

Abstract
Toxoplasma gondii adenosine kinase (EC.2.7.1.20) is the major route of adenosine metabolism in this parasite. The enzyme is significantly more active than any other enzyme of the purine salvage in T. gondii and has been established as a potential chemotherapeutic target for the treatment of toxoplasmosis. Several 6-benzylthioinosines have already been identified as subversive substrates of the T. gondii but not human adenosine kinase. Therefore, these compounds are preferentially metabolized to their respective nucleotides and become selectively toxic against the parasites but not its host. In the present study, we report the testing of the metabolism of several carbocyclic 6-benzylthioinosines, as well as their efficacy as anti-toxoplasmic agents in cell culture. All the carbocyclic 6-benzylthioinosine analogues were metabolized to their 5'-monophosphate derivatives, albeit to different degrees. These results indicate that these compounds are not only ligands but also substrates of T. gondii adenosine kinase. All the carbocyclic 6-benzylthioinosine analogues showed a selective anti-toxoplasmic effect against wild type parasites, but not mutants lacking adenosine kinase. These results indicate that the oxygen atom of the sugar is not critical for substrate-binding. The efficacy of these compounds varied with the position and nature of the substitution on their phenyl ring. Moreover, none of these analogues exhibited host toxicity. The best compounds were carbocyclic 6-(p-methylbenzylthio)inosine (IC50 = 11.9 μM), carbocyclic 6-(p-methoxybenzylthio)inosine (IC50 = 12.1 μM), and carbocyclic 6-(p-methoxycarbonylbenzylthio)inosine ( IC50 = 12.8 μM). These compounds have about a 1.5-fold better efficacy relative to their corresponding 6-benzylthioinosine analogues (Rais et al., Biochem Pharmacol 2005;69:1409-19). The results further confirm that T. gondii adenosine kinase is an excellent target for chemotherapy and that carbocyclic 6-benzylthioinosines are potential antitoxoplasmic agents.
Keywords: Toxoplasma, Adenosine kinase, Subversive substrates, Carbocyclic 6-benzylthioinosine analogues, Chemotherapy
1. Introduction
The parasitic protozoan, Toxoplasma gondii, is the etiologic agent for toxoplasmosis, a parasitic disease wide spread among various warm-blooded animals including man [1]. Approximately 25% of the people worldwide, including 10% of the population in the US [2], are seropositive to T. gondii. This high prevalence of toxoplasmosis is attributed to its efficient spread through the food chain. Consequently, T. gondii is an important cause of foodborne infection in humans [3]. Infection with T. gondii is asymptomatic (90% of cases) in the general population and typically results in a mild flu-like syndrome that resolves without the need for intervention [4]. By contrast, the disease represents a major health problem for immunocompromised individuals, such as AIDS patients [5,6], organ transplant recipient patients [7], cancer chemotherapy patients [8] and the unborn children of infected mothers [1,9,10]. In such cases, toxoplasmic encephalitis is recognized as the most common cause of intracerebral mass lesions in AIDS patients and possibly the most commonly recognized opportunistic infection of the central nervous system [5,9]. Congenital toxoplasmosis is as high as 1/1000 live births [9]. Effects range in severity from asymptomatic to stillbirth, with the most common ailments being retinochoroiditis, cerebral calcifications, psychomotor disorder or mental retardation, and severe brain damage [9]. Additionally, T. gondii has recently been recognized as an important cause of ocular disease in healthy adults [11,12]. Recent reports indicate that chronic T. gondii infection may be a predisposing factor for the development of schizophrenia [13-15], an effect that may be driven by inflammation in the CNS [16] .
Despite these tragic implications, the current therapy has not changed in the past few decades. The efficacy of the current therapy for toxoplasmosis (a combination of pyrimethamine and sulfadiazine) is limited, primarily by serious host toxicity and ineffectiveness against tissue cysts. Furthermore, as many as 50% of patients do not respond to therapy. In addition, prolonged exposure to this regimen induces serious host toxicity such as bone marrow suppression and severe skin rashes forcing the discontinuation of the therapy [5,9,17,18]. Other therapies, e.g. clindamycin, spiramycin or atovaquone, have been met with limited success, particularly in the long-term management of these patients. Hence, there is a critical need to develop new and effective drugs with low host toxicity for the acute and chronic management of toxoplasmosis.
Rational drug design is usually based on biochemical and physiological differences between the pathogen and the host. One potential target for chemotherapeutic intervention against T. gondii is purine metabolism. These parasites replicate rapidly and require large amounts of purines for the synthesis of their nucleic acids and other vital components. In contrast to their host, T. gondii are purine auxotrophs and must rely on the salvage of their purine requirements from the host [19,20 and references therein].
Another striking difference between toxoplasma and their host is the nature of adenosine salvage. Adenosine is preferentially incorporated into the parasite nucleotide pool by at least a 10-fold higher rate than any other purine nucleobase or nucleoside tested [21,22]. Furthermore, adenosine is directly phosphorylated to AMP, from which all other purine nucleotides can be synthesized to fulfill the parasite purine requirements. This reaction is catalyzed by the enzyme adenosine kinase (EC.2.7.1.20) which is almost 10 times more active than any other purine salvage enzyme in this parasite [22]. This contrasts sharply with most mammalian cells where adenosine is predominantly deaminated by adenosine deaminase (EC 3.5.4.4) to inosine, which is then cleaved by purine nucleoside phosphorylase (EC 2.4.2.1) to hypoxanthine as previously reviewed [6,7]. Neither of these two enzymes have any appreciable activity in T. gondii [8].
Structure-activity relationships [23-26], biochemical [24-31], metabolic [19,27-31], and molecular [32] investigations have demonstrated that the substrate specificity, as well as other characteristics of T. gondii adenosine kinase, differs significantly from those of the human enzyme, and have established the enzyme as an excellent potential chemotherapeutic target for the treatment of toxoplasmosis [19,20]. It was also demonstrated that 6-benzylthioinosine, among other 6-substituted purine nucleoside analogues, is a substrate for the parasite, but not human adenosine kinase [19,23,27-29]. Furthermore, 6-benzylthioinosine was shown to be metabolized preferentially to the nucleotide level and becomes selectively toxic to T. gondii, but not their host, thereby acting as a subversive substrate [19,23,27-29]. Therefore, modification of the chemical structure of 6-benzylthioinosine could further potentiate its anti-toxoplasmic efficacy.
