

Abstract
In the recent decades, oxidative stress has become focus of interest in most biomedical disciplines and many types of clinical research. Increasing evidence from research on several diseases show that oxidative stress is associated with the pathogenesis of diabetes, obesity, cancer, ageing, inflammation, neurodegenerative disorders, hypertension, apoptosis, cardiovascular diseases, and heart failure. Based on this research, the emerging concept is that oxidative stress is the “final common pathway”, through which risk factors of several diseases exert their deleterious effects. Oxidative stress causes a complex dysregulation of cell metabolism and cell-cell homeostasis. In this review, we discuss the role of oxidative stress in the pathogenesis of insulin resistance and beta-cell dysfunction. These are the two most relevant mechanisms in the pathophysiology of type 2 diabetes, and in the pathogenesis of diabetic vascular complications, the leading cause of death in diabetic patients.
Keywords: type 2 diabetes, diet, lifestyle, nutrition, physical activity, weight loss, glycemic control
Abbreviations: ADMA - asymmetric dimethylarginine; AGE - advanced glycation end products; AOC - antioxidant capacity; BH4 - tetrahydrobiopterin; DNA - deoxyribonucleic acid; ELISA - enzyme-linked immunosorbent assay; EPR - electron paramagnetic resonance; ESR - electron spin resonance; GAPDH - glyceraldehyde-3-phosphate dehydrogenase; GC-MS - gas chromatography-mass spectometry; GLUT4 - glucose transporter type 4; GSH - reduced glutathione; GSSG - oxidized glutathione; GTP - guanosine triphosphate; eNOS - endothelial nitric oxide synthase; iNOS - inducible nitric oxide synthase; nNOS - neuronal nitric oxide synthase; HIT-T15 cell - insulin-secreting hamster insulinoma tumor cell; H2O2 - hydrogen peroxide; HO• - hydroxyl radical; HPLC - high pressure liquid chromatography; IR - insulin receptor; IRS - insulin receptor substrate; JNK - c-Jun N-terminal kinase; LA - lipoic acid; M - magnetic spin quantum number of the spin state; MAP - mitogen-activated protein; MDA - malondialdehyde; NADPH - nicotinamide adenine dinucleotide phosphate; NO - nitric oxide; NO- - nitroxyl anion; NO2- - nitrite; NO3- - nitrate; NOx - nitrite + nitrate (index); NOS - nitric oxide synthase; O2- - superoxide anion; OONO- - peroxynitrite; PARP - poly (ADP-ribose) polymerase; PI 3-kinase - phosphoinositid-3-kinase; PKC - protein kinase C; ROM - reactive oxygen metabolites; ROS - reactive oxygen species; RNS - reactive nitrogen species; RS - reactive species; RSNO - S-nitrosothiol; SOD - superoxide dismutase; TAC - total antioxidant capacity; TNFα - tumor necrose factor alpha; UCP-1 - mitochondrial uncoupling protein 1; VEGF - vascular endothelial growth factor; ZDF - Zucker diabetic fatty
Reactive oxidative species (ROS) and reactive nitrogen species (RNS)
Biological systems living in aerobic conditions are exposed to oxidants, either generated intentionally or as byproducts. Generally, such oxidants occur in two categories, reactive oxygen and reactive nitrogen species (ROS and RNS, respectively). ROS is a collective term describing the chemical species that are formed upon incomplete reduction of oxygen. It includes superoxide anion (O2-), hydrogen peroxide (H2O2), and hydroxyl radical (HO•). In contrast, RNS refers to all oxidation states and reactive adducts of nitrogenous nitric oxide synthase (NOS) products, from nitric oxide (NO) to nitroxyl (NO-), S-nitrosothiol (RSNO), and peroxynitrite (OONO-), as product of the reaction between NO and O2- (Table 1) [1].
Table 1. Reactive oxygen species (ROS) and reactive nitrogen species (RNS) implicated in the pathogenesis of diabetes and its complications.
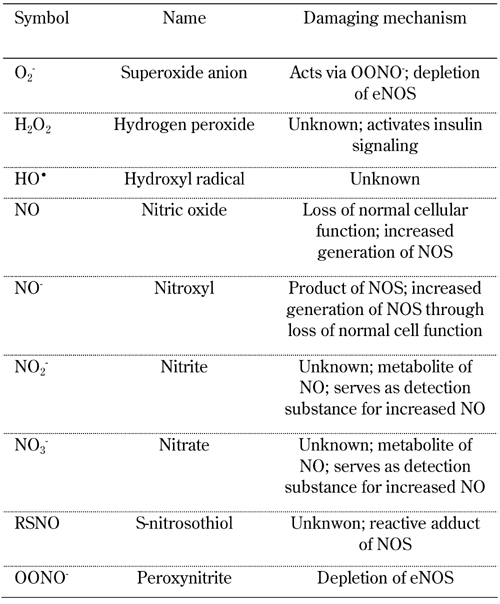
ROS and RNS have critical biological functions essential for normal physiology. Overproduction or deficiency of ROS and RNS may result in impaired homeostasis and associated pathology. In healthy organisms, oxidants are balanced by reductants (antioxidants). According to recent reports it is believed that almost all pathogenetic mechanisms involve a disequilibrium in the redox balance as "final common pathway" [1]. This review summarizes recent knowledge on the relevance of oxidative stress in the physiopatology of diabetes. Particular attention is given to NO. In this context, aspects of diabetes pathogenesis, complications, and therapy options are discussed (Figure 1).
Figure 1.
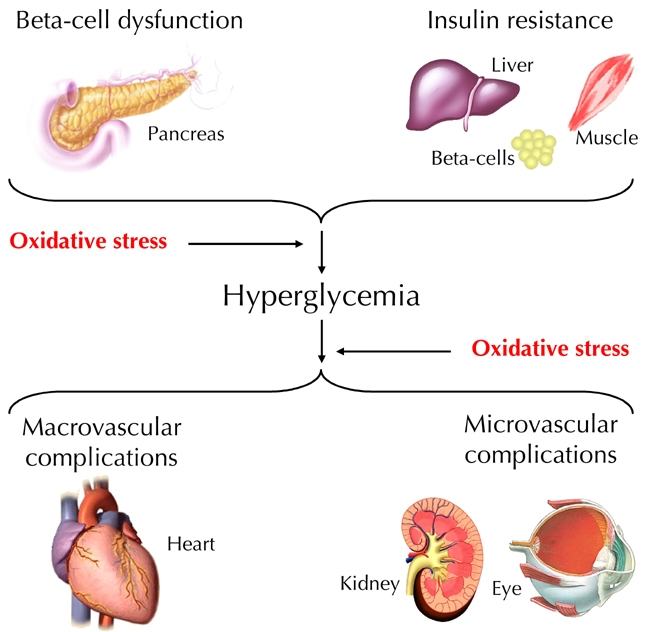
Oxidative stress is involved in the pathogenesis of diabetes and hyperglycemia-associated vascular complications.
Methods for detecting oxidative stress metabolites
In clinical practice, the analytical measurement of oxidative stress markers has been difficult. The difficulties are due to the short half-life of the majority of such compounds (few seconds), and the applicability of determination methods. Blood and urine samples are the eligible biological materials for oxidant and antioxidant status assessments in humans. However, some antioxidant enzymes and oxidative stress metabolites have also been determined in tissue extracts, especially in animal models [2, 3]. A series of methods including enzyme-linked immunosorbent assay (ELISA), high pressure liquid chromatography (HPLC), spectroscopy, gas chromatography-mass spectometry (GC-MS), immunoblotting, electroelution fractionation, isoelectric focusing, voltametry, and electron paramagnetic resonance (EPR) spectroscopy have been applied for the determination of oxidative stress metabolites [4-6]. This section briefly introduces the commonly used approaches to detect free radicals, specific reactive oxygen, nitrogen species, and antioxidant biomarkers, and discusses their reliability.
