

Abstract
T cell homeostasis is crucial for maintaining an efficient and balanced T cell immunity. The interaction between TCR and self peptide (sp) MHC ligands is known to be the key driving force in this process, and it is believed to be functionally and mechanistically different from that initiated by the antigenic TCR stimulation. Yet, very little is known about the downstream signaling events triggered by this TCR-spMHC interaction and how they differ from those triggered by antigenic TCR stimulation. In this study, we show that T cell conditional ablation of MEKK3, a Ser/Thr kinase in the MAPK cascade, causes a significant reduction in peripheral T cell numbers in the conditional knockout mice, but does not perturb thymic T cell development and maturation. Using an adoptive mixed transfer method, we show that MEKK3-deficient T cells are severely impaired in lymphopenia-induced cell proliferation and survival. Interestingly, the Ag-induced T cell proliferation proceeds normally in the absence of MEKK3. Finally, we found that the activity of ERK1/2, but not p38 MAPK, was attenuated during the lymphopenia-driven response in MEKK3-deficient T cells. Together, these data suggest that MEKK3 may play a crucial selective role for spMHC-mediated T cell homeostasis.
The number of peripheral T cells is always maintained at a fairly steady level throughout life via a process called T cell peripheral homeostasis (1-3). T cell homeostasis involves mechanisms to control peripheral T cell survival, proliferation, and apoptosis (1, 4, 5). In addition to maintaining a steady number of peripheral T cells, T cell homeostasis also controls T cell repertoire diversity; thus, this process plays a central role to maintain an effective and adequate adaptive immunity (6-8). More importantly, perturbation of normal T cell homeostasis has also been linked to various autoimmune conditions such as type I diabetes, systemic lupus erythematosus, and Sjorgen’s syndrome, etc. (9-12).
The basal proliferation of peripheral naive T cells, called homeostatic proliferation, is very slow in the absence of foreign Ags or microorganism infections. This process accelerates when the total number of T cells in the body is at low levels, known as lymphopenic condition (or lymphopenia). This condition occurs naturally during neonatal T cell development, or can be induced during chemotherapy, irradiation, or viral infection, and promotes T cell homeostatic proliferation in an attempt to restore normal size of the T cell pool (1). This process has been studied extensively using lymphopenic mice such as RAG1−/− or irradiated mice. When a small number of T cells are transferred into these mice, they undergo a vigorous proliferation known as lymphopenia-induced proliferation (LIP)3 (4, 13). LIP has been demonstrated to be a physiological process for maintaining T cell homeostasis (4, 14). Two discrete populations of proliferating T cells have been identified. One population is highly proliferative (known as fast proliferating (f.p.)) with more than seven cell divisions within 7 days after transfer. In contrast, the other one undergoes slower proliferation with only one to five cell divisions within the same period of time (15, 16). Careful analysis of these two populations suggests that the f.p. T cells are induced by either commensal bacteria Ags or high-affinity self Ags in a way that resembles T cell activation induced by foreign Ags (15, 16). In contrast, the slower proliferating population is considered to undergo homeostatic proliferation stimulated by weak self Ags (1, 15). The lymphopenic condition has been referred to sometimes as space, which indicates the resources required for T cell survival and proliferation (such as self peptides (sp) and IL-7) (6, 17). It has been proposed that competition among T cells for access to the limited resources (or space) is critical for T cell homeostasis (6).
T cell homeostasis is critically controlled by two different pathways. The first one requires TCR signals induced by the spMHC (5, 17, 18). In the absence of this TCR-spMHC interaction, T cell homeostatic processes, such as proliferation and survival, are severely impaired (19-22). This TCR-spMHC-mediated stimulation under the lymphopenic condition differs from that of Ag-mediated T cell activation (22-24). For instance, several T cell activation markers, such as CD62L, CD25, and CD69, remain unchanged in this process (25, 26). However, the memory T cell surface marker, CD44, is slowly up-regulated following homeostatic proliferation or LIP, suggesting that when naive T cells undergo LIP, there is a concomitant conversion to memory phenotype (23, 27, 28). The second signal is provided by common γ-chain cytokines such as IL-7 and/or IL-15 (29-33). These cytokines either work alone or together with the TCR-spMHC-mediated signals to regulate peripheral T cell homeostasis (32, 34).
The MAPK cascades are highly conserved intracellular signaling networks that are used by eukaryotic cells to transduce a wide spectrum of extracellular signals (35-37). MAPKs are activated through a three-kinase module, including a MAPK, a MAPK kinase, and a MAPK kinase kinase (MAP3K). Upon immune receptor stimulation or proinflammatory cytokine receptor stimulation, the MAPK pathways are rapidly induced to regulate the expression of those genes that are essential for both the innate and adaptive immune responses (37, 38). More than five mammalian MAPK cascades are known to coexist and function either simultaneously or sequentially in both immune and nonimmune cells. Yet, the precise mechanisms by which the MAPK pathways are activated and how individual MAPK cascade functions specifically in distinctive immune cells under various physiological settings remain to be fully understood.
MEKK3, a member of the MAP3K superfamily, shares a high homology with MEKK2, whereas its similarity with other MAP3Ks is limited to the kinase domain (39-43). Although most MAP3Ks are able to activate similar downstream MAPK cascades in vitro, including the JNKs, ERK1/2, p38, and ERK5 under certain conditions, the in vivo studies suggest that their specificities are more restricted toward certain downstream MAPK cascades in controlling unique cellular functions (35, 44, 45). MEKK3 is activated through a dimerization-induced autophosphorylation on a conserved serine residue in its activation loop (46, 47). Mouse genetic studies show that MEKK3 is essential for embryonic cardiovascular development (48, 49). In innate immune response, MEKK3 was found to mediate the proinflammatory cytokine and TLR-induced JNK and p38 MAPK activation in murine embryonic fibroblasts cells (50-53). However, very little is known about the role of MEKK3 in adaptive immune responses, and no in vivo study of MEKK3 in T cells has been done to date due to the early embryonic lethality of the germline MEKK3 knockout (KO) mice.
In this study, we have generated Mekk3 conditional KO mice with a specific ablation of MEKK3 in T cells. We found that deletion of MEKK3 in T cells did not significantly alter the thymic T cell development, but led to a peripheral T cell homeostasis defect. MEKK3 is critical for the proliferation of CD4 T cells and survival of both naive CD4 and CD8 T cells under lymphopenic conditions. This defect is partially due to a decreased active ERK1/2 level in MEKK3-deficient T cells under lymphopenic condition. Together, our study uncovered a critical role of MEKK3 in peripheral T cell homeostasis.
Materials and Methods
Targeting vector and generation of Mekk3 conditional KO mice
We used the same strategy and techniques described in our previous study (48, 54) to construct the conditional Mekk3-targeting vector and to generate the targeted mice. As illustrated in Fig. 1A, a Loxp sequence (from pBS246; Life Technologies) was subcloned into the XbaI site (a BamHI site is introduced at this location) in the intron upstream of the exon 15 that encodes aa 493–537 of MEKK3 in the kinase subdomains VII-VIII. Another Loxp was inserted at the XbaI site (destroyed after cloning) in the intron downstream from this exon, followed by a Frt-flanked Neor cassette (a gift from P. Zhang, Baylor College of Medicine, Houston, TX). The 8-kb XbaI-PstI genomic fragment of Mekk3 with the above Loxp sequences, the Frt-flanked Neor cassette, and a 1.6-kb XbaI-PstI genomic sequence as the short arm was ligated to a PGK-TK cassette, then subcloned into the pBluescript-SKII to create the conditional Mekk3-targeting vector. Locations of the Southern blot probe (shaded oval) and the P1 and P2 primers for PCR genotyping that amplify fragments with the sizes of 1392, 1499, and 579 bp for the wild-type (WT), the flox, and the Δflox allele, respectively, are indicated. The locations of the loxp (filled arrowhead) and Frt (open diamond) sequences in the targeting vector and the targeted locus are shown. The sizes of BamHI fragments that are detected by the Southern blot probe in the WT (17 kb), flox-Neor (10 kb), and flox (8 kb) alleles are also shown. ROSA-Flp indicates a knock-in mouse line with the Flp gene being knocked in to the rosa26 locus, whereas the Lck-Cre indicates a transgenic mouse line with a cre gene specifically controlled by a T cell-specific promoter, the Lck promoter. E14, E15, and E16 are exons of the Mekk3 gene where the targeting event occurs.
FIGURE 1.
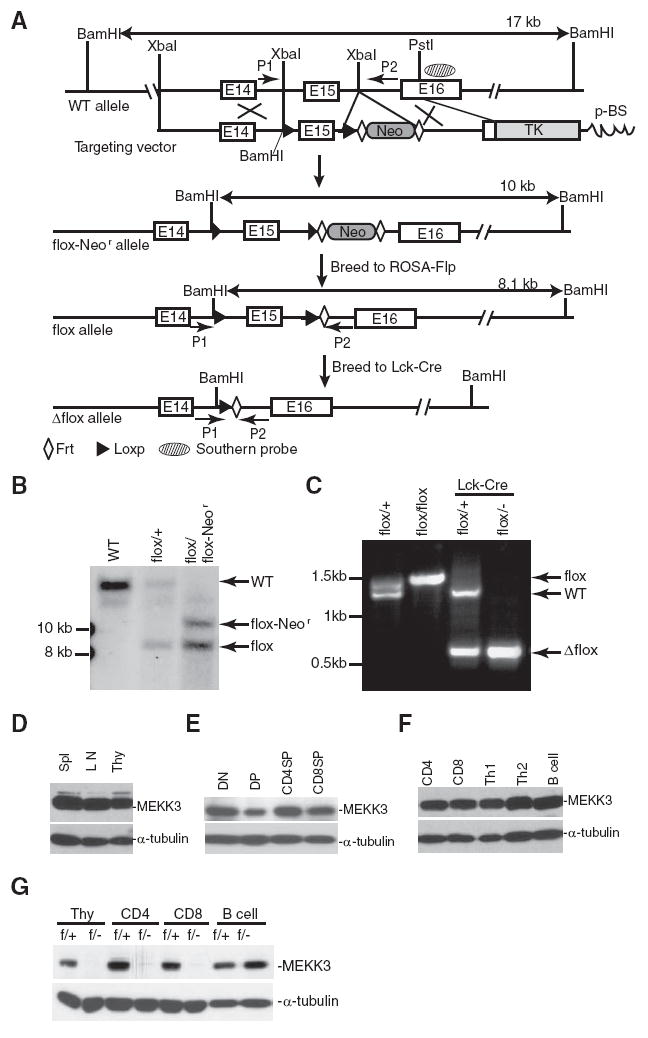
Conditional ablation of MEKK3 in T cell lineages. A, A diagram showing the Mekk3 gene locus (WT allele), the targeting vector, the Loxp sequence-targeted allele containing the neomycin resistant gene (flox-Neor allele), the Loxp sequence-targeted allele with the Neor gene deleted by ROSA-Flp (flox allele), and the T cell conditionally deleted allele by Lck-Cre (Δflox allele). See Materials and Methods for detail. B, Southern blot analysis of genomic DNAs isolated from the tails of a WT mouse, a heterozygous mouse with one WT allele and one flox allele (flox/+), and a heterozygous mouse with one flox-Neor and one flox allele (flox/flox-Neor), as indicated. The DNAs were digested with BamHI enzyme, and the Southern probe is shown in A above. C, PCR genotyping of the Mekk3 conditionally targeted mice. DNAs were prepared from total thymocytes isolated from Mekk3flox/+ (flox/+), Mekk3flox/− (flox/−), Lck-Cre-Mekk3flox/+, and Lck-Cre- Mekk3flox/− mice, as indicated. +, Indicates MEKK3 WT allele; −, indicates the Mekk3 germline KO allele generated from our previous study (48). (See supplemental Materials and Methods4 for detailed description of Southern blot and PCR genotyping.) D–F, Immunoblot analysis of MEKK3 expression in total lymphocyes isolated from spleen, LN, and thymus (D), or in purified subsets of thymocytes (E), or in purified naive CD4 T cells, CD8 T cells, in vitro differentiated Th1 and Th2 cells, and purified B cells (F), using an anti-MEKK3-specific mAb. The α-tubulin level was determined as loading controls. DN, DP, CD4SP, and CD8SP are DN, DP, CD4+CD8− SP, and CD8+CD4− SP thymocytes, respectively. G, Detection of MEKK3 deletion in T cells from the Mekk3 T-KO mice. Total thymocytes (Thy) or purified CD4, CD8, and B cells isolated from either NCL mice (f/+) or Mekk3 T-KO mice (f/−) were examined for MEKK3 expression by immunoblotting with an anti-MEKK3 mAb, as described above in D.