We have previously synthesized 6-benzylthioinosine analogues with various substitutions at their purine ring which increased the binding affinities of these analogues to T. gondii adenosine kinase [24,25] as well as their efficacy as anti-toxoplasmic agents [29,31]. As a part of our continuing effort to develop potential anti-toxoplasmic agents, we turned our attention to carbocyclic nucleosides, wherein an oxygen atom of the ribose ring is replaced by a methylene group. An advantage of the carbocyclic nucleosides is metabolic stability due to the absence of a typical glycosidic bond [33,34]. The carbocyclic nucleosides also have considerable effects on ring conformation and lipophilicity which may improve the therapeutic potency of carbocyclic nucleosides [33,34]. Specifically, we wanted to explore the structural effect of carbasugar moiety on the efficacy of 6-benzylthioinosine analogues. In the present study we report the testing of newly synthesized carbocyclic 6-benzylthioinosine analogues, with various substitutions at their phenyl ring as anti-toxoplasmic agents in cell culture.
2. Materials and methods
2.1. Chemicals
The carbocyclic 6-benzylthioinosine (1) analogues (2-21) were synthesized as previously described [26]. The chemical structures of these compounds are shown in Table 1. 6-benzylthioinosine was generously provided by Dr. Mohamed Nasr, Drug Development and Clinical Sciences Branch, NIAID. [5,6-13H]uracil was purchased from Moravek Biochemicals. RPMI-1640 medium from GIBCO BRL; penicillin G and streptomycin sulfate from Mediatech/Cellgro; FBS (fetal bovine serum) from HyClone Laboratories. All other chemicals and compounds were obtained from Sigma Chemical Co. or Fisher Scientific.
Table 1.
Chemical structure of carbocyclic 6-benzylthioinosine and its substituted analogues
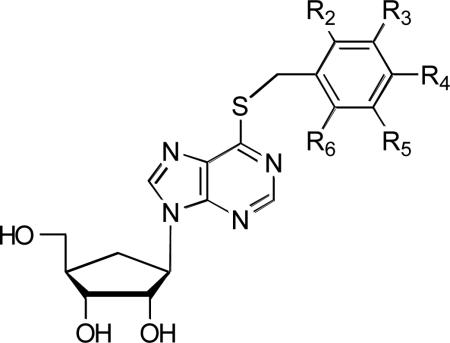 | |||||
|---|---|---|---|---|---|
| Compounds | R2 (ortho) | R3 (meta) | R4 (para) | R5 (meta) | R6 (ortho) |
| 1. Carbocyclic 6-(benzylthio)inosine | -H | -H | -H | -H | -H |
| 2. Carbocyclic 6-(o-fluorobenzylthio)inosine | -F | -H | -H | -H | -H |
| 3. Carbocyclic 6-(o-chlorobenzylthio)inosine | -Cl | -H | -H | -H | -H |
| 4. Carbocyclic 6-(o-methylbenzylthio)inosine | -CH3 | -H | -H | -H | -H |
| 5. Carbocyclic 6-(m-methylbenzylthio)inosine | -H | -CH3 | -H | -H | -H |
| 6. Carbocyclic 6-(m-trifluoromethylbenzylthio)inosine | -H | -CF3 | -H | -H | -H |
| 7. Carbocyclic 6-(m-nitrobenzylthio)inosine | -H | -NO2 | -H | -H | -H |
| 8. Carbocyclic 6-(p-fluorobenzylthio)inosine | -H | -H | -F | -H | -H |
| 9. Carbocyclic 6-(p-chlorobenzylthio)inosine | -H | -H | -Cl | -H | -H |
| 10. Carbocyclic 6-(p-bromobenzylthio)inosine | -H | -H | -Br | -H | -H |
| 11. Carbocyclic 6-(p-methylbenzylthio)inosine | -H | -H | -CH3 | -H | -H |
| 12. Carbocyclic 6-(p-nitrobenzylthio)inosine | -H | -H | -NO2 | -H | -H |
| 13. Carbocyclic 6-(p-cyanobenzylthio)inosine | -H | -H | -CN | -H | -H |
| 14. Carbocyclic 6-(p-methoxycarbonylbenzylthio)inosine | -H | -H | -CO2CH3 | -H | -H |
| 15. Carbocyclic 6-(p-methoxybenzylthio)inosine | -H | -H | -OCH3 | -H | -H |
| 16. Carbocyclic 6-(p-trifluoromethoxybenzylthio)inosine | -H | -H | -OCF3 | -H | -H |
| 17. Carbocyclic 6-(p-methylsulfonylbenzylthio)inosine | -H | -H | -SO2CH3 | -H | -H |
| 18. Carbocyclic 6-(o,p-dichlorobenzylthio)inosine | -Cl | -H | -Cl | -H | -H |
| 19. Carbocyclic 6-(m,p-dichlorobenzylthio)inosine | -H | -Cl | -Cl | -H | -H |
| 20. Carbocyclic 6-(o-chloro-o-fluorobenzylthio)inosine | -F | -H | -H | -H | -Cl |
| 21. Carbocyclic 6-(o,m,p-trimethylbenzylthio)inosine | -CH3 | -H | -CH3 | -H | -CH3 |
2.2. Maintenance of T. gondii
The RH and TgAK-3 strains of T. gondii were propagated by intraperitoneal passage in female CD 1 mice (20-25 g). RH is a wild type strain and TgAK-3 is a knockout mutant deficient in adenosine kinase [32]. Mice were injected i.p. with an inoculum (106 cells) of T. gondii contained in 0.2 mL of sterile PBS (phosphate buffered saline), pH 7.2. The parasites were harvested from the peritoneal cavity by injection, aspiration and reinjection of 3-5 mL of PBS (2-3 times). The peritoneal fluid was examined microscopically to determine the concentration of T. gondii and to ascertain the extent of contamination by host cells. Two-day transfers generally produce parasite preparations that contain very little contamination and have a viability of > 95%.
2.3. Preparation of parasites
When T. gondii were used for in vitro incorporation studies, the procedure was performed aseptically and the parasites were washed and resuspended in RPMI-1640 medium containing 100 units/mL penicillin G, 100 μg/mL streptomycin sulfate, and 3% FBS.
2.4. Evaluation of carbocyclic 6-benzylthioinosine analogues as alternative substrates for purified T. gondii adenosine kinase
The assay mixture contained 50 mM Tris-Cl, pH 7.5, 0.25 mM ATP; 5 mM MgCl2; 1 mM DTT, 25 mM NaF, 100 μM of the analogue under study, and 100 μL of purified recombinant enzyme prepared as previously described [35], in a final volume of 200 mL. Incubation was carried out overnight at 37°C and terminated by boiling in a water bath for 2 min followed by freezing at -20°C for at least 20 min. Precipitated proteins were removed by centrifugation and the substrate and products in the supernatant were separated by HPLC.
