

Abstract
The immune system has evolved to protect the host from a universe of pathogenic microbes that are themselves constantly evolving. The immune system also helps the host eliminate toxic or allergenic substances that enter through mucosal surfaces. Central to the immune system’s ability to mobilize a response to an invading pathogen, toxin or allergen is its ability to distinguish self from non-self. The host uses both innate and adaptive mechanisms to detect and eliminate pathogenic microbes. Both of these mechanisms include self-nonself discrimination. This overview identifies key mechanisms used by the immune system to respond to invading microbes and other exogenous threats and identifies settings in which disturbed immune function exacerbates tissue injury.
Keywords: Adaptive immunity, atopy, B cell, complement, costimulation, inflammation, innate immunity, superantigen, T cell, tolerance
Introduction
Humans and other mammals live in a world that is heavily populated by both pathogenic and non-pathogenic microbes, and that contains a vast array of toxic or allergenic substances that threaten normal homeostasis. The community of microbes includes both obligate pathogens, and beneficial, commensal organisms, which the host must tolerate and hold in check in order to support normal tissue and organ function. Pathogenic microbes possess a diverse collection of mechanisms by which they replicate, spread and threaten normal host functions. At the same time that the immune system is eliminating pathological microbes and toxic or allergenic proteins, it must avoid responses that produce excessive damage of self-tissues or that might eliminate beneficial, commensal microbes. Our environment contains a huge range of pathogenic microbes and toxic substances that challenge the host by a very broad selection of pathogenic mechanisms. It is not surprising, therefore, that the immune system uses a complex array of protective mechanisms to control and usually eliminate these organisms and toxins. A general feature of the immune system is that these mechanisms rely on detecting structural features of the pathogen or toxin that mark it as distinct from host cells. Such host-pathogen or host-toxin discrimination is essential to permit the host to eliminate the threat without damaging its own tissues.
The mechanisms permitting recognition of microbial, toxic, or allergenic structures can be broken down into two general categories: i) hard-wired responses that are encoded by genes in the host’s germ line and that recognize molecular patterns shared both by many microbes and toxins that are not present in the mammalian host; and ii) responses that are encoded by gene elements that somatically rearrange to assemble antigen-binding molecules with exquisite specificity for individual unique foreign structures. The first set of responses constitutes the innate immune response. Because the recognition molecules used by the innate system are expressed broadly on a large number of cells, this system is poised to act rapidly after an invading pathogen or toxin is encountered and thus constitutes the initial host response. The second set of responses constitutes the adaptive immune response. Because the adaptive system is composed of small numbers of cells with specificity for any individual pathogen, toxin or allergen, the responding cells must proliferate after encountering the antigen in order to attain sufficient numbers to mount an effective response against the microbe or the toxin. Thus, the adaptive response generally expresses itself temporally after the innate response in host defense. A key feature of the adaptive response is that it produces long-lived cells that persist in an apparently dormant state, but that can re-express effector functions rapidly after another encounter with their specific antigen. This provides the adaptive response with the ability to manifest immune memory, permitting it to contribute prominently to a more effective host response against specific pathogens or toxins when they are encountered a second time, even decades after the initial sensitizing encounter.
Discrimination of Self from Nonself
The immune system employs many potent effector mechanisms that have the ability to destroy a broad range of microbial cells and to clear a broad range of both toxic and allergenic substances. It is critical, therefore, that the immune response is able to avoid unleashing these destructive mechanisms against the mammalian host’s own tissues. The ability of the immune response to avoid damaging self-tissues is referred to as self-tolerance. Because failure of self-tolerance underlies the broad class of autoimmune diseases, this process has been extensively studied. It is now clear that mechanisms to avoid reaction against self-antigens are expressed in many parts of both the innate and the adaptive immune response. The mechanisms that underlie protection of normal self-tissues from immune damage will be discussed as each of the major effector arms of the host immune response is introduced.
Because an important aspect of the T cell arm of the immune system is to recognize host cells that are infected by viruses, intracellular bacteria or other intracellular parasites, T cells have evolved an elegant mechanism that recognizes foreign antigens together with self-antigens as a molecular complex (see ‘Antigen Recognition by T Lymphocytes’ below). This requirement that T cells recognize both self-structures and foreign antigens makes the need for these cells to maintain self-tolerance particularly important.