The first step was to create embryonic stem (ES) cells targeted on the Mekk3 locus with the two-loxp sequences flanking exon 15 (flox), but still containing the neomycin selection marker that we called the flox-Neor allele. ES cells were cultured, transfected with the targeting vector, and selected, as previously described (48, 54). The targeted ES cells were used to generate mice with germline transmission of this targeted allele. We bred these mice to a ROSA-Flp mouse line to delete the neomycin gene sandwiched between the two Frt sequences, resulting in the generation of the Loxp sequence knock-in mice that flank exon 15 (see diagram in Fig. 1A). We named the knock-in allele as the Mekk3 flox allele, and the knock-in mice as Mekk3flox/+ (one knock-in allele and one WT allele). Mekk3flox/flox (both knock-in alleles) mice are generated by breeding Mekk3flox/+ mice to each other. MEKK3 germline KO (Mekk3+/−) mice (48) were bred with Lck-Cre transgenic mice (The Jackson Laboratory) to obtain Lck-Cre-Mekk3+/− mice. These mice were bred with Mekk3flox/flox mice to obtain Lck-Cre-Mekk3f/− (Mekk3 T-KO, also called the f/− mice throughout this work) and Lck-Cre-Mekk3f/+ (normal control littermate (NCL), or f/+ mice throughout this work) mice. All of the mice described above were backcrossed with C57BL/6 mice (The Jackson Laboratory) for more than 10 generations to obtain a pure C57BL/6 background.
Mice
C57BL/6-Ly5.2 mice were obtained from The Jackson Laboratory. B6.PLThy1a/Cy and OT-II/Thy1.1 transgenic mice were obtained from T. Chi (Yale University, New Haven, CT). ROSA26-EYFP transgenic mouse is a gift from F. Costantin (Columbia University, New York, NY). OT-II transgenic mouse is a gift from D. Chen (M.D. Anderson Cancer Center, Houston, TX). OTII mice were bred with Mekk3f/f mice to generate OT-II-Mekk3f/f mice, which in turn bred to Lck-Cre-Mekk3+/− mice to generate OT-II-Lck-Cre-Mekk3f/− (OT-II Mekk3 T-KO mice) and OT-II- Lck-Cre-Mekk3f/+ (OT-II normal littermate control mice). MHC class II KO mice (homozygous null for H2-Ab1, H2-Aa, H2-Eb1, H2-Eb2, H2-Ea) were obtained from D. Rothstein (Yale University, New Haven, CT). All mice were on C57BL/6 background. All mice were used between 6 and 10 wk of age, and were housed in a pathogen-free animal care facility at M.D. Anderson Cancer Center or Yale University. All mouse experiments were approved by Institutional Animal Care and Use Committee of M.D. Anderson Cancer Center, and Yale University.
Cell preparation and Western blot analysis
Lymphocytes from thymus, spleen, and lymph nodes (LN) were lysed for 15 min on ice in cell lysis buffer (25 mM Tris-HCI (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 1 mM PMSF, 1 mM Na3VO4, and protease inhibitor mixture). Total cell lysates were resolved by 8 or 10% SDS-PAGE gels for immunoblot analysis with an anti-MEKK3 mAb or an anti-tubulin Ab (BD Biosciences).
Lymphocyte staining and flow cytometry
Lymphocytes were isolated from thymuses, spleens, and LN of 6- to 10-wk-old mice. For cell surface staining, cells were washed in FACS buffer (2% FCS PBS with 0.09% NaN3), and then incubated with one of the following Abs alone or in combination, as indicated: anti-CD3 PEcy5.5, anti-CD69 PE, anti-CD4 PECy7, anti-CD8 Pacific Blue, anti- CD62L allophycocyanin, anti-CD62L PE, anti-CD44 Alexa750 (eBioscience), anti-CD5 PE, and anti-CD127 PE (IL-7R) (BD Biosciences), in FACS buffer on ice for 30 min. Cells were then washed two more times with FACS buffer, and fixed in 1% paraformaldehyde in PBS before being analyzed with a LSRII machine (BD Biosciences). For intracellular staining of phospho-ERK1/2 and phospho-p38, cells were first stained with surface markers, as mentioned above, and then fixed with Fix/Perm Medium A (Caltag Laboratories) for 30 min at room temperature. After washing, cells were incubated with anti-phospho-ERK1/2 (1:50) and anti-phospho-p38 (1:50) (Cell Signaling Technology) in Fix/Perm Medium B (Caltag Laboratories) for 60 min at 4°C, followed by incubation with goat anti-rabbit Alexa647 (1:200; Molecular Probes) for 30 min at 4°C. For intracellular staining of Foxp3, a Foxp3-allophycocyanin staining kit (BD Biosciences) was used. For intracellular staining of Bcl-2, cells were fixed and permeabilized with Fix/Perm kits (BD Biosciences) before being stained with an anti-Bcl-2 PE Ab. Stained cells were analyzed by a LSR II machine (BD Biosciences). Postacquisition analysis was performed with FLowJo software (Tree Star).
T cell differentiation
Spleen CD4 T cells were first enriched by AutoMACS (Miltenyi Biotec), according to manufacturer’s instructions. The enriched CD4+ cells were stained with anti-CD4 FITC, anti-CD62L allophycocyanin, and anti-CD44 PE (BD Biosciences). CD4+CD62L+CD44low naive T cells were sorted on a BD FACS Aria cell sorter. Purified naive CD4 T cells were then stimulated with plate-bound anti-CD3 (1 μg/ml) plus 1 μg/ml anti-CD28 in 24-well plate, in the presence of 50 U/ml rIL-2 (PeproTech) in T cell medium (RPMI 1640, 10% FBS, 1× antibiotics (Sigma-Aldrich), and 50 μM 2-ME). For Th1 differentiation, anti-IL-4 (5 μg/ml; Pierce) and IL-12 (10 ng/ml; PeproTech) were added to the culture. For Th2 differentiation, anti-IFN-γ (5 μg/ml; Pierce) and IL-4 (10 ng/ml; PeproTech) were added to the culture. Cells were cultured for 7 days before harvesting for immunoblotting analysis.
T cell stimulation and proliferation
Thymocytes were stimulated with different concentrations of plate-bound anti-CD3 (clone 145-2C11). Cells were stimulated in 96-well plate at 2 × 105 cells/well in T cell medium (RPMI 1640, 10% FBS, 1× antibiotics, and 50 μM 2-ME). An anti-CD28 (clone 37.51) was added to the medium at 1 μg/ml when indicated. The plates were incubated in a 37°C CO2 incubator. Supernatant was collected at 24 h to measure the production of IL-2 by ELISA. For CD4 T cell stimulation, total splenocytes were used as APCs after depletion of T cells using CD90.2 positive selection kit (Stem-Cell Technologies). Purified CD4 T cells were mixed with APCs at 1:1 ratio. Mixed cells were plated onto 96-well plates at 2 × 105 cells/well in T cell medium in the presence of indicated concentrations of soluble anti-CD3 (clone 145-2C11) Ab. For proliferation assay, cells were pulsed with 1 μCi of [3H]thymidine (PerkinElmer) per well during the last 8 h of a 72-h culture period. The cells were then harvested with a Tomtec cell harvester and counted with a beta plate liquid scintillation counter. All results are expressed as means ± SDs of triplicate cultures.
CFSE labeling
Splenocytes/Lymphocytes (1 × 107 cells/ml) were labeled with 5 μM CFSE (Invitrogen) in 0.1% BSA/PBS for 15 min at 37°C. Labeling was terminated by adding 10 vol of T cell medium and washed twice. Labeled cells were resuspended in serum-free RPMI 1640 for i.v. injection, or in T cell medium for in vitro stimulation.
Mixed adoptive transfer
For lymphopenic induced proliferation assay, C57BL/6-Ly5.2 (CD45.1-positive) mice were sublethally irradiated (600 rad) 24 h before transfer. C57BL/6.PLThy1a/Cy (Thy1.1) and C57BL/6 (congenic for Thy1.2) LN cells were mixed at 1:1 ratio. Mixture was labeled with CFSE. Five million mixed LN cells were injected into sublethally irradiated recipient mice i.v. Three or 7 days after transfer, spleen and mesenteric LN were harvested from recipient mice for flow cytometry analysis. For T cell survival and homing experiments, CD4 single-positive (SP) thymocytes were purified by depleting of CD8-positive cells in the thymus using CD8+-positive selection beads (BD Biosciences). Purified CD4SP thymocytes from Mekk3 T-KO or its littermate control (Thy1.1−CD45.2+) were mixed with CD4SP thymocytes from Thy1.1 mice at ~1:1 ratio, respectively. Mixture was transferred into C57BL/6-Ly5.2 mice. One, 3, or 7 days after transfer, spleen and LN were harvested from recipient mice for flow cytometry analysis. For the mixed bone marrow transplant experiment, bone marrow cells from f/− (CD45.1−CD45.2+) mice were mixed with bone marrow cells from WT C57BL/6-ly5.2 (CD45.1+CD45.2−) mice, and transferred into lethally irradiated (1000 rad/10 Gy) C57BL/6-ly5.2 mice. Mice were sacrificed 8 wk later, and donor cells in the thymus and spleen were analyzed by FACS staining for congenic markers.