2.5. HPLC analysis of the products of the enzyme assays
Hewlett Packard 1050 HPLC systems equipped with autosamplers, autoinjectors, quaternary pumps, multiple wave length diode array base triple channel UV monitors were used. The systems are operated by computer programs which handle data analysis, comparison, and storage of data after each run. Nucleoside and nucleotide contents are analyzed using a Bio Basic AX Thermo Hypersil (25 cm × 0.46 cm, ODS 5 μm) reversed-phase column (Keystone Scientific, inc, Bellefonte, PA). A 100 μL sample is injected. Elution is carried out for 40 min with a flow rate of 1 mL/min starting with a 35 min linear gradient of Buffer A (50 mM ammonium acetate, 0.5% acetonitrile, pH 3.0), to Buffer B (50 mM ammonium acetate, 60% acetonitrile, pH 4.8). This was followed by a 5-min runtime of 100% Buffer A. The eluent is monitored at 254 nm and the λmax of the compound under study. Compounds are identified by retention time, coelution with authentic samples and/or absorbance ratio of the compound's λmax/254 nm. Mass spectrometry analyses were used to verify the identity of the products as described below.
2.6. Mass spectrometry analyses
Mass spectrometry analyses were performed on a Micromass Q-Tof 2 mass spectrometer (Micromass, Manchester, UK). The samples were evaporated to dryness and dissolved in 50/50 acetonitrile/water, containing 0.1% formic acid and injected into a 1 μL/minute isocratic flow of the same solvent. The flow was introduced into the Nano-LC interface of the mass spectrometer. The mass spectra (electrospray ionization) were recorded in the negative ion mode.
2.7. Evaluation of carbocyclic 6-benzylthioinosine and its analogues as potential anti-toxoplasmic agents in tissue culture
The wild type RH and adenosine kinase deficient mutant TgAK-3 [32] strains of T. gondii were used in these experiments. The adenosine kinase deficient mutant TgAK-3 was used as a control to verify that the promising drugs were metabolized by adenosine kinase in vivo. The effects of purine analogues as anti-toxoplasmosis agents were determined by measuring their ability to inhibit the replication of intracellular T. gondii in tissue culture, using monolayers of human foreskin fibroblasts (CRL-1634, American Type Culture Collection, Rockville, MD), grown for no more than 20 passages in RPMI 1640 medium [28,29]. The viability of intracellular parasites was evaluated by the selective incorporation of radiolabeled uracil into the nucleic acids of the parasites (minimum of triplicate assays) as previously described [27-29,31,36]. Briefly, confluent cells (4-5 day incubation) were cultured for 24 hrs in the 24-well flat bottom microtiter plates (~ 5 × 105/1 mL/well) and incubated at 37°C in 5% CO2, 95% air to allow the cells to attach. The medium was then removed and the cells were infected with isolated T. gondii in medium with 3% FBS (1 parasite/cell). After a 1 hr incubation, the cultures were washed with media with 10% FBS to remove extracellular parasites. FBS was maintained at a final concentration of 10%. Compounds were dissolved in 50% ethanol and then added to cultures of the parasite-infected cells to give a final concentration of 0, 5, 10, 25, and 50 μM. The final concentration of ethanol when the compounds were added to the wells was 5%. After an additional 18 hrs incubation the medium was replaced with 1 mL drug free media containing [5,6-13H]uracil (5 μCi/mL) and incubated for another 6 hrs after which the media was removed. The fibroblasts were then released from the wells by trypsinization with the addition of 200 μL trypsin/EDTA (2.5X) to each well. After 10 min incubation, 1 mL of ice cold 10% trichloroacetic acid (TCA) was added to each well. The plates were then placed on a shaker to insure the detachment of the cells. The suspended contents of each well was filtered through GF/A 2.4 cm glass microfiber filters (Whatman, Hillsboro, OR), which were pre-washed each with 1 mL double distilled H2O and dried. After filtration, the filters were washed with 10 mL of methanol, left to dry, then placed in scintillation vials containing 5 mL of Econo-Safe scintillation fluor (Research Products International Corp., Mount Prospect, IL), and radioactivity was counted using an LS5801 Beckman scintillation counter. The effect of the compounds on the growth of the parasite was estimated as a percent reduction in the uptake of radiolabeled uracil by treated parasites as compared to the untreated controls [27-29,31,36]. Radiolabel incorporation closely correlates with parasite growth [37]. The IC50 (the concentration causing 50% inhibition) values were estimated by a polynomial equation from dose response data from at least three separate experiments.
2.8. Host-toxicity of carbocyclic 6-benzylthioinosine and its analogues
Possible toxicity against the host cells by the same doses of the various analogues used in the above experiments was measured (minimum of triplicate assays) using a modification of the Microculture Tetrazolium (MTT) assay on uninfected monolayers of human foreskin fibroblasts (grown for no more than 20 passages) in RPMI 1640 medium [27-29,31,36]. Briefly, confluent cells were incubated for at least 24 hrs in 96-well flat bottom microtiterplates (~ 105/200 μL/well) at 37 °C in 5% CO2, 95% air to allow the cells to attach. The medium was then replaced with 200 μL of fresh medium. The appropriate concentration of the compounds was dissolved in 50 μL of medium, and added to each well to give the desired final concentrations. The cultures were then incubated for 48 hrs after which 50 μL of sterile MTT solution (2 mg/1 mL PBS) was added to each well. MTT solution was sterilized by filtration through 0.22 μm filters (Costar, Cambridge, MA). After 4 hrs incubation, the medium was removed and 100 μL of dimethylsulfoxide (DMSO) was added to each well and the plates were shaken gently for 2-3 min to dissolve the formed formazan crystals. The absorbance was measured at 540 nm using a computerized microtiterplate reader (Themomax, Molecular Devices).
