

SUMMARY
G protein-coupled receptors (GPCRs) are integral to cellular function in nearly all physiologic and many pathologic processes. GPCR signaling represents an intricate balance between receptor activation, inactivation (desensitization, internalization and degradation) and resensitization (recycling and de novo synthesis). Complex formation between phosphorylated GPCRs, arrestins and an ever-increasing number of effector molecules is known to regulate cellular function. Previous studies have demonstrated that, although N-formyl peptide receptor (FPR) internalization occurs in the absence of arrestins, FPR recycling is arrestin-dependent. Furthermore, FPR stimulation in the absence of arrestins leads to receptor accumulation in perinuclear endosomes and apoptosis. In this study, we show that the interaction of GPCR-bound arrestin with Adaptor Protein 2 (AP-2) is a critical anti-apoptotic event. In addition, AP-2 associates with the receptor-arrestin complex in perinuclear endosomes and is required for proper post-endocytic GPCR trafficking. Finally, we observed that depletion of endogenous AP-2 results in the initiation of apoptosis upon stimulation of multiple GPCRs including P2Y purinergic receptors and CXCR2 but not CXCR4. We propose a model in which the abnormal accumulation of internalized GPCR-arrestin complexes in recycling endosomes, resulting from defective arrestin-AP-2 interactions, leads to the specific initiation of aberrant signaling pathways and apoptosis.
Keywords: apoptosis, GPCR, FPR, arrestin, adaptor protein-2, post-endocytic trafficking, recycling
INTRODUCTION
G protein-coupled receptors (GPCR) are involved in virtually every aspect of human physiology including cardiovascular (1), immune (2) and neuronal systems (3). An important feature of GPCR signaling is the cycle of receptor activation, desensitization, internalization, down-regulation/degradation, recycling and resensitization. When these processes are interrupted, they can detrimentally affect cellular migration (4), proliferation and cell adhesion (5). In the case of the v2 vasopressin receptor, intracellular accumulation of the receptor through constitutive desensitization and internalization leads to nephrogenic diabetes insipidus (6).
GPCRs are activated by a myriad of ligands including amino acids and their derivatives, peptides, proteins, ions, lipids and photons. Ligand-bound GPCRs activate heterotrimeric G proteins, resulting in adenylyl cyclase and phospholipase activation, among other effector systems. Activated receptors are subsequently phosphorylated in complex patterns on intracellular domains by serine/threonine G protein-coupled receptor kinases (GRKs), which reduce receptor affinity for G proteins and increase receptor affinity for arrestins (7–10). Arrestin binding sterically blocks receptor-G protein interactions, thereby terminating G protein signaling, while simultaneously providing a scaffold to coordinate the recruitment of internalization machinery, leading to receptor sequestration (11–13). In this model, based primarily on studies of the β2-adrenergic receptor (β2-AR), adaptor protein (AP)-2 and clathrin bind the carboxy terminus of arrestin (14), translocating receptors to clathrin-coated pits for internalization. Arrestin-recruited Src then phosphorylates dynamin, which pinches off the plasma membrane invagination to form an endosome, whereupon arrestin dissociates from the receptor (15). Internalized β2-AR in the Rab5-containing early endosomal compartment is then sorted to lysosomes (via a Rab7-containing compartment) for degradation or to the cell surface (via Rab4-containing endosomes) (16).
This classic pathway of GPCR internalization and post-endocytic trafficking is, however, not observed with all GPCRs. For example, internalization of the m2-muscarinic acetylcholine receptor is not dependent on either arrestin or clathrin (17) and the N-formyl peptide receptor (FPR) internalizes in arrestin knockout cell lines, in which β2-AR internalization is completely inhibited (18, 19). In contrast to GPCRs that follow the rab4-mediated, rapid recycling pathway or the rab7-mediated, lysosomal degradation pathway (20), some GPCRs recycle via perinuclear endosomes, associated with Rab11. Such receptors include the FPR (18), v2 vasopressin receptor (21), somatostatin receptor 3 (22) and CXCR2 (23). In many cases, such GPCRs have been shown to recycle more slowly and form stable complexes with arrestins, resulting in prolonged endosome-associated arrestin (18, 24, 25). Although this stable association has been shown to be critical in prolonged ERK activation in the cytoplasm (25, 26), little is known regarding the role of arrestin in regulating intracellular trafficking of GPCRs, particularly via the perinuclear endosome pathway. Arrestin dissociation has however been implicated as an essential step in the recycling and resensitization of the bradykinin B2 receptor (27). Furthermore, in cells lacking both arrestin-2 and arrestin-3, recycling of the FPR is absent, resulting in receptor accumulation in perinuclear endosomes and suggesting a critical role for arrestin in recycling of the FPR from this compartment (18). Thus, arrestins appear to mediate multiple diverse aspects of GPCR trafficking.
Subsequent studies of FPR trafficking in arrestin-deficient cells revealed that, in addition to a recycling defect, cells also underwent rapid apoptosis via a cytochrome-dependent pathway (28). A requirement for receptor internalization was demonstrated using a signaling-competent, internalization-defective mutant of the FPR. Furthermore, apoptosis was prevented by ERK and other signaling inhibitors. This led to the hypothesis that the accumulation of ligand-activated FPR in recycling endosomes in the absence of arrestins results in aberrant signaling, which initiates apoptotic pathways culminating in caspase activation. Based on the fact that both the trafficking and signaling defects could be rescued by the re-introduction of either arrestin-2 or arrestin-3, we hypothesized that these two apparently distinct defects may be mechanistically linked. Because of the large number of arrestin-interacting proteins, we speculated that the absence of specific interactions might be responsible for the observed defects. Therefore, in this work, we undertook a mapping study to identify the sites within arrestin that when mutated, result in aberrant trafficking and signaling, culminating in apoptosis. Our results reveal novel mechanisms by which AP-2 specifically regulates FPR trafficking from recycling endosomes and prevents apoptosis.
RESULTS
Rescue of FPR-mediated Apoptosis by Arrestin-2 Domains
We have previously described a requirement for arrestins in preventing apoptosis resulting from activation of multiple GPCRs, including the FPR, CXCR2 and Angiotensin II (Type 1A) receptor (28). Arrestins also play a critical role in the proper post-endocytic intracellular trafficking of GPCRs such as the FPR, although FPR internalization per se does not require arrestin. To define the region(s) of arrestin-2 (arr2) responsible for preventing FPR-mediated apoptosis, we generated four constructs producing large fragments of arr2: amino acids 1–186, 177–418, 319–418 and 1–382. The structure of arr2 is composed of two domains of beta-pleated sheets (amino acids 1–172 and 180–352) and a C-terminal “tail” (amino acids 357–418) (29). Arr2-(319–418) contains the “tail” that acts as a dominant-negative construct inhibiting β2-AR internalization (30) whereas arr2-(1–382) is a truncated form of arr2 that has been previously described as constitutively active with respect to receptor binding (7, 8, 31, 32). Mouse embryonic fibroblasts (MEF) deficient in both arr2 and arr3, but stably expressing the FPR (arr2−/−/3−/− FPR MEF), were used to assess arr2 mutants without competition from endogenous arrestins. Arrestin-deficient MEF cells transiently transfected with the four GFP-fused arr2 fragments, wild type arr2 and empty GFP vector were assayed for apoptosis upon FPR stimulation by evaluation of cell rounding, previously shown to correlate absolutely with conventional apoptosis markers such as Annexin V/PI positivity and caspase activation (28). As shown in Fig. 1A, unstimulated cells (expressing GFP, wild type or mutant arrestins) did not exhibit apoptosis. On the contrary, 70–80% of ligand-stimulated cells expressing GFP alone exhibited an apoptotic phenotype, whereas cells expressing wild type arr2 were indistinguishable from unstimulated cells, as previously described (28). Furthermore, none of the four expressed arr2 domains were capable of preventing FPR-mediated apoptosis, although the arr2-(177–418) showed a small reduction in this parameter. Of the four arrestin mutants, arr2-(1–382) is the only domain that demonstrated association with the FPR upon stimulation (determined by confocal fluorescence microscopy, unpublished data). This is consistent with previous data that demonstrated colocalization of arr2-(1–382) with the FPR upon receptor activation (9) and binding to the FPR in reconstitution assays (8, 31). These results demonstrate that the binding of arr2 alone (i.e. arr2-(1–382)) is insufficient to prevent FPR-mediated apoptosis and furthermore that sequences within the carboxy terminus of arr2 (amino acids 383–418) are essential to prevent apoptosis.
Figure 1. Rescue of FPR-mediated apoptosis by arrestin-2 mutants.
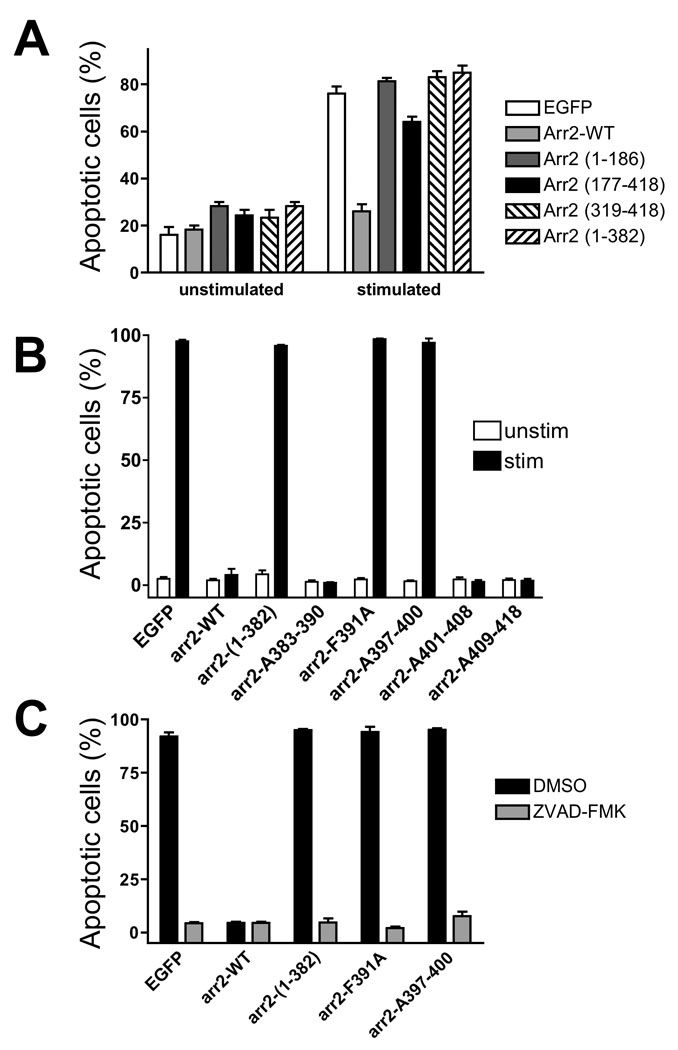
(A) Arr2−/−/3−/− FPR MEF cells were transiently transfected with wild type or arr2 domains fused to GFP. Cells were stimulated for 5 hours in SFM with 10nM fMLF or vehicle at 37°C. GFP-expressing cells were randomly viewed using phase-contrast microscopy and counted for normal morphology or cell rounding, indicative of apoptosis. Only wild type arr2 was capable of preventing FPR-mediated apoptosis. Data are expressed as mean +/− SEM rounded cells/GFP-expressing cell from three independent experiments. (B) Arr2 mutants within the carboxy terminus were assessed for fMLF-induced apoptosis. Arr2−/−/3−/− FPR cells transiently transfected with GFP-fused arrestin mutants were assayed for apoptosis. Cells were stained with PI and 100–300 GFP-expressing cells were viewed and scored for the presence of PI staining. Data are expressed as mean ± SEM PI positive/GFP cell from three independent experiments. (C) Reversal of FPR-mediated apoptosis by the caspase inhibitor ZVAD-FMK. Data are expressed as mean ± SEM from three independent experiments
Rescue of FPR-mediated Apoptosis by Arrestin-2 Tail Mutants
The tail of arr2 contains recognition sites for multiple adapter and signaling proteins, including clathrin (30, 33, 34), AP-2 (14, 29) and ERK (35), which phosphorylates a serine at position 412. Based on the crude mapping results (Fig. 1A), we hypothesized amino acids in arr2 carboxy terminus were responsible for suppressing FPR-mediated apoptosis. To test this hypothesis, we generated five mutants of arr2 using alanine-scanning/replacement mutagenesis of residues 383–390, 391, 397–400, 401–408 and 409–418 (in which the indicated amino acids were mutated to alanine residues). The arr2-F391A mutant has previously been described as reducing AP-2 binding (29) and the arr2-A397–400 mutant (heretowith referred to as arr2-4A) encompasses residues K397, M399 and K400 that have individually been shown to regulate AP-2 binding (14). GFP-fusions of the five arr2 tail mutants were tested for their ability to prevent FPR-mediated apoptosis as determined by propidium iodide staining, which provides a lower basal level of estimated apoptotic cells. Greater than 90% of GFP vector- and arr2-(1–382)-expressing cells were PI positive upon ligand (fMLF) stimulation (Fig. 1B), completely consistent with cell rounding results (Fig. 1A). Expression of wild type arr2 and mutants encompassing residues 383–390, 401–408 and 409–418 rescued cells from FPR-mediated apoptosis; however, the arr2-F391A and −4A mutants were completely ineffective in preventing apoptosis (Fig. 1B). To confirm that positive PI staining was the result of FPR-mediated apoptosis, empty GFP, wild type arr2, -(1–382), -F391A and −4A were treated with the pan-caspase inhibitor, zVAD-FMK, prior to stimulation. Transfected cells that underwent apoptosis in response to FPR stimulation failed to do so in the presence of zVAD-FMK (Fig. 1C), demonstrating that the PI staining in the presence of the arr2 mutants F391A and 4A represents FPR-mediated, caspase-dependent apoptosis consistent with our previous characterization of this apoptotic pathway (28).