In vitro activation-induced cell death and IL-7 protection assays
Thymocytes were seeded at 106 cells/well in 2 ml of T cell medium with 10% FBS into six-well plates precoated with 10 μg/ml control IgG or anti-CD3 (clone 2C11) Ab/ml and incubated for 12 and 24 h or with 0.5 μg of anti-Fas Ab (clone jo-2; BD Biosciences)/ml for 12 and 24 h. For IL-7-mediated protection assay, total splenocytes were seeded onto 24-well plates at 106 cells/well in 1 ml of T cell medium containing either no murine rIL-7 or with murine rIL-7 at 10 ng/ml (Peprotech). Cells were collected at indicated time points, and stained with annexin V-PE (BD Biosciences) and 7-aminoactinomycin D (7-AAD; eBioscience).
T cell-dendritic cell (DC) coculture
CFSE-labeled T cells (4 × 105) were mixed with bone marrow-derived dentritic cells (BMDCs) (2 × 106) in 24-well plates. BMDCs were generated and matured following a previously described method (55).
OVA immunization
CFSE-labeled OT-II T cells were transferred into C57BL/6-Ly5.2 mice i.p. and immunized 24 h later with an OVA/CFA mixture at the base of the tail. The OVA/CFA mixture was prepared by mixing OVA (1 mg/ml) and CFA (1 mg/ml) in PBS and vortexed for 4 h at room temperature. Each mouse received one single s.c. injection of 100 μl OVA/CFA mixture. Three days later, splenocytes were collected from the recipient mice for flow cytometry analysis.
Results
Generation of Mekk3 T cell conditional KO (T-KO) mice
We previously generated germline Mekk3 KO mice and demonstrated that MEKK3 is required for early embryonic development (48, 49). To investigate MEKK3 function in T cells, we generated Mekk3 floxed mice and Mekk3 T cell conditional KO mice (Fig. 1, A–C). MEKK3 is expressed in splenocytes, thymocytes, and LN (Fig. 1D), in FACS-sorted CD4−CD8− double-negative (DN), CD4+CD8+ double-positive (DP), CD4+CD8− CD4SP, and CD4−CD8+ CD8SP thymocytes (Fig. 1E), and in FACS-sorted peripheral CD4, CD8 T cells, in vitro differentiated Th1 and Th2 T cells, and B cells (Fig. 1F). However, the expression of MEKK3 was efficiently ablated in the thymocytes, and purified CD4−, CD8-T cells from Mekk3 T-KO mice, but not the NCL mice (Fig. 1G). In addition, MEKK3 expression in B cells from the Mekk3 T-KO mice was similar to that observed in the NCL mice. Furthermore, using a ROSA26R-EYFP reporter mouse, we confirmed that the cre gene was expressed only in T cell lineage in the Lck-Cre transgenic mice starting at the DN stage (56) (supplemental Fig. S1).4 Together, these data show that we successfully deleted MEKK3 in the T cell lineage in which MEKK3 is stably and ubiquitously expressed in both naive and differentiated effector T cells.
MEKK3 is not required for the development of major subsets of T cells in thymus
We first analyzed the thymus of Mekk3 T-KO mice. No difference in thymic cellularity was found between the Mekk3 T-KO and NCL mice (supplemental Table S1).4 Nor did we found any significant change in the number and the percentage of DN, DP, CD4SP, or CD8SP thymocytes (Fig. 2, A and B). Expression of other T cell markers, including CD3, CD69, and CD44, was also not affected (supplemental Fig. S2).4 The development of T regulatory cells (CD4+CD8−CD25+Foxp3+) also appeared normal in the Mekk3 T-KO mice (supplemental Fig. S3).4 Furthermore, we found no difference in TCR-induced cell proliferation and IL-2 production between the Mekk3 T-KO and NCL mice (Fig. 2, C and D). Finally, the TCR-, CD95-, and dexamethasone-induced cell death of thymocytes from the Mekk3 T-KO and NCL mice was also similar (Fig. 2E). Together, these results suggest that MEKK3 may not be required for thymic T cell development.
FIGURE 2.
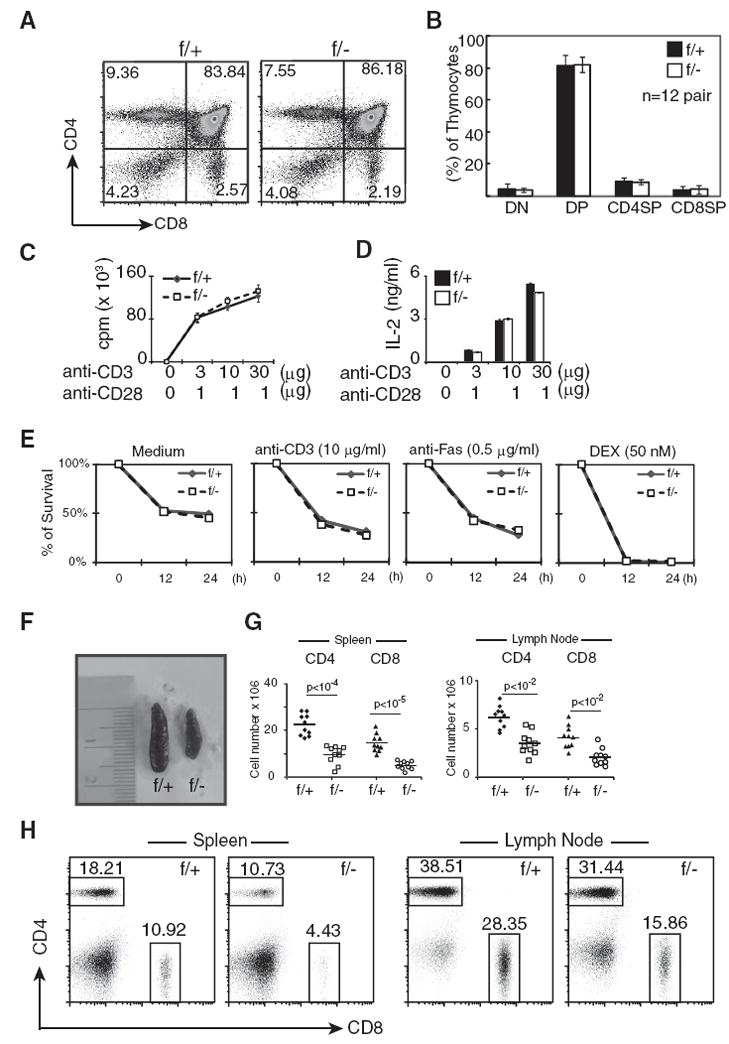
Analyses of T cell development in Mekk3 T-KO mice. A, Total thymocytes were prepared from a pair of 8-wk-old NCL (f/+) and Mekk3 T-KO (f/−) mice, and analyzed by multicolor flow cytometry using PerCP anti-CD4 and FITC anti-CD8 Abs. Numbers in the graphs show percentages of each gated subset of thymocytes. B, A bar graph shows the summary of the percentages of the DN, DP, CD4SP, and CD8SP thymocytes determined by flow cytometry described above in A. Data represent the mean ± SD of results from analyzing 12 pairs of 6- to 10-wk-old NCL (f/+) and Mekk3 T-KO (f/−) littermates. C and D, Thymocytes from NCL (f/+) and Mekk3 T-KO (f/−) mice were stimulated with indicated concentrations of plate-bound anti-CD3 Ab together with soluble anti-CD28 Ab. Cell proliferation (C) and IL-2 production (D) were determined by [3H]thymidine incorporation and an ELISA, respectively. E, Thymocytes from NCL (f/+) and Mekk3 T-KO (f/−) mice were either untreated (0 h) or treated with 10 μg/ml plate-bound anti-CD3 Ab, or 0.5 μg/ml anti-Fas Ab, or 50 nM dexamethasone (DEX) for 12 or 24 h, as indicated. The samples were stained for CD4, CD8, annexin V, and 7-AAD, and analyzed on gated CD4+CD8+ DP population by flow cytometry. The annexin V and 7-AAD DN cells are considered as viable cells. The relative survival rate is calculated as the percentage of remaining annexin V and 7-AAD DN cells after treatment normalized to that at time zero that arbitrarily set as 100%. Data are representative of four independent experiments. F, A picture showing the sizes of spleens from an 8-wk-old Mekk3 T-KO mouse (f/−) and its NCL (f/+). G, Scatterplots showing the total numbers of CD4 and CD8 T cells in spleen and LN from Mekk3 T-KO (f/−) and NCL (f/+) mice. Data shown are results from 10 pairs of 6- to 10-wk-old NCL (f/+) and Mekk3 T-KO mice (f/−). The p values were calculated using double-tail paired Student’s t test. H, Dot plots show flow cytometry analysis of CD4 and CD8 T cells from the spleen (top) and LN (bottom) of an 8-wk-old NCL (f/+) and a Mekk3 T-KO (f/−) mouse. The numbers next to the gated cells are percentages of CD4 or CD8 T cells in the spleen and LN. These numbers are used to calculate the total number of CD4 and CD8 T cells in spleen and LN shown in G above.
Reduction of peripheral T cell number in Mekk3 T-KO mice
We next examined the peripheral T cells from the Mekk3 T-KO mice. Anatomical analysis of Mekk3 T-KO mice revealed a striking reduction in the size of the spleen as compared with that of control littermates (Fig. 2F). This phenotype was accompanied by a significant reduction in total numbers of T cells. Total numbers of CD4 and CD8 T cells were significantly reduced (58 and 66% reduction for CD4 and CD8 T cells, respectively) (Fig. 2G). The percentages of CD4 and CD8 T cells in the Mekk3 T-KO mice were also reduced as compared with that from their WT littermates (Fig. 2H). Together, the above results suggest a role of the MEKK3 pathway in regulating peripheral T cell homeostasis.
MEKK3 is required for T cell homeostatic proliferation and survival
To test whether deletion of MEKK3 in T cells causes any homeostatic defect, we examined whether Mekk3 KO T cells were able to proliferate and expand in a lymphopenic environment. Same numbers of LN cells from either Mekk3 T-KO mice or NCL mice were mixed with WT Thy1.1 T cells and transferred into sublethally irradiated C57BL/6-Ly5.2 recipient mice (CD45.1) after being labeled with CFSE dye. Before transfer, the ratios of mixed donor T cells were verified by flow cytometry (Fig. 3, A and C). Seven days after transfer, the donor T cells from the spleen and LN were analyzed. The number of Mekk3 KO CD4 (Fig. 3B) and CD8 (Fig. 3D) T cells that were recovered from the spleen and LN of recipient mice was dramatically reduced as compared with the Thy1.1 T cells. In contrast, the number of the NCL T cells in the recipient spleen and LN remained comparable to that of the Thy1.1 T cells (Fig. 3, B and D). These results thus strongly suggest that MEKK3 plays a critical role in maintaining peripheral CD4 and CD8 T cell numbers in lymphopenic hosts, and the reduction of peripheral T cells in the Mekk3 T-KO mice may be due to an impaired homeostatic T cell survival.
FIGURE 3.