3. Results and Discussion
3.1. Evaluation of 6-benzylthioinosine analogues as alternative substrates for purified T. gondii adenosine kinase
Carbocyclic 6-benzylthioinosine (1) and its analogues (Table 1) were tested as alternative substrates of T. gondii adenosine kinase. HPLC analysis of the substrates and products of the enzyme assays demonstrated that carbocyclic 6-benzylthioinosine (1) and its analogues were converted to their respective nucleoside 5’-monophosphates by T. gondii adenosine kinase. Figure 1 shows the reversed-phase HPLC profile of the metabolism of one of these analogues, carbocyclic 6-(p-fluorobenzylthio)inosine (8), to its 5’-monophosphate derivative. In the control (Panel A) carbocyclic 6-(p-fluorobenzylthio)inosine (8) eluted at 23.8 min and there was no traces of its 5’-monophosphate derivative. In the experimental assay (Panel B), carbocyclic 6-(p-fluorobenzylthio)inosine (8) eluted at 24.6 min and its 5’-monophosphate derivative appeared as a new peak at 21.2 min. As expected, carbocyclic 6-(p-fluorobenzylthio)inosine (8) and its 5’-monophosphate derivative had higher absorbance at 289 nm, the λmax of carbocyclic 6-(p-fluorobenzylthio)inosine, than at 254 nm. Time course studies showed that there was a decrease in the amount of carbocyclic 6-(p-fluorobenzylthio)inosine with time accompanied by an increase in the formation of carbocyclic 6-(p-fluorobenzylthio)inosine 5’-monophosphate. The other analogues have similar metabolic profiles although the retention time and amount of nucleoside 5’-monophosphates synthesized varied between different compounds. The chemical structures of the metabolites as the respective nucleoside 5’-monophosphates of the compounds studied were verified by Mass spectrometry. Figure 2 shows the mass spectrum of carbocyclic 6-(p-fluorobenzylthio)inosine 5’-monophosphate and its (M-H) ion at 471.08. The metabolism of carbocyclic 6-benzylthioinosine (1) and its analogues, to their respective nucleoside 5’-monophosphates indicates that these compounds are alternate substrates of T. gondii adenosine kinase. Therefore, apparent Ki values presented in Table 2 are equal to apparent Km values [38]. These results demonstrate that the oxygen atom of the ribose ring is not critical for the carbocyclic 6-(benzylthio)inosines to act as substrates for T. gondii adenosine kinase.
Figure 1.

The reversed-phase HPLC profile of the metabolism of carbocyclic 6-(p-fluorobenzylthio)inosine to its 5’-monophosphate. (A) Controls, carbocyclic 6-(p-fluorobenzylthio)inosine in reaction mixture after incubation without the toxoplasma adenosine kinase. (B) Experimental, carbocyclic 6-(p-fluorobenzylthio)inosine after incubation with the toxoplasma adenosine kinase. In both profiles (A) and (B), the above panel shows the profile monitored at 254 nm and the lower panel shows the profile monitored at 289 nm, the λmax of carbocyclic 6-(p-fluorobenzylthio)inosine.
Figure 2.

Negative electrospray mass spectrum of carbocyclic 6-(p-fluorobenzylthio)inosine 5’-monophosphate showing the (M-H) ion at 469.2
Table 2.
Binding affinity (apparent Ki) of carbocyclic 6-benzylthioinosine analogues to Toxoplasma gondii adenosine kinase and the effect of different doses on host-toxicity and percent survival of wild type (RH) and adenosine kinase deficient (TgAK-3) strains of the parasite grown in human skin fibroblasts in culture.
| Compound | Ki [μM]a | Infection | Concentration [μM] |
IC50 [μM] | ||||
|---|---|---|---|---|---|---|---|---|
| 0 | 5 | 10 | 25 | 50 | ||||
| 1. Carbocyclic 6-(benzylthio)inosine | 105 ± 12 | Wild Type | 100 | 84.7 | 42.7 | 4.9 | 0.0 | 19.1 ± 0.4 |
| TgAK-3 | 100 | 100 | 100 | 100 | 100 | |||
| None | 100 | 100 | 100 | 100 | 94.3 | |||
| 2. Carbocyclic 6-(o-fluorobenzylthio)inosine | 55 ± 10 | Wild Type | 100 | 98.6 | 93.7 | 39.2 | 12.7 | 38.9 ± 15.6 |
| TgAK-3 | 100 | 100 | 100 | 100 | 100 | |||
| None | 100 | 100 | 100 | 100 | 85.8 | |||
| 3. Carbocyclic 6-(o-chlorobenzylthio)inosine | 24 ± 5.4 | Wild Type | 100 | 79.5 | 48.8 | 9.8 | 2.2 | 20.0 ± 2.3 |
| TgAK-3 | 100 | 100 | 100 | 100 | 100 | |||
| None | 100 | 100 | 100 | 100 | 100 | |||
| 4. Carbocyclic 6-(o-methylbenzylthio)inosine | 25 ± 3.3 | Wild Type | 100 | 82.7 | 45.4 | 7.6 | 0.0 | 19.7 ± 2.8 |
| TgAK-3 | 100 | 100 | 100 | 100 | 100 | |||
| None | 100 | 100 | 100 | 100 | 99.2 | |||
| 5. Carbocyclic 6-(m-methylbenzylthio)inosine | 27 ± 4.2 | Wild Type | 100 | 88.3 | 64.7 | 13.7 | 1.2 | 23.8 ± 1.1 |
| TgAK-3 | 100 | 100 | 100 | 100 | 100 | |||
| None | 100 | 100 | 100 | 100 | 96.1 | |||
| 6. Carbocyclic 6-(m-trifluoromethylbenzylthio)inosine | 22 ± 1.3 | Wild Type | 100 | 79.0 | 47.1 | 6.3 | 1.0 | 19.1 ± 1.4 |
| TgAK-3 | 100 | 100 | 100 | 100 | 100 | |||
| None | 100 | 100 | 100 | 100 | 90.6 | |||