A dynamic indicator of oxidative stress in vivo is the quantification of redox state of the GSH/GSSG pool in tissue and/or plasma. This can be determined biochemically [4], or by HPLC according to the method described by Jones [7]. A direct measurement of ROS, including free radicals and peroxides, is difficult to perform due to their biochemical instability. Electron spin resonance (ESR) spectroscopy, also known as electron paramagnetic resonance (EPR) spectroscopy, is the only analytical approach that enables direct detection of free radicals such as NO, superoxide, and hydroxyl radical [8]. With a limited sensitivity of 10-9 M, EPR is capable of detecting free radical-derived species produced during oxidative and inflammatory injury, including ascorbyl radical, tocopheroxyl radical, and heme-nitrosyl complexes [9].
Other methods have been developed to indirectly detect oxidant/free radical formation in vitro and in vivo. Lipid peroxidation is one of the most widely used indicators for determining the cellular oxidant status. However, the method is non-specific, relying on the detection of substances such as malondialdehyde (MDA) or other reactive aldehydes generated from the in vivo or ex vivo decomposition of lipid peroxidation products [10]. HPLC [11] and several commercially available ELISA kits have been developed to quantify other attractive markers and indicators of lipid peroxidation such as F2-isoprostanes [12]. Recently, a new method of measuring reactive oxygen metabolites (ROM) in the blood was developed, the so-called d-ROM test. It is based on the free radical analytical system that determines mainly lipid hydroperoxides in serum, the main component of ROM [13]. Despite its oxidant power, hydroperoxide is relatively stable in the blood. Therefore, it is easily detectable.
NO laboratory measurement is extremely difficult due to the short half-life and the very low concentration in biological fluids. Nitrite (NO2-) and nitrate (NO3-) are the most stable metabolites of endogenous NO. They are accessible by quantitative analysis, so that determination of these inorganic metabolites in blood and urine turned out to be the most suitable method to assess indirect quantification of NO production in vivo. Mass-spectrometry-based methods are presently the most accurate methods for quantitative determination of nitrate and nitrite in biological fluids. Also, other analytical methods, such as gas and liquid chromatography and electrophoresis, are also applied effectively [14-18].
Commercially available kits are based on the "old" Griess reaction (Roche Diagnostics GmbH, Mannheim, Germany). These kits allow the spectrophotometric detection of nitrate and nitrite. Due to their simplicity, rapidity, and cheapness they have been broadly applied in clinical practice. However, it is well known that pre-analytical, and analytical, factors can interfere with the analysis of nitrite and nitrate by Griess assay. Many studies have shown that parallel comparison of nitrite plus nitrate (NOx) quantification by different methods produced diverging values and discrepancies principally due to methodological problems [19, 20]. One of the most important factors interfering with the Griess reaction is the protein content in biological samples. Other factors responsible for inaccurate determination of NOx levels are i) turbidity resulting from aggregated or precipitated proteins, ii) conversion of nitrite to S-nitroso compounds by reduced thiols, and iii) other reactions with tyrosine and tryptophan that create colored products. Recently, it has been demonstrated that the Griess assay, supplemented with an appropriate deproteinization procedure of the serum sample, can be successfully applied for NO metabolite measurement. This procedure allows to obtain NOx levels in agreement with CG-MS and HPLC [21].
At present, there is increasing interest to investigate correlations between NO production and asymmetric dimethylarginine (ADMA), an endogenous nitric oxide synthase inhibitor. The existing methods for ADMA determination are HPLC, liquid chromatography-mass spectrometry (LC-MS), and GC-MS [22]. Recently, the performance of a new ELISA assay kit for determination of ADMA was evaluated against a reversed phase HPLC method. Despite differing principles, both methods showed a good correlation [23].
Antioxidant capacity (AOC) includes endogenous compounds (bilirubin, uric acid, superoxide dismutases, catalase, glutathione peroxidase, etc.), and exogenous compounds (carotenoids, tocopherols, ascorbate, bioflavonoids, etc.) [24]. These antioxidant species are subdivided into three categories: 1. enzyme systems, 2. small molecules, and 3. proteins. The total antioxidant capacity (TAC) measures either the combination of both small molecule antioxidants and proteins, or small molecules alone. An enhanced chemiluminescence assay is commonly used to measure the non-enzymatic TAC, but it is expensive and time-consuming. Therefore, commercial colorimetric kit assays are frequently preferred [25]. Measurement of AOC enzyme activity (catalase, superoxide dismutases, glutathione peroxidise) is usually determined using commercial kits based on different spectrophotometric reactions. Finally, most of serum AOC-related indirect biochemical markers as bilirubin, total cholesterol, low density lipoprotein cholesterol, high density lipoprotein cholesterol, triglyceride, alanine aminotransferase, blood urea nitrogen, iron, and uric acid are nowadays measured by well-established routine procedures based on automated systems.
ROS and RNS in physiology and pathophysiology
Production of ROS and RNS occurs in response to extracellular and intracellular stimuli. Extracellular stimuli act through plasma membrane receptors and include TNFα, hormones, and growth factors, including platelet-derived growth factor, epithelial growth factor, and insulin. Intracellular stimuli include nutrients [26, 27]. nicotinamide adenine dinucleotide phosphate (NADPH) oxidase [28], NOS [29], and mitochondrial electron transfer [30]. These are the most relevant sources of reactive species (RS), including ROS and RNS. They can react with multiple cellular components (proteins, lipids, nucleic acids), and generate reversible or irreversible oxidative modifications. They also activate various signaling cascades, some of which are designated for sensing and responding to “stress” like the mitogen-activated protein (MAP) kinase family or c-Jun N-terminal kinase (JNK) [31].
Functional effects of RS ranges from physiological to pathophysiological aspects. Vascular tone [32], cell adhesion [33], immune responses [34], growth factors, and hormone action [35], are all examples of RS participation in normal physiology. On the other hand, a causative role of RS has been implicated in ageing [36], cancer [37], atherosclerosis [38], neurodegenerative diseases [39], obesity, and diabetes [40]. RS involvement in normal physiological processes involves carefully regulated production in a tight spatial-temporal manner, causing reversible oxidative modifications. Pathophysiological processes mediated by RS are more likely to involve irreversible modifications of cellular components such as proteins, lipids, or DNA.
Oxidative stress and insulin signal transduction
In vitro and molecular studies
High concentrations of H2O2 activate insulin signaling and induce typical metabolic actions of insulin. This result could be considered as the first experimental documentation on the link between ROS and insulin [41]. In particular, H2O2 employs the same pathway as insulin and causes downstream propagation of the signal yielding typical metabolic actions of insulin. H2O2 increases glucose uptake in adipocytes and muscles [42], and also stimulates GLUT4 translocation and lipid synthesis in adipocytes [43].
Insulin receptor substrate (IRS) proteins are effectors for tyrosine kinase activity of the insulin receptor (IR) upon insulin binding. These proteins are involved in a critical step of insulin signaling. In normal-unstimulated state, insulin signaling molecules are distributed between the cytosol and internal membrane pools. Whereas upon insulin stimulation, tyrosine residues on IR and IRS are phosphorylated through activated insulin receptor kinase. This leads to the recruitment of PI 3-kinase to the plasma membrane and to internal membrane pools. Subsequently, the activation of small GTPase Rac induces cytoskeletal reorganization that propagates the insulin signals. Finally, this culminates into typical metabolic effects of insulin such as increased glucose uptake. Under conditions of increased oxidative stress, stress-responsive signaling cascades are activated, such as the MAP kinase cascades. This leads to increased Ser/Thr phosphorylation of IRS molecules. Modified IRS molecules are released from internal membrane pools and are subjected to increased protein degradation. In these conditions, insulin fails to elicit normal metabolic effects. This is because IRS molecules are decreased in content and cannot be normally tyrosine phosphorylated when hyperphosphorylated on certain Ser/Thr residues [44]. Note that, receptor substrates are able to directly modify the expression of glucose/metabolic genes such as GLUT4 [45] and adiponectin [46].
