP (Figure S2). The integrity of the diiron site in the absence of FMN is supported by the crystal structure of Tm FDP (1VME) which has no FMN bound and shows a diiron site structure and overall protein fold very similar to those observed in the crystal structure of Mt FDP (PDB ID 1YCF) (Figure 1&S3). This structural equivalence has been confirmed by a crystal structure of the Tm deflavo-FDP prepared in our lab (A. Taylor, P. J. Hart, J. Caranto, D. M. Kurtz, Jr., unpublished). As anticipated, the deflavo-FDP has no detectable multi-turnover O2R or NOR activity (Figure S2), but the deflavo-FDP can be quantitatively reconstituted with FMN while retaining full iron occupancy. The reflavinated protein shows full restoration of O2R and NOR activity (Figure S2).
UV-vis spectral characterization of the reaction of reduced deflavo-FDP with NO
Reduction of deflavo-FDP with dithionite results in the loss of broad absorption features between 350 and 420 nm that originate from carboxylate and bridging oxo/hydroxo-to-iron(III) LMCT transitions (Figure 2), and presumably results in diferrous deflavo-FDP. Addition of up to 1 equiv NO to reduced deflavo-FDP produces a pale yellow color associated with absorption bands at 420, 455, and 638 nm (Figure 2&S4). These absorption features are characteristic of nitrosyl-to-iron LMCT transitions of non-heme {FeNO}7 complexes with O/N ligands (Table 1) (21–23, 32–36). At 1 equiv NO per diiron site, the 420/455-nm absorption corresponds to a ε420/455 nm ~ 1,000 M−1 cm−1 which is a good match for non-heme {FeNO}7 species in proteins and synthetic model (Table 1). The high absorbance at 420/455 nm reached with 1 equiv NO also suggests that the diiron(II) site binds NO with high affinity. We refer to this 1 added NO:diiron complex as deflavo-FDP(NO). Further addition of NO, up to 2 equiv, results in a significant loss of the nitrosyl-to-iron LMCT bands in favor of absorption increases below 420 nm (Figure 2&S4). These absorption changes suggest that, above 1 equiv NO added per diiron site promotes the formation of oxidized, presumably diferric, deflavo-FDP. However, this conversion is incomplete, and further NO addition results in increased absorptions at 455 nm and 638 nm (Figure 2&S4). These rising absorption features at high NO concentrations are assigned to a deflavo-FDP(NO)2 complex that forms in parallel with oxidized deflavo-FDP (see below).
Figure 2.

UV-vis spectra of oxidized (orange), dithionite reduced (black), and reduced deflavo-FDP after addition of 1 equiv (red), 2 equiv (green) and 1 atm NO (blue) at room temperature (protein concentration = 147 μM in 50 mM MOPS pH 7.4; extinction coefficient per diiron site). Also shown is the difference spectrum [1-atm NO spectrum] – 0.65 [oxidized spectrum] (black dashed line).
Table 1.
Spectroscopic properties of non-heme ferrous-nitrosyl complexes containing O/N ligands.
| {FeNO}7 species | λmax (nm) (ε in M−1 cm−1)a | ν(Fe-NO) (Δ15N) (cm−1) | δ(Fe-N-O) (Δ15N) (cm−1) | ν(N-O) (Δ15N) (cm−1) | Ref. |
|---|---|---|---|---|---|
| Deflavo-FDP-(NO) | 420, 455(1000) | 451 (−9) | not obs. | this work | |
| Deflavo-FDP-(NO)2 | 455 (1000) | 459 (−7) | 444 (−8) | 1749 (−30) | this work |
| Wild-type R2-(NO)2 | 450 (760) | 445 (−7) | 434 (−9) | 1742 (−29) | (21, 23) |
| D84E-R2-(NO)2 | 450 (1200) | 454 (−8) | 442 (−8) | 1735 (−30) | (23) |
| Hemerythrin-(NO) | 408 (1000) | 433 (−6) | 421 (−6) | not obs. | (35) |
| Superoxide reductase-(NO) | 475 (530) | 475 (−7) | 1721 (−31) | (52) | |
| Fe(EDTA)-(NO) | 434 (900) | 496 (−4) | 493 (−10) | 1776 (−37) | this work and (40, 53) |
| [Fe2(NO)2(μ-XDK)(μ- O2CPh)(ImH)2(O2CPh)-(MeOH)] | 450 (1,000) | n.r.b | n.r.b | n.r.b | (23) |
| Fe2(NO)2(Et-HPTB)(O2CPh)(BF4)2 | n.r.b,c. | n.r.b | n.r.b | 1785 | (47) |
per iron.
not reported.
obscured by ligand absorptions.
The NO adducts that form at high vs low added NO:protein ratios can be distinguished by their sensitivity towards dithionite. Specifically, when the deflavo-FDP(NO) complex is exposed to a 2-fold molar excess of dithionite at room temperature, the nitrosyl-to-iron LMCT transitions are bleached in a monotonous fashion and with a slow apparent t1/2 ~ 15 min (Figure S5A). In contrast, the 455-nm absorption due to the deflavo-FDP(NO)2 complex formed with excess NO decays upon exposure to dithionite within the manual mixing time (Figure S5B). This rapid bleaching is accompanied by the slower reduction of the oxidized diiron sites; the residual absorption that decays at an even slower rate is likely to correspond to a small population of mixed-valent FeIIIFeII sites (Figure S5B). This interpretation is supported by the UV-vis spectral changes observed for the reaction of 2 equiv dithionite with oxidized deflavo-FDP (Figure S5C). In this experiment, the spectrum obtained immediately after mixing is distinct from that of the oxidized deflavo-FDP, and the relative absorbance loss near 400 nm and gain at ~500 nm can be assigned to mixed-valent diiron contributions; subsequent traces show a slow bleaching of these visible absorptions that is equivalent to the later changes observed during dithionite reduction of the deflavo-FDP(NO)2/oxidized deflavo-FDP mixture. Taken together, these data suggest that the bleaching of the deflavo-FDP(NO) complex by dithionite reflects the slow NO off-rate from the mononitrosyl complex as dithionite scavenges free NO from solution. The faster reaction of the deflavo-FDP(NO)2 with dithionite could reflect either a faster NO off rate or super-reduction of the diiron-dinitrosyl complex.2 The fraction of oxidized deflavo-FDP present in the mixture produced by exposure of the reduced protein to high NO is re-reduced slowly by dithionite with formation of mixed-valent and diferrous states.