MEKK3 is required for LIP and survival. A, Lymphocytes from LN prepared from NCL (f/+) or Mekk3 T-KO (f/−) were mixed at ~1:1 ratio with the lymphocytes from the Thy1.1 congenic C57BL/6 mice. The numbers shown in the graphs are the percentages of the NCL CD4 T cells, Mekk3 KO CD4 T cells, and their respectively mixed Thy1.1 CD4 T cells, as determined by multicolor flow cytometry before transfer. The mixed cells were also labeled with 5 μM CFSE before transfer. B, Multicolor flow cytometry analyses of the donor CD4 T cells from the spleens and LN of recipient mice at day 7 after transfer using Abs against CD45.2, CD4, CD8, and Thy1.1. The CD45.2-positive cells (donor cells) were gated and analyzed for CD4+/Thy1.1− and CD4+/Thy1.1+ populations, as indicated (upper graphs). The numbers next to the gated cells indicate the relative percentages of donor NCL CD4 T cells, the Mekk3 KO CD4 T cells, and the cotransferred Thy1.1 CD4 T cells. The gated CD4+/Thy1.1− and CD4+/Thy1.1+ populations from the upper panels were further analyzed for CFSE dye dilution, as shown by histograms in the middle panels. The solid line shows the CFSE dilution of Thy1.1 CD4 T cells, whereas the shadowed areas are for the cotransferred NCL or Mekk3 KO T cells. Bottom two graphs, Show the expanded CFSE dilution peaks of Mekk3 KO T cells (f/−) from spleen and LN, as indicated by the arrows. The f.p. cells are indicated. C, Same as A, except the staining was for CD8 T cells. D, Same as B, except the staining was for CD8 T cells. E, Analysis of CFSE dilution of donor CD4 T cells under lymphopenic condition at days 3 and 5. Donor T cells were mixed, labeled with CFSE, transferred to irradiated recipient mice, and analyzed at day 3 or 5, as indicated after transfer and as described above in A and B. The numbers show the relative percentages of the cells divided more than once. F, Bone marrow cells isolated from Mekk3 T-KO and WT control (C57BL/6-Ly5.2) mice were mixed at roughly 1:1 ratio before being transferred into lethally irradiated C57BL/6-Ly5.2 mice. Eight weeks later, the ratios of WT (CD45.1+CD45.2−) and Mekk3 KO T cells (KO) (CD45.1−CD45.2+) were analyzed by flow cytometry on gated thymic CD4SP, CD8SP, and DP cells, and splenic CD4, CD8, and non-T (CD4−CD8−) cells, respectively, as indicated. The numbers in the graphs show the percentages of the gated cells in each analysis.
The proliferation of donor T cells was assessed by CFSE dilution. Whereas the WT CD4 T cells divided similarly to the cotransferred Thy1.1 counterparts, interestingly, the slower proliferating population, but not the f.p. population, of the Mekk3 KO CD4 T cells divided much less in the recipient as compared with its cotransferred Thy1.1 counterparts (Fig. 3B, lower graphs). Up to four distinct CFSE peaks were observed in the slow proliferating Thy1.1 CD4 T cells or NCL CD4 T cells (39% of the cells that divided once, 21% divided twice, and 6% divided three times, respectively, for the Thy1.1 CD4 T cells). In contrast, only one CFSE dilution peak containing ~20% of the cells could be detected in the slow proliferating Mekk3 KO CD4 T cells. This reduced CFSE dilution in Mekk3 KO CD4 T cells was observed as early as day 3, and became clearer at day 5 after transfer (Fig. 3E). The Mekk3 KO CD8 T cells, however, appeared to be able to divide as well as the WT T cells (Fig. 3D, bottom graphs).
To further confirm that the defects in peripheral homeostasis of Mekk3 KO CD4 and CD8 T cells are intrinsic to the Mekk3 gene deletion, we conducted mixed bone marrow transfer experiments. As shown in Fig. 3F, we found that in the presence of WT T cell competition, the recovery of Mekk3 KO CD4 and CD8 T cells was significantly reduced in the periphery, but not in the thymus, consistent with our conclusion that MEKK3 is required for peripheral T cell homeostasis.
MEKK3 is not required for Ag- or anti-TCR/CD3-induced proliferation
In the above adoptive transfer experiments, we found that the f.p. subset of CD4 T cells appeared not requiring MEKK3 for proliferation in contrast to the slower proliferating subset (Fig. 3B, lower graphs). It is believed that the T cell subsets that undergo slower proliferation are responding to the homeostatic/lymphopenic signals, whereas the subset of CD4 T cells that undergo fast proliferation is induced by either commensal bacteria Ags or by high-affinity self Ags. Some of those cells could also be effector memory cells (1, 4).
These results, therefore, seem to suggest that MEKK3 may be required only for homeostatic proliferation signals, but not for antigenic stimulation. Indeed, when we examined the purified Mekk3 KO CD4 T cells isolated from either the spleen or LN for their responses to anti-TCR/CD3 stimulation, we found no difference between the NCL and Mekk3 KO T cells (Fig. 4, A and B). We further examined the induction of Mekk3 KO CD4 T cell proliferation by foreign Ag stimulation using the Mekk3 KO OVA-specific OT-II TCR-transgenic CD4 T cells in vivo and in vitro. We found that Ag OVA induced similar T cell proliferation of both WT and Mekk3 KO OT-II T cells (Fig. 4, C and D). These results thus indicate that the MEKK3 pathway may be specifically used by T cells for the homeostatic proliferation, but not Ag-driven proliferation.
FIGURE 4.
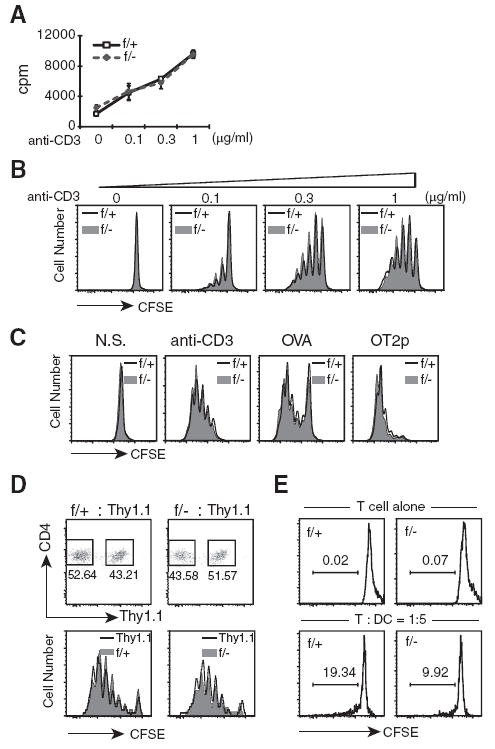
MEKK3 is not required for Ag-induced proliferation. A, Naive CD4 T cells (CD4+/CD62L+) were purified by FACS sorting from either NCL (f/+) or Mekk3 T-KO (f/−) mice. Equal numbers of each sorted cells were stimulated for 3 days in triplicates with different concentrations of plate-bound anti-CD3 Ab, as indicated. [3H]Thymidine was added during the last 12 h of culture before harvesting. Average [3H]thymidine incorporation was determined by scintillation counting. The data shown are a representative of three independent experiments from three pairs of NCL and Mekk3 T-KO mice. B, The purified naive CD4 T cells were labeled with 5 μM CFSE before being stimulated with either no Ab, or with 0.1, 0.3, and 1 μg of anti-CD3 Ab in the presence of T cell-depleted C57BL/6 splenocytes as APCs. CD4 T cells were analyzed 3 days later for CFSE dilution. The solid lines in the graphs show CFSE dilutions of NCL CD4 T cells (f/+), whereas the shaded areas represent the CFSE dilutions of Mekk3 KO CD4 T cells (f/−). C, Splenocytes from NCL (f/+) and Mekk3 T-KO (f/−) OT-II transgenic mice were labeled with CFSE and stimulated with anti-CD3 Ab (1 μg/ml), OVA (20 μg/ml), or OVA323–339 peptide (OT2p, ISQAVHAAHAEINEAGR) (5 μg/ml), as indicated. CFSE dilution was analyzed by flow cytometry on gated CD4+TCRvβ5+ cells at day 3. D, CD4 T cells from NCL (f/+) and Mekk3 T-KO (f/−) OT-II transgenic mice were mixed at 1:1 ratio with CD4 T cells from Thy1.1/OT-II transgenic mice, and then labeled with CFSE. Ten million mixed OTII cells were transferred via i.v. into C57BL/6-Ly5.2 recipient mice. The recipient mice were immunized with OVA 24 h later, and splenocytes from the recipient mice were collected for flow cytometry analysis at day 3. The CD4+CD45.2+TCRvβ5+ positive cells (donor cells) were gated and analyzed for Thy1.1− and Thy1.1+ populations, as indicated (upper graphs). The numbers next to the gated cells show the relative percentages of the donor NCL CD4 T cells, Mekk3 KO CD4 T cells, and the cotransferred Thy1.1 OT-II T cells. The gated CD4+/Thy1.1− and CD4+/Thy1.1+ populations from the upper panels were further analyzed for CFSE dilution, as shown by histograms in the lower panels. The solid line shows the CFSE dilution of Thy1.1 CD4 T cells, whereas the shadowed areas are for the cotransferred NCL or Mekk3 KO OT-II T cells. Data are representative of three independent experiments with three pairs of recipients. E, CFSE-labeled f/+ or f/− CD4 T cells (4 × 105) were cultured in 24-well plates alone or with BMDCs (2 × 106). Cell proliferation was assayed at day 5 by CFSE dilution on gating CD4+ T cells. The numbers in the graphs show the percentages of CD4 T cells that were divided in coculture condition. In the absence of DCs, no cell division was detected in either f/+ or f/− CD4 T cells.
To further test whether MEKK3 may be required for spMHC-driven homeostatic proliferation, we used a DC-T cell coculture assay described previously for non-Ag-driven T cell proliferation in vitro by syngeneic DCs (25). As shown in Fig. 4E, when cocultured with syngeneic DCs, the percentage of dividing WT CD4 T cells (19.32%) was almost double the percentage of dividing Mekk3 KO T cells (9.92%). In the absence of syngeneic DCs, neither WT nor Mekk3 KO CD4 T cells were able to proliferate. These results thus suggest that MEKK3 is required for mediating the spMHC-induced TCR signals for T cell homeostasis. Consistent with these results, we found that when WT or Mekk3 KO CD4 T cells were transferred into the MHC class II-deficient mice, similar numbers of WT and Mekk3 KO donor T cells were recovered (supplemental Fig. S4).4
MEKK3 regulates CD44 expression in naive CD4 T cells
It has been previously shown that when naive CD4 T cells undergo a homeostatic/lymphopenic proliferation, there is a concurrent up-regulation of CD44 expression from low to medium-high (23, 57). This concomitant CD44 up-regulation was not accompanied by up-regulation of other T cell activation markers such as CD69 and CD25, or down-regulation of CD62L (25, 26), suggesting that this CD44 up-regulation is a process of T cell homeostasis response. We thus examined the CD44 levels in naive (CD62Lhigh) CD4 and CD8 T cells from the spleens of NCL and Mekk3 T-KO mice. Whereas we did not find any significant differences in the expression of T cell surface activation markers such as CD69, CD25, CD62L, and CD5 between NCL and Mekk3 KO CD4 T cells (supplemental Fig. S5 and S6),4 the levels of CD44 expression on the surface of Mekk3 KO naive CD4 cells were consistently reduced (Fig. 5, A–C). When CD62Lhigh CD4 naive T cells were arbitrarily divided into CD44low, CD44medium, or CD44high subsets (Fig. 5A), Mekk3 T-KO mice had significantly more CD62LhighCD44low, but much less CD62LhighCD44medium CD4 T cells when compared with the NCL mice (Fig. 5, A and B). Because up-regulation of CD44 expression in naive CD4 T cells is suggested to associate with homeostatic proliferation of CD4 T cells in the periphery, these results further support a role of MEKK3 in CD4 T cell homeostasis. Interestingly, analysis of CD8 T cells from the NCL and Mekk3 T-KO mice (supplemental Fig. S7)4 did not show any difference in CD44 expression, indicating that MEKK3 is differentially involved in CD4 and CD8 T cell homeostasis.