Animal studies
The major source of oxidative stress in obese and/or diabetic subjects seems to be the liver and adipose tissue [47]. In obese mice, an increased H2O2 generation by adipose tissue could be observed prior to diabetes onset [48]. This event was accompanied by decreased mRNA levels of SOD, catalase, and glutathione peroxidise. Developing diabetes in these mice mildly exaggerated these alterations, which remained unobservable in other tissues. Obesity in combination with the related insulin resistance are frequently associated with increased accumulation of lipids (triglycerides) in the liver. Increased numbers of lipid peroxidation markers have been observed in the liver, in animal models of diabetes and obesity [49]. Oxidation-induced disruption of cellular redistributed signaling molecules in response to insulin stimulation was associated with impaired insulin action. An in vivo model of oxidative stress provided support for this notion. In this model, oxidative stress was induced in rats using an inhibitor of glutathione biosynthesis enzyme. The consequential drop in tissue levels of glutathione, a major cellular antioxidant, increased markers of oxidative stress and impaired glucose homeostasis in vivo.
Human studies
A large number of studies have evidenced the pivotal role of oxidative stress in insulin resistance states such as metabolic syndrome, obesity, and type 2 diabetes [50-52]. Decreased antioxidant capacity, increased production of ROS with oxidation products of lipids, DNA, and proteins have been reported in plasma, urine, and various tissues, suggesting systemic and organ-specific oxidative stress. Recent evidence for systemic oxidative stress includes the detection of increased circulating and urinary levels of the lipid peroxidation product F2-isoprostane (8-epi-prostaglandin F2α) in both type 1 and type 2 diabetic patients [53, 54], and in obesity. Remarkably, this marker correlated with blood glucose levels and glucose variability [55], and responded to antidiabetic intervention [54]. In particular, our group has shown the involvement of oxidative stress in the early phases of type 1 diabetes, with an impaired balance between NO, serum uric acid (a potent antioxidant), and asymmetric-dimethyl-L-arginine (ADMA) [56]. As described above, ROS and RNS are able to directly modify the expression of adiponectin. Secreted almost exclusively from adipocytes, it is inversely correlated with fat mass in obesity and with its associated cardiovascular risk. Note that, plasma and urinary lipid peroxidation markers indicative of systemic oxidative stress correlated with lower circulating adiponectin levels.
Oxidative stress and the beta-cell
Glucotoxicity and lipotoxicity are diabetes-related phenomena that generate oxidative stress. They are implicated in the pathogenesis of beta-cell dysfunction [57]. Hyperglycemia and hyperlipidemia follow the primary pathogenesis of diabetes and exert additional toxic effects on beta-cells. There is evidence resulting from in vitro and in vivo studies that glucose and lipids are harmful to beta-cells. Interestingly, some studies reported that lipotoxicity only occurs in the presence of concomitant elevated glucose levels [58, 59]. Consequently, hyperglycemia might be a prerequisite for negative effects of lipotoxicity. Therefore, the term glucolipotoxicity may be a better term than lipotoxicity to describe the baneful relationship between lipids and beta-cell function.
In vitro/molecular studies
Robertson et al. have demonstrated how insulin gene expression, insulin content, and glucose-induced insulin secretion were progressively and drastically compromised over time when beta-cell lines (HIT-T15 cells) were exposed to high glucose concentrations [60]. Decreased levels of insulin mRNA, insulin content, and insulin release were regarded as evidence for glucotoxic effects on beta-cells caused by chronic exposure to high glucose concentrations. Evidence that glucotoxicity is related to oxidative stress stems from early reports that the antioxidants, N-acetylcysteine and aminoguanidine, protect HIT-T15 cells and isolated islets against adverse effects from exposure to high glucose concentrations [61, 62]. Using N-acetylcysteine, or aminoguanidine, in cells cultured for many passages under high glucose conditions, it is possible to observe an antioxidant drug concentration-related preservation of insulin promoter activity and levels of insulin mRNA [63]. A prolonged in vitro exposure of isolated islets or insulin-secreting cells to elevated levels of fatty acids is associated with inhibited glucose-induced insulin secretion, impaired insulin gene expression, and induction of cell death by apoptosis. Note that, most glucolipotoxicity-related effects on beta-cells involve the generation of oxidative stress and inflammation.
Antioxidative therapy in animal studies
The db/db mouse is a well-accepted genetic rodent model of type 2 diabetes carrying a mutation in the leptin receptor gene. Kaneto et al. have shown in an in vivo study that N-acetylcysteine was effective in enhancing intra-peritoneal glucose tolerance in db/db mice [64]. The authors observed that N-acetylcysteine improved insulin secretion, moderately decreased blood glucose levels, enhanced beta-cell mass, and decreased apoptosis in the diabetic mice. They also reported preserved amounts of insulin content, and insulin mRNA.
Other successful attempts to ameliorate diabetes and oxidative stress in animal models have involved the use of vitamin E in Goto-Kakizaki rats [65] and gliclazide [66], an oral hypoglycemic agent with antioxidant properties. Zucker diabetic fatty (ZDF) rats carry a mutation in the leptin receptor gene and become obese and diabetic with age. Their treatment, with either N-acetylcysteine or aminoguanidine, prevented rise in plasma ROS and attenuated the degree of hyperglycemia, whilst minimizing glycosuria [63]. Regarding lipotoxicity, Sako et al. have demonstrated that a 48-h perfusion of intralipid, or oleate, impairs glucose-induced insulin secretion in normal rats [67].
Human studies
A large number of clinical studies have shown that diabetes is associated with higher-than-normal oxidative stress. However, it is difficult to demonstrate a direct relation between hyperglycemia, hyperlipidemia, oxidative stress, and beta-cell dysfunction in humans, as there is no “gold-standard” procedure to test the relation. Nonetheless. Del Guerra et al. examined islets isolated from pancreata of 13 type 2 diabetic cadaveric organ donors [68]. They observed high levels of oxidative stress markers, low levels of glucose-induced insulin secretion, and reduced insulin mRNA. In another study, arginine-induced insulin secretion was improved in a type 2 diabetic patient following 28 days of oral N-acetylcysteine treatment [69]. Recently, Cusi et al. have shown that acipimox, a drug reducing circulating fatty acid levels, ameliorates insulin secretion in subjects with familiar history of diabetes [70].
Oxidative stress and diabetic vascular complications
Role of ROS and RNS in diabetic vascular dysfunction
Cardiovascular risk factors promote the production of ROS. Imbalance between endogenous oxidants and antioxidants results in oxidative stress, a condition that contributes to impaired NO bioavailability and vascular dysfunction. NO is a gaseous lipophilic free radical generated by three distinct isoforms of NOS, type 1 or neuronal (nNOS), type 2 or inducible (iNOS), and type 3 or endothelial NOS (eNOS). Expression of eNOS is altered in many types of cardiovascular disease such as atherosclerosis, diabetes, and hypertension. Atherosclerosis is considered as the underlying pathology of cardiovascular diseases such as peripheral vascular disease, stroke, and coronary heart disease.