Previous results have demonstrated that receptor internalization, which occurs via arrestin-independent mechanisms, is essential for FPR-mediated apoptosis (28). Because AP-2 is required for the internalization of certain GPCRs, we hypothesized that overexpression of arr2 mutants might inhibit FPR internalization, thereby preventing FPR-mediated apoptosis. To examine this, we measured FPR internalization for each arrestin mutant. We determined that the extent of FPR internalization varied between 65–80%, with no significant difference between the extents or rates (data not shown) of internalization for any of the arrestin constructs expressed, demonstrating that rescue of FPR-mediated apoptosis by arrestin mutants is not due to inhibition of FPR internalization. In addition, internalization of the F391A mutant is consistent with our previous studies showing that FPR internalization is independent of clathrin-dependent mechanisms (36).
Arrestin-2 Mutants that Fail to Rescue FPR-mediated Apoptosis Accumulate in Perinuclear Endosomes
As we have previously observed that, in arrestin-deficient cells, FPR-mediated apoptosis is associated with defective intracellular trafficking, this raises the question as to whether the arr2-F391A and arr2-4A mutants allow normal trafficking of the FPR and to what extent they remain associated with the receptor as it traffics intracellularly. Localization of the FPR (visualized using an Alexa633 labeled N-formyl-Leucyl-Leucyl-Phenylalanyl-Leucyl-Tyrosinyl-Lysine ligand (633-6pep)) and arr2 mutants (tagged with monomeric red fluorescent protein, mRFP1 (37)) was determined using confocal fluorescence microscopy. The perinuclear endosomal recycling compartment was visualized using a GFP fusion of Rab11 (which has also been shown to localize to the Golgi compartment (38)). In unstimulated cells, Rab11 is located in a perinuclear region and arr2 (either wild type or mutant) is dispersed throughout the cytoplasm. Activation of the FPR with fluorescent ligand (633-6pep) in the absence of arr2 (empty mRFP1 vector) resulted in accumulation of the FPR in perinuclear endosomes, exhibiting substantial overlap with Rab11 (Fig. 2). Expression of wild type arr2, while also leading to localization of the FPR in perinuclear endosomes, resulted in a greater proportion of non-perinuclear vesicles containing FPR and arrestin, consistent with normal trafficking and recycling of the FPR (18). To control for the possible mistrafficking of arrestin-fluorescent protein fusions (due to potential dimerization for example), we cotransfected arrestin2-GFP with arrestin2-mRFP (note that Aequorea GFP and Discosoma RFP are distinct proteins that do not cross-dimerize, and that mRFP was specifically engineered not to dimerize with itself). Internalized ligand-bearing vesicles showed a perfect concordance between the GFP and mRFP arrestin fusion proteins, indicating normal binding and trafficking interactions (data not shown).
Figure 2. Arrestin-2 mutants incapable of rescuing apoptosis accumulate in recycling endosomes.
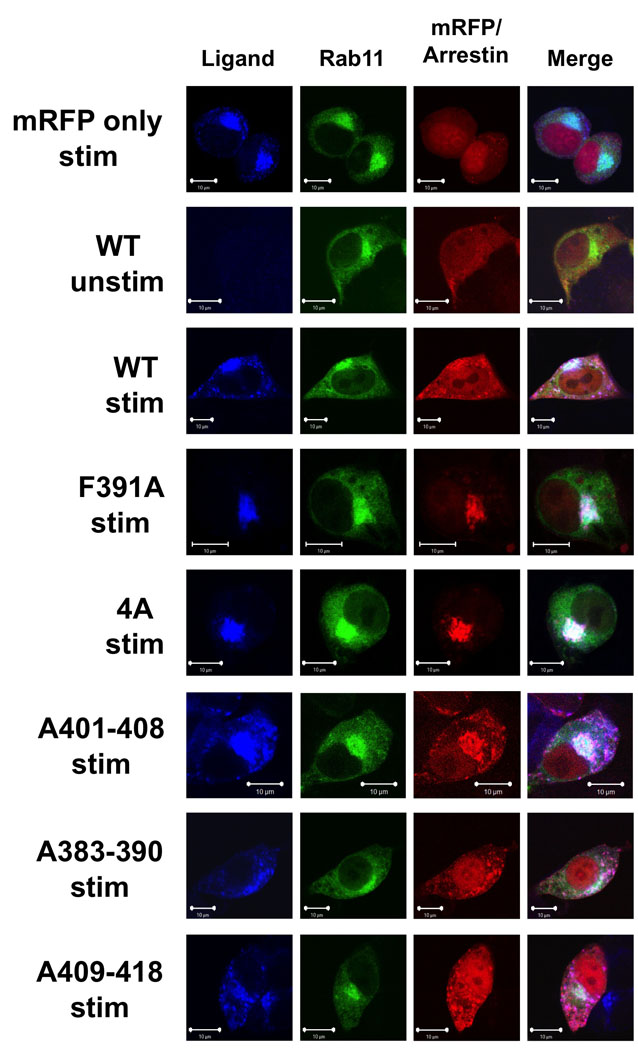
Arr2−/−/3−/− FPR MEF cells were transiently transfected with GFP-fused Rab11 and mRFP-fused arrestins (wild type, arr2-F391A, arr2-4A, arr2-A401-408, arr2-A383-390 and arr2-A409-418). Cells were stimulated with the 633-6pep for 1 hour at 37°C and imaged by confocal fluorescence microscopy. In the absence of arrestin (mRFP only) or the presence of arr2-F391A or arr2-4A, the FPR-ligand complex accumulated extensively in the Rab11 compartment. In cells expressing wild type arr2, arr2-A401-408, arr2-A383-390 and arr2-A409-418, the receptor is also present in cytoplasmic vesicles. Representative images are shown from three independent experiments. Scale bars, 10µm.
The non-perinuclear complexes of FPR-633-6pep and arr2 observed following internalization of receptor represent both newly internalized as well as recycling endosomes (returning to the plasma membrane) based on the two following independent results. First, following a 10 min stimulation of arrestin-deficient cells with 633-6pep, followed by 50 min chase in the absence of ligand, the FPR is not seen in non-perinuclear vesicles, as it is in the presence of arrestins (data not shown). The presence of the FPR in vesicles after 50 min of agonist depletion in wild type cells suggests that this population represents recycling receptor, since no such vesicles are seen in the arrestin-deficient cells. Second, FPR internalization experiments performed with 10nM fMLF (the concentration of 633-6pep used in imaging experiments) in the presence of wild type arr2, reveal that the FPR achieves an equilibrium within 10 min wherein only approximately 25–30% of the total receptor is internalized (compared to 1 µM fMLF, where 75–80% of the FPR is internalized, data not shown). This equilibrium with less total internalized receptor suggests that recycling is taking place under these conditions.
Localization results for the mutant arrestins paralleled their apoptotic phenotype. Expression of both the arr2-F391A and −4A mutants (which did not rescue FPR-mediated apoptosis) resulted in accumulation of the FPR with the associated mutant arrestin in perinuclar endosomes (Fig. 2, note also the more rounded morphology of non-dividing cells undergoing apoptosis, cf. arr2-F391A and −4A vs. WT stimulated). All other mutants (which did rescue the apoptotic phenotype) produced FPR trafficking patterns indistinguishable from wild type arr2 (Fig. 2). These results provide additional support for the link between FPR trafficking defects and the initiation of FPR-mediated apoptosis observed in the absence of arrestins as well as in the presence of the arr2-F391A and −4A mutants.
Previous reports have demonstrated that recycling of the FPR is impaired in the absence of arrestins and that this phenotype is rescued by reconstitution with wild type arr2 (18). In the current study, arr2−/−/3−/− FPR MEF cells transfected with EGFP alone recycled little internalized FPR (<3%) whereas reconstitution with wild type arr2 significantly increased the amount of FPR recycled to 15% of the internalized receptor. Expression of either the F391A or 4A mutants yielded no increase in recycling of the FPR (<3%), confirming that the accumulation of the FPR in recycling endosomes corresponds to a lack of recycling.
Arrestin-2 Mutants that Support FPR-mediated Apoptosis Exhibit Altered Associations with AP-2
The region of arr2 encompassing amino acids 391–400 has previously been associated with altered binding to AP-2 (14, 29). To better understand the relationship between AP-2 association and FPR trafficking, we used confocal fluorescence microscopy to track the interaction of AP-2 with the FPR-arr2 complex. In unstimulated cells, AP-2 is distributed throughout the cytoplasm as well as in puncta at the plasma membrane (Fig. 3). In arr2−/−/3−/− FPR MEF cells, following 60 min of stimulation with 633-6pep in the absence of transfected arrestins, the FPR accumulates in a perinuclear region with Rab11 (Fig. 3, cf. Fig. 2). This observation and the lack of AP-2 association with the FPR in non-perinuclear endosomes at 15 and 30 minutes in the absence of arrestins (Suppl. Fig. 1) are consistent with a requirement for arr2 in the recruitment of AP-2. Upon expression of wild type arr-2 in the arr2−/−/3−/− FPR cells, ligand stimulation results in the accumulation of AP-2 with the 633-6pep-FPR/arr2 complex in the perinuclear region at 60 min. However, AP-2 is not observed to associate with non-perinuclear endosomes containing FPR-arr2 complexes, suggesting that AP-2 is not associated with the complex after exiting perinuclear endosomes. At 15 minutes, almost no AP-2 was seen associated with FPR-arr2 complexes in perinuclear endosomes. It is not until 30 minutes that significant levels of AP-2 are concentrated with the FPR and wild type arr2 in perinuclear endosomes, suggesting that AP-2 does not associate with FPR-arr2 complexes prior to internalization.