The picture that emerges from these experiments is that the deflavo-FDP diferrous site binds one NO molecule with relatively high affinity before further reaction with excess NO to produce a mixture of oxidized deflavo-FDP and unreactive {FeNO}7 centers. Presuming the as-isolated spectrum is equivalent to the post-NO treated oxidized spectrum, we subtracted its contribution to the absorption spectra; the remaining spectra correspond to the fraction of non-heme {FeNO}7 complexes with absorption bands at 455 and 638 nm (Figure 2). Using this analysis, the fraction of oxidized protein resulting from the excess NO treatment was consistently reproducible for individual deflavo-FDP preparations, typically ranging from 60 to 70%, reaching 40% for one of the preparations (Figure S6). The origin of this variability is currently unclear. The remaining absorbance at 455 nm corresponds to a molar extinction coefficient of ~1,000 M−1cm−1 per iron. This value is again in good agreement with other non-heme {FeNO}7 complexes (Table 1) and suggests that the diiron sites that do not proceed to the diiron(III) state instead form stable diferrous-dinitrosyl [{FeNO}7]2 clusters, as previously observed with reduced R2 exposed to excess NO (21, 23).
FTIR evidence for N2O production
Consistent with the preceding UV-vis absorption analysis, FTIR spectroscopy shows production of N2O from the reaction of excess NO with reduced deflavo-FDP. Specifically, we detect the N-N stretching mode of N2O at 2330 cm−1 in the FTIR spectra of deflavo-FDP samples and compare these measurements with data for myoglobin and terminal oxidases as negative and positive controls, respectively (Figure S7). The intensity of the FTIR band at 2330 cm−1 upon completion of the reaction reveals that ~0.7 equiv of N2O is formed per deflavo-FDP diiron sites, which closely matches the estimated fraction of oxidized diferric deflavo-FDP from UV-vis absorption spectroscopy. A comparable FTIR experiment with fully reduced reflavinated FDP exposed to excess NO from NONOate shows ~2.0 equiv N2O produced per FDP diiron site (Figure S7), which confirms that all reducing equivalents are used to produce N2O.
EPR characterization of the reaction of reduced deflavo-FDP with NO
As established earlier for native FDPs (3, 30, 37), the oxidized diiron site of deflavo-FDP is EPR silent as bridging solvent and carboxylate ligands promote antiferromagnetic coupling of the two nonheme iron(III) ions. The dithionite-reduced deflavo-FDP is also EPR silent, as expected for a fully reduced diiron(II) site with no evidence for the formation of mixed valent Fe(II)•Fe(III) cluster, as observed previously in redox titration of FDPs (30, 37). Addition of 1 equiv of NO results in the appearance of a broad axial EPR signal centered at g ~ 2 which we assign to an S = 1/2 diiron-mononitrosyl species produced by antiferromagnetic coupling of an S = 2 Fe(II) to an S = 3/2 {FeNO}7 species (Figure 3). This EPR spectrum is reminiscent of the axial signals of hemerythrin-NO adducts with g// and g⊥ near 2.8 and 1.8, respectively (35, 38), although the anisotropic components of deflavo-FDP(NO) are more difficult to pinpoint since the splitting is small and the g strain on both g// and g⊥ are relatively large. Double integrations of the EPR signal of deflavo-FDP(NO) against Cu(II)EDTA standards indicate that the axial g ~ 2 signal represents 0.75 spin per diiron site. This deviation from stoichiometry is easily explained by some dissociation of NO from deflavo-FDP(NO) complex prior to freezing the EPR tubes since the complex is prepared without NO excess; a slight overshot in the NO addition, above 1 equiv, will also lower the deflavo-FDP(NO) content to produce a fraction of diferric deflavo-FDP. As expected from the UV-vis experiments, the g ~ 2 EPR signal from the diiron-mononitrosyl complex disappears upon addition of 2 equiv NO, concomitant with the appearance of new EPR resonances near g ~ 4 (Figure 3).
Figure 3.

EPR spectra of oxidized deflavo-FDP (top trace), dithionite-reduced deflavo-FDP (second trace), and reduced deflavo-FDP after the addition of 1 equiv (third trace), 2 equiv (forth trace) and 0.05 atm NO headspace (bottom trace) at 4.2 K. Conditions: protein concentration, 100 μM in 50 mM MOPS pH 7.4; microwave frequency, 9.66 GHz; microwave power, 2 mW; modulation amplitude, 10 G.
These EPR features at g ~ 4 are consistent with rhombic EPR signatures from S = 3/2 {FeNO}7 species (39–40). Comparing double integrations of these signals with those of Fe(II)EDTA(NO) standards shows that less than 3% of the total iron content contribute to this regions of the EPR spectra in deflavo-FDP samples treated with 2 equiv NO, but that this value rises to ~30% of the total iron when the free NO concentration is increased to 100 μM with an 0.05 atm NOg headspace (Figure 3). Further increases in free NO concentration do not affect the intensity of the g ~ 4 signals (data not shown). The multiplicity of g ~ 4 resonances is readily assigned to two distinct S = 3/2 {FeNO}7 species, a more rhombic one with g = 4.37, 3.68, 2.00 and another with g = 4.14, 3.93, 2.0 (Figure 3). While the more rhombic signal represents a minor species at all NO concentrations, the more axial species becomes prominent at the higher NO concentration. The quantitation of the g ~ 4 signals matches well with the ~30% of diferrous-dinitrosyl [{FeNO}7]2 sites estimated from the UV-vis analysis. The EPR data, thus, imply that the remaining 30% of deflavo-FDP diiron sites form {FeNO}7 centers that are magnetically uncoupled. This observation is in striking contrast with the magnetically coupled, EPR-silent diferrous-dinitrosyl [{FeNO}7]2 sites of R2(NO)2 (21, 23).