FIGURE 5.
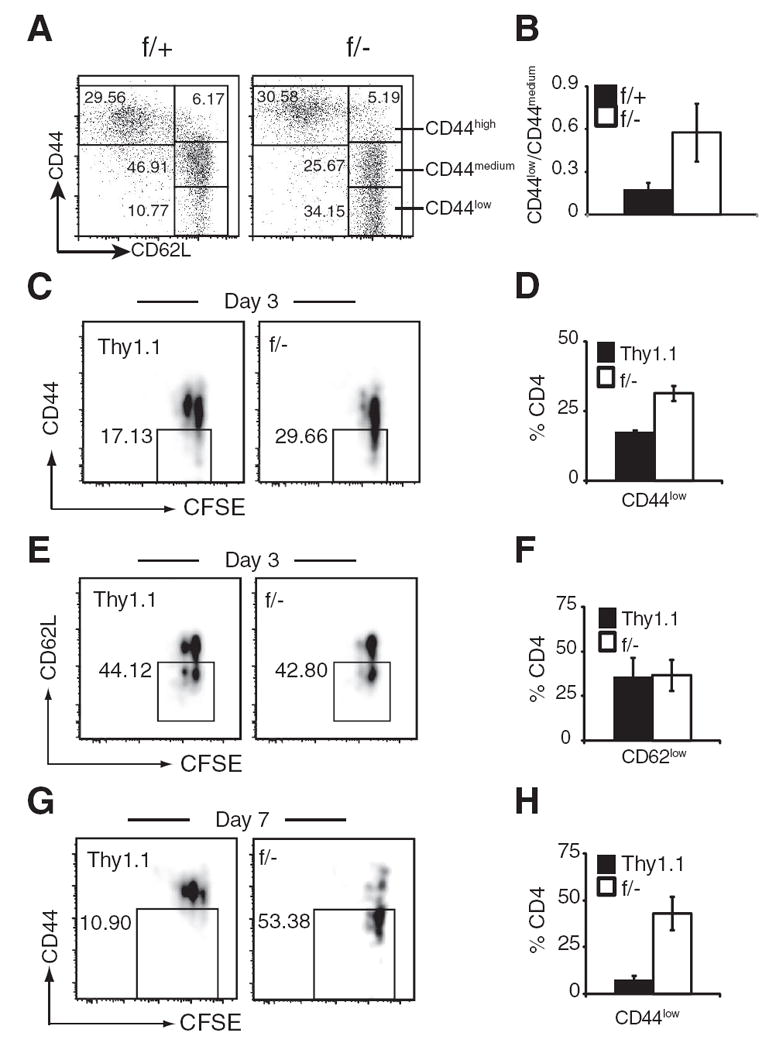
MEKK3 regulates naive CD4 T cell CD44 expression. A, Spleen CD4 T cells from NCL (f/+) and Mekk3 T-KO mice (f/−) were analyzed for their CD44 and CD62L expression by flow cytometry. The plots shown are for gated CD4 T cells. The numbers in the figure show percentages of the gated CD4 T cells. The CD62Lhigh population was divided into three subpopulations designated as CD44high, CD44medium, and CD44low based on their CD44 levels, as indicated. B, A bar graph shows the average ratios of CD44low to CD44medium naive (CD62Lhigh) CD4 T cells from NCL (f/+) and Mekk3 T-KO (f/−) mice. Data represent mean ±SD of the results from analysis of 11 pairs of 6- to 10-wk-old mice. C and H, Analysis of CD44 expression in donor CD4 T cells from the recipient mice. C, E, and G, CFSE-labeled Thy1.1 LN T cells were mixed with Mekk3 KO T cells (f/−) at ~1:1 ratio and transferred into sublethally irradiated C57BL/6-Ly5.2 mice, as described in Fig. 3. Recipient mice were analyzed at day 3 or 7, as indicated. CD44 (C and G) expression, CD62L (E) expression, and CFSE dye dilution of the transferred (donor) CD4 T cells (by gating on the CD4+CD45.2+ donor population) are shown. Thy1.1 and Mekk3 KO donor T cells are distinguished by gating on Thy1.1+ and Thy1.1− (f/−) populations. The numbers next to the gated cells in the plots are percentages of CD44low or CD62Llow cells, respectively. D, F, and H, Bar graphs show the percentages of CD44low (D and H) and CD62Llow (F) T cells to the total donor Thy1.1 (■) or Mekk3 KO (f/−) T cells (□), as indicated. Data are representative of three independent experiments, with three pairs of recipient mice in each experiment.
We next examined up-regulation of CD44 expression in naive CD4 T cells transferred to lymphopenic mice using the same mixed transfer protocol described above. As shown in Fig. 5, C and D, 3 days after transfer, the percentage of Mekk3 KO CD44low T cells that had not been divided (no CFSE dilution) was higher (29.66%) than that of WT CD4 T cells (17.13%). In contrast, at this time point, we did not observe any difference in CD62L expression between the control and Mekk3 KO CD4 T cells (Fig. 5, E and F). Furthermore, 7 days posttransfer, the percentage of Mekk3 KO CD44low cells was even higher, whereas almost all WT cells up-regulated their CD44 level (Fig. 5, G and H).
MEKK3 is not required for T cell trafficking/homing
In addition to the defects in proliferation, problems in trafficking and homing to secondary lymphoid organs may also account for the decreased numbers of Mekk3 KO CD4 and CD8 T cells in the spleens and LN of the recipient mice described above. To address this possibility, we conducted the mixed transfer experiment and determined the recovery of Mekk3 KO T cells at early time points (24–72 h) following the transfer. To avoid the contribution of cell number change due to cell proliferation, nonirradiated mice were used as recipient hosts because LIP does not occur in nonlymphopenic host. As shown in Fig. 6, A and B, there was no difference in trafficking or homing of Mekk3 KO CD4 T cells to spleen at 24 and 72 h. As expected, no proliferation of either WT or Mekk3 KO T cells was observed, as indicated by the lack of CFSE dilution (Fig. 6B). Similarly, no difference in trafficking and homing to LN was detected between NCL and Mekk3 KO T cells either (data not shown). Similar results were found for CD8 T cells (supplemental Fig. S8).4 Thus, these results indicate that MEKK3 may not play a role in T cell trafficking or homing to the secondary lymphoid organs.
FIGURE 6.
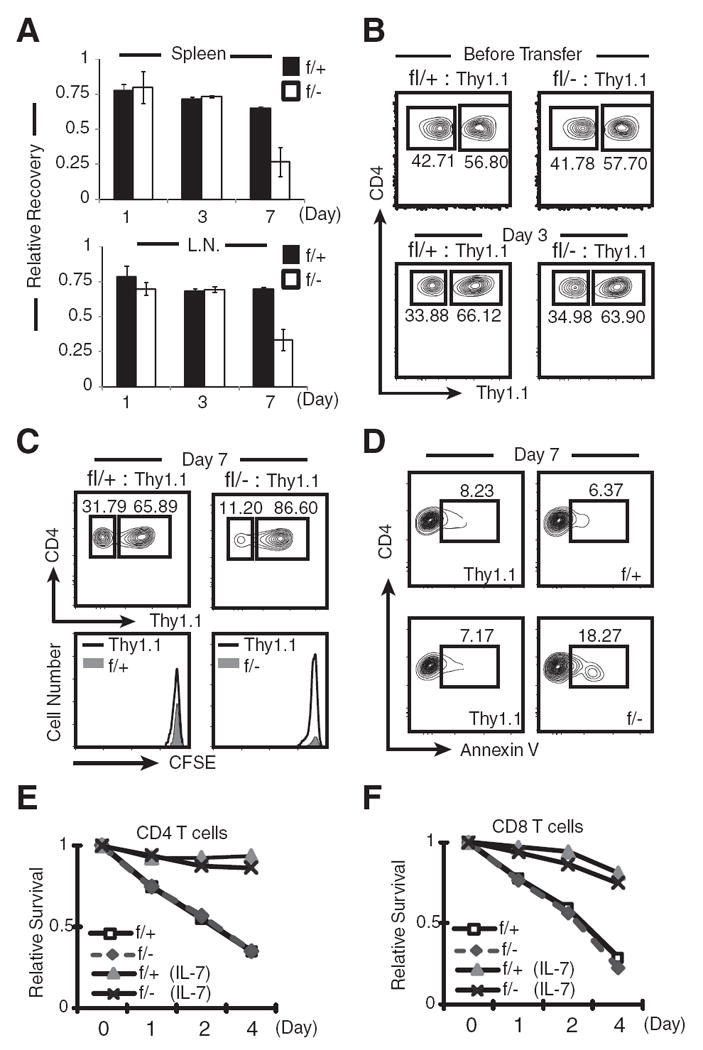
MEKK3 is not required for T cell homing and IL-7-mediated T cell survival. A, Purified CD4+ SP thymocytes from Thy1.1 mice were mixed at ~1:1 ratio with purified CD4+ SP thymocytes from Mekk3 T-KO (f/−) or NCL (f/+) mice, respectively. The mixed cells were transferred into nonirradiated C57BL/6-Ly5.2 mice, as described in Fig. 3. The donor CD4 T cells (CD45.2+) in the spleen and LN were analyzed by flow cytometry at days 1, 3, and 7, respectively, for their recovery relative to the cotransferred Thy1.1 CD4 T cells. The relative recovery is calculated by the following formula: For f/− cells, ([(f/−)%/(Thy1.1)%] after transfer)/([(f/−)%/(Thy1.1)%] before transfer); for f/+ cells, ([(f/+)%/(Thy1.1)%] after transfer)/([(f/+)%/(Thy1.1)%] before transfer). B and C, Flow cytometry graphs show the percentages of NCL (f/+) or Mekk3 KO (f/−) donor CD4 T cells relative to the cotransferred Thy1.1 CD4 T cells before transfer, at day 3 (B), and day 7 (C) after transfer. The CFSE labeling at day 7 was also shown (C). D, Flow cytometry plots show the percentages of the apoptotic cells in adoptively transferred Thy1.1, NCL (f/+), and Mekk3 KO (f/−) CD4 T cells, as determined by annexin V staining. The cells shown in the plots are gated on donor CD4 T cells (CD45.2+). The Thy1.1+ and Thy1.1− (f/+ or f/−) cells are also shown. Numbers shown in the plots are percentages of annexin V-positive cells. E and F, Spleen CD4 (E) or CD8 (F) T cells were cultured with 10 ng/ml IL-7 or with medium alone for 1, 2, and 4 days before being assessed for their survival by 7-AAD and annexin V double staining. The 7-AAD and annexin V DN cells at the beginning of culture were set arbitrarily as 1. Relative survival rate was calculated as the percentage of 7-AAD and annexin V DN cells at each time point normalized to day 0 (beginning of culture), which was set as 100%.