General Features of Innate and Adaptive Immunity
Broadly defined, the innate immune system includes all aspects of the host’s immune defense mechanisms that are encoded in their mature functional forms by the germ-line genes of the host. These include physical barriers, such as epithelial cell layers that express tight cell-cell contacts (tight junctions, cadherin-mediated cell interactions, and others), the secreted mucus layer that overlays the epithelium in the respiratory, gastrointestinal and genitourinary tracts, and the epithelial cilia that sweep away this mucus layer permitting it to be constantly refreshed after it has been contaminated with inhaled or ingested particles. The innate response also includes soluble proteins and bioactive small molecules that are either constitutively present in biological fluids (such as the complement proteins, defensins, and ficolins1–3) or that are released from cells as they are activated (including cytokines that regulate the function of other cells, chemokines that attract inflammatory leukocytes, lipid mediators of inflammation, reactive free radical species, and bioactive amines and enzymes that also contribute to tissue inflammation). Lastly, the innate immune system includes membrane bound receptors and cytoplasmic proteins that bind molecular patterns expressed on the surfaces of invading microbes. Some aspects of the innate host defenses are constitutively active (such as the mucociliary blanket overlying many epithelia), and others are activated following interactions of host cells or host proteins with chemical structures that are characteristic of invading microbes but that are absent from host cells.
Unlike the innate mechanisms of host defense, the adaptive immune system manifests exquisite specificity for its target antigens. Adaptive responses are based primarily on the antigen-specific receptors expressed on the surfaces of T- and B-lymphocytes. Unlike the germ-line-encoded recognition molecules of the innate immune response, the antigen-specific receptors of the adaptive response are encoded by genes that are assembled by somatic rearrangement of germ-line gene elements to form intact T cell receptor (TCR) and immunoglobulin (B cell antigen receptor; Ig) genes. The assembly of antigen receptors from a collection of a few hundred germ-line-encoded gene elements permits the formation of millions of different antigen receptors, each with potentially unique specificity for a different antigen. The mechanisms governing the assembly of these B and T cell antigen receptors and assuring the selection of a properly functioning repertoire of receptor-bearing cells from the huge randomly generated potential repertoire will be introduced below and discussed in more detail in chapters 3 and 4.
The innate and adaptive immune systems are often described as contrasting, separate arms of the host response; however, they usually act together, with the innate response representing the first line of host defense, and with the adaptive response becoming prominent after several days, as antigen-specific T and B cells have undergone clonal expansion. Components of the innate system contribute to activation of the antigen-specific cells. Additionally, the antigen-specific cells amplify their responses by recruiting innate effector mechanisms to bring about the complete control of invading microbes. Thus, while the innate and adaptive immune responses are fundamentally different in their mechanisms of action, synergy between them is essential for an intact, fully effective immune response.
Cellular Elements of the Immune Response
An intact immune response includes contributions from many subsets of leukocytes. The different leukocyte subsets can be discriminated morphologically by a combination of conventional histological stains, and by analysis of the spectrum of glycoprotein differentiation antigens that are displayed on their cell membranes. These differentiation antigens are detected by their binding of specific monoclonal antibodies. These cell phenotype-determining antigens are assigned cluster of differentiation (CD) numbers. There are currently over 350 defined CD antigens. Updates are issued by Human Cell Differentiation Molecules (HCDM), an organization that organizes periodic Human Leukocyte Differentiation Antigen (HLDA) workshops at which newly identified cell surface molecules are defined and registered. The next HLDA workshop (HLDA9) will be held in Barcelona, Spain, and the summary of authorized CD molecules will be published at http://www.hcdm.org/.
Mature, circulating leukocytes differentiate from hematopoietic stem cells (Figure 1). These stem cells can be recognized by their own spectrum of defining cell surface antigens and can be purified from bone marrow, peripheral blood, and the placenta.4 The recognition that pluripotent hematopoietic stem cells can be purified in substantial quantities has accelerated progress in hematopoietic cell transplantation and provides considerable promise for somatic cell-based gene therapy. Progress in the field of stem cell therapy is described in chapter 30.
Figure 1. Hematopoietic Stem Cell-Derived Cell Lineages.