| 7. Carbocyclic 6-(m-nitrobenzylthio)inosine | 27 ± 2.7 | Wild Type | 100 | 84.4 | 50.9 | 4.2 | 0.0 | 19.7 ± 0.2 |
| TgAK-3 | 100 | 100 | 100 | 100 | 100 | |||
| None | 100 | 100 | 100 | 100 | 100 | |||
| 8. Carbocyclic 6-(p-fluorobenzylthio)inosine | 32 ± 8.3 | Wild Type | 100 | 89.2 | 61.5 | 12.4 | 3.4 | 23.2 ± 1.3 |
| TgAK-3 | 100 | 100 | 100 | 100 | 100 | |||
| None | 100 | 100 | 100 | 100 | 100 | |||
| 9. Carbocyclic 6-(p-chlorobenzylthio)inosine | 16 ± 2.2 | Wild Type | 100 | 72.9 | 11.9 | 0.0 | 0.0 | 14.5 ± 1.3 |
| TgAK-3 | 100 | 100 | 100 | 100 | 100 | |||
| None | 100 | 100 | 100 | 100 | 100 | |||
| 10. Carbocyclic 6-(p-bromobenzylthio)inosine | 21 ± 3.7 | Wild Type | 100 | 77.1 | 47.3 | 1.2 | 1.0 | 18.3 ± 1.3 |
| TgAK-3 | 100 | 100 | 100 | 100 | 100 | |||
| None | 100 | 100 | 100 | 100 | 100 | |||
| 11. Carbocyclic 6-(p-methylbenzylthio)inosine | 7.5 ± 0.6 | Wild Type | 100 | 43.4 | 2.54 | 0.0 | 0.0 | 11.9 ± 0.4 |
| TgAK-3 | 100 | 99.0 | 100 | 100 | 100 | |||
| None | 100 | 100 | 100 | 100 | 100 | |||
| 12. Carbocyclic 6-(p-nitrobenzylthio)inosine | 94 ± 15 | Wild Type | 100 | 100 | 97.6 | 44.2 | 16.8 | 42.8 ± 5.5 |
| TgAK-3 | 100 | 100 | 100 | 100 | 100 | |||
| None | 100 | 100 | 100 | 100 | 100 | |||
| 13. Carbocyclic 6-(p-cyanobenzylthio)inosine | 129 ± 25 | Wild Type | 100 | 100 | 100 | 48.8 | 17.8 | 49.0 ± 10.3 |
| TgAK-3 | 100 | 100 | 100 | 100 | 100 | |||
| None | 100 | 100 | 100 | 100 | 100 | |||
| 14. Carbocyclic 6-(p-methoxycarbonylbenzylthio)inosine | 41 ± 3.2 | Wild Type | 100 | 53.9 | 6.3 | 1.5 | 0.0 | 12.8 ± 0.3 |
| TgAK-3 | 100 | 100 | 100 | 100 | 100 | |||
| None | 100 | 100 | 99.4 | 88.7 | 87.1 | |||
| 15. Carbocyclic 6-(p-methoxybenzylthio)inosine | 62 ± 4.7 | Wild Type | 100 | 48.3 | 1.9 | 0.0 | 0.0 | 12.1 ± 0.3 |
| TgAK-3 | 100 | 98.4 | 100 | 100 | 100 | |||
| None | 100 | 100 | 100 | 94.3 | 82.1 | |||
| 16. Carbocyclic 6-(p-trifluoromethoxybenzylthio)inosine | 66 ± 7.2 | Wild Type | 100 | 87.3 | 53.6 | 8.1 | 0.0 | 21.0 ± 1.7 |
| TgAK-3 | 100 | 100 | 100 | 100 | 100 | |||
| None | 100 | 100 | 98.2 | 96.7 | 89.3 | |||
| 17. Carbocyclic 6-(p-methylsulfonylbenzylthio)inosine | 74 ± 7.8 | Wild Type | 100 | 66.8 | 21.2 | 0.0 | 0.0 | 14.6 ± 1.1 |
| TgAK-3 | 100 | 100 | 100 | 100 | 100 | |||
| None | 100 | 100 | 100 | 100 | 100 | |||
| 18. Carbocyclic 6-(o,p-dichlorobenzylthio)inosine | 61 ± 7.9 | Wild Type | 100 | 84.4 | 45.1 | 0.0 | 0.0 | 18.6 ± 0.9 |
| TgAK-3 | 100 | 100 | 100 | 100 | 100 | |||
| None | 100 | 100 | 100 | 100 | 90.6 | |||
| 19. Carbocyclic 6-(m,p-dichlorobenzylthio)inosine | 17 ± 4.1 | Wild Type | 100 | 59.7 | 17.8 | 8.3 | 1.5 | 14.4 ± 0.6 |
| TgAK-3 | 100 | 100 | 100 | 100 | 100 | |||
| None | 100 | 100 | 100 | 100 | 73.1 | |||
| 20. Carbocyclic 6-(o-chloro-o-fluorobenzylthio)inosine | 31 ± 4.2 | Wild Type | 100 | 56.3 | 15.6 | 3.7 | 0.0 | 13.6 ± 0.2 |
| TgAK-3 | 100 | 100 | 100 | 100 | 100 | |||
| None | 100 | 100 | 100 | 100 | 100 | |||
| 21. Carbocyclic 6-(o,m,p-trimethylbenzylthio)inosine | 63 ± 6.9 | Wild Type | 100 | 71.0 | 32.7 | 0.0 | 0.0 | 16.1± 1.2 |
| TgAK-3 | 100 | 100 | 100 | 100 | 100 | |||
| None | 100 | 100 | 100 | 90.6 | 93.2 | |||
| 22. 6-Benzylthioinosine | 14 ± 2.4 | Wild Type | 100 | 92.7 | 45.8 | 7.6 | 0.0 | 20.4 ± 1.2 |
| TgAK-3 | 100 | 100 | 100 | 100 | 100 | |||
| None | 100 | 100 | 99.7 | 99.4 | 98.8 | |||
| 23. Sulfadiazine | Wild Type | 100 | 92.9 | 58.0 | 53.4 | 46.3 | 27.3 ± 3.3 | |
| None | 100 | 98.2 | 99.8 | 99.8 | 102 | |||
| 24. Pyrimethamine | Wild Type | 100 | 98.9 | 55.3 | 25.1 | 23.2 | 16.1 ± 2.5 | |
| None | 100 | 101 | 100 | 108 | 108 | |||
Host toxicity of uninfected cells was measured by MTT method in at least 2 independent experiments each of 3 replica as previously described [24-29,31].
Percent survival of parasites was measured by incorporation of [5,6-13H]uracil in at least 2 independent experiments of 3 replica each as previously described [24-29,31].
Data from reference [26].
3.2. Evaluation of anti-toxoplasmic activity and host-toxicity in tissue culture
Carbocyclic 6-benzylthioinosine (1) and its analogues (2-21) were evaluated as potential anti-toxoplasmic agents against wild type (RH) and adenosine kinase deficient (TgAK-3) strains of T. gondii grown in human foreskin fibroblasts in culture. As a positive control, pyrimethamine and sulfadiazine, the standard chemotherapeutic agents used in the treatment of toxoplasmosis, were also evaluated. The inhibitory efficacies of these compounds (estimated as IC50) on the growth of toxoplasma were measured using uracil uptake assays. Uracil uptake assays are highly specific to T. gondii as mammalian cells do not incorporate uracil into their nucleoside/nucleotide pool or nucleic acids [27-29,31,36,37]. Therefore, an exponential increase in radiolabel incorporation closely correlates with the exponential growth of the parasite [37].