RR and FTIR characterization of reduced deflavo-FDP-NO complexes
The deflavo-FDP(NO)2 complex resulting from reaction of reduced protein with excess NO is sufficiently stable to allow extended acquisition times for optimal RR spectral characterization. As expected, the RR spectra obtained with 458-nm laser excitation revealed enhancement of vibrational modes characteristic of the {FeNO}7 unit (Figure 4 and Table 1). A band at 459 cm−1 that downshifts to 452 (−7) and 447 (−12) cm−1 with 15NO and 15N18O, respectively, is assigned to the ν(Fe-NO) mode. In the high frequency region, a band at 1749 cm−1 downshifts to 1719 (−30) and 1679 (−70) cm−1 with 15NO and 15N18O, respectively (Figure 4) and is readily assigned to the ν(N-O) mode. In addition to these two Fe-N-O vibrations, another isotope-sensitive RR band is observed at 903 cm−1 and shifts to 888 (−15) and 879 (−24) cm−1 with 15NO and 15N18O, respectively. Comparable RR signals in the 900-cm−1 region for the NO adduct of R2 were assigned to a combination of bending and stretching Fe-N-O modes (23). An equivalent RR band and isotope dependence are observed for the mononuclear complex, Fe(II)EDTA(NO) (Figure S8), which rules out some alternative assignments, such as a bound hyponitrite (41), for this mid-frequency vibration. All of these assignments are consistent with those in Table 1 for {FeNO}7 units in other proteins and synthetic complexes, including the deduced Fe-N-O bending/stretching combination modes.
Figure 4.

Room temperature RR spectra of reduced deflavo-FDP (protein concentration ~ 1 mM in 50 mM MOPS pH 7.4) before (green) and after exposure to excess 14NO (top black), 15NO (top red), and 15N18O (top blue). The three bottom spectra are “NO complexes” minus “reduced” difference spectra color-coded as for the top spectra. The spectral subtractions were normalized on the 1003-cm−1 band from Phe; the sharp 980-cm−1 band originates from sulfate byproduct of dithionite oxidation.
The [{FeNO}7]2 complex of deflavo-FDP can also be characterized by light-induced FTIR difference spectroscopy at low temperature. The “dark” minus “illuminated” difference spectra obtained at 30 K show a single ν(N-O) at 1751 cm−1 that shifts to 1721 (−30) cm−1 with 15NO (Figure 5). These frequencies are in good agreement with those observed in the RR spectra and support the notion that the two {FeNO}7 units experience similar environments within the protein matrix. Nitrosyl ligands, when photo-dissociated from metal centers at cryogenic temperatures, typically remain as free NO molecules trapped with the protein pocket, and typically exhibit very weak ν(N-O) modes between 1850 and 1860 cm−1 (23, 28, 42). Such vibrations should appear as negative band(s) in the FTIR difference spectra, but so far, we have not detected such signals in a reproducible fashion in the light-induced difference spectra of deflavo-FDP(NO)2. The empty FMN-binding site may provide a more expansive pocket with multiple docking sites for the dissociated NO, which may broaden these signals beyond detection.
Figure 5.

FTIR difference spectra (“dark” minus “illuminated”) of deflavo-FDP(14NO)2 (black) and deflavo-FDP(15N O)2 (grey) obtained below 30 K (protein concentration ~ 1.2 mM).
The mononitrosyl deflavo-FDP(NO) complex forms without appreciable fractions of the dinitrosyl complex only when using ≤1 equiv NO:diiron site. Under these conditions significant dissociation and degassing of free NO can take place in RR capillaries. Nevertheless, RR spectra obtained with acquisition time of a few minutes reveal vibrational modes similar to those of the [{FeNO}7]2 complex, but at lower energy (Figure 6). A broad RR band centered at 451 cm−1 downshifts to 442(−9) cm−1 and 436(−15) cm−1 with 15NO and 15N18O, respectively.3 Changes in half-widths and bandshapes with different NO-isotopes in deflavo-FDP(NO) suggest that this broad band may be composed of more than one unresolved components and may reflect overlapped bending and stretching modes of the Fe-N-O unit. Due to the low signal-to-noise ratio of these RR spectra, no other vibrational modes could be clearly identified. As expected, additions of excess NO to the mononitrosyl complexes produce RR spectra equivalent to those of deflavo-FDP(NO)2 (data not shown). Low-temperature FTIR photolysis experiments carried out to further characterized the deflavo-FDP(NO) complex were unsuccessful.
Figure 6.

Room temperature RR difference spectra (“NO complexes” minus “reduced protein”) of deflavo-FDP(NO) formed with 14NO (top trace), 15NO (middle trace), and 15N18O (bottom trace) (protein concentration ~ 1 mM in 50 mM MOPS pH 7.4).
DISCUSSION
The heterologously expressed Tm FDP has full diiron site but only partial FMN occupancies. We found that the as-isolated protein has a high O2R activity relative to its NOR activity. The O2R activity is consistent with its upregulation in T. maritima exposed to oxidative conditions (29). However, the relatively low NOR activity of the heterologously expressed Tm FDP is still significantly above background. Whether this NOR activity is physiologically relevant in Thermotoga maritima remains to be determined, but in other organism, FDPs appear to confer both oxidative and nitrosative stress protection in vivo (43–44).
In an attempt to distinguish between NOR mechanisms of FDPs described in the Introduction, we prepared Tm deflavo-FDP and characterized its reaction with NO. In agreement with the deposited X-ray crystal structure (and our own), our results demonstrate that the FMN cofactor can be removed from Tm FDP without irreversible disruption of the protein structure and specifically, that the diiron site remains intact in the FMN-free protein. As expected, the FMN-free FDP is catalytically inactive, but can be readily and quantitatively reconstituted with FMN, while retaining full iron occupancy. The reflavinated protein regained O2R and NOR activities that were indistinguishable from the initially isolated protein on an FMN basis.