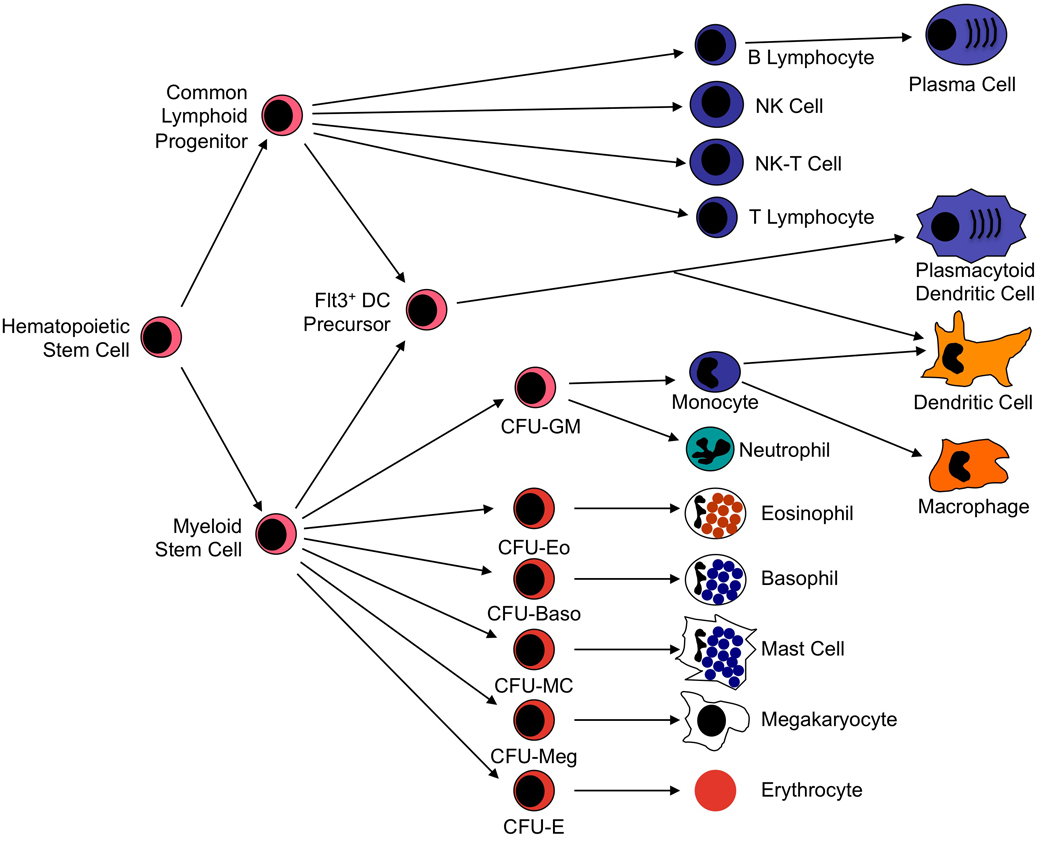
Pluripotent hematopoietic stem cells differentiate in bone marrow into common lymphoid or common myeloid progenitor cells. Lymphoid stem cells give rise to B cell, T cell, and NK cell lineages. Myeloid stem cells give rise to a second level of lineage specific colony form unit (CFU) cells that go on to produce neutrophils, monocytes, eosinophils, basophils, mast cells, megakaryocytes, and erythrocytes. Monocytes differentiate further into macrophages in peripheral tissue compartments. Dendritic cells (DC) appear to develop primarily from a DC precursor that is distinguished by its expression of the Flt3 receptor. This precursor can derive from either lymphoid or myeloid stem cells and gives rise to both classical DC and plasmacytoid DC. Classical DC can also derive from differentiation of monocytoid precursor cells. Modified with permission from Huston.114
Formation of the full complement of immune system cells begins when a pluripotent hematopoietic stem cell differentiates into the common myeloid progenitor cell or the common lymphoid progenitor. The common lymphoid progenitor differentiates further into the four major populations of mature lymphocytes: B cells, T cells, natural killer (NK) cells, and NK-T cells. These lymphocyte subsets can be discriminated by surface phenotype. B cells are phenotypically defined by their expression of the B cell receptor for antigen, membrane anchored Ig. Subsets of B cells have been defined that differ in the types of antigen to which they respond and in the types of antibody they produce. T cells are defined by their cell surface expression of the TCR, a transmembrane heterodimeric protein that binds processed antigen displayed by antigen presenting cells (APC). As will be discussed below, T cells exist in several functionally significant subtypes and subsets of those types. NK cells are defined morphologically as large granular lymphocytes. They are distinguished by their lack of either TCR or surface Ig. They recognize their virus-infected or tumor cell targets using a complex collection of activating and inhibitory cell surface receptors.5 And NK-T cells share characteristics of both NK cells and T cells.6
Myeloid stem cells (also termed common myeloid progenitors) give rise to the several different forms of granulocytes, to megakaryocytes and platelets, and to erythrocytes. Cells of the granulocyte lineage that play prominent immune functions include neutrophils, monocytes, macrophages, eosinophils, basophils, and mast cells. In some mammals, platelets also release immunologically significant mediators that expand their repertoire beyond their role in hemostasis. The immune functions of the classical granulocytes have been inferred from the immunologically active molecules they produce and from their accumulation in specific pathological conditions. For example, neutrophils produce large quantities of reactive oxygen species that are cytotoxic to bacterial pathogens. They also produce enzymes that appear to participate in tissue remodeling and repair following injury. Neutrophils accumulate in large quantities at sites of bacterial infection and tissue injury and possess prominent phagocytic capabilities that permit them to sequester microbes and particulate antigens internally where they can be destroyed and degraded. Thus, it is clear that they play a major role in clearance of microbial pathogens and repair of tissue injury.7 More recently, however, neutrophils have been recognized to produce substantial amounts of the cytokines Tumor Necrosis Factor (TNF) and interleukin (IL)-12 as well as certain chemokines. This supports an additional immunoregulatory role of neutrophils.