The results in Table 2 show that all the carbocyclic 6-benzylthioinosines were effective against infection with the wild type (RH) T. gondii in a dose-dependent manner. Moreover, none of the active compounds were effective against infection with the adenosine kinase deficient strain (TgAK-3). This is consistent with the expected role of these carbocyclic analogues in preferentially being metabolized by T. gondii adenosine kinase to toxic nucleotides which is involved in the selective death of the parasite. It should be noted that the lack of sensitivity of TgAK-3 to these purine nucleoside analogues is not due to a lower growth rate of the mutant. TgAK-3 is much more aggressive in its growth than the RH wild type in both tissue culture and animals [29]. Therefore, it can be concluded that the presence of T. gondii adenosine kinase is a requirement for the carbocyclic 6-benzylthioinosines to exert their anti-toxoplasmic effect. Consequently, these compounds are substrates for T. gondii adenosine kinase in vivo as was the case with the in vitro enzyme assays [26]. These results confirm further the HPLC analyses which demonstrated that these carbocyclic 6-benzylthioinosines are converted to their respective 5'-monophosphate by T. gondii adenosine kinase, which is a prerequisite for these analogues to exert their anti-toxoplasmic effect.
In general, the newly synthesized carbocyclic 6-(benzylthio)inosines showed potent anti-toxoplasmic efficacy with IC50 values ranging from 11.9 to 49.0 μM. It is noteworthy that the unsubstituted carbocyclic analogue 1 (IC50 = 19.1 μM) exhibited potency close to that of 6-benzylthioinosine (IC50 = 20.4 μM) in the cell culture evaluation (Table 2) despite its attenuated binding affinity (Km = 105 μM) relative to 6-benzylthioinosine (Km = 14.3 μM). Nevertheless, the degree of anti-toxoplasmic efficacy of these carbocyclic analogues (Table 2) appears to correlate well with their Km values (Table 2) which, in turn, were influenced by the nature and position of substituents on the 6-benzyl group. The relative differences between the enzymatic binding affinity and the cellular activity (Table 2) could be attributed to differences in the transport of the carbocyclic 6-(benzylthio)inosines into the cell.
Single substitutions at the ortho or meta position did not enhance anti-toxopalsmic efficacy over that of the unsubstituted carbocyclic 6-(benzylthio)inosine (1). On the other hand, the nature of the substituent at the para position seems to have a substantial impact on the anti-toxoplasmic efficacy of the carbocyclic compounds. Carbocyclic 6-(p-methylbenzylthio)inosine (11, 11.9 μM), carbocyclic 6-(p-methoxybenzylthio)inosine (15, 12.1 μM) and carbocyclic 6-(p-methoxycarbonylbenzylthio)inosine (14, 12.8 μM) were the best compounds and their potencies were better than that of the clinically used drugs, pyrimethamine (IC50 = 16.1 μM) and sulfadiazine (IC50 = 27.3 μM). On the other hand, replacing the p-hydrogen with a cyano or nitro substituent resulted in a decreased binding affinity, as observed with carbocyclic 6-(p-cyanobenzylthio)inosine (13, 49 μM) and carbocyclic 6-(p-nitrobenzylthio)inosine (12, 43 μM) which were the weakest ligands in this carbocyclic series. The meta-para di-substitutions (carbocyclic 6-(m,p-dichlorobenzylthio)inosine, 19) and the di-ortho substitutions (carbocyclic 6-(o,p-dichlorobenzylthio)inosine, 18) were more favorable than the ortho-meta di-substitutions(e.g. carbocyclic 6-(o,p-dichlorobenzylthio)inosine, 18) in agreement with their binding affinities. Triple substitutions with a methyl group as in carbocyclic 6-(o,m,p-trimethylbenzylthio)inosine (21, 16.1 μM) showed increased anti-toxoplasmic potency in comparison to a single methyl substitution at the meta (5, 23.8 μM) position but decreased in activity in comparison to a single methyl substitution at the para (11, 11.9 μM) position.
These results illustrate that the nature and position of the substituent(s) on the aromatic ring of the benzylthio group seem to have distinct influences on the binding affinity of carbocyclic 6-benzylthioinosine. These results have revealed a different pattern of structure-efficacy relationships than that observed with 6-benzylthioinosine [24,27,29] and 7-deaza-6-benzylthioinosines [25,31]. The p-cyano derivatives of 6-(benzylthio)inosine [24,29] and 7-deaza-6-(benzylthio)inosine [25,31] were among the best ligands of T. gondii adenosine kinase while the carbocyclic nucleoside with the same substituent (carbocyclic 6-(p-cyanobenzylthio)inosine, 13) was the weakest compound of all the carbocyclic nucleosides tested. These results are attributed to the fact that the binding affinity for T. gondii adenosine kinase is sensitive to the sugar motif conformational distribution at the enzyme binding site [26]. The carbasugar moiety slightly changed the overall orientation of these analogues inside the binding site and thus carbocyclic nucleosides with the functionalized aromatic group are recognized by the enzyme active site in a slightly different manner than do the corresponding ribonucleosides [26].
Table 2 also shows that none of the compounds had effects on the survival of uninfected host cells, indicating that host toxicity is of little concern for these compounds. The lack of hosttoxicity is due to at least two factors. First, 6-substituted compounds are not active substrates for mammalian adenosine kinase [24,27,29]. Thus, no toxic nucleotides are formed in the absence of the parasite enzyme. Secondly, the newly synthesized compounds are analogues of p-nitrobenzylthioinosine (NBMPR), a known inhibitor of nucleoside transport in mammalian cells. Indeed, our previous studies have demonstrated that NBMPR is transported and metabolized only by cells infected with wild type toxoplasma [28]. On the other hand, NBMPR is neither transported nor metabolized by uninfected host cells or cells infected with mutant parasites lacking the adenosine/purine transporter [28]. It was concluded that the presence of a functional toxoplasma adenosine/purine transporter and adenosine kinase are prerequisites for the transport and metabolism of the 6-substituted compounds by infected cells [28]. Therefore, the newly synthesized compounds may act similarly to NBMPR and not gain entry to uninfected host cells to exert toxic side-effects.
It is clear from the present study that analogues of carbocyclic 6-benzylthioinosine have anti-toxoplasmic effects and that they work by a different mechanism than the other known anti-toxoplasmic drugs. The mechanism and/or targets of toxicities of carbocyclic 6-benzylthioinosine and its analogues are not known at the present time. However, such toxicities are clearly mediated by the nucleotides of these analogues since no toxicities were observed in the absence of adenosine kinase and lack of nucleotide synthesis. The nucleotides of carbocyclic 6-benzylthioinosine and its analogues could interfere with, among other things, other steps of purine salvage, nucleic acids and/or protein synthesis as well as reactions carried out by protein kinases leading to the death of the parasite. Experiments are currently being conducted in our laboratories to address these questions.
In summary, the present results demonstrate that the incorporation of the carbocyclic motif led to improved anti-toxoplasmic efficacy of several of the 6-benzylthioinosines by about 1.5-fold [29 and Table 2]. Carbocyclic 6-benzylthioinosine and its analogues exhibited selective anti-toxoplasmic effects in cell culture. The efficacy of these compounds as selective anti-toxoplasmic agents varied according to the nature and position of various substituents on the phenyl ring of the benzyl group. The ortho or meta substitutions appeared to be less important for anti-toxoplasmic activity, while the para substitutions impacted more the cellular activity of such analogues as compared to the parent compound carbocyclic 6-benzylthioinosine (1).