Stoichiometric addition of 1 NO:diiron site to reduced deflavo-FDP results in the formation of a stable deflavo-FDP(NO) complex. The UV-vis absorption spectrum of this complex with bands at 420, 455, and 638 nm is characteristics of non-heme {FeNO}7 complexes containing oxygen and nitrogen ligands. The calculated extinction coefficient of the chromophoric species (ε420/455 nm = ~1,000 mM−1 cm−1) is in good agreement with values reported for other proteins and synthetic {FeNO}7 complexes. Such {FeNO}7 complexes are best described as S = 3/2 species with a high-spin iron(III) (S = 5/2) antiferromagnetically coupled to a NO− (S = 1) and show characteristic EPR signals around g ~ 4 (43–44). In the deflavo-FDP(NO) complex, antiferromagnetic coupling between the {FeNO}7 S = 3/2 and the second iron(II) S = 2 produces an S = 1/2 [FeII•{FeNO}7] species that exhibits a broad g ~ 2 signal, as previously observed in the NO adduct of hemerythrin (43–44). The double integration of this EPR signal suggests that the stoichiometric addition of NO nearly quantitatively converts the diiron(II) to [FeII•{FeNO}7] sites. To our knowledge this is the first example of a diferrous-mononitrosyl complex formed in a diiron(II) protein or synthetic complex where the two metals share the same ligand sets.
The near-stoichiometric formation of the diferrous-mononitrosyl complex implies that, at least in deflavo-FDP, the first NO molecule binds with higher affinity than that of the second NO. This order of NO affinities seems in conflict with the cooperativity in NO binding inferred from the sigmoidal Michaelis-Menten plots for NOR activities of some other FDPs (3, 31, 45). The relatively low NOR activity of flavinated Tm FDP may reflect a lack of cooperativity in NO binding. Relative NOR vs O2R activities are also variable among FDPs for unknown reasons (1–6, 46).
Further addition of NO up to 2 equiv and beyond results in a change in the UV-vis absorption spectrum and the loss of the broad g = 2 EPR signal, consistent with NO reacting with the diferrous-mononitrosyl site. The product of this reaction is not homogenous: up to 70% of the diiron sites are catalytically competent and produce N2O, as revealed by FTIR, while the remaining diiron sites form magnetically uncoupled {FeNO}7 S = 3/2 species with characteristic g ~ 4 EPR signatures. Quantitative analyses of the UV-vis, FTIR, and EPR data all support these relative proportions for a given deflavo-FDP preparation, even though some variability in proportions of the two diiron products was observed from preparation to preparation.
The lack of magnetic coupling between the two {FeNO}7 centers of deflavo-FDP(NO)2 requires a major disruption of the efficient exchange coupling pathway provided by the oxo/hydroxo solvent bridge between the iron centers in the [FeIII•FeIII], mixed-valent [FeII•FeIII] diiron sites, and in the diiron-mononitrosyl complex [FeII•{FeNO}7] (3, 30, 37). Presumably, the solvent bridge is lost upon formation of the diferrous-dinitrosyl complex, but the bridging Asp carboxylate present in both reduced and oxidized FDP diiron sites may or may not remain in the diferrous-dinitrosyl complex. To our knowledge, there has been no report of synthetic analogs containing a carboxylate-bridged diiron site without at least one other supporting bridge. The only well-characterized, carboxylate-bridged, non-heme, O/N-ligated diferrous-dinitrosyl synthetic complex, Fe2(NO)2(Et-HPTB)(O2CPh)(BF4)2, includes an additional bridging alkoxo ligand (47). This complex is EPR silent due to antiferromagnetic coupling between the two {FeNO}7 centers (47). In the case of the diferrous-dinitrosyl adduct of ribonucleotide reductase R2 protein, the antiferromagnetic coupling between the two irons (and EPR silent character) may be due to the presence of both bidentate and monodentate carboxylate bridges, as observed in the diferrous R2 protein (15). Dinuclear Mn(II) sites in both proteins and synthetic complexes containing a bidentate carboxylate as the only bridging ligand have reportedly shown no evidence of magnetic exchange coupling (48–49). A single bridging carboxylate with anti-syn configuration can accommodate metal-metal distance beyond 6.3 Å (50–51), and the two {FeNO}7 units could move further apart if the bridging carboxylate group adopt a non-bridging conformation. We view these data as evidence of conformational flexibility at the diiron site since uncoupling of the two irons at the active site is fully reversible and is not associated with iron loss. Indeed, the [{FeNO}7]2 species can be re-oxidized after exchanging the sample headspace, first with Ar then with O2, re-reduced, and re-exposed to NO to regenerate the same fraction of [{FeNO}7]2 species, with no loss of iron (data not shown). Similarly, two consecutive redox-cyclings of deflavo-FDP(NO)2 do not decrease the steady-state O2R or NOR activity observed after FMN addition.
The vibrational characterization of the uncoupled [{FeNO}7]2 reveals a single set of Fe-N-O vibrations, suggesting that the individual {FeNO}7 units adopt equivalent configurations. The Fe-N-O vibrations of deflavo-FDP(NO)2 are similar to those observed previously in R2(NO)2 (23), which is not unexpected since the endogenous iron ligands are comparable in both proteins. Our vibrational characterization of deflavo-FDP(NO) is less complete than for the dinitrosyl complex, but the ν(Fe-NO) mode is only ~8 cm−1 lower than in the [{FeNO}7]2 complex.
The reactions of reduced deflavo-FDP with NO based on our results are summarized in Scheme 3.
Scheme 3.