MEKK3 is required for survival of naive T cells
Although Mekk3 KO CD4 T cells were able to home to spleen and LN between day 1 and 3, at day 7 posttransfer, there was a significant reduction of Mekk3 KO CD4 T cells as compared with the cotransferred WT Thy1.1 cells (Fig. 6, A and C). Because neither WT nor Mekk3 KO T cells proliferated under this condition, as indicated by the lack of CFSE dilution, this result indicated that MEKK3 is required for naive CD4 T cell survival in the periphery. Consistent with this hypothesis, a significant increase in the numbers of Mekk3 KO CD4 T cells that had undergone apoptosis as compared with the control cotransferred T cells was detected at day 7 after transfer (Fig. 6D). We also examined the survival of donor CD8 T cells in lymphorepleted hosts (nonirradiated hosts) and found that Mekk3 is required for CD8 T cell survival (supplemental Fig. S8).4 Together with the results from Fig. 3, these data demonstrate that MEKK3 is required for survival of naive CD4 and CD8 T cells.
MEKK3 is not required for IL-7-dependent naive T cell survival
IL-7 was shown to play a key role in protecting naive T cells from dying in the periphery under homeostatic conditions (30, 32, 33). We did not find any significant differences in the expression of IL-7R between NCL and Mekk3 KO CD4 T cells (supplemental Fig. S6).4 To further determine whether the IL-7 signaling is MEKK3 dependent, we comparatively examined the survival of control WT T cells and Mekk3 KO CD4 T cells (Fig. 6E) and CD8 T cells (Fig. 6F), either in the absence or presence of IL-7 for 4 days. In the absence of IL-7, Mekk3 KO T cells died at a rate identical with that of WT T cells. Addition of IL-7 to the culture rescued both WT and Mekk3 KO CD4 and CD8 T cells from dying to similar levels (Fig. 6, E and F). These results thus suggest that MEKK3 plays a critical role in maintaining the peripheral T cell survival in an IL-7-independent manner.
MEKK3-dependent lymphopenic proliferation and survival require ERK1/2, but not p38 MAPKs
MEKK3 has been shown to be a potent upstream activating kinase for several MAPKs, including JNK, ERK1/2, p38, and ERK5, in innate immune cells and also in nonimmune cells, but its role as an activator of MAPKs in T cells has not been fully studied. To determine whether MEKK3 is involved in activating MAPKs during T cell homeostasis, we examined ERK1/2 and p38 activation in T cells undergoing homeostatic proliferation. We stained the mixed WT and Mekk3 KO T cells isolated from the recipient mice 5 days after transfer with either an anti-p-ERK1/2 or an anti-p-p38 Ab, and analyzed the levels of active ERK1/2 or p38 by multicolor flow cytometry. As shown in Fig. 7, there was a clear reduction in active ERK1/2 in the Mekk3 KO CD4 (Fig. 7A) and CD8 (Fig. 7B) T cells as compared with that in the control cotransferred WT T cells. Under the same staining condition, very little difference in the level of active p38 was detected between the WT and Mekk3 KO T cells. This result demonstrates that MEKK3 is essential to activate ERK1/2 pathway in response to homeostatic cues. Given the well-documented function of ERK1/2 in regulating cell proliferation and survival, it also suggests that the MEKK3-mediated proliferation and survival of T cells in lymphopenic host are through the activation of ERK1/2 pathway.
FIGURE 7.
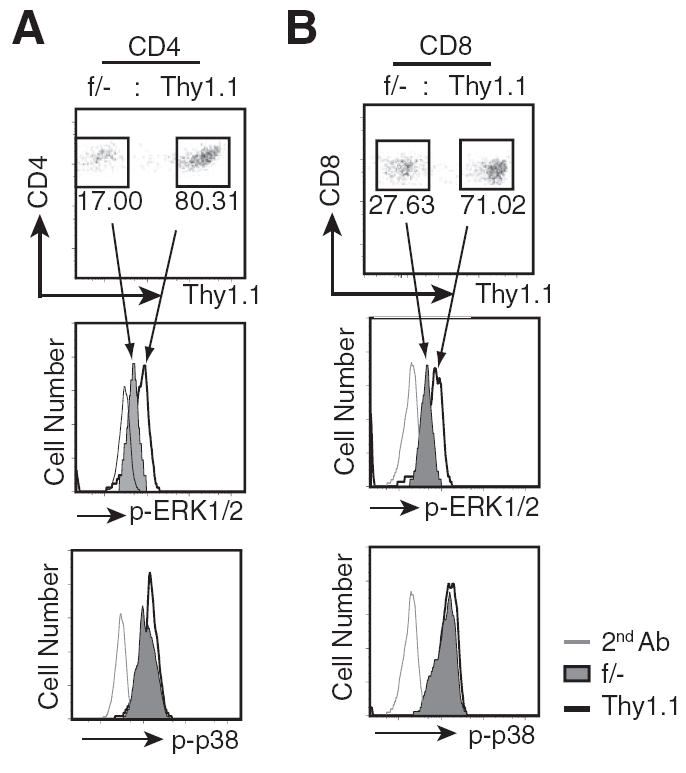
MEKK3 is required for ERK1/2, but not p38 activation in lymphopenic host. A, Splenocytes prepared from the Thy1.1 and the Mekk3 T-KO (f/−) mice were mixed at ~1:1 ratio, CFSE labeled, and transferred into sublethally irradiated C57BL/6-Ly5.1 mice, as described in Fig. 3. The donor CD4 T cells from the spleen of recipient mice were isolated at day 5 after transfer and analyzed for the pERK1/2 and p-p38 level by flow cytometry. The donor T cells were first gated for their CD45.2+ expression, and were further analyzed for the Thy1.1 marker. The numbers shown in the plots (upper panel) are percentages of donor Thy1.1 and Mekk3 KO (f/−) CD4 T cells, respectively. The gated cells in the upper plot (indicated by the arrowed lines) were further analyzed for their p-ERK1/2 and p-p38 activity (histograms in the middle and lower panels). The grey lines show the second Ab staining, the solid lines show the staining for Thy1.1 cells, and the shadowed areas show the staining for Mekk3 KO CD4 T cells, respectively. B, Same as A, except for CD8 T cells. Data are representative of three independent experiments, with three recipient mice in each experiment.
Discussion
T cell homeostasis is a critical process for maintaining a balanced and effective adaptive immunity. Although it has been known for quite some time that both TCR-mediated recognition of spMHC ligands and cytokines are important regulators of T cell homeostasis, the intracellular signal transduction pathways that are responsible for mediating these homeostatic signals are not fully understood. In our present study, we demonstrated that MEKK3, a highly conserved member of MAP3K, is selectively used by T cells to mediate the spMHC-induced TCR signals.
MEKK3 is a ubiquitously expressed Ser/Thr protein kinase in both immune and nonimmune cells. In the T cell lineage, we show that it is expressed as early as the DN stage. Except in DP thymocytes, where its expression seems reduced to some extent, MEKK3 is highly expressed in all other subsets of both naive and differentiated CD4 and CD8 T cells. Whereas these expression results strongly suggest an important role of MEKK3 in T cell function, it should be noticed that MEKK2, a closely related homologue of MEKK3, also is coexpressed with MEKK3 in the same cells (data not shown). This suggests that MEKK2 may compensate for some of the MEKK3 functions in Mekk3 KO T cells, and thus may partially explain the relatively normal thymus development in Mekk3 T-KO mice. Although all the major subsets of thymocytes are relatively unchanged in Mekk3 T-KO mice, we found a small, but seemingly consistent accumulation of DN3 (CD4−/CD8−CD25−/CD44+) subset of thymocytes (data not shown). In fact, this is the stage when a major deletion of MEKK3 by the Lck-Cre begins (supplemental Fig. S1).4 Accumulation of DN3 cells has been suggested to indicate an insufficient pre-TCR signal at this stage (58). We do not know whether this mild defect in Mekk3 T-KO mice is due to the inefficient Mekk3 deletion at this time, or is due to the compensation of MEKK2 expression. Nevertheless, the MEKK3-deficient thymocytes seem to be able to develop through this stage and undergo normal DP and CD4 and CD8 SP cell development.
Despite relatively normal thymus development, the CD4 and CD8 T cells in the spleen and LN of Mekk3 T-KO mice were found significantly reduced. These results lead us to believe that the MEKK3-mediated signaling pathway in T cells may be a critical regulatory circuit for T cell homeostasis. Indeed, by using a mixed adoptive transfer protocol, we have provided strong evidence that MEKK3 is absolutely required for peripheral T cell homeostasis. When the MEKK3-deficient CD4 and CD8 T cells were adoptively cotransferred with the congenic Thy1.1 CD4 and CD8 T cells into lymphopenic recipient mice, they were severely impaired in their ability to undergo homeostasis in lymphopenic hosts as compared with the cotransferred Thy1.1 T cells. In contrast, the T cells from the NCL mice (Lck-Cre-Mekk3f/+) proliferate and survive to the same extent as the Thy1.1 T cells. Interestingly, the reduction of CD4 and CD8 T cells is accompanied sometimes by the reduction of total splenocyte number, resulting in a visibly smaller spleen in size (Fig. 2F). Because Mekk3 is only deleted in T cells, these results may suggest that defective T cell function may also impair the homeostasis of other immune cells such as DCs, B cells, or stromal fibroblastic reticular cells in the periphery.
Although our results show that both CD4 and CD8 T cells require MEKK3 for peripheral homeostasis, the function of MEKK3 in these two subsets of T cells may differ. For CD4 T cells, we found that both LIP and T cell survival were severely impaired. In contrast, CD8 T cells did proliferate in the lymphopenic hosts. We do not know what accounts for this difference, but it has been known that CD8 T cells generally proliferate more vigorously under lymphopenic condition (34, 59). It is also possible that MEKK3 may be dispensable for the proliferation program in CD8 T cells. Alternatively, other MAP3Ks such as MEKK2 may partially compensate the lost of function of MEKK3 in these cells. However, despite their ability to proliferate, Mekk3 KO CD8 T cells, like Mekk3 KO CD4 T cells, were unable to maintain peripheral homeostasis, suggesting that both CD4 and CD8 T cells require MEKK3 for survival.
TCR interaction with spMHC and common γ-chain cytokines such as IL-7 and IL-15 are two essential factors required for peripheral T cell homeostasis. In our study, we found that MEKK3 may not be required for IL-7-mediated survival response because IL-7 appears to protect both the WT and Mekk3 KO T cells equally well in vitro. In this regard, we did not find any significant difference in expression of prosurvival molecule Bcl-2 between the WT and Mekk3 KO T cells (supplemental Fig. S6).4 This seems to be consistent with our unpublished result showing that MEKK3 is not strongly activated by IL-7 in T cells. In contrast, MEKK3 is activated by TCR stimulation, indicating that MEKK3 may be required to transduce the TCR signals (data not shown). The requirement of MEKK3 in the TCR signal transduction is strongly supported by the observation that in Mekk3 KO T cells, the activity of a known MEKK3 target, ERK1/2 MAPK, was attenuated under the lymphopenic conditions (Fig. 7).