Figure 3. Arrestin-2 mutants differentially interact with AP-2.
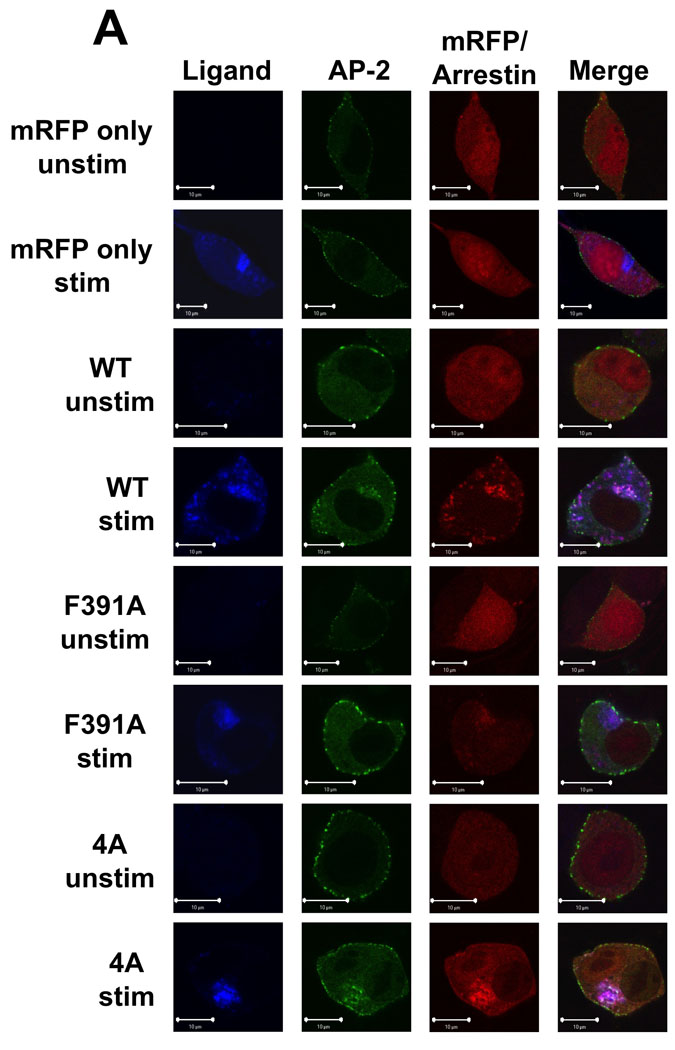
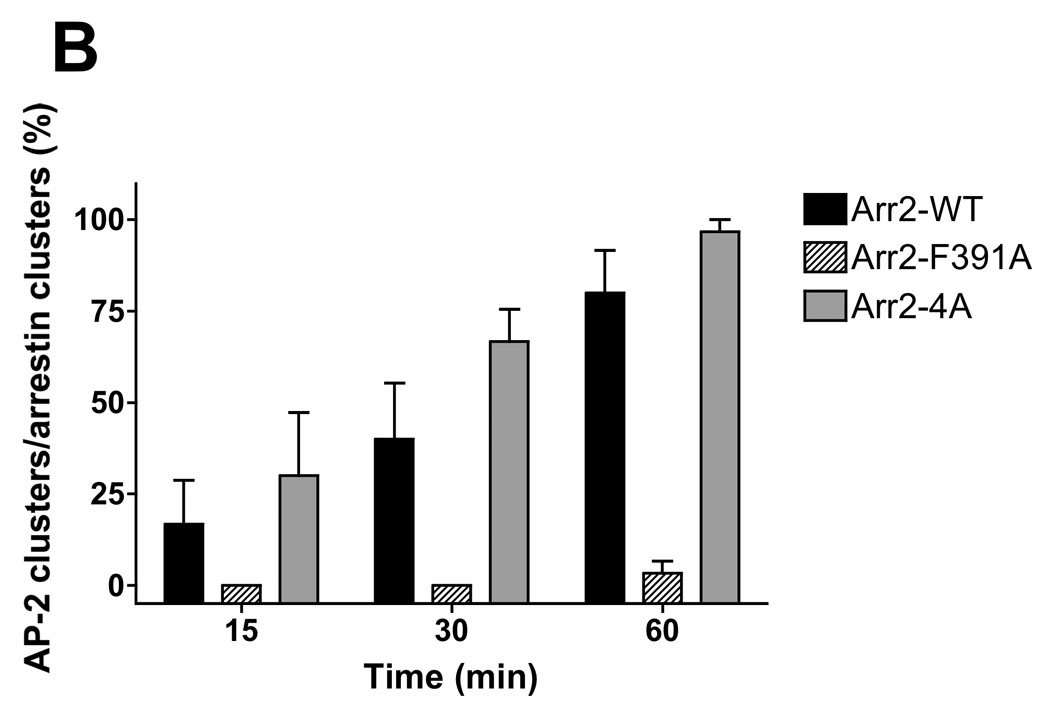
(A) Arr2−/−/3−/− FPR MEF cells were transiently transfected with mRFP-fused arrestins (wild type, arr2-F391A and arr2-4A) and the GFP-fused α subunit of AP-2. Cells were subsequently stimulated with 633-6pep at 37°C for the indicated time and imaged by confocal fluorescence microscopy. In cells expressing wild type arr2 or arr2-4A, following 60 min stimulation, AP-2 is localized with the FPR and arrestin in perinuclear endosomes. However, in cells lacking arrestins or expressing arr2-F391A, no AP-2 is associated with internalized receptor. Representative images are shown from three independent experiments. Scale bars, 10µm. See Suppl. Fig. 1 for images of additional time points. (B) The rate of AP-2 association with arrestin was determined by viewing individual cells and scoring as to whether or not perinuclear AP-2 clusters were present in a given cell containing perinuclear arrestin clusters. Data were normalized to the arr2-4A response at 60 minutes. Cells were counted from three independent experiments and data are expressed as the percentage of cells displaying colocalized AP-2/arrestin clusters, mean +/− SEM.
Expression of the F391A and 4A arrestin mutants yield substantially different results with respect to AP-2 association. In the presence of the arr2-F391A mutant, AP-2 showed no association with the 633-6pep-FPR/arr2-F391A complex at any of the time points (Fig. 3A and Suppl. Fig. 1). This is consistent with published reports that demonstrate decreased AP-2 binding to arr2-F391A (14). However, while we hypothesized that the same would be true of the arr2-4A mutant, this was not the case. Following stimulation for 60 minutes, AP-2 showed strong colocalization with the 633-6pep-FPR/arr2-4A complex, in many cases stronger than that observed with wild type arr2. A similar association of AP-2 with the 633-6pep-FPR/arr2-4A complex was also observed following 15 and 30 minutes stimulation. Our results suggest that the arr2-4A mutant, in contrast to the arr2-F391A mutant, is capable of binding AP-2. The major difference between wild type arr2 and the arr2-4A mutant is that there is little non-perinuclear 633-6pep-FPR/arr2-4A complex (particularly at 60 min) suggesting an accumulation of this particular complex in recycling endosomes.
To examine the rate of association of AP-2 with FPR/arrestin complexes, we measured AP-2 colocalization with FPR/arr2 complexes over time by visually scoring the fraction of cells in which AP-2 was colocalized with perinuclear FPR/arr2 complexes (Fig. 3B). At 15 minutes, 20–30% of cells show AP-2 to be clustered with arrestin when either wild type arr2 or arr2-4A is expressed. The percentage of cells showing colocalization increased at the 30 and 60 minute time points for both wild type arr2 (~35 and ~80%, respectively) and arr2-4A (~60 and ~95%, respectively). The arr2-F391A mutant however showed essentially no AP-2 localization at any time point.
To confirm the above results and validate our use of a GFP-tagged α subunit of AP-2, we examined U937 cells that express both endogenous arrestins (arr2 and arr3) and were stably transfected to express the FPR (U937 FPR). U937 cells are a promonocytic cell line used extensively as a model for FPR function (39, 40). Confocal immunofluorescence microscopy of U937 FPR cells transiently expressing Rab11-GFP and stained with antibodies against endogenous AP-2 confirms our results regarding the subcellular localization of AP-2 (Suppl. Fig. 2A). In unstimulated cells, Rab11-GFP is localized to the perinuclear region while AP-2 shows puncta at the cell membrane with limited cytosolic staining. Upon stimulation of the FPR with Alexa 546-6pep (546-6pep) for 30 min, ligand-FPR complexes colocalize with endogenous AP-2 in perinuclear endosomes. In addition, ligand-receptor complexes exist outside the perinuclear region, but with little AP-2 staining, consistent with the idea that AP-2 associates primarily with the FPR at perinuclear endosomes. Alternatively, these vesicles may represent recycling endosomes emanating from the perinuclear endosomal recycling compartment (see AP-1 results below). To confirm that transient expression of Rab11-GFP did not alter the localization of AP-2, we also stained untransfected U937 cells with antibodies against endogenous AP-2, confirming the perinuclear colocalization of AP-2 with ligand upon FPR stimulation (Suppl. Fig. 3A, cf. Suppl. Figs. 2A at 30 min).
To extend our microscopy results, we immunoprecipitated FLAG-tagged arrestins and assessed AP-2 binding over a time course of FPR activation (Fig. 4A). Western blotting with antibodies directed against the β subunit of AP-2 (the subunit that directly binds wild type arr2 (41)) did not detect any β-adaptin following immunoprecipitation with anti-FLAG antibodies using cell lysates from fMLF-stimulated, arrestin-deficient cells. Upon expression of FLAG-tagged wild type arr2, β-adaptin was detected in the immunoprecipitate as early as 5 minutes after ligand stimulation (quantitated in Fig. 4B), but not in unstimulated cells. Although there was virtually no detectable binding of AP-2 to the F391A mutant (particularly at 30 and 60 min), AP-2 bound to the 4A mutant to a much greater extent than wild type arr2 (~6-fold increase).
Figure 4. Immunoprecipitation of AP-2 with arrestin-2 mutants.