The observations made in our present [Table 2] and past studies [19-31] can provide important guidance to further development of anti-toxoplasmic agents. We have previously shown that the inclusion of the 7-deaza motif into the structure of various 6-benzylthioinosine analogues increased the binding affinity of the new analogues [24, 25]. For example, the inclusion of the 7-deaza motif into 6-(p-methylbenzylthio)inosine increased the relative binding affinity of the resulting 7-deaza-6-(p-methylbenzylthio)inosines) by 2.8-fold [24,25]. Considering that inclusion of the carbocyclic motif into 6-(p-methylbenzylthio)inosine also enhanced the binding of 6-(p-methylbenzylthio)inosine (11) by 3.7-fold [24 and Table 2], it is suggested that if both the deaza- and carbocyclic motifs were combined into the structures of certain 6-benzylthioinosine analogues (e.g. synthesis of carbocyclic 7-deaza-6-(p-methylbenzylthio)inosines) may yield better subversive substrates as potential chemotherapeutic agents. This will also further contribute to the development of T. gondii adenosine kinase as a target for the chemotherapy of toxoplasmosis and possibly other related pathogenic protozoa
Acknowledgments
This research was supported by the U.S. Public Health Service Grant AI-52838 from the National Institute of Health. We thank Mudar Al Safarjalani, P.E. for the computer programming and estimations of the IC50 values, and Marion Kirk for Mass spectrometry analyses. The mass spectrometer was purchased by Grant S10RR13795 and UAB Health Services Foundation General Endowment Fund.
Abbreviations used
- FBS
fetal bovine serum
- MTT
microculture tetrazolium test
- NBMPR
nitrobenzylthioinosine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dubey JP, Beattie CP. Toxoplasmosis of Animals and Man. CRC Press; Boca Raton, FL: 1988. [Google Scholar]
- 2.Jones JL, Kruszon-Moran D, Sanders-Lewis K, Wilson M. Toxoplasma gondii infection in the United States, 1999-2004, Decline from the prior decade. Am. J. Trop. Med. Hyg. 2007;77:405–410. [PubMed] [Google Scholar]
- 3.Mead PS, Slutsker L, Dietz V, McCaig LF, Bresee JS, Shapiro C, Griffin PM, Tauxe RV. Food-related illness and death in the United States. Emerg. Infect. Dis. 1999;5:607–625. doi: 10.3201/eid0505.990502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peterson E. Epidemiology, diagnostics, and chemotherapy. In: Ajioka JW, Soldati D, editors. Toxoplasma: Molecular and Cellular Biology. Horizon Bioscience; Norfolk, UK: 2007. pp. 37–58. [Google Scholar]
- 5.Luft BJ. Toxoplasma gondii. In: Walzer PD, Genta RM, editors. Parasitic Infections in the Compromised Host. Marcel Dekker; New York: 1989. pp. 179–279. [Google Scholar]
- 6.Luft BJ, Hafner R, Korzun AH, Leport C, Antoniskis D, Bosler EM, Bourland DD, Uttamchandani R, Fuhrer J, Jacobson J, Morlat P, Vilde J, Remington JS. Toxoplasmic encephalitis in patients with the acquired immunodeficiency syndrome. N. Engl. J. Med. 1993;329:995–1000. doi: 10.1056/NEJM199309303291403. [DOI] [PubMed] [Google Scholar]
- 7.Israelski DM, Remington JS. Toxoplasmosis in the non-AIDS immunocompromised host. Curr. Clin. Top. Infect. Dis. 1993;13:322–356. [PubMed] [Google Scholar]
- 8.Israelski DM, Remington JS. Toxoplasmosis in patients with cancer. Clin. Infect. Dis. 1993a;17(Suppl 2):S423–35. doi: 10.1093/clinids/17.supplement_2.s423. [DOI] [PubMed] [Google Scholar]
- 9.Remington JS, McLeond R, Desmonts G. Toxoplasmosis. In: Remington JS, Klein JO, editors. Infectious Diseases of the Fetus and Newborn Infant. W.B. Saunders Company; Philadelphia: 1995. pp. 140–267. [Google Scholar]
- 10.Wong S, Remington JS. Toxoplasmosis in pregnancy. Clin Infect Dis. 1994;18:853–862. doi: 10.1093/clinids/18.6.853. [DOI] [PubMed] [Google Scholar]
- 11.Holland GN. Ocular toxoplasmosis: a global reassessment. Part I: epidemiology and course of disease. Am. J. Ophthalmol. 2003;136:973–988. doi: 10.1016/j.ajo.2003.09.040. [DOI] [PubMed] [Google Scholar]
- 12.Holland GN. Ocular toxoplasmosisi: a global reassessment. Part II: disease manifestations and management. Am. J. Ophthalmol. 2004;137:1–17. [PubMed] [Google Scholar]
- 13.Mortensen PB, Norgaard-Pederson B, Waltoft BL, Sorensen TL, Hougaard D, Yolken RH. Early infections of Toxoplasma gondii and later development of schizophrenia. Schiz. Bull. 2007;33:741–744. doi: 10.1093/schbul/sbm009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Torrey EF, Bartko JJ, Lun ZR, Yolken RH. Antibodies to Toxoplasma gondii in patients with schizophrenia: a meta-analysis. Schiz. Bull. 2007;33:719–736. doi: 10.1093/schbul/sbl050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Niebuhr DW, Millikan AM, Cowan DN, Yolken RH, Yuanzhang L, Weber NS. Selected infectious agents and risk of schizophrenia among U.S. military personnel. Am. J. Psychiatry. 2008;165:99–106. doi: 10.1176/appi.ajp.2007.06081254. [DOI] [PubMed] [Google Scholar]
- 16.Schwarcz R, Hunter CA. Toxoplasma gondii and Schizophrenia: Linkage through astrocytederived kynurenic acid. Schiz. Bull. 2007;33:652–653. doi: 10.1093/schbul/sbm030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Subauste CS, Remington JS. Immunity to Toxoplasma gondii. Curr Opin Immunol. 1993;5:532–537. doi: 10.1016/0952-7915(93)90034-p. [DOI] [PubMed] [Google Scholar]