In vitro/molecular studies
The pathology of atherosclerosis is complex and involves structural elements of the arterial wall, platelets, leukocytes, and inflammatory cells such as monocytes and macrophages [71, 72]. The endothelium is a dynamic interface between the arterial wall and the circulating cells. Therefore, endothelial dysfunction accounts for one of the primary causes of atherosclerosis. Since the endothelium is the major source of NO in the vasculature, loss of normal cellular function can result in altered NO synthesis. The endothelium provides a constitutive supply of NO from eNOS, and under certain conditions (e.g. inflammation) it can produce excessive NO from the inducible isoform iNOS [73]. Therefore, regulation of NOS is central in the development and progression of atherosclerosis.
Diabetes complications are characterized by endothelial (vascular) dysfunction. A number of studies have suggested that ROS play an important role in the pathogenesis of diabetic vascular dysfunction as the pathways of diabetic complications have a close relationship to oxidative stress. In particular, increased glucose leads to increased mitochondrial formation of ROS. Superoxide is a ROS that produces peroxynitrite when reacting with NO. Peroxynitrite induces cellular damage through depletion of the co-factor of the endothelial isoform of NOS (eNOS), tetrahydrobiopterin (BH4). The pathway of peroxynitrite-mediated injury involves DNA strand breakage, activation of the nuclear enzyme poly (ADP-ribose) polymerase (PARP), and inhibition of glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Also, it activates the "classic" pathways of diabetic complications, including: i) the polyol pathway, ii) the advanced glycation end products (AGE) pathway, iii) the protein kinase C (PKC) pathway, and iv) the hexosamine pathway. PARP activation can also upregulate various proinflammatory pathways which leads to pathological modifications in adhesion molecule expression, angiogenesis, and other processes.
Recently, two other aspects have been related to glucose-induced oxidative stress: 1. "glucose variability" and 2. "glucose memory". Regarding 1. Several studies suggested that intermittent low and high glucose conditions are even more deleterious to endothelial cell function than a steady, constant increase of glucose [74]. These conditions also induce endothelial cells to enter into a proinflammatory state, and this state is associated with the upregulation of various adhesion molecules and proinflammatory cytokines [74]. Some of the pathways implicated in these exacerbated cellular responses, involve activation of PKC, NADPH oxidases, and mitochondrial oxidants. Regarding 2. It has been shown in culture and in diabetic rats that endothelial cells exhibit a persistence or 'memory' of induced basement membrane mRNA expression long after normalization of high glucose levels [75]. This finding suggests that glucose induces long-lasting deleterious effects that persist beyond the period of hyperglycemia.
Animal studies
Animal models of diabetes are associated with reduced bioavailability of NO and impaired endothelium-dependent relaxation [76]. The latter is abrogated after acute incubation with superoxide dismutase, a superoxide scavenger [77]. Consequently, studies involving streptozotocin-induced diabetic rats, showed that diabetes-induced increase in both retinal vascular endothelial growth factor (VEGF) concentrations and lipid peroxidation could be prevented by antioxidant treatment [78]. These results suggest a major role for NO in destruction by O2- in diabetes-associated vascular dysfunction. Interestingly, eNOS knockout mice exhibited accelerated diabetic nephropathy [79], supporting a role for deficient eNOS-derived NO production in the pathogenesis of diabetic nephropathy. Moreover, diabetic animals exhibited increased mRNA and protein for eNOS [80]. Whereas, other studies reported reduced cGMP formation. Interestingly, eNOS knockout mice exhibited accelerated diabetic nephropathy [79], supporting a role for deficient eNOS-derived NO production in the pathogenesis of diabetic nephropathy.
iNOS is very relevant to diabetic pathophysiology. Recent reports reveal that decreased expression of eNOS accompanies increased expression of iNOS and nitrotyrosine during the progression of diabetes in rats [81]. This finding suggests that induction of iNOS in cardiovascular tissues is dependent on the duration of diabetes and contributes significantly to depressed responses to vasoactive agents.
Human studies
In vivo studies revealed that oxidative stress due to hyperglycemia occurs before late complications become clinically evident [56]. This finding suggests that oxidative stress plays a crucial role in the pathogenesis of late diabetic complications. It has also been documented in human studies that endothelial cells in diabetes fail to produce sufficient amount of NO and fail to relax in response to endothelium-dependent vasorelaxants (e.g. acetylcholine, bradykinin, shear stress, etc.) [82]. Further clinical data have demonstrated that rapid glycemic swings are associated with an exacerbated degree of oxidant production in human diabetes [55], and are deleterious to the endothelial function of type 2 diabetic patients [83]. Overall, these data outline the importance of steady glucose control and the potential involvement of oxidative and nitrosative stress in the pathogenesis of complications due to poorly controlled diabetes.
Diabetic subjects have reduced antioxidant capacity which could favor oxidative stress. A decline in important cellular antioxidant defense mechanisms, including the glutathione redox system and vitamin C-vitamin E cycle, significantly increases the susceptibility to oxidative stress. Thus, attempts have been made to reduce oxidative stress-dependent cellular changes in patients with diabetes by supplementation with naturally occurring antioxidants, especially vitamins E and C. In this regard, lipoic acid (LA) levels are reduced in diabetic patients. LA has long been used for the treatment of diabetic neuropathy. Recent evidence indicates that it increases insulin sensitivity in patients with type 2 diabetes [84]. LA is a naturally occurring antioxidant and participates in establishing a cellular antioxidant network by raising intracellular glutathione levels.
Therapeutic approaches to reduce oxidative stress
There are two main approaches to modulate oxidative stress: 1. preventing ROS/RNS generation (cellular systems and animal studies), and 2. enhancing antioxidant defense (cellular systems, animal and human studies). At cellular level, it is possible to inhibit several sources of oxidative stress (e.g. mitochondrial uncoupling proteins 1 (UCP1), NADPH oxidases, iNOS) or to enhance antioxidant defense (e.g. by lipoic acid, vitamin C and E, GSH, increasing intracellular catalase, or SOD activity).
Regarding animal studies, transgenic mice overexpressing UCP1 in adipose tissue or muscle were found to be protected from obesity and its metabolic consequences [85, 86]. Whereas, in vivo administration of apocynin, a pharmacological inhibitor of the NADPH oxidase system, decreased adipose tissue ROS generation and lipid peroxidation products in a genetic mouse model of obesity and diabetes, and decreased glucose and insulin levels [48]. Different antioxidants, including vitamins C and E, lipoic acid, and flavanoids, have been shown to attenuate different markers of systemic oxidative stress in a variety of experimental animal models of obesity and diabetes. They are also able to improve insulin and glucose levels, and reduce micro and macrovascular dysfunction.
Human intervention trials are constricted to antioxidant defense increase by antioxidant supplementation. Enhanced insulin sensitivity has been demonstrated by treatment with LA [84], vitamin E [87], and vitamin C [88] in small-sized, short-term trials. However, these results have not been confirmed in other studies. Late diabetes complications associate with metabolic control. They do not seem to be positively affected by antioxidant therapy. As a consequence, despite some supporting evidence for the ability to improve insulin action with antioxidants, current clinical guidelines do not recommend antioxidant supplementation for the general population of persons with impaired insulin action and type 2 diabetes.