Like neutrophils, monocytes and macrophages are also highly phagocytic for microbes and particles that have been marked for clearance by binding Ig and/or complement. They appear to be mobilized shortly after the recruitment of neutrophils and they persist for long periods at sites of chronic inflammation and infection. In addition to participating in acute inflammatory responses, they are prominent in granulomatous processes throughout the body. They use production of nitric oxide as a major mechanism for killing microbial pathogens, and also produce large amounts of cytokines such as IL-12 and interferon (IFN)-γ giving them a regulatory role in adaptive immune responses. Depending on the nature of activating signals that are present when macrophages differentiate from immature precursor cells and when they receive their first activation signal, macrophages can adopt one of several phenotypes.8 Classically activated macrophages produce large amounts of IFN-γ, IL-6, IL-12, and TNF and express potent pro-inflammatory and anti-bacterial activities. Alternatively activated macrophages are induced by IL-4, IL-10, or IL-13, especially in the presence of glucocorticoid hormones and express anti-inflammatory functions through their own production of IL-10, the IL-1 receptor antagonist, and transforming growth factor β (TGFβ).9 It is likely that further study will identify additional functional macrophage subsets, establishing additional ways in which these innate immune system cells serve fundamental immunoregulatory functions.
Eosinophils are readily recognized by their prominent cytoplasmic granules that contain toxic molecules and enzymes that are particularly active against helminths and other parasites. The production of eosinophils from the bone marrow and their survival in peripheral tissues are enhanced by the cytokine IL-5, making them prominent cells in most allergic responses.10 Basophils and mast cells are morphologically similar cells that represent distinct lineages. By virtue of the cell surface expression of high affinity receptors for IgE (FcεRI), they are key initiators of immediate hypersensitivity responses and the host response to helminthic parasites, releasing histamine and other preformed mediators from their granules and producing important quantities of lipid mediators that stimulate tissue inflammation, edema, and smooth muscle contraction. Recent studies have demonstrated that in addition to their role in immediate hypersensitivity responses, mast cells play prominent roles in the host response to bacterial infection as well. Importantly, mast cells and, more prominently, basophils can release substantial amounts of IL-4, suggesting that they can play important roles in the induction of allergic immune responses.11
Phagocytic cells of the monocyte/macrophage lineage also play key roles in the adaptive immune response by taking up microbial antigens, processing them by proteolysis to peptide fragments, and presenting them in forms that can activate T responses. Additional cells in this lineage include Langerhans cells in the epidermis, Kupffer cells in the liver, and microglial cells in the central nervous system. The most potent types of APC are the broad class of dendritic cells that are present in most tissues of the body and concentrated in the secondary lymphoid tissues.12 All of these cells express both class I and class II major histocompatibility complex (MHC) molecules that are used to permit recognition of processed antigen by the TCR on T cells (see below). All MHC bearing cells appear to have the potential to express APC function if stimulated appropriately. In addition to the conventional dendritic cells described above, which have been thought to be derived from myeloid precursor cells (Figure 1), a second type of dendritic cell is recognized. These cells are designated plasmacytoid dendritic cells because of their histological morphology. They can produce very high levels of type I interferon and are thought to play special roles in antiviral host defense and autoimmunity.13 Recent studies of dendritic cell differentiation indicate that both myeloid stem cells and common lymphoid progenitors can give rise to both conventional dendritic cells and plasmacytoid dendritic cells, most likely through a dendritic cell precursor that is defined by its expression of the fms-like tyrosine kinase receptor-3 (Flt3).14, 15
Antigen Recognition by T Lymphocytes/Major Histocompatibility Molecules