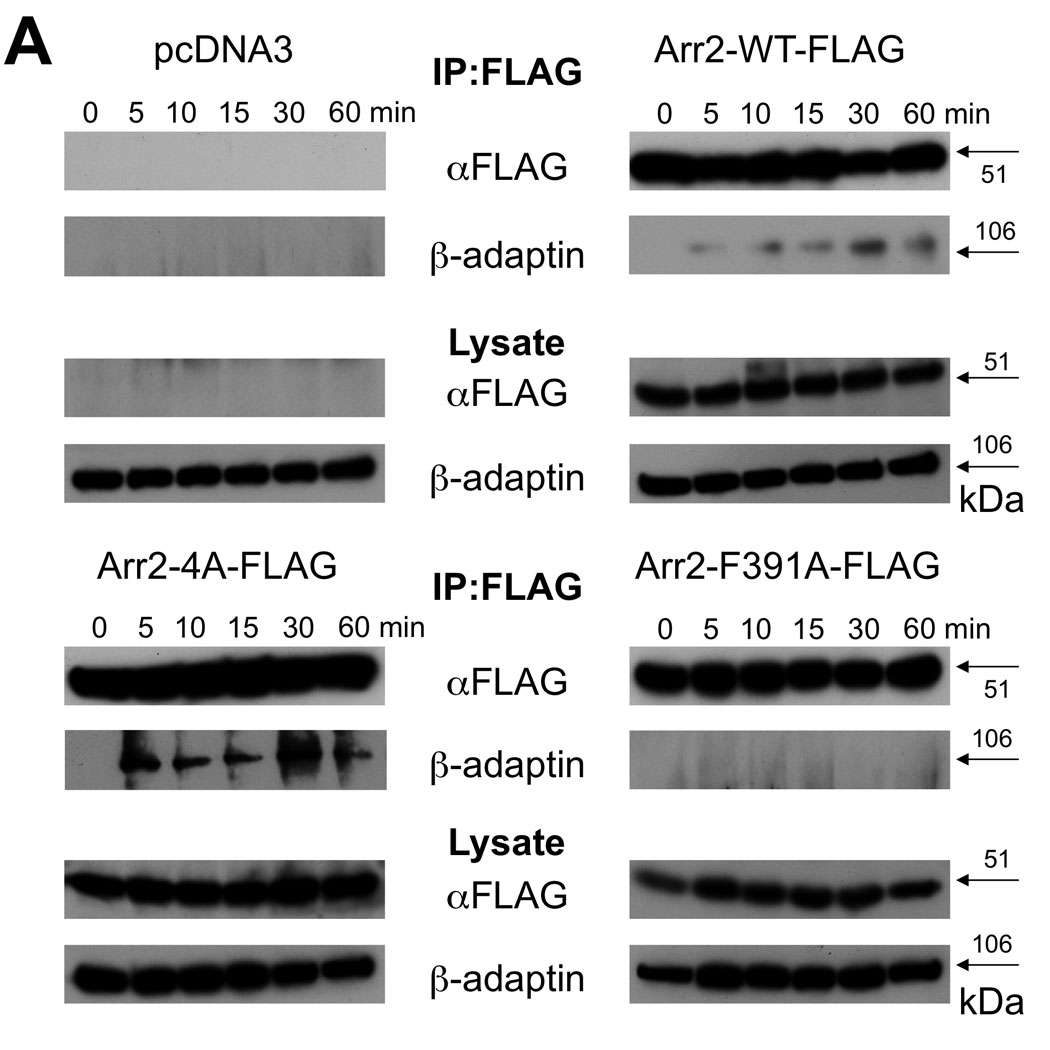
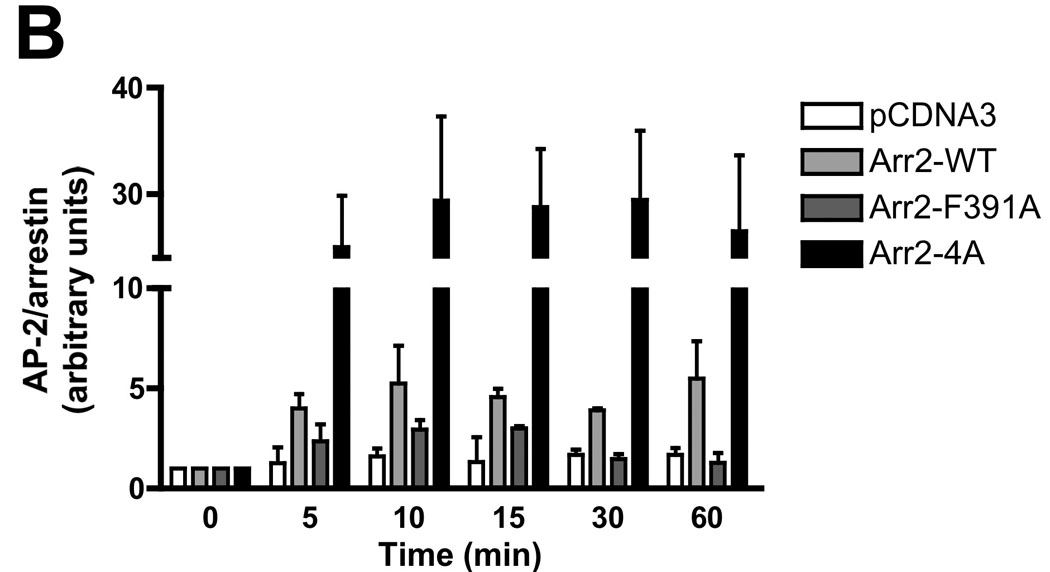
(A) Arr2−/−/3−/− FPR MEF cells were transiently transfected with FLAG-tagged arrestins. Cells were stimulated for the indicated times with 10nM fMLF and lysed. Lysates were immunoprecipitated with anti-FLAG antibodies, resolved by SDS-PAGE and blotted for FLAG-tagged arrestins and the β-adaptin subunit of AP-2. Representative blots are shown. (B) Quantitation of immunoprecipitated proteins by optometric density. Data are expressed as mean +/− SEM of β-adaptin/arrestin intensity ratios and normalized to respective zero time points. Data are from three independent experiments.
The FPR Displays Differential Arrestin-dependent Trafficking Patterns with AP-1
AP-1 has been localized to the TGN and endosomes of cells and is required for trafficking of proteins to and from these compartments (42). In addition, the β subunit of AP-1 shows significant overall homology to the β subunit of AP-2 with amino acids shown to be necessary for arr-2 binding to β2-adaptin (i.e. E849, Y888 and E902) (43) being absolutely conserved in β1-adaptin (44). To determine whether AP-1 might also play a role in FPR trafficking, we examined the localization of the FPR, arrestins and AP-1 in arr2−/−/3−/− FPR MEF cells using confocal fluorescence microscopy. In unstimulated cells, AP-1 was localized primarily in a perinuclear region, but was also present in the cytoplasm. When the FPR was stimulated in arrestin-deficient cells, receptor accumulated in perinuclear endosomes overlapping substantially with AP-1 (Fig. 5A). Upon stimulation of cells expressing wild type arr2, the FPR-arr2 complexes were also colocalized with AP-1 in the perinuclear region. However, arr2 was also observed outside this region in cytoplasmic vesicles in association with the FPR and AP-1. This suggests that upon internalization, the FPR traffics to perinuclear endosomes where it associates with AP-1, which may escort receptor back to the cell surface. The fact that arrestin appears to mediate these late trafficking events indicates a role for arrestin in receptor trafficking at a much later stage than previously thought. In contrast to wild type arr2, the F391A and 4A mutants both accumulated with the FPR in AP-1-positive endosomes in the perinuclear region with no receptor, arrestin or AP-1 observed outside this cellular compartment, consistent with the lack of recycling. To further assess AP-1 trafficking with FPR/arr2 complexes, we quantitated the number of cells that showed AP-1 puncta outside of the perinuclear region (Fig. 5B). Only when wild type arr2 is present is AP-1 observed on vesicles outside the perinuclear region following FPR activation. Finally, pulse-chase experiments (10 min pulse with 633-6pep, 50 min chase) using arr2−/−/3−/− FPR cells show that AP-1 is colocalized with 633-6pep-FPR-wild type arr2 in cytoplasmic vesicles following the 50 min chase, but not in early endosomes immediately following internalization (10 min pulse with no chase, Suppl. Fig. 4).
Figure 5. FPR-arrestin-2 trafficking and binding with AP-1.
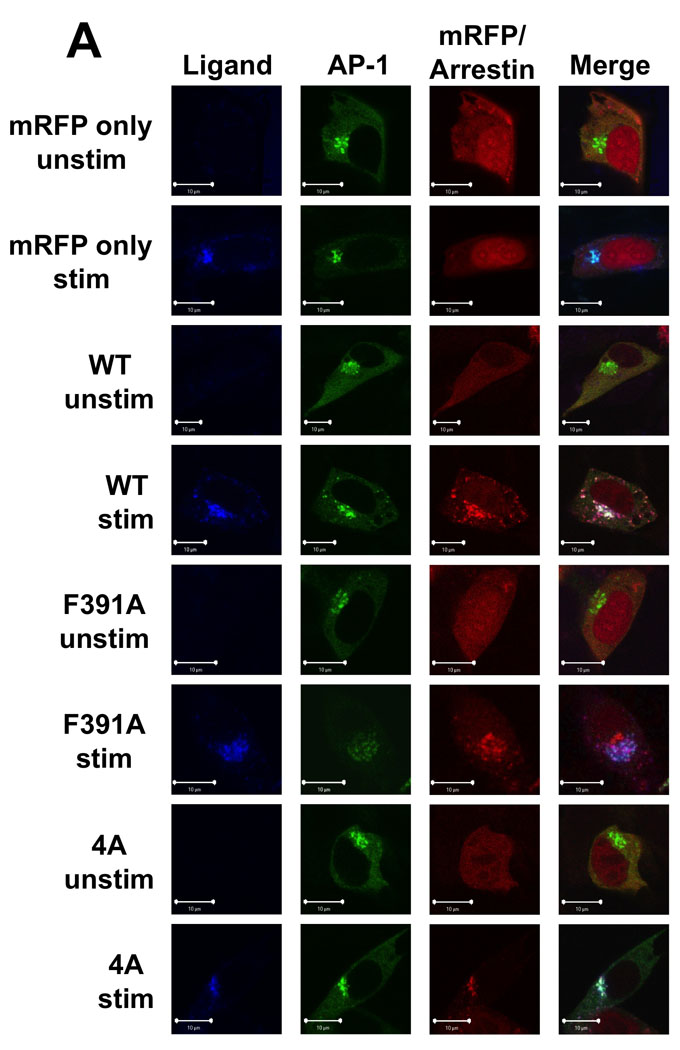
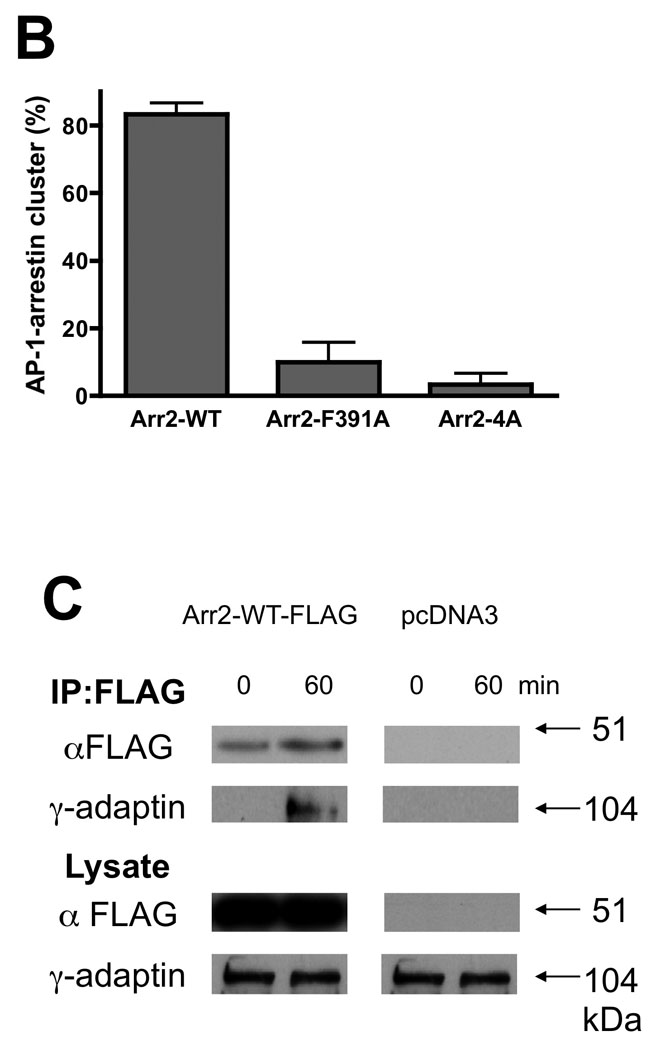
(A) Arr2−/−/3−/− FPR MEF cells were transiently transfected with mRFP-fused arrestins (wild type, arr2-F391A and arr2-4A) and the GFP-fused γ subunit of AP-1. Cells were then stimulated with 10nM 633-6pep ligand, fixed and viewed by confocal fluorescence microscopy. In all cases, after 60min stimulation, FPR-arrestin complexes were associated with AP-1. Note that only with the stimulated cells expressing wild type arr2 are cytoplasmic vesicles containing AP-1, FPR and arrestin observed outside the perinuclear recycling compartment. Representative images are shown from three independent experiments. Scale bars, 10µm. (B) AP-1 differentially associates with non-perinuclear arrestin clusters. Association was determined by viewing cells with non-perinuclear arrestin clusters and scoring whether or not AP-1 clusters colocalized. Data are expressed as mean +/− SEM for >30 cells from three independent experiments. (C) Arr2 and AP-1 interact in response to FPR activation. Arr2−/−/3−/− FPR cells were transiently transfected with arr2-WT-FLAG or empty vector and immunoprecipitated with anti-FLAG antibodies after FPR activation. Protein was resolved by SDS-PAGE and blotted as indicated. Representative blots from three independent experiments are shown.