Conclusions
A large body of evidence suggests that oxidative stress has a key role in the pathogenesis of diabetes and its complications. Clearly, both insulin resistance and beta-cell dysfunction, two central events in the pathophysiology of type 2 diabetes, have been linked to a redox unbalance. At the same time, oxidative stress has been implicated in the pathogenesis of diabetes-related vascular complications. Oxidative stress in diabetic vascular disease (i.e., a relative overproduction of ROS), contributes markedly to endothelial dysfunction. It is mainly caused by an imbalance between the activity of endogenous pro-oxidative enzymes (such as NADPH oxidase, xanthine oxidase, and mitochondrial respiratory chain) and antioxidative enzymes (such as superoxide dismutase, glutathione peroxidase, heme oxygenase, and catalase) in favor of the former. In the state of oxidative stress, the production of ROS exceeds the available antioxidant defense systems. As a consequence, bioactivity of NO, a paracrine factor that controls the vascular tone, inhibits platelet function, prevents adhesion of leukocytes, and reduces proliferation of the intima (anti-atherosclerotic mechanism). A dominant mechanism reducing the bioavailability of vascular NO relates to its rapid oxidative inactivation by the ROS O2-. In addition, there is evidence that persisting oxidative stress renders eNOS dysfunctional such that it does not produce NO any longer, but O2- instead.
These data support the concept that an antioxidant therapy may be of great interest to diabetic patients. To test this hypothesis, several studies have been carried out, but unfortunately, produced conflicting results. Several explanations have been proposed to justify such results. Interestingly, recent studies underline the importance of early hyperglycemia-induced oxidative stress treatment, because the redox unbalance can determine epigenetic changes that persist during normoglycemia (metabolic memory) [89, 90]. As a consequence, antioxidant treatment could be of great help in the first stages of vascular dysfunction. Our hope is that new initiatives to study the mechanisms causing the generation of oxidative stress in diabetes may lead to the discovery and evaluation of new antioxidant molecules that could inhibit the diabetes-related oxidative stress mechanisms.
Disclosures (conflict of interests statement): The authors report no conflict of interests.
References
- 1.D'Autreaux B, Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol. 2007;8(10):813–824. doi: 10.1038/nrm2256. [DOI] [PubMed] [Google Scholar]
- 2.Ibrahim W, Tatumi V, Yeh CC, Hong CB, Chow CK. Effects of dietary carnosine and vitamin E on antioxidant and oxidative status of rats. Int J Vitam Nutr Res. 2008;78(4-5):230–237. doi: 10.1024/0300-9831.78.45.230. [DOI] [PubMed] [Google Scholar]
- 3.Valdivia PA, Zenteno-Savin T, Gardner SC, Aguirre AA. Basic oxidative stress metabolites in eastern Pacific green turtles (Chelonia mydas agassizii) Comp Biochem Physiol C Toxicol Pharmacol. 2007;146(1-2):111–117. doi: 10.1016/j.cbpc.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 4.Camera E, Picardo M. Analytical methods to investigate glutathione and related compounds in biological and pathological processes. J Chromatogr B Analyt Technol Biomed Life Sci. 2002;781(1-2):181–206. doi: 10.1016/s1570-0232(02)00618-9. [DOI] [PubMed] [Google Scholar]
- 5.Tarpey MM, Wink DA, Grisham MB. Methods for detection of reactive metabolites of oxygen and nitrogen: in vitro and in vivo considerations. Am J Physiol Regul Integr Comp Physiol. 2004;286(3):R431–R444. doi: 10.1152/ajpregu.00361.2003. [DOI] [PubMed] [Google Scholar]
- 6.Palmieri B, Sblendorio V. Oxidative stress tests: overview on reliability and use. Part I Eur Rev Med Pharmacol Sci. 2007;11(5):309–342. [PubMed] [Google Scholar]
- 7.Jones DP. Redox potential of GSH/GSSG couple: assay and biological significance. Methods Enzymol. 2002;348:93–112. doi: 10.1016/s0076-6879(02)48630-2. [DOI] [PubMed] [Google Scholar]
- 8.Dambrova M, Baumane L, Kalvinsh I, Wikberg JE. Improved method for EPR detection of DEPMPO-superoxide radicals by liquid nitrogen freezing. Biochem Biophys Res Commun. 2000;275:895–898. doi: 10.1006/bbrc.2000.3387. [DOI] [PubMed] [Google Scholar]
- 9.Laurindo FR, Pedro MD Barbeiro HV, Pileggi F, Carvalho MH, Augusto O, da Luz PL. Vascular free radical release ex vivo and in vivo evidence for a flow-dependent endothelial mechanism. Circ Res. 1994;74:700–709. doi: 10.1161/01.res.74.4.700. [DOI] [PubMed] [Google Scholar]
- 10.Pryor WA, Stanley JP, Blair E. Autoxidation of polyunsaturated fatty acids: II. A suggested mechanism for the formation of TBA-reactive materials from prostaglandin-like endoperoxides. Lipids. 1976;11:370–379. doi: 10.1007/BF02532843. [DOI] [PubMed] [Google Scholar]
- 11.Wallin B, Rosengren B, Shertzer HG, Camejo G. Lipoprotein oxidation and measurement of thiobarbituric acid reacting substances formation in a single microtiterplate: its use for evaluation of antioxidants. Anal Biochem. 1993;208:10–15. doi: 10.1006/abio.1993.1002. [DOI] [PubMed] [Google Scholar]
- 12.Roberts LJ, Morrow JD. Measurement of F(2)-isoprostanes as an index of oxidative stress in vivo. Free Radic Biol Med. 2000;28:505–513. doi: 10.1016/s0891-5849(99)00264-6. [DOI] [PubMed] [Google Scholar]
- 13.Aberti A, Bolognini L, Carratelli M, Della Bona MA, Mavviantelli D. Assessing oxidative stress with the D-Roms test. Some mechanistic consideration; Proceedings of the SFRR Summer Meeting; 1997. pp. 82–83. [Google Scholar]
- 14.Ellis G, Adatia I, Yazdanpanah M, Makela SK. Nitrite and nitrate analyses: a clinical biochemistry perspective. Clin Biochem. 1998;31(4):195–220. doi: 10.1016/s0009-9120(98)00015-0. [DOI] [PubMed] [Google Scholar]
- 15.Tsikas D. Methods of quantitative analysis of the nitric oxide metabolites nitrite and nitrate in human biological fluids. Free Radic Res. 2005;39(8):797–815. doi: 10.1080/10715760500053651. [DOI] [PubMed] [Google Scholar]
- 16.Wennmalm A, Benthin G, Edlund A, Jungersten L, Kieler-Jensen N, Lundin S, Westfelt UN, Petersson AS, Waagstein F. Metabolism and excretion of nitric oxide in humans. An experimental and clinical study. Circ Res. 1993;73(6):1121–1127. doi: 10.1161/01.res.73.6.1121. [DOI] [PubMed] [Google Scholar]
- 17.Leone AM, Francis PL, Rhodes P, Moncada S. A rapid and simple method for the measurement of nitrite and nitrate in plasma by high performance capillary electrophoresis. Biochem Biophys Res Commun. 1994;200(2):951–957. doi: 10.1006/bbrc.1994.1542. [DOI] [PubMed] [Google Scholar]