- 18.Wong S, Remington JS. Biology of Toxoplasma gondii. AIDS. 1993;7:299–316. doi: 10.1097/00002030-199303000-00001. [DOI] [PubMed] [Google Scholar]
- 19.el Kouni MH. Potential chemotherapeutic targets in the purine metabolism of parasites. Pharmacol Therapeut. 2003;99:283B309. doi: 10.1016/s0163-7258(03)00071-8. [DOI] [PubMed] [Google Scholar]
- 20.el Kouni MH. Adenosine metabolism in Toxoplasma gondii: Potential targets for chemotherapy. Curr Pharm Des. 2007;13:581–597. doi: 10.2174/138161207780162836. [DOI] [PubMed] [Google Scholar]
- 21.Pfefferkorn ER, Pfefferkorn LC. The biochemical basis for resistance to adenine arabinoside in a mutant of Toxoplasma gondii. J Parasitol. 1978;64:486–492. [PubMed] [Google Scholar]
- 22.Krug EC, Marr JJ, Berens RL. Purine Metabolism in Toxoplasma gondii. J Biol Chem. 1989;264:10601–10607. [PubMed] [Google Scholar]
- 23.Iltzsch MH, Uber SS, Tankersley KO, el Kouni MH. Structure activity relationship for the binding of nucleoside ligands to adenosine kinase from Toxoplasma gondii. Biochem Pharmacol. 1995;49:1501–12. doi: 10.1016/0006-2952(95)00029-y. [DOI] [PubMed] [Google Scholar]
- 24.Yadav V, Chu CK, Rais RH, Al Safarjalani ON, Guarcello V, Naguib FNM, el Kouni MH. Synthesis, biological activity and molecular modeling of 6-benzylthioinosine analogues as subversive substrates of Toxoplasma gondii adenosine kinase. J Med Chem. 2004;47:1987–96. doi: 10.1021/jm030537y. [DOI] [PubMed] [Google Scholar]
- 25.Kim YA, Sharon A, Chu CK, Rais RH. Al Safarjalani ON, Naguib FNM, el Kouni MH. Structure-activity relationships of 7-deaza-6-benzylthioinosine analogues as ligands of Toxoplasma gondii adenosine kinase. J Med Chem. 2008;51:3934–3945. doi: 10.1021/jm800201s. [DOI] [PubMed] [Google Scholar]
- 26.Kim YA, Rawal RK, Yoo J, Sharon A, Jha AK, Chu CK, Rais RH, Al Safarjalani ON, Naguib FNM, el Kouni MH. Structure-activity relationships of carbocyclic 6-benzylthioinosine analogues as subversive substrates of Toxoplasma gondii adenosine kinase. Bioorganic Med Chem. doi: 10.1016/j.bmc.2010.04.003. in Press. [DOI] [PubMed] [Google Scholar]
- 27.el Kouni MH, Guarcello V, Al Safarjalani ON, Naguib FNM. Metabolism and selective toxicity of 6-nitrobenzylthioinosine in Toxoplasma gondii. Antimicrob Aagents Chemother. 1999;43:2437–2443. doi: 10.1128/aac.43.10.2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Al Safarjalani ON, Naguib FNM, el Kouni MH. Uptake of nitrobenzylthioinosine and purine β-l-nucleosides by intracellular Toxoplasma gondii. Antimicrob Agents Chemother. 2003;47:3247–3251. doi: 10.1128/AAC.47.10.3247-3251.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rais RH, Al Safarjalani ON, Yadav V, Guarcello V, Kirk M, Chu CK, Naguib FNM, el Kouni MH. 6-Benzylthioinosine analogues as subversive substrate of Toxoplasma gondii adenosine kinase: activities and selective toxicities. Biochem Pharmacol. 2005;69:1409–19. doi: 10.1016/j.bcp.2005.02.017. [DOI] [PubMed] [Google Scholar]
- 30.Gupte A, Buolamwini JK, Yadav V, Chu CK, Naguib FNM, el Kouni MH. 6-Benzylthioinosine analogues: promising anti-toxoplasmic agents as inhibitors of the mammalian nucleoside transporter ENT1 (es). Biochem Pharmacol. 2005;71:69–73. doi: 10.1016/j.bcp.2005.10.031. [DOI] [PubMed] [Google Scholar]
- 31.Al Safarjalani ON, Rais RH, Kim YA, Chu CK, Naguib FNM, el Kouni MH. 7-Deaza-6-benzylthioinosine analogues as subversive substrate of Toxoplasma gondii adenosine kinase: Activities and selective toxicities. Biochem Pharmacol. 2008;76:958–966. doi: 10.1016/j.bcp.2008.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sullivan WJ, Chiang CW, Wilson CM, Naguib FNM, el Kouni MH, Donald RGK, Roos DS. Insertional tagging and cloning of at least two loci associated with resistance to adenine arabinoside in Toxoplasma gondii and cloning of the adenosine kinase locus. Mol Biochem Parasitol. 1999;103:1–14. doi: 10.1016/s0166-6851(99)00114-0. [DOI] [PubMed] [Google Scholar]
- 33.Herdewijn P, De Clercq E, Balzarini J, Vanderhaeghe H. Synthesis and antiviral activity of the carbocyclic analogues of (E)-5-(2-halovinyl)-2'-deoxyuridines and (E)-5-(2-halovinyl)-2'-deoxycytidines. J Med Chem. 1985;28:550–555. doi: 10.1021/jm50001a003. [DOI] [PubMed] [Google Scholar]
- 34.Ferrero M, Gotor V. Biocatalytic selective modifications of conventional nucleosides, carbocyclic nucleosides, and C-nucleosides. Chem Rev. 2000;100:4319–4348. doi: 10.1021/cr000446y. [DOI] [PubMed] [Google Scholar]
- 35.Zhang Y, el Kouni MH, Ealick SE. Structure of Toxoplasma gondii adenosine kinase in complex with an ATP analog at 1.1 Å resolution. Acta Cryst D. 2006;D62:142–5. doi: 10.1107/S090744490503430X. [DOI] [PubMed] [Google Scholar]
- 36.Kim YA, Sharon A, Chu CK, Rais RH, Al Safarjalani ON, Naguib FNM, el Kouni MH. Synthesis, biological evaluation and molecular modeling studies of N6-benzyladenosine analogues as potential anti-toxoplasma agents. Biochem Pharmacol. 2007;73:1558–1572. doi: 10.1016/j.bcp.2007.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pfefferkorn ER, Pfefferkorn LC. Specific labeling of intracellular Toxoplasma gondii with uracil. J Protozool. 1977;4:449–453. doi: 10.1111/j.1550-7408.1977.tb04774.x. [DOI] [PubMed] [Google Scholar]
- 38.Cha S. Kinetics of enzyme reactions with competing alternative substrates. Mol Pharmacol. 1968;4:621–629. [PubMed] [Google Scholar]