In order to confirm that the GFP-fused β subunit of AP-1 accurately reflects endogenous AP-1 trafficking, we again used confocal immunofluorescence microscopy of U937 FPR cells transiently transfected only with Rab11-GFP (Suppl. Figs. 2B and 3A). In unstimulated cells, AP-1 is predominantly colocalized with perinuclear Rab11 with minimal non-perinuclear staining. Upon stimulation of the FPR with 546-6pep, ligand-receptor complexes colocalize with Rab11 and AP-1 (as well as AP-2, Suppl. Fig. 3B) in the perinuclear region. In addition, ligand-receptor complexes found in non-perinuclear endosomes (lacking Rab11) also colocalize significantly with AP-1 consistent with recycling of the FPR to the plasma membrane.
Based on the colocalization of the FPR, arrestin and AP-1 upon receptor activation, we sought to determine whether arrestin also forms a complex with AP-1. Arr2−/−/3−/− FPR cells were transiently transfected with arr2-WT-FLAG and stimulated prior to immunoprecipitation with anti-FLAG antibodies. AP-1 was detected in the immunoprecipitates by blotting for the γ subunit of AP-1. In the absence of arrestins, AP-1 was not detected in anti-FLAG immunoprecipitates (Fig. 5C). However, in the presence of arr2-WT-FLAG, the γ subunit of AP-1 was detected in the anti-FLAG immunoprecipitates in a stimulation-dependent manner, indicating an association between the FPR-arrestin complex and AP-1.
AP-2 Regulates FPR Recycling and GPCR-mediated Apoptosis
In order to determine explicitly whether AP-2 and AP-1 regulate FPR recycling and GPCR-mediated apoptosis in general, we used siRNAs to knockdown the µ1A and µ2 subunits of AP-1 and AP-2, respectively in U937 FPR cells (Fig. 6A). Knockdown of µ1A and µ2 subunit expression was >90% in both cases. In addition, expression of the β subunit of AP-2 and γ subunit of AP-1 was decreased as well, but not to the same extent (~70% and ~50% respectively, unpublished data). Although the rate of FPR internalization was not affected by knockdown of either AP-1 or AP-2 (half time for internalization: control, 1.4+/−0.2 min; AP-1, 1.5+/−0.2 min; AP-2, 1.6+/−0.2 min), the extent of FPR internalization was modestly reduced upon AP-2 knockdown (control, 75+/−2%; AP-1, 82+/−3%; AP-2, 57+/−5%). In contrast, knockdown of AP-2 produced a significant decrease of approximately 40% in FPR recycling, whereas knockdown of AP-1 produced no effect (Fig. 6B). Visualization of FPR trafficking in AP-2 siRNA-treated U937 cells also demonstrated a partial accumulation of the FPR in perinuclear endosomes, whereas AP-1 siRNA treatment yielded a wild type intracellular FPR distribution (Suppl. Fig. 3C). To determine whether the observed reduction in FPR recycling resulted in an alteration in cell survival, U937 FPR cells treated with siRNA for AP-1 or AP-2 were stimulated with ligand and evaluated for apoptosis. Only stimulated cells treated with siRNA directed against AP-2 showed a profound apoptotic response with over 90% of the cells undergoing apoptosis (Fig. 6C). Pretreatment with the caspase inhibitor ZVAD-FMK completely blocked the ligand-dependent apoptosis. Finally, we asked whether this induction of apoptosis was unique to the FPR or occurred with other GPCRs. To test this, U937 cells stably transfected with either CXCR2 or CXCR4 were treated with AP-2 siRNA and stimulated with the appropriate ligand. As a control for an endogenously expressed receptor, we also stimulated the U937 FPR cells with ATP, which is known to activate P2Y purinergic receptors on these cells (45). Ligand dependent apoptosis was observed in both the CXCR2-expressing cells as well as the U937 FPR cells stimulated with either fMLF or ATP (Fig. 6D). However, ligand stimulation of CXCR4-expressing cells had no effect. These results mirror our previous findings in arr2−/−/3−/− FPR MEF cells, where stimulation of FPR and CXCR2 but not CXCR4 induced apoptosis (28). These results demonstrate that many but not all GPCRs are capable of initiating apoptosis in the absence of a functional arrestin-AP-2 interaction.
Figure 6. GPCR-mediated apoptosis and recycling upon adaptor protein depletion.
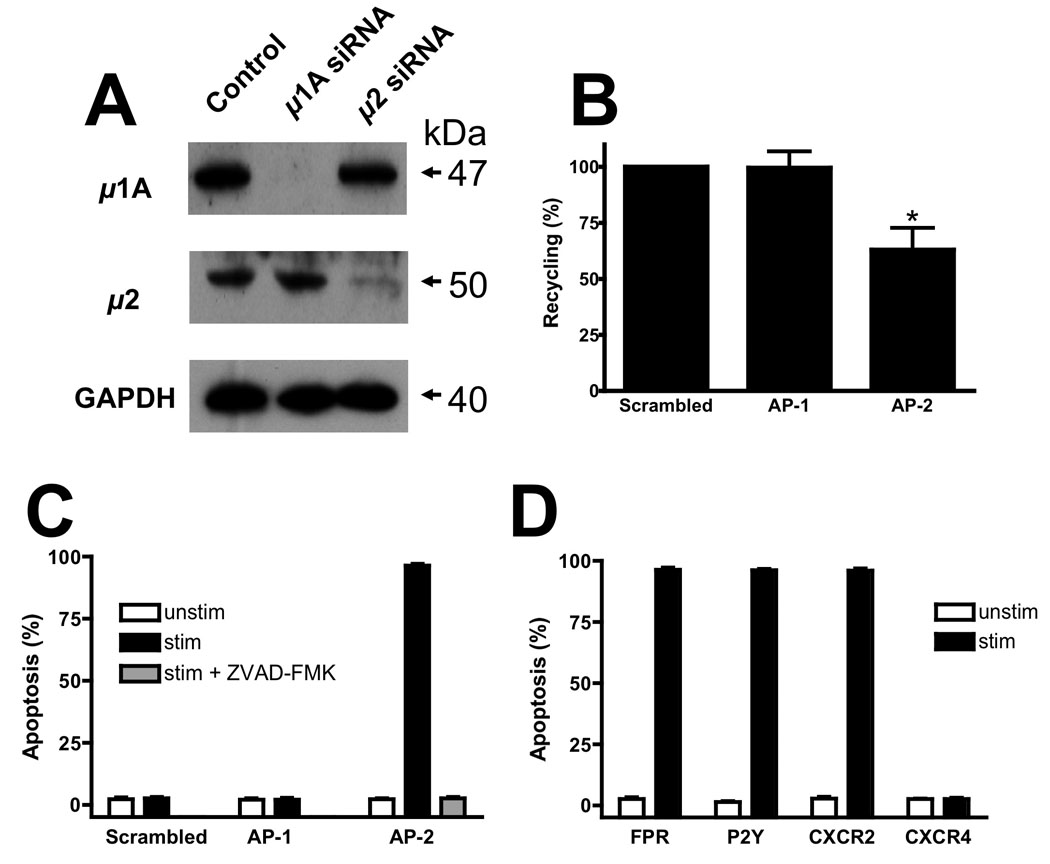
(A) U937 FPR cells were electroporated with control, AP-1 or AP-2 siRNA. A fraction of the electroporated cells were harvested before experimentation and lysed for evaluation of knockdown efficiency. Lysates were resolved by SDS-PAGE and blotted with the indicated antibodies. Representative blots are shown from three experiments. (B) U937 FPR cells were treated with the indicated siRNA and assayed for FPR recycling. FPR recycling was normalized to that of the control siRNA transfected cells. Control cells internalized 75% of the surface FPR and recycled ~50% of the internalized receptor. Data are expressed as mean+/− SEM FPR recycling/internalization and are representative of three independent experiments. *p<0.05 (C) U937 FPR cells were treated with the indicated siRNA and assayed for FPR-mediated apoptosis using PI staining. Cellular depletion of AP-2 using siRNA against the µ2-subunit resulted in significant ligand-dependent apoptosis. Depletion of AP-1 or use of control siRNA had no effect. Caspase inhibition with ZVAD-FMK inhibited FPR-mediated apoptosis in AP-2 depleted cells. Data are expressed as mean+/− SEM and are representative of three independent experiments. (D) U937 cells stably transfected with the FPR, CXCR2 or CXCR4 were transfected with siRNA against AP-2 and assayed for ligand-dependent apoptosis (using fMLF, IL-8 and SDF-1, respectively). FPR-expressing cells were also stimulated with ATP to activate endogenous P2Y purinergic receptors. Data are expressed as mean+/− SEM and are representative of three independent experiments.
DISCUSSION
In summary, we have demonstrated that the ligand-dependent accumulation of GPCR-arrestin complexes in perinuclear recycling endosomes leads to the rapid initiation of apoptosis. Furthermore, receptor accumulation and apoptosis were induced by the disruption of the arrestin-AP-2 interaction, either by a single point mutation in arr2 (F391A) or by depleting AP-2 levels. It is interesting to note that stabilization of the arr2-AP-2 complex in the 4A mutant also affected receptor trafficking and induced apoptosis, suggesting that cyclic binding and release of AP-2 is necessary for proper trafficking. This is also the first report to describe a role for AP-2 in the recycling of internalized GPCRs. Although depletion of AP-1, best known for mediating vesicle traffic between the Golgi compartment and endosomes, did not prevent receptor recycling, it was found to be associated with receptor-arrestin complexes in perinuclear endosomes and non-perinuclear endosomes emanating from the recycling compartment, suggesting a possible role in GPCR re-expression. This observation is consistent with reported roles for AP-1 in the formation of recycling vesicles from internalized receptors (46–48). As signaling inhibitors have been shown to inhibit FPR-mediated apoptosis, our results suggest that inhibition of proper FPR trafficking also alters the spatial control of GPCR signaling complexes leading to the initiation of apoptosis within the cell. This is supported by results that show spatial control of GPCRs can induce or limit their potential to initiate cellular signaling pathways (49).