Reduction of NO to N2O by the reduced diiron site in the absence of FMN (pathway A in Scheme 3), is consistent with the hyponitrite mechanism in Scheme 2. While Scheme 2, as suggested by the computational study by Siegbahn and coworkers (25), indicates that the second NO reacts with the {FeNO}7 species rather than the second iron(II), a magnetically coupled diferrous-dinitrosyl could be a transient intermediate in pathway A and in the hyponitrite mechanism. Thus, super-reduction of {FeNO}7 units to nitroxyl {FeN(H)O}8 species does not represent an absolute requisite step in the NOR reaction pathway in FDPs. The parallel pathway B in Scheme 3 leading to a stable magnetically uncoupled diferrous-dinitrosyl may be prevented in the flavinated enzyme via steric constraints. Alternatively, turnover of an [{FeNO}7]2 complex in FDP may require reduction by the proximal FMN cofactor, as proposed in the “super-reduction” mechanism (Scheme 2) (5, 11). Uncoupling of the two {FeNO}7 units may also permit a reaction pathway in the flavinated enzyme where each iron center functions independently to form mononuclear hyponitrite complexes, thereby maximizing scavenging efficiency under high NO stress conditions. The results of this study provide a clear framework for further investigation of this unique family of enzymes.
Supplementary Material
Acknowledgments
We thank Drs. Adeniki Otoikhian and Ninian Blackburn for help with ICP-OES, and Dr. James Whittaker for the use of his helium cryostat.
Footnotes
Abbreviations: FDP, flavodiiron protein; FMN, flavin mononucleotide; Mt, Moorella thermoacetica; Tm, Thermotoga maritima; MMOH, hydroxylase component of soluble methane monooxygenase; Δ9D, Δ9-stearoyl-acyl carrier protein desaturase; EPR, electron paramagnetic resonance; RR, resonance Raman; FTIR, Fourier transform infra-red; Rd, rubredoxin; NROR, NADH:rubredoxin oxidoreductase; XDK, m-xylylenediamine bis(Kemp’s triacid)imide; MOPS, 3-(N-morpholino)propanesulfonic acid; O/N, oxygen/nitrogen; Et-HPTB, N,N,N′,N′-tetrakis(N-ethyl-2-benzimidazolylmethyl)-1,3-diaminopropane); TMPD, N,N,N′,N′,-tetramethyl-p-phenylenediamine.
To our knowledge, [S = 3/2 {FeNO}7]/{FeN(H)O}8 reduction potentials have not been reported. The reduction potential for NO/3NO− and NO/1HNO couples are −0.8 V and −1.7 V, respectively. In iron-porphyrins (54), heme proteins (55), and octahedral non-heme iron complexes (56), {FeNO}7 species are S = 1/2 systems with reduction potentials between ca −0.6 and −1.4 V. While these reduction potentials suggest that those of S = 3/2 {FeNO}7 species to be inaccessible to dithionite, the{FeNO}6 species nitroprusside ion, [Fe(CN)5(NO)]2−, was recently shown to be reducible to the {FeN(H)O}8 species by two equivalents of dithionite in aqueous solution at pH 10 (57).
The impact of the 18O-labeling on the observed frequency may reflect contribution from Fe-N-O bending and/or one-body stretching motions of the NO group with regard to the iron.
SUPPORTING INFORMATION AVAILABLE: O2R and NOR activity assays of as-isolated and deflavo-FDPs; UV-vis absorption spectra of as-isolated, deflavo and reflavinated Tm FDPs; view of superposition of Mt FDP and Tm FDP protein backbones; UV-vis titrations of reduced deflavo-FDP with NO; UV-vis monitoring of the reaction of dithionite with deflavo-FDP(NO), deflavo-FDP(NO)2, and oxidized deflavo-FDP; FTIR monitoring of N2O production; RR spectra of FeII(EDTA)NO. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported in part by the National Institute of Health (P.M.-L., GM74785; D.M.K. Jr., GM040388) and a Vertex pharmaceutical scholarship for T.H.
References
- 1.Gardner AM, Helmick RA, Gardner PR. Flavorubredoxin, an inducible catalyst for nitric oxide reduction and detoxification in Escherichia coli. J Biol Chem. 2002;277:8172–8177. doi: 10.1074/jbc.M110471200. [DOI] [PubMed] [Google Scholar]
- 2.Gomes CM, Giuffre A, Forte E, Vicente JB, Saraiva LM, Brunori M, Teixeira M. A novel type of nitric-oxide reductase. Escherichia coli flavorubredoxin. J Biol Chem. 2002;277:25273–25276. doi: 10.1074/jbc.M203886200. [DOI] [PubMed] [Google Scholar]
- 3.Silaghi-Dumitrescu R, Coulter ED, Das A, Ljungdahl LG, Jameson GN, Huynh BH, Kurtz DM., Jr A flavodiiron protein and high molecular weight rubredoxin from Moorella thermoacetica with nitric oxide reductase activity. Biochemistry. 2003;42:2806–2815. doi: 10.1021/bi027253k. [DOI] [PubMed] [Google Scholar]
- 4.Saraiva LM, Vicente JB, Teixeira M. The role of the flavodiiron proteins in microbial nitric oxide detoxification. Adv Microb Physiol. 2004;49:77–129. doi: 10.1016/S0065-2911(04)49002-X. [DOI] [PubMed] [Google Scholar]
- 5.Kurtz DMJ. Flavo-diiron enzymes: nitric oxide or dioxygen reductases? Dalton Trans. 2007:4115–4121. [Google Scholar]