A major challenge faced by the immune system is to identify host cells that have been infected by microbes that then use the cell to multiply within the host. Simply recognizing and neutralizing the microbe in its extracellular form does not effectively contain this type of infection. The infected cell that serves as a factory for production of progeny microbes must be identified and destroyed. In fact, if the immune system were equally able to recognize extracellular microbes and microbially infected cells, a microbe that managed to generate large amounts of extracellular organisms or antigen might overwhelm the recognition capacity of the immune system, allowing the infected cells to avoid immune recognition. A major role of the T cell arm of the immune response is to identify and destroy infected cells. T cells can also recognize peptide fragments of antigens that have been taken up by APC through the process of phagocytosis or pinocytosis. The way the immune system has evolved to permit T cells to recognize infected host cells is to require that the T cell recognize both a self-component and a microbial structure. The elegant solution to the problem of recognizing both a self-structure and a microbial determinant is the family of MHC molecules. MHC molecules (also called the human leukocyte-associated [HLA] antigens) are cell surface glycoproteins that bind peptide fragments of proteins that either have been synthesized within the cell (class I MHC molecules) or that have been ingested by the cell and proteolytically processed (class II MHC molecules).
Class I MHC Molecules
There are three major HLA class I molecules, designated HLA-A, -B, and -C, each encoded by a distinct gene. The class I HLA molecules are cell surface heterodimers consisting of a polymorphic transmembrane 44-kd α-chain (also designated the class I heavy chain) associated with the 12-kd non-polymorphic β2-microglobulin (β2m) protein.16 The α-chain determines whether the class I molecule is an HLA-A, -B, or –C molecule. The HLA-A, -B, and -C α-chain genes are encoded within the MHC on chromosome 6 (Figure 2), and the β2-microglobulin gene is encoded on chromosome 15. The α-chain gene encodes three extracellular domains (designated α1, α2, and α3), a transmembrane domain and a short intracellular domain that anchors the protein in the cell membrane. The α3 domain consists of 5 antiparallel β-strands that form an immunoglobulin-type fold (Figure 3). The α1 and α2 domains each encode an α-helix and several β-strands. The α1 and α2 domains associate with each other with their β-strands forming a platform on which the two α-helices rest. This forms a groove in which antigenic peptides can bind. This complex of class I MHC molecule and antigenic peptide produces a composite structure that is the molecular target of the TCR. The TCR contacts both the antigenic peptide and the flanking α-helices. The TCR has no measurable affinity for the antigenic peptide alone, and very low affinity for MHC molecules containing other peptides. These observations form the molecular basis for the phenomenon of ‘MHC restriction’ described by studies of Zinkernagel and Doherty, in which they recognized that T cells could only recognize their specific antigen when it was presented in association with a specific self-MHC molecule.17
Figure 2. Molecular Map of the Human Major Histocompatibility Complex.

The human MHC, designated HLA, is encoded on the short arm of chromosome 6. The locations of the major HLA and related genes are shown above a scale showing approximate genetic distances in kilobase pairs of DNA (kbp). The genes encoding the Class I HLA heavy chains (shown in blue) are clustered at the telomeric end of the complex. The genes encoding the Class II HLA α and β chains (shown in green) plus the genes encoding the LMP1/2, TAP1/2, and Tapascin (TAPBP) molecules (shown in orange) are clustered at the centromeric end of the complex. In between the Class I and the Class II genes are additional genes designated Class III (shown in red). These include genes encoding the cytochrome P450 21-hydroxylase (CYP21B), an inactive cytochrome P450 pseudogene (CYP21Ps), complement components C4, C2 and factor B (Bf), tumor necrosis factor (TNF), and the two lymphotoxin chains (LTA, LTB). There are two isoforms of complement C4 designated C4A and C4B. C4A interacts more efficiently with macromolecules containing free amino groups (protein antigens), whereas C4B interacts more efficiently with macromolecules containing free hydroxyl groups (glycoproteins and carbohydrates). There are genes encoding two additional HLA Class I-like molecules designated MICA and MICB (shown in purple) located between the Class III genes and the classical Class I genes. Non-functional pseudogenes are shown in gray and further designated by italics.