The F391A arrestin mutant was previously shown to exhibit decreased binding to the β subunit of AP-2 through in vitro binding assays while its overexpression in cells inhibited internalization of the β2-AR (29). While the 4A mutant has not been previously reported, the mutation and characterization of individual amino acids within this site (K397, M399 and K400) have been described (14). These individual mutants showed similar binding properties with the β subunit of AP-2 and produced similar effects on β2-AR internalization as the F391A mutant. While the F391A mutant did not significantly colocalize or co-immunoprecipitate with AP-2, the 4A mutant interacted strongly with AP-2 upon FPR activation. The reason for increased AP-2 binding to an arrestin mutant whose component mutations show decreased binding is unclear, but may be due to conformational changes within the protein that alter the binding properties of this specific motif or secondary binding sites. Similar to our arrestin 4A mutant, arrestin mutants I386A and V387A show 5-fold enhanced binding to AP-2 whereas an F388A substitution prevents binding (50). Furthermore, the AP-2 mutant R879A shows enhanced (10-fold) binding to arr2 (51). Together, these results demonstrate that this interaction can be both positively and negatively modulated by mutations in both arrestin and AP-2. The inhibitory effect of increased AP-2 binding by the 4A mutant suggests that appropriate cycling of AP-2 with arrestin (association followed by dissociation) is required for the exit of the FPR from recycling endosomes. This idea is supported by evidence demonstrating Src activity as a necessary component for AP-2/arrestin dissociation and internalization of the β2-AR (41).
GPCR-arrestin interactions have also been shown to initiate apoptosis in the Drosophila eye, where alterations in the processing of rhodopsin, through diverse molecular mechanisms such as deletion of one of the two visual arrestins, leads to light-dependent retinal degeneration (52). Furthermore, loss-of-function mutants in a Ca2+-dependent serine/threonine phosphatase (rdgC) as well as an eye-specific phospholipase (norpA), which activates rdgC, result in the formation of stable internalized rhodopsin-arrestin complexes that initiate apoptosis in photoreceptor cells resulting in retinal degeneration (53, 54). Interestingly, in the norpA Drosophila mutant, disruption of arrestin/AP-2 binding through the introduction of arrestin point mutants rescued apoptosis by preventing rhodopsin internalization (52). Support for the conservation of this visual pathway has recently been reported in mice. A mutant opsin (K296E), associated with autosomal dominant retinitis pigmentosa, is hyperphosphorylated and its excessive association with arrestin results in the mislocation and accumulation of the complex to the inner segments (55). These results bear many similarities to our results in non-visual systems. First of all, we have previously reported that ligand-dependent GPCR-mediated apoptosis in arrestin-deficient cells requires receptor internalization (28). Second, in this study, we demonstrated that the accumulation of GPCR-arrestin complexes in recycling endosomes initiates rapid apoptosis. However, in contrast to the Drosophila system, AP-2 plays a critical role in mediating the appropriate trafficking of GPCRs out of recycling endosomes, thus preventing the initiation of apoptosis. Thus, our results demonstrate that arrestins likely play a critical role in the maintenance of cell viability in all eukaryotic cells.
Based on the results of this study, we propose a new model of FPR internalization and recycling. In this model, following ligand binding the FPR can internalize in an arrestin-independent manner. Before, during or after internalization, the FPR binds to arrestin, resulting in the generation of FPR/arrestin complexes in early endosomes. At some point during its trafficking to perinuclear recycling compartment, the FPR-arrestin complex recruits AP-2. Within or as it exits this compartment, the FPR/arrestin complex releases AP-2, whereupon AP-1 associates with the FPR/arrestin complex. Along the path to the cell surface the complex dissociates and the FPR is dephosphorylated. Finally, the FPR completes its return to the cell surface in a resensitized form ready to continue signaling.
In this report, we have described a novel mechanism for GPCR post-endocytic trafficking and recycling. This is the first report that identifies a definitive role for arrestin in the late stages of GPCR trafficking as compared to other GPCRs, where arrestins are involved in the earliest events of GPCR trafficking (i.e. internalization). In addition, the observation that FPR-mediated apoptosis occurs under conditions where arrestin mutants are bound to the FPR demonstrates that the receptor-initiated apoptotic signaling in arrestin-deficient cells is not purely a result of receptor activation in the absence of arrestins and furthermore confirms that receptor-arrestin accumulation in recycling endosomes results in aberrant signaling that leads to apoptosis. With this report of novel roles for arrestins and adaptor proteins in the trafficking and signaling of GPCRs, new avenues for the targeting of GPCR function are presented that may lead to the development of therapeutic interventions for disease states.
MATERIALS AND METHODS
Reagents, Plasmids and Mutagenesis
All reagents are from Sigma unless otherwise noted. With the exception of the arr2-F391A mutant (a gift from Jeffrey Benovic), regions of arr2 were mutated by site-directed mutagenesis and cloned into EGFP-N1 vector or mRFP1 vector using standard subcloning procedures and HindIII/ApaI restriction sites. These constructs were amplified using PCR with primers that created HindIII/ApaI restriction sites and subcloned as describe above. Arr2-WT-FLAG was created by digesting Arr2-WT-GFP at the ApaI/NotI restriction sites to remove GFP and inserting a linker that contained the FLAG sequence. Arr2-F391A- and arr2-4A–FLAG were constructed by digesting with HindIII/ApaI sites and subcloning the ~1300bp fragment into the HindIII/ApaI restriction sites of Arr2-WT-FLAG. All mutants were confirmed by DNA sequencing. Rab11-GFP was a gift from Angela Wandinger-Ness. GFP-fused α subunit of AP-2 and GFP-fused γ subunit of AP-1 were gifts from Lois Greene (56).
Cell culture and Transfection
Arr2−/−/3−/− FPR cells were grown in DMEM with 10% fetal bovine serum, 100 units/mL penicillin and 100units/mL streptomycin at 37°C and 5% CO2. U937 cells were grown in RPMI with 10% fetal bovine serum, 100 units/mL penicillin and 100units/mL streptomycin at 37°C and 5% CO2. Transient transfections of mouse embryonic fibroblasts were performed with Lipofectamine 2000 according to manufacturer’s instructions. siRNA transfection of U937 FPR cells was performed using siPORT Electroporation Buffer (Ambion) with a Genepulser Xcell (Bio Rad) according to manufacturer instructions. Cells were transfected with 20µg of siRNA twice at 72 hour intervals and assayed 72 hours after the second transfection. Cell survival was measured using Trypan Blue and was >95% for all transfections. siRNAs used for depletion of µ1A (AP-1) and µ2 (AP-2) were as previously described (57). mRNA gene target sequences for siRNA design were as follows: µ1A—GGCAUCAAGUAUCGGAAGA; µ2—GUGGAUGCCUUUCGGGUCA. A nonfunctional siRNA (Ambion) was used as a control.
Apoptosis
For cell rounding assays, arr2−/−/3−/− FPR cells transiently transfected with GFP-fused arrestins were plated on 12mm-glass coverslips, incubated overnight and serum-starved for 30 minutes. Cells were incubated in serum-free medium (SFM) or were stimulated with 10nM fMLF in SFM 5 hours at 37°C. Cells were washed twice with PBS, fixed with 2% paraformaldehyde and mounted using Vectashield. Slides were viewed in a blinded manner by phase-contrast microscopy and random fields were evaluated (at least five) until 100–300 GFP-expressing cells were assayed. GFP-expressing cells were counted “positive” for cell death if they rounded (spherical and refractile cells with no extensions). Data are expressed as a percentage of rounded/GFP-expressing cells. For propidium iodide iodide staining, arr2−/−/3−/− FPR cells were transfected as above. Stimulated MEF and U937 cells were stained with 100ng/mL propidium iodide in SFM at room temperature for 5–10 minutes, washed twice with PBS, fixed with 2% paraformaldehyde and mounted using Vectashield. Slides were viewed in a blinded manner by fluorescence microscopy and random fields were evaluated (at least five) until 100–300 cells (GFP-expressing for MEFs) were evaluated. Cells were counted “positive” for cell death if they were stained with propidium iodide. Data are expressed as a percentage of PI-positive/GFP-expressing cells for MEFs and percentage of PI-positive cells for U937 cells.
FPR Internalization
Arr2−/−/3−/− FPR cells transiently transfected with GFP-fused arrestins were harvested by trypsinization. U937 FPR cells were electroporated with siRNAs and harvested by centrifugation. Cells were resuspended in SFM and stimulated for 2, 5, 10, 20 and 30 minutes with 1µM fMLF. Cells were then washed extensively with cold SFM to remove excess unlabelled ligand. Remaining receptors on the cell surface were labeled with 10nM 633-6pep. Cells were then assayed by flow cytometry using a Becton-Dickinson FACSCalibur. Cells (100,000) were gated for live cells using forward and side scatter parameters. GFP-fused arrestin mutant-expressing cells were subsequently gated using FL-1 and the mean channel fluorescence (MCF) was determined in FL-4 to monitor cell surface expression of the FPR. Non-specific binding was determined by labeling arr2−/−/3−/− cells not expressing the FPR with 633-6pep and assaying as described. Non-specific binding was subtracted before further calculations. MCF from unstimulated cells represented 100% FPR cell surface expression. Cell surface expression from stimulated cells was calculated by dividing the MCF following treatment by the MCF from unstimulated cells. Internalization data were then plotted using GraphPad Prism to calculate maximum internalization extents using a singe exponential decay. Statistical analysis was performed using Student’s t-test.
Confocal Fluorescence Microscopy
Arr2−/−/3−/− FPR cells or U937 cells were transiently transfected with mRFP-fused arrestins, GFP-fused Rab11, GFP-fused α subunit of AP-2 or GFP-fused γ subunit of AP-1. Arr2−/−/3−/− FPR cells were plated on 25mm-glass coverslips and grown overnight. U937 FPR cells were harvested by centrifugation. Cells were serum-starved for 30 minutes and stimulated with 10 nM 633- or 546-6pep in SFM for indicated time at 37°C. Coverslips or harvested cells were rinsed with PBS, fixed with 2% paraformaldehyde at room temperature and mounted with Vectashield. For experiments using antibody staining, after fixation cells were incubated in 0.02% Saponin/3% BSA for 15–20 minutes at room temperature. Cells were washed and incubated in 1:100 primary antibody (100/3 for γ1-adaptin antibody or AP-6 for α-adaptin (Affinity Bioreagents)) in 3% NGS for 30 minutes at room temperature. After further washing, cells were incubated in 1:200 secondary antibody (Cy5-conjugated Donkey Anti-Mouse (Jackson Immunoresearch)) in 3% NGS for 30 minutes at room temperature. Cells were then washed and mounted using Vectashield. Fluorescence images were acquired using a Zeiss LSM 510 inverted laser scanning microscope equipped with He-Ne and Kr-Ar lasers. To assess the extent of colocalization, cells with arrestin clusters were viewed and scored for the presence of any corresponding AP-2 or AP-1 clusters, respectively. Data were expressed as percentage of cells (mean +/− SEM) with colocalized clusters from >25 cells/experiment. Values were normalized to the wild type arrestin response at 60 minutes.
Cell Lysis and Immunoprecipitation
Arr2−/−/3−/− FPR MEF cells transiently transfected with FLAG-tagged arrestins were grown to confluence. Cells were serum-starved for 30 min and stimulated for the designated times with 10nM fMLF in SFM at 37°C. Medium was removed and cold co-IP lysis buffer (1 mL 1% v/v TX-100, 150mM NaCl, 10mM Tris-HCl pH 7.4 supplemented with protease inhibitor cocktail (Calbiochem)) was added immediately. Lysates were collected, incubated on ice for 30 min, and centrifuged at maximum speed for 30 min at 4°C. An aliquot from each tube was set aside for determining pre-immunoprecipitation levels of proteins by Western blot. For immunoprecipitations, 25µL of protein A Sepharose (Pierce) was washed three times with co-IP lysis buffer. Beads were rotated for at least one hour at 4°C in 250µL of co-IP lysis buffer with 1:1000 M2 Anti-FLAG antibody. Beads were washed with co-IP lysis buffer and lysates were added and rotated overnight at 4°C. The following day, beads were washed again with co-IP lysis buffer, 40µL of 2X Sample Buffer was added and immunoprecipitated proteins released by boiling for 5 minutes.