- 6.Di Matteo A, Scandurra FM, Testa F, Forte E, Sarti P, Brunori M, Giuffre A. The O2-scavenging flavodiiron protein in the human parasite Giardia intestinalis. J Biol Chem. 2008;283:4061–4068. doi: 10.1074/jbc.M705605200. [DOI] [PubMed] [Google Scholar]
- 7.Wasser IM, de Vries S, Moënne-Loccoz P, Schroder I, Karlin KD. Nitric oxide in biological denitrification: Fe/Cu metalloenzyme and metal complex NOx redox chemistry. Chem Rev. 2002;102:1201–1234. doi: 10.1021/cr0006627. [DOI] [PubMed] [Google Scholar]
- 8.Moënne-Loccoz P. Spectroscopic characterization of heme iron-nitrosyl species and their role in NO reductase mechanisms in diiron proteins. Natl Prod Rep. 2007;24:610–620. doi: 10.1039/b604194a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Watmough NJ, Field SJ, Hughes RJ, Richardson DJ. The bacterial respiratory nitric oxide reductase. Biochem Soc Trans. 2009;37:392–399. doi: 10.1042/BST0370392. [DOI] [PubMed] [Google Scholar]
- 10.Frazao C, Silva G, Gomes CM, Matias P, Coelho R, Sieker L, Macedo S, Liu MY, Oliveira S, Teixeira M, Xavier AV, Rodrigues-Pousada C, Carrondo MA, Le Gall J. Structure of a dioxygen reduction enzyme from Desulfovibrio gigas. Nat Struct Biol. 2000;7:1041–1045. doi: 10.1038/80961. [DOI] [PubMed] [Google Scholar]
- 11.Silaghi-Dumitrescu R, Kurtz DM, Jr, Ljungdahl LG, Lanzilotta WN. X-ray crystal structures of Moorella thermoacetica FprA. Novel diiron site structure and mechanistic insights into a scavenging nitric oxide reductase. Biochemistry. 2005;44:6492–6501. doi: 10.1021/bi0473049. [DOI] [PubMed] [Google Scholar]
- 12.Nordlund P, Sjöberg B-M, Eklund H. Three-dimensional structure of the free radical protein of ribonucleotide reductase. Nature. 1990;345:593–598. doi: 10.1038/345593a0. [DOI] [PubMed] [Google Scholar]
- 13.Voegtli WC, Khidekel N, Baldwin J, Ley BA, Bollinger JM, Rosenzweig AC. Crystal structure of the ribonucleotide reductase R2 mutant that accumulates a mu-1,2-peroxodiiron(III) intermediate during oxygen activation. J Am Chem Soc. 2000;122:3255–3261. [Google Scholar]
- 14.Baldwin J, Voegtli WC, Khidekel N, Moënne-Loccoz P, Krebs C, Pereira AS, Ley BA, Huynh BH, Loehr TM, Riggs-Gelasco PJ, Rosenzweig AC, Bollinger JM., Jr Rational reprogramming of the R2 subunit of Escherichia coli ribonucleotide reductase into a self-hydroxylating monooxygenase. J Am Chem Soc. 2001;123:7017–7030. doi: 10.1021/ja002114g. [DOI] [PubMed] [Google Scholar]
- 15.Voegtli WC, Sommerhalter M, Saleh L, Baldwin J, Bollinger JM, Jr, Rosenzweig AC. Variable coordination geometries at the diiron(II) active site of ribonucleotide reductase R2. J Am Chem Soc. 2003;125:15822–15830. doi: 10.1021/ja0370387. [DOI] [PubMed] [Google Scholar]
- 16.Rosenzweig AC, Frederick CA, Lippard SJ, Nordlund P. Crystal structure of a bacterial non-heme iron hydroxylase that catalyzes the biological oxidation of methane. Nature. 1993;366:537–543. doi: 10.1038/366537a0. [DOI] [PubMed] [Google Scholar]
- 17.Rosenzweig AC, Nordlund P, Takahara PM, Frederick CA, Lippard SJ. Geometry of the soluble methane monooxygenase catalytic diiron center in two oxidation states. Chem Biol. 1995;2:409–418. [PubMed] [Google Scholar]
- 18.Whittington DA, Lippard SJ. Crystal structures of the soluble methane monooxygenase hydroxylase from Methylococcus capsulatus (Bath) demonstrating geometrical variability at the dinuclear iron active site. J Am Chem Soc. 2001;123:827–38. doi: 10.1021/ja003240n. [DOI] [PubMed] [Google Scholar]
- 19.Lindqvist Y, Huang W, Schneider G, Shanklin J. Crystal structure of delta9 stearoyl-acyl carrier protein desaturase from castor seed and its relationship to other di-iron proteins. EMBO J. 1996;15:4081–4092. [PMC free article] [PubMed] [Google Scholar]
- 20.Moche M, Shanklin J, Ghoshal A, Lindqvist Y. Azide and acetate complexes plus two iron-depleted crystal structures of the di-iron enzyme delta9 stearoyl-acyl carrier protein desaturase. Implications for oxygen activation and catalytic intermediates. J Biol Chem. 2003;278:25072–25080. doi: 10.1074/jbc.M301662200. [DOI] [PubMed] [Google Scholar]
- 21.Haskin CJ, Ravi N, Lynch JB, Münck E, Que L., Jr Reaction of NO with the reduced R2 protein of ribonucleotide reductase from Escherichia coli. Biochemistry. 1995;34:11090–11098. doi: 10.1021/bi00035a014. [DOI] [PubMed] [Google Scholar]
- 22.Coufal DE, Tavares P, Pereira AS, Hyunh BH, Lippard SJ. Reactions of nitric oxide with the reduced non-heme diiron center of the soluble methane monooxygenase hydroxylase. Biochemistry. 1999;38:4504–4513. doi: 10.1021/bi9823378. [DOI] [PubMed] [Google Scholar]
- 23.Lu S, Libby E, Saleh L, Xing G, Bollinger JM, Jr, Moënne-Loccoz P. Characterization of NO adducts of the diiron center in protein R2 of Escherichia coli ribonucleotide reductase and site-directed variants. Implications for the O2-activation mechanism. J Biol Inorg Chem. 2004;9:818–827. doi: 10.1007/s00775-004-0582-8. [DOI] [PubMed] [Google Scholar]