Although defects in proliferation and survival of Mekk3 KO T cells are most likely the major causes for their defects in maintaining their peripheral numbers, other possibilities, such as defects in their ability to traffic and home to the secondary lymphoid organs, may also lead to the decrease in the peripheral T cell numbers. However, this seems not the case, because similar numbers of the Mekk3 KO T cells or the NCL T cells and the cotransferred Thy1.1 T cells were recovered within the first 3 days of transfer under nonlymphopenic conditions in which neither WT nor Mekk3 KO T cells underwent proliferation. Furthermore, we found that when cocultured with syngeneic DCs in vitro in the absence of Ag, the Mekk3 KO CD4 T cells also proliferated poorly as compared with the control WT T cells (Fig. 4E). These results thus strongly suggest that MEKK3 is required for transducing the TCR signals induced by spMHC for both CD4 and CD8 T cells to undergo homeostatic/lymphopenic proliferation and survival. Because homeostatic/lymphopenic proliferation and survival of naive T cells may impact the memory T cell development, it would be interesting to test whether there is a defect in the development of memory T cells in Mekk3 T-KO mice in the future.
One surprising result from this study is that the Ag-induced CD4 T cell proliferation does not require MEKK3. We tested it in several ways by using either plate-bound anti-CD3 Ab, soluble anti-CD3 Ab in the presence of T-depleted spleen APCs, or the exogenous Ag OVA (or OVA peptide) to activate MEKK3-deficient OT-II transgenic T cells. In all these cases, the purified naive Mekk3 KO CD4 T cells displayed the same level of proliferation as the CD4 T cells from NCL mice. Consistently, the f.p. population in the LIP assay ( f.p. in Fig. 3B), whose proliferation is believed to be controlled by high-affinity commensal bacterial Ags or high-affinity self Ags, was not affected by MEKK3 deletion. These results suggest an interesting possibility that the MEKK3 pathway may be selectively used by T cells to transduce relatively weak TCR signals initiated by the spMHC ligands in noninfectious conditions. To date, there is no well-established system for studying the difference in TCR response triggered by self Ags and foreign Ags. Our Mekk3 conditional KO mice are likely to be a useful tool for further dissecting the signaling events that are induced by the spMHC ligands.
What are the underlying mechanisms that may account for the difference between the spMHC ligand-induced T cell responses vs the foreign Ag-induced T cell responses? It is known that the homeostatic proliferation induced by spMHC ligands usually does not alter the expression of several classical T cell activation markers such as CD69, CD25, and CD62L. However, it does up-regulate the T cell activation marker CD44 that is more often used as a marker for memory T cells. It was shown that under lymphopenic conditions, CD44 is up-regulated after one round of LIP (14). However, whether CD44 up-regulation is also associated with normal physiological T cell homeostasis remains unclear. In our study, we found that in Mekk3 T-KO mice, there is a significant increase of CD44low naive CD4 T cells (CD62Lhigh) (Fig. 5). This result suggests that the CD44 level in naive CD4 T cell population may be gradually up-regulated under the slow process of homeostasis. This process seems to require MEKK3 function because, as shown in our mixed transfer experiments, the Mekk3 KO CD4 T cells had the reduced ability to up-regulate their CD44 level, and were also unable to proliferate under these conditions as compared with their cotransferred counterparts (Fig. 5). It is unclear at the moment whether it is the inability of Mekk3 KO T cells to proliferate that prevents the CD44 up-regulation or the result of the defects in CD44 up-regulation that impairs Mekk3 KO T cells’ ability to proliferate. Our results support the notion that CD44 expression is associated with the process of T cell homeostasis in CD4 T cells. It should be noted though that the CD44 expression in Mekk3 KO CD8 T cells was not affected. We do not know the underlying reason for this difference between the CD4 and CD8 T cells. However, given the differential requirement for MEKK3 for lymphopenia/homeostasis-induced proliferation between CD4 and CD8 T cells, this result seems to link the cell proliferation potential with the ability of CD44 up-regulation.
How would the T cell homeostasis defects in Mekk3 T-KO mice affect the overall adaptive immunity in these mice, and how would alteration in T cell numbers affect the repertoire of peripheral T cells? It would be easy to predict that the Mekk3 T-KO mice would have a reduced immune response due to its reduced peripheral T cell numbers. However, because the Ag-mediated proliferation of Mekk3 KO T cells remains intact, the Mekk3 KO mice may be able to partially overcome the deficit in the number of peripheral T cells. Instead, the generation and maintenance of memory pool of peripheral T cells may be affected. It is possible that MEKK3 is also required for maintaining memory T cell homeostasis. Finally, because the Mekk3 T-KO mice are chronically lymphopenic, it is possible that these mice may select certain high-affinity autoreactive T cells that may lead to the development of autoimmune diseases. Future studies in these areas with our Mekk3 T-KO mice should prove to be fruitful.
Supplementary Material
Acknowledgments
We thank Drs. Frank Costantin, Christopher B. Wilson, Tian Chi, Chen Dong, Susan Kaech, David Rothstein, and Pumin Zhang for sharing the following reagents: Thy1.1 OTII-transgenic mice, OTII-transgenic mice, ROSA26-EYFP mice, CD4-Cre mice, MHC class II KO mice, and Frt-loxp-Neor plasmid. We thank Dr. Xiao-Feng F. Qin for help with the staining of T regulatory cells, and Dr. Susan Kaech and Timothy Hand for advice on mixed transfer experiments. We also thank Drs. Jianzhu Chen, Susan Kaech, and Adam Lazorchak for critically reading the manuscript and for insightful suggestions. In addition, we thank Dou Liu, Adam Lazorchak, and other members in the Su laboratory for helpful suggestions and technical help.
Footnotes
This work was supported in part by National Institutes of Health Grant AI 063348 (to B.S.), GM 59638 (to Y.Z.), and American Heart Association Grant 0765060Y (to V.F.). X.C. is a recipient of Gershon/Trudeau Fellowship from Immunobiology at Yale University.
Abbreviations used in this paper: LIP, lymphopenia-induced proliferation; 7-AAD, 7-aminoactinomycin D; BMDCs, bone marrow-derived dentritic cells; DN, double negative; DP, double positive; ES, embryonic stem; f.p., fast proliferating; KO, knockout; LN, lymph node; MAP3K, MAPK kinase kinase; NCL, normal control littermate; sp, self peptide; SP, single positive; WT, wild type.
The online version of this article contains supplemental material.
Disclosures The authors have no financial conflict of interest.
References
- 1.Sprent J, Cho JH, Boyman O, Surh CD. T cell homeostasis. Immunol Cell Biol. 2008;86:312–319. doi: 10.1038/icb.2008.12. [DOI] [PubMed] [Google Scholar]
- 2.Goldrath AW, Bevan MJ. Selecting and maintaining a diverse T-cell repertoire. Nature. 1999;402:255–262. doi: 10.1038/46218. [DOI] [PubMed] [Google Scholar]
- 3.Freitas AA, Rocha B. Population biology of lymphocytes: the flight for survival. Annu Rev Immunol. 2000;18:83–111. doi: 10.1146/annurev.immunol.18.1.83. [DOI] [PubMed] [Google Scholar]
- 4.Grossman Z, Min B, Meier-Schellersheim M, Paul WE. Concomitant regulation of T-cell activation and homeostasis. Nat Rev Immunol. 2004;4:387–395. doi: 10.1038/nri1355. [DOI] [PubMed] [Google Scholar]
- 5.Surh CD, Sprent J. Homeostatic T cell proliferation: how far can T cells be activated to self-ligands? J Exp Med. 2000;192:F9–F14. doi: 10.1084/jem.192.4.f9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mahajan VS, Leskov IB, Chen JZ. Homeostasis of T cell diversity. Cell Mol Immunol. 2005;2:1–10. [PubMed] [Google Scholar]
- 7.Tanchot C, Rosado MM, Agenes F, Freitas AA, Rocha B. Lymphocyte homeostasis. Semin Immunol. 1997;9:331–337. doi: 10.1006/smim.1997.0090. [DOI] [PubMed] [Google Scholar]
- 8.Freitas A, Chen J. Introduction: regulation of lymphocyte homeostasis. Microbes Infect. 2002;4:529–530. doi: 10.1016/s1286-4579(02)01568-x. [DOI] [PubMed] [Google Scholar]
- 9.Gleeson PA, Toh BH, van Driel IR. Organ-specific autoimmunity induced by lymphopenia. Immunol Rev. 1996;149:97–125. doi: 10.1111/j.1600-065x.1996.tb00901.x. [DOI] [PubMed] [Google Scholar]
- 10.Sakaguchi N, Miyai K, Sakaguchi S. Ionizing radiation and autoimmunity: induction of autoimmune disease in mice by high dose fractionated total lymphoid irradiation and its prevention by inoculating normal T cells. J Immunol. 1994;152:2586–2595. [PubMed] [Google Scholar]
- 11.Williams KM, Hakim FT, Gress RE. T cell immune reconstitution following lymphodepletion. Semin Immunol. 2007;19:318–330. doi: 10.1016/j.smim.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schulze-Koops H. Lymphopenia and autoimmune diseases. Arthritis Res Ther. 2004;6:178–180. doi: 10.1186/ar1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bourgeois C, Stockinger B. T cell homeostasis in steady state and lymphopenic conditions. Immunol Lett. 2006;107:89–92. doi: 10.1016/j.imlet.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 14.Min B, Sempowski GD, Paul WE. Neonates support “homeostatic” proliferation. Adv Exp Med Biol. 2002;512:91–95. [PubMed] [Google Scholar]
- 15.Kieper WC, Troy A, Burghardt JT, Ramsey C, Lee JY, Jiang HQ, Dummer W, Shen H, Cebra JJ, Surh CD. Recent immune status determines the source of antigens that drive homeostatic T cell expansion. J Immunol. 2005;174:3158–3163. doi: 10.4049/jimmunol.174.6.3158. [DOI] [PubMed] [Google Scholar]