- 18.Everett SA, Dennis MF, Tozer GM, Prise VE, Wardman P, Stratford MR. Nitric oxide in biological fluids: analysis of nitrite and nitrate by high-performance ion chromatography. J Chromatogr A. 1995;706(1-2):437–442. doi: 10.1016/0021-9673(95)00078-2. [DOI] [PubMed] [Google Scholar]
- 19.Tsikas D, Gutzki FM, Rossa S, Bauer H, Neumann C, Dockendorff K, Sandmann J, Frolich JC. Measurement of nitrite and nitrate in biological fluids by gas chromatography-mass spectrometry and by the Griess assay: problems with the Griess assay - solutions by gas chromatography-mass spectrometry. Anal Biochem. 1997;244(2):208–220. doi: 10.1006/abio.1996.9880. [DOI] [PubMed] [Google Scholar]
- 20.Becker AJ, Uckert S, Tsikas D, Noack H, Stief CG, Frolich JC, Wolf G, Jonas U. Determination of nitric oxide metabolites by means of the Griess assay and gas chromatography-mass spectrometry in the cavernous and systemic blood of healthy males and patients with erectile dysfunction during different functional conditions of the penis. Urol Res. 2000;28(6):364–369. doi: 10.1007/s002400000141. [DOI] [PubMed] [Google Scholar]
- 21.Romitelli F, Santini SA, Chierici E, Pitocco D, Tavazzi B, Amorini AM, Lazzarino G, Di Stasio E. Comparison of nitrite/nitrate concentration in human plasma and serum samples measured by the enzymatic batch Griess assay, ion-pairing HPLC and ion-trap GC-MS: the importance of a correct removal of proteins in the Griess assay. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;851(1-2):257–267. doi: 10.1016/j.jchromb.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 22.Schwedhelm E. Quantification of ADMA: analytical approaches. Vasc Med. 2005;10(Suppl 1):S89–S95. doi: 10.1177/1358836X0501000113. [DOI] [PubMed] [Google Scholar]
- 23.Siroka R, Trefil L, Rajdl D, Racek J, Cibulka R. Asymmetric dimethylarginine - comparison of HPLC and ELISA methods. Chromatogr B Analyt Technol Biomed Life Sci. 2007;850(1-2):586–587. doi: 10.1016/j.jchromb.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 24.Borek C. Dietary antioxidants and human cancer. Integr Cancer Ther. 2004;3:333–341. doi: 10.1177/1534735404270578. [DOI] [PubMed] [Google Scholar]
- 25.Said TM, Kattal N, Sharma RK, Sikka SC, Thomas AJ Jr, Mascha E, Agarwal A. Enhanced chemiluminescence assay vs. colorimetric assay for measurement of the total antioxidant capacity of human seminal plasma. J Androl. 2003;24(5):676–680. doi: 10.1002/j.1939-4640.2003.tb02726.x. [DOI] [PubMed] [Google Scholar]
- 26.Kanda M, Ihara Y, Murata H, Urata Y, Kono T, Yodoi J, Seto S, Yano K, Kondo T. Glutaredoxin modulates platelet-derived growth factor-dependent cell signaling by regulating the redox status of low molecular weight protein-tyrosine phosphatase. J Biol Chem. 2006;281:28518–28528. doi: 10.1074/jbc.M604359200. [DOI] [PubMed] [Google Scholar]
- 27.Sundaresan M, Yu ZX, Ferrans VJ, Irani K, Finkel T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science. 1995;270:296–299. doi: 10.1126/science.270.5234.296. [DOI] [PubMed] [Google Scholar]
- 28.Bedrad K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 29.Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang DX, Gutterman DD. Mitochondrial reactive oxygen species-mediated signaling in endothelial cells. Am J Physiol Heart Circ Physiol. 2007;292:H2023–H2031. doi: 10.1152/ajpheart.01283.2006. [DOI] [PubMed] [Google Scholar]
- 31.Evans JL, Goldfine ID, Maddux BA, Grodsky GM. Oxidative stress and stress-activated signaling pathways: a unifying hypothesis of type 2 diabetes. Endocr Rev. 2002;23:599–622. doi: 10.1210/er.2001-0039. [DOI] [PubMed] [Google Scholar]
- 32.Gutterman DD, Miura H, Liu Y. Redox modulation of vascular tone: focus of potassium channel mechanisms of dilation. Arterioscler Thromb Vasc Biol. 2005;25:671–678. doi: 10.1161/01.ATV.0000158497.09626.3b. [DOI] [PubMed] [Google Scholar]
- 33.Chiarugi P, Pani G, Giannoni E, Taddei L, Colavitti R, Raugei G, Symons M, Borrello S, Galeotti T, Ramponi G. Reactive oxygen species as essential mediators of cell adhesion: the oxidative inhibition of a FAK tyrosine phosphatase is required for cell adhesion. J Cell Biol. 2003;161:933–944. doi: 10.1083/jcb.200211118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grisham MB. Reactive oxygen species in immune responses. Free Radic Biol Med. 2004;36:1479–1480. doi: 10.1016/j.freeradbiomed.2004.03.022. [DOI] [PubMed] [Google Scholar]
- 35.Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 36.Lee HC, Wei YH. Oxidative stress, mitochondrial DNA mutation, and apoptosis in aging. Exp Biol Med. 2007;232:592–606. [PubMed] [Google Scholar]
- 37.Storz P. Reactive oxygen species in tumor progression. Front Biosci. 2005;10:1881–1896. doi: 10.2741/1667. [DOI] [PubMed] [Google Scholar]
- 38.Madamanchi NR, Vendrov A, Runge MS. Oxidative stress and vascular disease. Arterioscler Thromb Vasc Biol. 2005;25:29–38. doi: 10.1161/01.ATV.0000150649.39934.13. [DOI] [PubMed] [Google Scholar]
- 39.Knight JA. Reactive oxygen species and the neurodegenerative disorders. Ann Clin Lab Sci. 1997;27:11–25. [PubMed] [Google Scholar]
- 40.Bashan N, Kovsan J, Kachko L, Ovadia H, Rudich A. Positive and negative regulation of insulin signaling by reactive oxygen and nitrogen species. Physiol Rev. 2009;89:27–71. doi: 10.1152/physrev.00014.2008. [DOI] [PubMed] [Google Scholar]
- 41.Czech MP, Lawrence JC Jr, Lynn WS. Evidence for the involvement of sulfhydryl oxidation in the regulation of fat cell hexose transport by insulin. Proc Natl Acad Sci USA. 1974;71:4173–4177. doi: 10.1073/pnas.71.10.4173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Higaki Y, Mikami T, Fujii N, Hirshman MF, Koyama K, Seino T, Tanaka K, Goodyear LJ. Oxidative stress stimulates skeletal muscle glucose uptake through a phosphatidylinositol-3-kinasedependent pathway. Am J Physiol Endocrinol Metab. 2008;294:E889–E897. doi: 10.1152/ajpendo.00150.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.May JM, de Haen C. The insulin-like effect of hydrogen peroxide on pathways of lipid synthesis in rat adipocytes. J Biol Chem. 1979;254:9017–9021. [PubMed] [Google Scholar]
- 44.Powell DJ, Hajduch E, Kular G, Hundal HS. Ceramide disables 3-phosphoinositide binding to the pleckstrin homology domain of protein kinase B (PKB)/Akt by a PKCzeta-dependent mechanism. Mol Cell Biol. 2003;23:7794–7808. doi: 10.1128/MCB.23.21.7794-7808.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pessler D, Rudich A, Bashan N. Oxidative stress impairs nuclear proteins binding to the insulin responsive element in the GLUT4 promoter. Diabetologia. 2001;44:2156–2164. doi: 10.1007/s001250100024. [DOI] [PubMed] [Google Scholar]