Western Blotting
Lysates or immunoprecipitates were resolved with SDS-PAGE. Proteins were transferred to PVDF membranes and blocked for 1 hour with 5% dry milk in TBS-T. Blotting was carried out using 1:1000 dilutions of biotinylated M2 mouse anti-FLAG antibody, M2 mouse Anti-FLAG, mouse anti-β-adaptin antibody (BD Biosciences), mouse 100/3 to γ1-adaptin antibody, rabbit RY/1 to µ1A (gift from L. Traub), rabbit R11–29 to µ2 ((58), gift from J. Bonifacino) and 1:4000 mouse anti-GAPDH antibody (Chemicon) in 5% dry milk in TBS-T overnight at 4°C. Blots were washed with TBS-T and incubated with 1:2500 HRP-streptavidin (in 5% BSA in TBS-T), 1:2500 HRP rabbit anti-mouse antibody or 1:2500 HRP goat anti-rabbit antibody (in 5% Dry Milk in TBS-T) at room temperature for 1 or 3 hours, respectively. Bands were visualized using SuperSignal West Pico Chemiluminescent Substrate (Pierce). Densitometry was performed using Quantity One (BioRad) and ratios expressed as mean ratio +/− SEM adaptin/arrestin immunoprecipitated and normalized to zero time points for each vector expressed.
FPR Recycling
Arr2−/−/3−/− FPR cells transiently transfected with GFP-fused arrestins or U937 FPR cells transfected with siRNA were harvested and resuspended in SFM. An aliquot was removed to measure total cell surface receptor. The remaining cells were stimulated with 1µM fMLF in SFM at 37°C for 1 hour and were then washed extensively to remove excess unlabelled ligand. Half the remaining cells were resuspended in pre-warmed SFM for 30 min at 37°C (20 min for U937 FPR cells) to allow the FPR to recycle. The other half was kept on ice to measure post-internalization cell surface receptor levels. All aliquots were then resuspended in SFM containing 10nM 633-6pep and assayed by flow cytometry using a Becton-Dickinson FACSCalibur. For analysis, assayed cells were gated for live cells using forward and side scatter parameters. These cells were then gated using FL-1 for GFP-fused arrestin mutant expression and the mean channel fluorescence was measured in FL-4 to monitor cell surface expression of the FPR. Non-specific binding was determined by labeling arrestin knockout cell lines (or U937 cells lines) that did not express FPR and was subtracted from all values. To account for differences in total recycling that could be due to differences in the initial extent of internalization, the fraction of recycled FPR (starting from the final internalization time point) was divided by the fraction of internalized FPR. Data are expressed as a percentage of recycled receptor /internalized receptor.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Charlotte Vines and Angela Wandinger-Ness for helpful comments. This work was funded by NIH grants AI36357 and GM68901 to E.R.P. and pre-doctoral fellowship BC030217 from the Department of Defense Breast Cancer Research Program to B.M.W. Flow cytometry data and confocal images in this study were generated in the Flow Cytometry and Fluorescence Microscopy Facilities, respectively, at the University of New Mexico Health Sciences Center, which received support from NCRR 1 S10 RR14668, NSF MCB9982161, NCRR P20 RR11830, NCI P30 CA118100, NCRR S10 RR19287, NCRR S10 RR016918, the University of New Mexico Health Sciences Center, and the University of New Mexico Cancer Center.
Glossary
Abbreviations used
- AP-1
adaptor protein-1 complex
- AP-2
adaptor protein-2 complex
- Arr2
arrestin-2
- Arr2−/−/3−/− FPR MEF
arrestin-2−/−/-3−/− knockout MEF cells stably expressing the FPR
- fMLF
N-formyl-Methionyl-Leucyl-Phenylalanine
- FPR
N-formyl peptide receptor
- GPCR
G protein-coupled receptor
- MEF
mouse embryonic fibroblast
- PI
propidium iodide
- SFM
serum-free medium (DMEM or RPMI)
- 6pep
N-formyl-Leucyl-Leucyl-Phenylalanyl-Leucyl-Tyrosinyl-Lysine
- U937 FPR
U937 cells stably expressing the FPR
REFERENCES
- 1.Penela P, Murga C, Ribas C, Tutor AS, Peregrin S, Mayor F., Jr Mechanisms of regulation of G protein-coupled receptor kinases (GRKs) and cardiovascular disease. Cardiovasc Res. 2006;69(1):46–56. doi: 10.1016/j.cardiores.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 2.Lombardi MS, Kavelaars A, Heijnen CJ. Role and modulation of G protein-coupled receptor signaling in inflammatory processes. Crit Rev Immunol. 2002;22(2):141–163. [PubMed] [Google Scholar]
- 3.Premont RT. Once and future signaling: G protein-coupled receptor kinase control of neuronal sensitivity. Neuromolecular Med. 2005;7(1–2):129–147. doi: 10.1385/NMM:7:1-2:129. [DOI] [PubMed] [Google Scholar]
- 4.Moratz C, Harrison K, Kehrl JH. Role of RGS proteins in regulating the migration of B lymphocytes. Arch Immunol Ther Exp (Warsz) 2004;52(1):27–35. [PubMed] [Google Scholar]
- 5.Luttrell DK, Luttrell LM. Not so strange bedfellows: G-protein-coupled receptors and Src family kinases. Oncogene. 2004;23(48):7969–7978. doi: 10.1038/sj.onc.1208162. [DOI] [PubMed] [Google Scholar]
- 6.Barak LS, Oakley RH, Laporte SA, Caron MG. Constitutive arrestin-mediated desensitization of a human vasopressin receptor mutant associated with nephrogenic diabetes insipidus. Proc Natl Acad Sci U S A. 2001;98(1):93–98. doi: 10.1073/pnas.011303698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Key TA, Bennett TA, Foutz TD, Gurevich VV, Sklar LA, Prossnitz ER. Regulation of formyl peptide receptor agonist affinity by reconstitution with arrestins and heterotrimeric G proteins. J Biol Chem. 2001;276(52):49204–49212. doi: 10.1074/jbc.M109475200. [DOI] [PubMed] [Google Scholar]
- 8.Key TA, Foutz TD, Gurevich VV, Sklar LA, Prossnitz ER. N-formyl peptide receptor phosphorylation domains differentially regulate arrestin and agonist affinity. J Biol Chem. 2003;278(6):4041–4047. doi: 10.1074/jbc.M204687200. [DOI] [PubMed] [Google Scholar]
- 9.Key TA, Vines CM, Wagener BM, Gurevich VV, Sklar LA, Prossnitz ER. Inhibition of chemoattractant N-formyl peptide receptor trafficking by active arrestins. Traffic. 2005;6(2):87–99. doi: 10.1111/j.1600-0854.2004.00248.x. [DOI] [PubMed] [Google Scholar]
- 10.Tobin AB, Butcher AJ, Kong KC. Location, location, location…site-specific GPCR phosphorylation offers a mechanism for cell-type-specific signalling. Trends Pharmacol Sci. 2008;29(8):413–420. doi: 10.1016/j.tips.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DeFea KA. Stop that cell! Beta-arrestin-dependent chemotaxis: a tale of localized actin assembly and receptor desensitization. Annu Rev Physiol. 2007;69:535–560. doi: 10.1146/annurev.physiol.69.022405.154804. [DOI] [PubMed] [Google Scholar]
- 12.DeWire SM, Ahn S, Lefkowitz RJ, Shenoy SK. Beta-arrestins and cell signaling. Annu Rev Physiol. 2007;69:483–510. doi: 10.1146/annurev.physiol.69.022405.154749. [DOI] [PubMed] [Google Scholar]
- 13.Premont RT, Gainetdinov RR. Physiological roles of G protein-coupled receptor kinases and arrestins. Annu Rev Physiol. 2007;69:511–534. doi: 10.1146/annurev.physiol.69.022405.154731. [DOI] [PubMed] [Google Scholar]
- 14.Kim YM, Benovic JL. Differential roles of arrestin-2 interaction with clathrin and adaptor protein 2 in G protein-coupled receptor trafficking. J Biol Chem. 2002;277(34):30760–30768. doi: 10.1074/jbc.M204528200. [DOI] [PubMed] [Google Scholar]
- 15.Ahn S, Maudsley S, Luttrell LM, Lefkowitz RJ, Daaka Y. Src-mediated tyrosine phosphorylation of dynamin is required for beta2-adrenergic receptor internalization and mitogen-activated protein kinase signaling. J Biol Chem. 1999;274(3):1185–1188. doi: 10.1074/jbc.274.3.1185. [DOI] [PubMed] [Google Scholar]
- 16.Cao TT, Deacon HW, Reczek D, Bretscher A, von Zastrow M. A kinase-regulated PDZ-domain interaction controls endocytic sorting of the beta2-adrenergic receptor. Nature. 1999;401(6750):286–290. doi: 10.1038/45816. [DOI] [PubMed] [Google Scholar]
- 17.Delaney KA, Murph MM, Brown LM, Radhakrishna H. Transfer of M2 muscarinic acetylcholine receptors to clathrin-derived early endosomes following clathrin-independent endocytosis. J Biol Chem. 2002;277(36):33439–33446. doi: 10.1074/jbc.M205293200. [DOI] [PubMed] [Google Scholar]
- 18.Vines CM, Revankar CM, Maestas DC, LaRusch LL, Cimino DF, Kohout TA, Lefkowitz RJ, Prossnitz ER. N-formyl peptide receptors internalize but do not recycle in the absence of arrestins. J Biol Chem. 2003;278(43):41581–41584. doi: 10.1074/jbc.C300291200. [DOI] [PubMed] [Google Scholar]
- 19.Kohout TA, Lin FS, Perry SJ, Conner DA, Lefkowitz RJ. beta-Arrestin 1 and 2 differentially regulate heptahelical receptor signaling and trafficking. Proc Natl Acad Sci U S A. 2001;98(4):1601–1606. doi: 10.1073/pnas.041608198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rosenfeld JL, Knoll BJ, Moore RH. Regulation of G-protein-coupled receptor activity by rab GTPases. Receptors Channels. 2002;8(2):87–97. [PubMed] [Google Scholar]
- 21.Innamorati G, Le Gouill C, Balamotis M, Birnbaumer M. The long and the short cycle. Alternative intracellular routes for trafficking of G-protein-coupled receptors. J Biol Chem. 2001;276(16):13096–13103. doi: 10.1074/jbc.M009780200. [DOI] [PubMed] [Google Scholar]
- 22.Kreuzer OJ, Krisch B, Dery O, Bunnett NW, Meyerhof W. Agonist-mediated endocytosis of rat somatostatin receptor subtype 3 involves beta-arrestin and clathrin coated vesicles. J Neuroendocrinol. 2001;13(3):279–287. doi: 10.1046/j.1365-2826.2001.00630.x. [DOI] [PubMed] [Google Scholar]
- 23.Fan GH, Lapierre LA, Goldenring JR, Richmond A. Differential regulation of CXCR2 trafficking by Rab GTPases. Blood. 2003;101(6):2115–2124. doi: 10.1182/blood-2002-07-1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oakley RH, Laporte SA, Holt JA, Barak LS, Caron MG. Molecular determinants underlying the formation of stable intracellular G protein-coupled receptor-beta-arrestin complexes after receptor endocytosis. J Biol Chem. 2001;276(22):19452–19460. doi: 10.1074/jbc.M101450200. [DOI] [PubMed] [Google Scholar]