- 16.Gudmundsdottir H, Turka LA. A closer look at homeostatic proliferation of CD4+ T cells: costimulatory requirements and role in memory formation. J Immunol. 2001;167:3699–3707. doi: 10.4049/jimmunol.167.7.3699. [DOI] [PubMed] [Google Scholar]
- 17.Jameson SC. Maintaining the norm: T-cell homeostasis. Nat Rev Immunol. 2002;2:547–556. doi: 10.1038/nri853. [DOI] [PubMed] [Google Scholar]
- 18.Surh CD, Sprent J. Regulation of mature T cell homeostasis. Semin Immunol. 2005;17:183–191. doi: 10.1016/j.smim.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 19.Stefanova I, Dorfman JR, Germain RN. Self-recognition promotes the foreign antigen sensitivity of naive T lymphocytes. Nature. 2002;420:429–434. doi: 10.1038/nature01146. [DOI] [PubMed] [Google Scholar]
- 20.Polic B, Kunkel D, Scheffold A, Rajewsky K. How αβ T cells deal with induced TCRα ablation. Proc Natl Acad Sci USA. 2001;98:8744–8749. doi: 10.1073/pnas.141218898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martin B, Becourt C, Bienvenu B, Lucas B. Self-recognition is crucial for maintaining the peripheral CD4+ T-cell pool in a nonlymphopenic environment. Blood. 2006;108:270–277. doi: 10.1182/blood-2006-01-0017. [DOI] [PubMed] [Google Scholar]
- 22.Ernst B, Lee DS, Chang JM, Sprent J, Surh CD. The peptide ligands mediating positive selection in the thymus control T cell survival and homeostatic proliferation in the periphery. Immunity. 1999;11:173–181. doi: 10.1016/s1074-7613(00)80092-8. [DOI] [PubMed] [Google Scholar]
- 23.Clarke SR, Rudensky AY. Survival and homeostatic proliferation of naive peripheral CD4+ T cells in the absence of self peptide:MHC complexes. J Immunol. 2000;165:2458–2464. doi: 10.4049/jimmunol.165.5.2458. [DOI] [PubMed] [Google Scholar]
- 24.Moses CT, Thorstenson KM, Jameson SC, Khoruts A. Competition for self ligands restrains homeostatic proliferation of naive CD4 T cells. Proc Natl Acad Sci USA. 2003;100:1185–1190. doi: 10.1073/pnas.0334572100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ge Q, Palliser D, Eisen HN, Chen J. Homeostatic T cell proliferation in a T cell-dendritic cell coculture system. Proc Natl Acad Sci USA. 2002;99:2983–2988. doi: 10.1073/pnas.052714199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Theofilopoulos AN, Dummer W, Kono DH. T cell homeostasis and systemic autoimmunity. J Clin Invest. 2001;108:335–340. doi: 10.1172/JCI12173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Min B, Yamane H, Hu-Li J, Paul WE. Spontaneous and homeostatic proliferation of CD4 T cells are regulated by different mechanisms. J Immunol. 2005;174:6039–6044. doi: 10.4049/jimmunol.174.10.6039. [DOI] [PubMed] [Google Scholar]
- 28.Voehringer D, Liang HE, Locksley RM. Homeostasis and effector function of lymphopenia-induced “memory-like” T cells in constitutively T cell-depleted mice. J Immunol. 2008;180:4742–4753. doi: 10.4049/jimmunol.180.7.4742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fry TJ, Mackall CL. The many faces of IL-7: from lymphopoiesis to peripheral T cell maintenance. J Immunol. 2005;174:6571–6576. doi: 10.4049/jimmunol.174.11.6571. [DOI] [PubMed] [Google Scholar]
- 30.Schluns KS, Kieper WC, Jameson SC, Lefrancois L. Interleukin-7 mediates the homeostasis of naive and memory CD8 T cells in vivo. Nat Immunol. 2000;1:426–432. doi: 10.1038/80868. [DOI] [PubMed] [Google Scholar]
- 31.Maraskovsky E, O’Reilly LA, Teepe M, Corcoran LM, Peschon JJ, Strasser A. Bcl-2 can rescue T lymphocyte development in interleukin-7 receptor-deficient mice but not in mutant rag-1−/− mice. Cell. 1997;89:1011–1019. doi: 10.1016/s0092-8674(00)80289-5. [DOI] [PubMed] [Google Scholar]
- 32.Tan JT, Dudl E, LeRoy E, Murray R, Sprent J, Weinberg KI, Surh CD. IL-7 is critical for homeostatic proliferation and survival of naive T cells. Proc Natl Acad Sci USA. 2001;98:8732–8737. doi: 10.1073/pnas.161126098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vivien L, Benoist C, Mathis D. T lymphocytes need IL-7 but not IL-4 or IL-6 to survive in vivo. Int Immunol. 2001;13:763–768. doi: 10.1093/intimm/13.6.763. [DOI] [PubMed] [Google Scholar]
- 34.Cho JH, Boyman O, Kim HO, Hahm B, Rubinstein MP, Ramsey C, Kim DM, Surh CD, Sprent J. An intense form of homeostatic proliferation of naive CD8+ cells driven by IL-2. J Exp Med. 2007;204:1787–1801. doi: 10.1084/jem.20070740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 36.Cobb MH. MAP kinase pathways. Prog Biophys Mol Biol. 1999;71:479–500. doi: 10.1016/s0079-6107(98)00056-x. [DOI] [PubMed] [Google Scholar]
- 37.Su B, Karin M. Mitogen-activated protein kinase cascades and regulation of gene expression. Curr Opin Immunol. 1996;8:402–411. doi: 10.1016/s0952-7915(96)80131-2. [DOI] [PubMed] [Google Scholar]
- 38.Dong C, Davis RJ, Flavell RA. MAP kinases in the immune response. Annu Rev Immunol. 2002;20:55–72. doi: 10.1146/annurev.immunol.20.091301.131133. [DOI] [PubMed] [Google Scholar]
- 39.Blank JL, Gerwins P, Elliott EM, Sather S, Johnson GL. Molecular cloning of mitogen-activated protein/ERK kinase kinases (MEKK) 2 and 3: regulation of sequential phosphorylation pathways involving mitogen-activated protein kinase and c-Jun kinase. J Biol Chem. 1996;271:5361–5368. doi: 10.1074/jbc.271.10.5361. [DOI] [PubMed] [Google Scholar]
- 40.Cheng J, Yang J, Xia Y, Karin M, Su B. Synergistic interaction of MEK kinase 2, c-Jun N-terminal kinase (JNK) kinase 2, and JNK1 results in efficient and specific JNK1 activation. Mol Cell Biol. 2000;20:2334–2342. doi: 10.1128/mcb.20.7.2334-2342.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Deacon K, Blank JL. Characterization of the mitogen-activated protein kinase kinase 4 (MKK4)/c-Jun NH2-terminal kinase 1 and MKK3/p38 pathways regulated by MEK kinases 2 and 3: MEK kinase 3 activates MKK3 but does not cause activation of p38 kinase in vivo. J Biol Chem. 1997;272:14489–14496. doi: 10.1074/jbc.272.22.14489. [DOI] [PubMed] [Google Scholar]
- 42.Ellinger-Ziegelbauer H, Brown K, Kelly K, Siebenlist U. Direct activation of the stress-activated protein kinase (SAPK) and extracellular signal-regulated protein kinase (ERK) pathways by an inducible mitogen-activated protein kinase/ERK kinase kinase 3 (MEKK) derivative. J Biol Chem. 1997;272:2668–2674. doi: 10.1074/jbc.272.5.2668. [DOI] [PubMed] [Google Scholar]
- 43.Xia Y, Wu Z, Su B, Murray B, Karin M. JNKK1 organizes a MAP kinase module through specific and sequential interactions with upstream and downstream components mediated by its amino-terminal extension. Genes Dev. 1998;12:3369–3381. doi: 10.1101/gad.12.21.3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- 45.Su B. Linking stress to immunity? Nat Immunol. 2005;6:541–542. doi: 10.1038/ni0605-541. [DOI] [PubMed] [Google Scholar]
- 46.Cheng J, Yu L, Zhang D, Huang Q, Spencer DM, Su B. Dimerization through the catalytic domain is essential for MEKK2 activation. J Biol Chem. 2005;280:13477–13482. doi: 10.1074/jbc.M414258200. [DOI] [PubMed] [Google Scholar]
- 47.Zhang D, Facchinetti V, Wang X, Huang Q, Qin J, Su B. Identification of MEKK2/3 serine phosphorylation site targeted by the Toll-like receptor and stress pathways. EMBO J. 2006;25:97–107. doi: 10.1038/sj.emboj.7600913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang J, Boerm M, McCarty M, Bucana C, Fidler IJ, Zhuang Y, Su B. Mekk3 is essential for early embryonic cardiovascular development. Nat Genet. 2000;24:309–313. doi: 10.1038/73550. [DOI] [PubMed] [Google Scholar]
- 49.Deng Y, Yang J, McCarty M, Su B. MEKK3 is required for endothelium function but is not essential for tumor growth and angiogenesis. Am J Physiol. 2007;293:C1404–C1411. doi: 10.1152/ajpcell.00058.2007. [DOI] [PubMed] [Google Scholar]
- 50.Huang Q, Yang J, Lin Y, Walker C, Cheng J, Liu ZG, Su B. Differential regulation of interleukin 1 receptor and Toll-like receptor signaling by MEKK3. Nat Immunol. 2004;5:98–103. doi: 10.1038/ni1014. [DOI] [PubMed] [Google Scholar]
- 51.Blonska M, Shambharkar PB, Kobayashi M, Zhang D, Sakurai H, Su B, Lin X. TAK1 is recruited to TNF-α receptor 1 in a rip-dependent manner and cooperates with MEKK3 leading to NF-κB activation. J Biol Chem. 2005;280:43056–43063. doi: 10.1074/jbc.M507807200. [DOI] [PubMed] [Google Scholar]
- 52.Yang J, Lin Y, Guo Z, Cheng J, Huang J, Deng L, Liao W, Chen Z, Liu Z, Su B. The essential role of MEKK3 in TNF-induced NF-κB activation. Nat Immunol. 2001;2:620–624. doi: 10.1038/89769. [DOI] [PubMed] [Google Scholar]
- 53.Qin J, Yao J, Cui G, Xiao H, Kim TW, Fraczek J, Wightman P, Sato S, Akira S, Puel A, et al. TLR8-mediated NF-κB and JNK activation are TAK1-independent and MEKK3-dependent. J Biol Chem. 2006;281:21013–21021. doi: 10.1074/jbc.M512908200. [DOI] [PubMed] [Google Scholar]
- 54.Guo Z, Clydesdale G, Cheng J, Kim K, Gan L, McConkey DJ, Ullrich SE, Zhuang Y, Su B. Disruption of Mekk2 in mice reveals an unexpected role for MEKK2 in modulating T-cell receptor signal transduction. Mol Cell Biol. 2002;22:5761–5768. doi: 10.1128/MCB.22.16.5761-5768.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, Muramatsu S, Steinman RM. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony- stimulating factor. J Exp Med. 1992;176:1693–1702. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, Costantini F. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol. 2001;1:4. doi: 10.1186/1471-213X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Min B, McHugh R, Sempowski GD, Mackall C, Foucras G, Paul WE. Neonates support lymphopenia-induced proliferation. Immunity. 2003;18:131–140. doi: 10.1016/s1074-7613(02)00508-3. [DOI] [PubMed] [Google Scholar]
- 58.Michie AM, Zuniga-Pflucker JC. Regulation of thymocyte differentiation: pre-TCR signals and β-selection. Semin Immunol. 2002;14:311–323. doi: 10.1016/s1044-5323(02)00064-7. [DOI] [PubMed] [Google Scholar]
- 59.Kamimura D, Bevan MJ. Naive CD8+ T cells differentiate into protective memory-like cells after IL-2 anti IL-2 complex treatment in vivo. J Exp Med. 2007;204:1803–1812. doi: 10.1084/jem.20070543. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.