- 24.Enemark JH, Feltham RD. Principles of structure, bonding, and reactivity for metal nitrosyl complexes. Coord Chem Rev. 1974;13:339–406. [Google Scholar]
- 25.Blomberg LM, Blomberg MR, Siegbahn PE. Theoretical study of the reduction of nitric oxide in an A-type flavoprotein. J Biol Inorg Chem. 2007;12:79–89. doi: 10.1007/s00775-006-0166-x. [DOI] [PubMed] [Google Scholar]
- 26.Blomberg ML, Blomberg MRA, Siegbahn PEM. A theoretical study of nitric oxide reductase activity in a ba3-type heme-copper oxidase. Biochim Biophys Acta. 2006;1757:31–46. doi: 10.1016/j.bbabio.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 27.Hayashi T, Lin IJ, Chen Y, Fee JA, Moënne-Loccoz P. Fourier transform infrared characterization of a CuB-nitrosyl complex in cytochrome ba3 from Thermus thermophilus: relevance to NO reductase activity in heme-copper terminal oxidases. J Am Chem Soc. 2007;129:14952–14958. doi: 10.1021/ja074600a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hayashi T, Lin MT, Ganesan K, Chen Y, Fee JA, Gennis RB, Moënne-Loccoz P. Accommodation of two diatomic molecules in cytochrome bo: insights into NO reductase activity in terminal oxidases. Biochemistry. 2009;48:883–890. doi: 10.1021/bi801915r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Le Fourn C, Fardeau ML, Ollivier B, Lojou E, Dolla A. The hyperthermophilic anaerobe Thermotoga maritima is able to cope with limited amount of oxygen: insights into its defence strategies. Environ Microbiol. 2008;10:1877–1887. doi: 10.1111/j.1462-2920.2008.01610.x. [DOI] [PubMed] [Google Scholar]
- 30.Vicente JB, Testa F, Mastronicola D, Forte E, Sarti P, Teixeira M, Giuffrè A. Redox properties of the oxygen-detoxifying flavodiiron protein from the human parasite Giardia intestinalis. Arch Biochem Biophys. 2009;488:9–13. doi: 10.1016/j.abb.2009.06.011. [DOI] [PubMed] [Google Scholar]
- 31.Hillmann F, Riebe O, Fischer RJ, Mot A, Caranto JD, Kurtz DM, Jr, Bahl H. Reductive dioxygen scavenging by flavo-diiron proteins of Clostridium acetobutylicum. FEBS Lett. 2009;583:241–245. doi: 10.1016/j.febslet.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arciero DM, Orville AM, Lipscomb JD. [17O]Water and nitric oxide binding by protocatechuate 4,5-dioxygenase and catechol 2,3-dioxygenase. Evidence for binding of exogenous ligands to the active site Fe2+ of extradiol dioxygenases. J Biol Chem. 1985;260:14035–14044. [PubMed] [Google Scholar]
- 33.Arciero DM, Lipscomb JD. Binding of 17O-labeled substrate and inhibitors to protocatechuate 4,5-dioxygenase-nitrosyl complex. Evidence for direct substrate binding to the active site Fe2+ of extradiol dioxygenases. J Biol Chem. 1986;261:2170–2178. [PubMed] [Google Scholar]
- 34.Nocek JM, Kurtz DM, Jr, Pickering RA, Doyle MP. Oxidation of deoxyhemerythrin to semi-methemerythrin by nitrite. J Biol Chem. 1984;259:12334–8. [PubMed] [Google Scholar]
- 35.Nocek JM, Kurtz DM, Jr, Sage JT, Xia YM, Debrunner P, Shiemke AK, Sanders-Loehr J, Loehr TM. Nitric oxide adducts of the binuclear iron site of hemerythrin: spectroscopy and reactivity. Biochemistry. 1988;27:1014–1024. doi: 10.1021/bi00403a026. [DOI] [PubMed] [Google Scholar]
- 36.Chen VJ, Orville AM, Harpel MR, Frolik CA, Surerus KK, Münck E, Lipscomb JD. Spectroscopic studies of isopenicillin N synthase. A mononuclear nonheme Fe2+ oxidase with metal coordination sites for small molecules and substrate. J Biol Chem. 1989;264:21677–21681. [PubMed] [Google Scholar]
- 37.Vicente JB, Teixeira M. Redox and spectroscopic properties of the Escherichia coli nitric oxide-detoxifying system Involving flavorubredoxin and Its NADH-oxidizing redox partner. J Biol Chem. 2005;280:34599–34608. doi: 10.1074/jbc.M506349200. [DOI] [PubMed] [Google Scholar]
- 38.Nocek JM, Kurtz DMJ, Sage JT, Debrunner PG, Maroney MJ, Que LJ. Nitric oxide adduct of the binuclear iron center in deoxyhemerythrin from Phascolopsis gouldii. Analogue of a putative intermediate in the oxygenation reaction. J Am Chem Soc. 1985;107:3382–3384. [Google Scholar]
- 39.Westre TE, Di Cicco A, Filipponi A, Natoli CR, Hedman B, Solomon EI, Hodgson KO. Determination of the Fe-N-O angle in {FeNO}7 complexes using multiple-scattering EXAFS analysis by GNXAS. J Am Chem Soc. 1994;116:6757–6768. [Google Scholar]
- 40.Brown CA, Pavlosky MA, Westre TE, Zhang Y, Hedman B, Hodgson KO, Solomon EI. Spectroscopic and theoretical description of the electronic structure of S=3/2 iron-nitrosyl complexes and their relation to O2 activation by nonheme iron enzyme active sites. J Am Chem Soc. 1995;117:715–732. [Google Scholar]
- 41.Varotsis C, Ohta T, Kitagawa T, Soulimane T, Pinakoulaki E. The structure of the hyponitrite species in a heme Fe-Cu binuclear center. Angew Chem Int Ed Engl. 2007;46:2210–2214. doi: 10.1002/anie.200602963. [DOI] [PubMed] [Google Scholar]