- 25.Tohgo A, Choy EW, Gesty-Palmer D, Pierce KL, Laporte S, Oakley RH, Caron MG, Lefkowitz RJ, Luttrell LM. The stability of the G protein-coupled receptor-beta-arrestin interaction determines the mechanism and functional consequence of ERK activation. J Biol Chem. 2003;278(8):6258–6267. doi: 10.1074/jbc.M212231200. [DOI] [PubMed] [Google Scholar]
- 26.Tohgo A, Pierce KL, Choy EW, Lefkowitz RJ, Luttrell LM. beta-Arrestin scaffolding of the ERK cascade enhances cytosolic ERK activity but inhibits ERK-mediated transcription following angiotensin AT1a receptor stimulation. J Biol Chem. 2002;277(11):9429–9436. doi: 10.1074/jbc.M106457200. [DOI] [PubMed] [Google Scholar]
- 27.Simaan M, Bedard-Goulet S, Fessart D, Gratton JP, Laporte SA. Dissociation of beta-arrestin from internalized bradykinin B2 receptor is necessary for receptor recycling and resensitization. Cell Signal. 2005;17(9):1074–1083. doi: 10.1016/j.cellsig.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 28.Revankar CM, Vines CM, Cimino DF, Prossnitz ER. Arrestins block G protein-coupled receptor-mediated apoptosis. J Biol Chem. 2004;279(23):24578–24584. doi: 10.1074/jbc.M402121200. [DOI] [PubMed] [Google Scholar]
- 29.Milano SK, Pace HC, Kim YM, Brenner C, Benovic JL. Scaffolding functions of arrestin-2 revealed by crystal structure and mutagenesis. Biochemistry. 2002;41(10):3321–3328. doi: 10.1021/bi015905j. [DOI] [PubMed] [Google Scholar]
- 30.Krupnick JG, Santini F, Gagnon AW, Keen JH, Benovic JL. Modulation of the arrestin-clathrin interaction in cells. Characterization of beta-arrestin dominant-negative mutants. J Biol Chem. 1997;272(51):32507–32512. doi: 10.1074/jbc.272.51.32507. [DOI] [PubMed] [Google Scholar]
- 31.Potter RM, Key TA, Gurevich VV, Sklar LA, Prossnitz ER. Arrestin variants display differential binding characteristics for the phosphorylated N-formyl peptide receptor carboxyl terminus. J Biol Chem. 2002;277(11):8970–8978. doi: 10.1074/jbc.M111086200. [DOI] [PubMed] [Google Scholar]
- 32.Kovoor A, Celver J, Abdryashitov RI, Chavkin C, Gurevich VV. Targeted construction of phosphorylation-independent beta-arrestin mutants with constitutive activity in cells. J Biol Chem. 1999;274(11):6831–6834. doi: 10.1074/jbc.274.11.6831. [DOI] [PubMed] [Google Scholar]
- 33.Goodman OB, Jr, Krupnick JG, Gurevich VV, Benovic JL, Keen JH. Arrestin/clathrin interaction. Localization of the arrestin binding locus to the clathrin terminal domain. J Biol Chem. 1997;272(23):15017–15022. doi: 10.1074/jbc.272.23.15017. [DOI] [PubMed] [Google Scholar]
- 34.Krupnick JG, Goodman OB, Jr, Keen JH, Benovic JL. Arrestin/clathrin interaction. Localization of the clathrin binding domain of nonvisual arrestins to the carboxy terminus. J Biol Chem. 1997;272(23):15011–15016. doi: 10.1074/jbc.272.23.15011. [DOI] [PubMed] [Google Scholar]
- 35.Lin FT, Krueger KM, Kendall HE, Daaka Y, Fredericks ZL, Pitcher JA, Lefkowitz RJ. Clathrin-mediated endocytosis of the beta-adrenergic receptor is regulated by phosphorylation/dephosphorylation of beta-arrestin1. J Biol Chem. 1997;272(49):31051–31057. doi: 10.1074/jbc.272.49.31051. [DOI] [PubMed] [Google Scholar]
- 36.Gilbert TL, Bennett TA, Maestas DC, Cimino DF, Prossnitz ER. Internalization of the human N-formyl peptide and C5a chemoattractant receptors occurs via clathrin-independent mechanisms. Biochemistry. 2001;40(12):3467–3475. doi: 10.1021/bi001320y. [DOI] [PubMed] [Google Scholar]
- 37.Campbell RE, Tour O, Palmer AE, Steinbach PA, Baird GS, Zacharias DA, Tsien RY. A monomeric red fluorescent protein. Proc Natl Acad Sci U S A. 2002;99(12):7877–7882. doi: 10.1073/pnas.082243699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen W, Feng Y, Chen D, Wandinger-Ness A. Rab11 is required for trans-golgi network-to-plasma membrane transport and a preferential target for GDP dissociation inhibitor. Mol Biol Cell. 1998;9(11):3241–3257. doi: 10.1091/mbc.9.11.3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Browning DD, Pan ZK, Prossnitz ER, Ye RD. Cell type- and developmental stage-specific activation of NF-kappaB by fMet-Leu-Phe in myeloid cells. J Biol Chem. 1997;272(12):7995–8001. doi: 10.1074/jbc.272.12.7995. [DOI] [PubMed] [Google Scholar]
- 40.Hsu MH, Chiang SC, Ye RD, Prossnitz ER. Phosphorylation of the N-formyl peptide receptor is required for receptor internalization but not chemotaxis. J Biol Chem. 1997;272(47):29426–29429. doi: 10.1074/jbc.272.47.29426. [DOI] [PubMed] [Google Scholar]
- 41.Fessart D, Simaan M, Laporte SA. c-Src regulates clathrin adapter protein 2 interaction with beta-arrestin and the angiotensin II type 1 receptor during clathrin- mediated internalization. Mol Endocrinol. 2005;19(2):491–503. doi: 10.1210/me.2004-0246. [DOI] [PubMed] [Google Scholar]
- 42.Robinson MS. Adaptable adaptors for coated vesicles. Trends Cell Biol. 2004;14:167–174. doi: 10.1016/j.tcb.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 43.Laporte SA, Miller WE, Kim KM, Caron MG. beta-Arrestin/AP-2 interaction in G protein-coupled receptor internalization: identification of a beta-arrestin binging site in beta 2-adaptin. J Biol Chem. 2002;277(11):9247–9254. doi: 10.1074/jbc.M108490200. [DOI] [PubMed] [Google Scholar]
- 44.Lundmark R, Carlsson SR. The beta-appendages of the four adaptor-protein (AP) complexes: structure and binding properties, and identification of sorting nexin 9 as an accessory protein to AP-2. Biochem J. 2002;362(Pt 3):597–607. doi: 10.1042/0264-6021:3620597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cowen DS, Berger M, Nuttle L, Dubyak GR. Chronic treatment with P2-purinergic receptor agonists induces phenotypic modulation of the HL-60 and U937 human myelogenous leukemia cell lines. J Leukoc Biol. 1991;50(2):109–122. doi: 10.1002/jlb.50.2.109. [DOI] [PubMed] [Google Scholar]
- 46.Deneka M, Neeft M, Popa I, van Oort M, Sprong H, Oorschot V, Klumperman J, Schu P, van der Sluijs P. Rabaptin-5alpha/rabaptin-4 serves as a linker between rab4 and gamma(1)-adaptin in membrane recycling from endosomes. Embo J. 2003;22(11):2645–2657. doi: 10.1093/emboj/cdg257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pagano A, Crottet P, Prescianotto-Baschong C, Spiess M. In vitro formation of recycling vesicles from endosomes requires adaptor protein-1/clathrin and is regulated by rab4 and the connector rabaptin-5. Mol Biol Cell. 2004;15(11):4990–5000. doi: 10.1091/mbc.E04-04-0355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Popa I, Deneka M, van der Sluijs P. Expression and properties of the Rab4, Rabaptin-5alpha, AP-1 complex in endosomal recycling. Methods Enzymol. 2005;403:526–540. doi: 10.1016/S0076-6879(05)03046-6. [DOI] [PubMed] [Google Scholar]
- 49.Daaka Y, Luttrell LM, Ahn S, Della Rocca GJ, Ferguson SS, Caron MG, Lefkowitz RJ. Essential role for G protein-coupled receptor endocytosis in the activation of mitogen-activated protein kinase. J Biol Chem. 1998;273(2):685–688. doi: 10.1074/jbc.273.2.685. [DOI] [PubMed] [Google Scholar]
- 50.Burtey A, Schmid EM, Ford MG, Rappoport JZ, Scott MG, Marullo S, Simon SM, McMahon HT, Benmerah A. The conserved isoleucine-valine-phenylalanine motif couples activation state and endocytic functions of beta-arrestins. Traffic. 2007;8(7):914–931. doi: 10.1111/j.1600-0854.2007.00578.x. [DOI] [PubMed] [Google Scholar]
- 51.Schmid EM, Ford MG, Burtey A, Praefcke GJ, Peak-Chew SY, Mills IG, Benmerah A, McMahon HT. Role of the AP2 beta-Appendage Hub in Recruiting Partners for Clathrin-Coated Vesicle Assembly. PLoS Biol. 2006;4(9) doi: 10.1371/journal.pbio.0040262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Orem NR, Xia L, Dolph PJ. An essential role for endocytosis of rhodopsin through interaction of visual arrestin with the AP-2 adaptor. J Cell Sci. 2006;119(Pt 15):3141–3148. doi: 10.1242/jcs.03052. [DOI] [PubMed] [Google Scholar]
- 53.Kiselev A, Socolich M, Vinos J, Hardy RW, Zuker CS, Ranganathan R. A molecular pathway for light-dependent photoreceptor apoptosis in Drosophila. Neuron. 2000;28(1):139–152. doi: 10.1016/s0896-6273(00)00092-1. [DOI] [PubMed] [Google Scholar]
- 54.Alloway PG, Howard L, Dolph PJ. The formation of stable rhodopsin-arrestin complexes induces apoptosis and photoreceptor cell degeneration. Neuron. 2000;28(1):129–138. doi: 10.1016/s0896-6273(00)00091-x. [DOI] [PubMed] [Google Scholar]
- 55.Chen J, Shi G, Concepcion FA, Xie G, Oprian D, Chen J. Stable rhodopsin/arrestin complex leads to retinal degeneration in a transgenic mouse model of autosomal dominant retinitis pigmentosa. J Neurosci. 2006;26(46):11929–11937. doi: 10.1523/JNEUROSCI.3212-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wu X, Zhao X, Puertollano R, Bonifacino JS, Eisenberg E, Greene LE. Adaptor and clathrin exchange at the plasma membrane and trans-Golgi network. Mol Biol Cell. 2003;14(2):516–528. doi: 10.1091/mbc.E02-06-0353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Janvier K, Bonifacino JS. Role of the endocytic machinery in the sorting of lysosome-associated membrane proteins. Mol Biol Cell. 2005;16(9):4231–4242. doi: 10.1091/mbc.E05-03-0213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Aguilar RC, Ohno H, Roche KW, Bonifacino JS. Functional domain mapping of the clathrin-associated adaptor medium chains mu1 and mu2. J Biol Chem. 1997;272(43):27160–27166. doi: 10.1074/jbc.272.43.27160. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.