- 46.Kamigaki M, Sakaue S, Tsujino I, Ohira H, Ikeda D, Itoh N, Ishimaru S, Ohtsuka Y, Nishimura M. Oxidative stress provokes atherogenic changes in adipokine gene expression in 3T3-L1 adipocytes. Biochem Biophys Res Commun. 2006;339:624–632. doi: 10.1016/j.bbrc.2005.11.059. [DOI] [PubMed] [Google Scholar]
- 47.Bakker SJ, IJzerman RG, Teerlink T, Westerhoff HV, Gans RO, Heine RJ. Cytosolic triglycerides and oxidative stress in central obesity: the missing link between excessive atherosclerosis, endothelial dysfunction, and beta-cell failure? Atherosclerosis. 2000;148(1):17–21. doi: 10.1016/s0021-9150(99)00329-9. [DOI] [PubMed] [Google Scholar]
- 48.Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, Nakayama O, Makishima M, Matsuda M, Shimomura I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest. 2004;114:1752–1761. doi: 10.1172/JCI21625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Svegliati-Baroni G, Candelaresi C, Saccomanno S, Ferretti G, Bachetti T, Marzioni M, De Minicis S, Nobili L, Salzano R, Omenetti A. et al. A model of insulin resistance and nonalcoholic steatohepatitis in rats: role of peroxisome proliferator-activated receptor-alpha and n-3 polyunsaturated fatty acid treatment on liver injury. Am J Pathol. 2006;169:846–860. doi: 10.2353/ajpath.2006.050953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Atabek ME, Vatansev H, Erkul I. Oxidative stress in childhood obesity. J Pediatr Endocrinol Metab. 2004;17:1063–1068. doi: 10.1515/jpem.2004.17.8.1063. [DOI] [PubMed] [Google Scholar]
- 51.Baynes JW. Role of oxidative stress in development of complications in diabetes. Diabetes. 1991;40:405–412. doi: 10.2337/diab.40.4.405. [DOI] [PubMed] [Google Scholar]
- 52.Block G, Dietrich M, Norkus EP, Morrow JD, Hudes M, Caan B, Packer L. Factors associated with oxidative stress in human populations. Am J Epidemiol. 2002;156:274–285. doi: 10.1093/aje/kwf029. [DOI] [PubMed] [Google Scholar]
- 53.Davi G, Chiarelli F, Santilli F, Pomilio M, Vigneri S, Falco A, Basili S, Ciabattoni G, Patrono C. Enhanced lipid peroxidation and platelet activation in the early phase of type 1 diabetes mellitus: role of interleukin-6 and disease duration. Circulation. 2003;107:3199–3203. doi: 10.1161/01.CIR.0000074205.17807.D0. [DOI] [PubMed] [Google Scholar]
- 54.Davi G, Ciabattoni G, Consoli A, Mezzetti A, Falco A, Santarone S, Pennese E, Vitacolonna E, Bucciarelli T, Costantini F. et al. In vivo formation of 8-iso-prostaglandin F2alpha and platelet activation in diabetes mellitus: effects of improved metabolic control and vitamin E supplementation. Circulation. 1999;99:224–229. doi: 10.1161/01.cir.99.2.224. [DOI] [PubMed] [Google Scholar]
- 55.Monnier L, Mas E, Ginet C, Michel F, Villon L, Cristol JP, Colette C. Activation of oxidative stress by acute glucose fluctuations compared with sustained chronic hyperglycemia in patients with type 2 diabetes. JAMA. 2006;295(14):1681–1687. doi: 10.1001/jama.295.14.1681. [DOI] [PubMed] [Google Scholar]
- 56.Pitocco D, Zaccardi F, Di Stasio E, Romitelli F, Martini F, Scaglione GL, Speranza D, Santini S, Zuppi C, Ghirlanda G. Role of asymmetric-dimethyl-l-arginine (ADMA) and nitrite/nitrate (NOx) in the pathogenesis of oxidative stress in female subjects with uncomplicated type 1 diabetes mellitus. Diabetes Res Clin Pract. 2009;86(3):173–176. doi: 10.1016/j.diabres.2009.09.019. [DOI] [PubMed] [Google Scholar]
- 57.Poitout V, Robertson RP. Glucolipotoxicity: fuel excess and beta-cell dysfunction. Endocr Rev. 2008;29(3):351–366. doi: 10.1210/er.2007-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.El-Assaad W, Buteau J, Peyot ML, Nolan C, Roduit R, Hardy S, Joly E, Dbaibo G, Rosenberg L, Prentki M. Saturated fatty acids synergize with elevated glucose to cause pancreatic beta-cell death. Endocrinology. 2003;144:4154–4163. doi: 10.1210/en.2003-0410. [DOI] [PubMed] [Google Scholar]
- 59.Harmon JS, Gleason CE, Tanaka Y, Poitout V, Robertson RP. Antecedent hyperglycemia, not hyperllipidemia, is associated with increased islet triacylglycerol content and decreased insulin gene mRNA level in Zucker diabetic fatty rats. Diabetes. 2001;50:2481–2486. doi: 10.2337/diabetes.50.11.2481. [DOI] [PubMed] [Google Scholar]
- 60.Robertson RP, Zhang HJ, Pyzdrowski KL, Walseth TF. Preservation of insulin mRNA levels and insulin secretion in HIT cells by avoidance of chronic exposure to high glucose concentrations. J Clin Invest. 1992;90:320–325. doi: 10.1172/JCI115865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kaneto H, Fujii J, Myint T, Miyazawa N, Islam KN, Kawasaki Y, Suzuki K, Makamura M, Tatsumi H, Yamasaki Y, Taniguchi N. Reducing sugars trigger oxidative modification and apoptosis in pancreatic beta-cells by provoking oxidative stress through the glycation reaction. Biochem J. 1996;320:855–863. doi: 10.1042/bj3200855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tajiri Y, Moller C, Grill V. Long term effects of aminoguanidine on insulin release and biosynthesis: evidence that the formation of advanced glycosylation end products inhibits beta-cell function. Endocrinology. 1997;138:273–280. doi: 10.1210/endo.138.1.4851. [DOI] [PubMed] [Google Scholar]
- 63.Tanaka Y, Gleason CE, Tran PO, Harmon JS, Robertson RP. Prevention of glucose toxicity in HIT-T15 cells and Zucker diabetic fatty rats by antioxidants. Proc Natl Acad Sci USA. 1999;96:10857–10862. doi: 10.1073/pnas.96.19.10857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kaneto H, Kajimoto Y, Miyagawa J, Matsuoka T, Fujitani Y, Umayahara Y, Hanafusa T, Matsuzawa Y, Yamasaki Y, Hori M. Beneficial effects of antioxidants in diabetes: possible protection of pancreatic beta-cells against glucose toxicity. Diabetes. 1999;48:2398–2406. doi: 10.2337/diabetes.48.12.2398. [DOI] [PubMed] [Google Scholar]
- 65.Ihara Y, Toyokuni S, Uchida K, Odaka H, Tanaka T, Ikeda H, Hiai H, Seino Y, Yamada Y. Hyperglycemia causes oxidative stress in pancreatic beta-cells of GK rats, a model of type 2 diabetes. Diabetes. 1999;48:927–932. doi: 10.2337/diabetes.48.4.927. [DOI] [PubMed] [Google Scholar]
- 66.Kimoto K, Suzuki K, Kizaki T, Hitomi Y, Ishida H, Katsuta H, Itoh E, Ookawara T, Suzuki K, Honke K, Ohno H. Gliclazide protects pancreatic beta-cells from damage by hydrogen peroxide. Biochem Biophys Res Commun. 2003;303:112–119. doi: 10.1016/s0006-291x(03)00310-3. [DOI] [PubMed] [Google Scholar]