- 42.Miller LM, Pedraza AJ, Chance MR. Identification of conformational substates involved in nitric oxide binding to ferric and ferrous myoglobin through difference Fourier transform infrared spectroscopy (FTIR) Biochemistry. 1997;36:12199–12207. doi: 10.1021/bi962744o. [DOI] [PubMed] [Google Scholar]
- 43.Wildschut JD, Lang RM, Voordouw JK, Voordouw G. Rubredoxin:oxygen oxidoreductase enhances survival of Desulfovibrio vulgaris Hildenborough under microaerophilic conditions. J Bacteriol. 2006;188:6253–6260. doi: 10.1128/JB.00425-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rodrigues R, Vicente JB, Félix R, Oliveira S, Teixeira M, Rodrigues-Pousada C. Desulfovibrio gigas flavodiiron protein affords protection against nitrosative stress In vivo. J Bacteriol. 2006;188:2745–2751. doi: 10.1128/JB.188.8.2745-2751.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Silaghi-Dumitrescu R, Ng KY, Viswanathan R, Kurtz DM., Jr A flavo-diiron protein from Desulfovibrio vulgaris with oxidase and nitric oxide reductase activities. Evidence for an in vivo nitric oxide scavenging function. Biochemistry. 2005;44:3572–3579. doi: 10.1021/bi0477337. [DOI] [PubMed] [Google Scholar]
- 46.Victor BL, Baptista AM, Soares CM. Dioxygen and nitric oxide pathways and affinity to the catalytic site of rubredoxin:oxygen oxidoreductase from Desulfovibrio gigas. J Biol Inorg Chem. 2009;14:853–862. doi: 10.1007/s00775-009-0497-5. [DOI] [PubMed] [Google Scholar]
- 47.Feig AL, Bautista MT, Lippard SJ. A carboxylate-bridged non-heme diiron dinitrosyl complex. Inorg Chem. 1996;35:6892–6898. doi: 10.1021/ic960552b. [DOI] [PubMed] [Google Scholar]
- 48.Adams H, Bailey NA, Debaecker N, Fenton DE, Kanda W, Latour JM, Okawa H, Sakiyama H. A dinuclear (mu-carboxylato)manganese(II) complex derived from a macrocyclic ligand: a structural model for active sites in natural systems. Angew Chem Int Ed Engl. 1997;33:2535–2537. [Google Scholar]
- 49.Samples CR, Howard T, Raushel FM, DeRose VJ. Protonation of the binuclear metal center within the active site of phosphotriesterase. Biochemistry. 2005;44:11005–11013. doi: 10.1021/bi0506270. [DOI] [PubMed] [Google Scholar]
- 50.Lis T, Matuszewski J. Manganese(II) malonate dihydrate: a reinvestigation. Acta Crystallogr B. 1979;35:2212–2214. [Google Scholar]
- 51.Rodriguez-Martin Y, Hernandez-Molina M, Sanchiz J, Ruiz-Perez C, Lloret F, Julve M. Crystal structure and magnetic properties of two- and three-dimensional malonato-bridged manganese(II) complexes. Dalton trans. 2003:2359–2365. [Google Scholar]
- 52.Clay MD, Cosper CA, Jenney FE, Jr, Adams MW, Johnson MK. Nitric oxide binding at the mononuclear active site of reduced Pyrococcus furiosus superoxide reductase. Proc Natl Acad Sci U S A. 2003;100:3796–801. doi: 10.1073/pnas.0636858100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Orville AM, Lipscomb JD. Simultaneous binding of nitric oxide and isotopically labeled substrates or inhibitors by reduced protocatechuate 3,4-dioxygenase. J Biol Chem. 1993;268:8596–8607. [PubMed] [Google Scholar]
- 54.(a) Olson LW, Schaeper D, Lançon D, Kadish KM. Characterization of several novel iron nitrosyl porphyrins. J Am Chem Soc. 1982;104:2042–2044. [Google Scholar]; (b) Lançon D, Kadish KM. Electrochemical and spectral characterization of iron mono- and dinitrosyl porphyrins. J Am Chem Soc. 1983;105:5610–5617. [Google Scholar]; (c) Pellegrino J, Bari SE, Bikiel DE, Doctorovich F. Successful stabilization of the elusive species {FeNO}8 in a heme model. J Am Chem Soc. 2009;132:989–995. doi: 10.1021/ja905062w. [DOI] [PubMed] [Google Scholar]
- 55.(a) Lin R, Farmer PJ. The HNO adduct of myoglobin: synthesis and characterization. J Am Chem Soc. 2000;122:2393–2394. [Google Scholar]; (b) Kumar MR, Pervitsky D, Chen L, Poulos T, Kundu S, Hargrove MS, Rivera EJ, Diaz A, Colon JL, Farmer PJ. Nitrosyl hydroxide (HNO) as an O2 analogue: long-lived HNO adducts of ferrous globins. Biochemistry. 2009;48:5018–5025. doi: 10.1021/bi900122r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.(a) Hauser C, Glaser T, Bill E, Weyhermüller T, Wieghardt K. The electronic structures of an isostructural series of octahedral nitrosyliron complexes {Fe-NO}6,7,8 elucidated by Mössbauer spectroscopy. J Am Chem Soc. 2000;122:4352–4365. [Google Scholar]; (b) Serres RG, Grapperhaus CA, Bothe E, Bill E, Weyhermüller T, Neese F, Wieghardt K. Structural, spectroscopic, and computational study of an octahedral, non-heme {Fe-NO}6–8 series: [Fe(NO)(cyclam-ac)]2+/+/0. J Am Chem Soc. 2004;126:5138–5153. doi: 10.1021/ja030645+. [DOI] [PubMed] [Google Scholar]
- 57.Montenegro AC, Amorebieta VT, Slep LD, Martin DF, Roncaroli F, Murgida DH, Bari SE, Olabe JA. Three redox states of nitrosyl: NO+, NO•, and NO−/HNO interconvert reversibly on the same pentacyanoferrate(II) platform. Angew Chem Int Ed. 2009;48:4213–4216. doi: 10.1002/anie.200806229. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.