- 67.Sako Y, Grill VE. A 48-hour lipid infusion in the rat timedependently inhibits glucose-induced insulin secretion and beta-cell oxidation through a process likely coupled to fatty acid oxidation. Endocrinology. 1990;127:1580–1589. doi: 10.1210/endo-127-4-1580. [DOI] [PubMed] [Google Scholar]
- 68.Del Guerra S, Lupi R, Marselli L, Masini M, Bugliani M, Sbrana S, Torri S, Pollera M, Boggi U, Mosca F, Del Prato S, Marchetti P. Functional and molecular defects of pancreatic islets in human type 2 diabetes. Diabetes. 2005;54:727–735. doi: 10.2337/diabetes.54.3.727. [DOI] [PubMed] [Google Scholar]
- 69.Robertson R, Zhou H, Zhang T, Harmon JS. Chronic oxidative stress as a mechanism for glucose toxicity of the beta cell in type 2 diabetes. Cell Biochem Biophys. 2007;48(2-3):139–146. doi: 10.1007/s12013-007-0026-5. [DOI] [PubMed] [Google Scholar]
- 70.Cusi K, Kashyap S, Gastaldelli A, Bajaj M, Cersosimo E. Effects on insulin secretion and insulin action of a 48-h reduction of plasma free fatty acids with acipimox in nondiabetic subjects. Am J Physiol Endocrinol Metab. 2007;292(6):E1775–E1781. doi: 10.1152/ajpendo.00624.2006. [DOI] [PubMed] [Google Scholar]
- 71.Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105(9):1135–1143. doi: 10.1161/hc0902.104353. [DOI] [PubMed] [Google Scholar]
- 72.Weber C, Zernecke A, Libby P. The multifaceted contributions of leukocyte subsets to atherosclerosis: lessons from mouse models. Nat Rev Immunol. 2008;8(10):802–815. doi: 10.1038/nri2415. [DOI] [PubMed] [Google Scholar]
- 73.MacNaul KL, Hutchinson NI. Differential expression of iNOS and cNOS mRNA in human vascular smooth muscle cells and endothelial cells under normal and inflammatory conditions. Biochem Biophys Res Commun. 1993;196(3):1330–1334. doi: 10.1006/bbrc.1993.2398. [DOI] [PubMed] [Google Scholar]
- 74.Piconi L, Quagliaro L, Da Ros R, Assaloni R, Giugliano D, Esposito K, Szabo C, Ceriello A. Intermittent high glucose enhances ICAM-1, VCAM-1, E-selectin and interleukin-6 expression in human umbilical endothelial cells in culture: the role of poly(ADP-ribose) polymerase. J Thromb Haemost. 2004;2(8):1453–1459. doi: 10.1111/j.1538-7836.2004.00835.x. [DOI] [PubMed] [Google Scholar]
- 75.Cagliero E, Maiello M, Boeri D, Roy S, Lorenzi M. Increased expression of basement membrane components in human endothelial cells cultured in high glucose. J Clin Invest. 1988;82(2):735–738. doi: 10.1172/JCI113655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Durante W, Sen AK, Sunahara FA. Impairment of endothelium-dependent relaxation in aortae from spontaneously diabetic rats. Br J Pharmacol. 1988;94(2):463–468. doi: 10.1111/j.1476-5381.1988.tb11548.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rosen P, Ballhausen T, Bloch W, Addicks K. Endothelial relaxation is disturbed by oxidative stress in the diabetic rat heart: influence of tocopherol as antioxidant. Diabetologia. 1995;38(10):1157–1168. doi: 10.1007/BF00422364. [DOI] [PubMed] [Google Scholar]
- 78.Obrosova IG, Minchenko AG, Marinescu V, Fathallah L, Kennedy A, Stockert CM, Frank RN, Stevens MJ. Antioxidants attenuate early up regulation of retinal vascular endothelial growth factor in streptozotocin-diabetic rats. Diabetologia. 2001;44(9):1102–1110. doi: 10.1007/s001250100631. [DOI] [PubMed] [Google Scholar]
- 79.Zhao HJ, Wang S, Cheng H, Zhang MZ, Takahashi T, Fogo AB, Breyer MD, Harris RC. Endothelial nitric oxide synthase deficiency produces accelerated nephropathy in diabetic mice. J Am Soc Nephrol. 2006;17(10):2664–2669. doi: 10.1681/ASN.2006070798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pieper GM, Siebeneich W, Moore-Hilton G, Roza AM. Reversal by L-arginine of a dysfunctional arginine/nitric oxide pathway in the endothelium of the genetic diabetic BB rat. Diabetologia. 1997;40(8):910–915. doi: 10.1007/s001250050767. [DOI] [PubMed] [Google Scholar]
- 81.Nagareddy PR, Xia Z, McNeill JH, MacLeod KM. Increased expression of iNOS is associated with endothelial dysfunction and impaired pressor responsiveness in streptozotocin-induced diabetes. Am J Physiol Heart Circ Physiol. 2005;289(5):H2144–H2152. doi: 10.1152/ajpheart.00591.2005. [DOI] [PubMed] [Google Scholar]
- 82.Avogaro A, Fadini GP, Gallo A, Pagnin E, de Kreutzenberg S. Endothelial dysfunction in type 2 diabetes mellitus. Nutr Metab Cardiovasc Dis. 2006;16(Suppl 1):S39–S45. doi: 10.1016/j.numecd.2005.10.015. [DOI] [PubMed] [Google Scholar]
- 83.Ceriello A, Esposito K, Piconi L, Ihnat MA, Thorpe JE, Testa R, Boemi M, Giugliano D. Oscillating glucose is more deleterious to endothelial function and oxidative stress than mean glucose in normal and type 2 diabetic patients. Diabetes. 2008;57(5):1349–1354. doi: 10.2337/db08-0063. [DOI] [PubMed] [Google Scholar]
- 84.Evans JL, Goldfine ID. Alpha-Lipoic acid: a multi-functional antioxidant that improves insulin sensitivity in patients with type 2 diabetes. Diabetes Technol Ther. 2000;2:401–413. doi: 10.1089/15209150050194279. [DOI] [PubMed] [Google Scholar]
- 85.Kopecky J, Clarke G, Enerback S, Spiegelman B, Kozak LP. Expression of the mitochondrial uncoupling protein gene from the aP2 gene promoter prevents genetic obesity. J Clin Invest. 1995;96:2914–2923. doi: 10.1172/JCI118363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li B, Nolte LA, Ju JS, Han DH, Coleman T, Holloszy JO, Semenkovich CF. Skeletal muscle respiratory uncoupling prevents diet-induced obesity and insulin resistance in mice. Nat Med. 2000;6:1115–1120. doi: 10.1038/80450. [DOI] [PubMed] [Google Scholar]
- 87.Paolisso G, Di Maro G, Galzerano D, Cacciapuoti F, Varricchio G, Varricchio M, D'Onofrio F. Pharmacological doses of vitamin E and insulin action in elderly subjects. Am J Clin Nutr. 1994;59:1291–1296. doi: 10.1093/ajcn/59.6.1291. [DOI] [PubMed] [Google Scholar]
- 88.Hirashima O, Kawano H, Motoyama T, Hirai N, Ohgushi M, Kugiyama K, Ogawa H, Yasue H. Improvement of endothelial function and insulin sensitivity with vitamin C in patients with coronary spastic angina: possible role of reactive oxygen species. J Am Coll Cardiol. 2000;35:1860–1866. doi: 10.1016/s0735-1097(00)00616-1. [DOI] [PubMed] [Google Scholar]
- 89.Ceriello A. Hypothesis: the "metabolic memory", the new challenge of diabetes. Diabetes Res Clin Pract. 2009;86(Suppl 1):S2–S6. doi: 10.1016/S0168-8227(09)70002-6. [DOI] [PubMed] [Google Scholar]
- 90.El-Osta A, Brasacchio D, Yao D, Pocai A, Jones PL, Roeder RG, Cooper ME, Brownlee M. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J Exp Med. 2008;205(10):2409–2417. doi: 10.1084/jem.20081188. [DOI] [PMC free article] [PubMed] [Google Scholar]