Figure 3. Structure of HLA Molecules.

Molecular models derived from crystal structures of class I (A–C) and class II (D–F) HLA molecules. A, the class I α1, α2, and α3 domains are shown (light blue) in non-covalent association with the β2m molecule. Coils represent α-helices, and broad arrows represent β-strands. Anti-parallel β-strands interact to form β-sheets. The α-helices in the α1 and α2 domains form the sides and floor of a groove that binds processed antigenic peptides (yellow). The transmembrane and intracytoplasmic portions of the heavy chain are not shown. B, top view of the α1 and α2 domains displaying the antigenic peptide in a molecular complex for recognition by the TCR of a CD8+ T cell (recognition site outlined by pink rectangle). C, side view of the α1 and α2 domains highlighting the TCR contact points on both the α-helices and antigenic peptide. D, side view of the HLA class II molecule showing the α chain (light blue) and the β chain (dark blue). In the class II protein, the peptide-binding groove is made of α helices in both the α1 and β1 domains and a β-sheet formed again by both the α1 and β1 domains. E, top view of the both the α1 and β1 domains and the processed antigenic peptide fragment as they would be seen by the TCR of a CD4+ T cell. F, side view highlighting the α1 and β1 domains and the antigenic peptide. Modified with permission from Bjorkman.16
A key biological consequence of requiring the T cell to recognize antigenic peptides only when they are bound in the groove of an HLA molecule is that this permits the T cell to ignore free extracellular antigen, and to focus rather on cells that contain the antigen. In the case of cells that are infected by a pathogenic microbe, this permits the T cells to focus their response on the infected cells. The α3 domain of the class I heavy chain interacts with the CD8 molecule on cytolytic T cells. This restricts recognition of antigenic peptides that are presented in class I HLA molecules to CD8+ cytolytic T cells. The binding of CD8 expressed by the T cell to the α3 domain of the class I molecule expressed by the APC strengthens the interaction of the T cell with the APC and helps assure that full activation of the T cell occurs.18
A prominent characteristic of HLA molecules is their structural polymorphism. As of October 2009, the ImMunoGeneTics HLA Database (http://www.ebi.ac.uk/imgt/hal/atats.html) recognized over 650 alleles at the HLA-A locus, over 1,000 alleles at the HLA-B locus, and over 350 alleles at the HLA-C locus. This polymorphism is largely in amino acids located in the floor and sides of the peptide binding groove, resulting in different peptide binding specificity of different class I alleles. The fact that there are three distinct HLA class I genes and that each is highly polymorphic means that all individuals in the population who are heterozygous at these loci have 6 distinct peptide binding grooves. Since each class I protein can bind many different peptides, having 6 peptide binding molecules results in the ability to bind a very diverse collection of antigenic peptides. Furthermore, on a population level, the diversity of peptide binding motifs is huge. Mutations in microbial antigens may permit the microbe to avoid binding (and, consequently, recognition) by a few HLA class I alleles, but no mutations will permit the microbe to avoid recognition broadly through the population.
Generally, the antigenic peptides that are found bound in the peptide binding groove of the HLA class I molecules are derived from proteins synthesized within the cell bearing the class I molecules. They are, consequently, described as ‘endogenous’ antigens. The molecular machinery that generates peptide fragments from intracellular proteins and directs them into the grooves of the class I molecules is increasingly well understood (Figure 4). Peptide fragments are generated from cellular proteins by the action of the proteasome, a proteolytic factory composed of over 25 subunits.19 Proteasomes are expressed constitutively in all cell types where they function in cellular homeostasis. Stimulation of cells with IFN-γ activates them for the production of antigenic peptide fragments that can be presented in HLA class I molecules. This activation induces the production of a variant of the proteasome termed the ‘immunoproteasome.’ Two of the subunits of the constitutively expressed proteasome are replaced in the immunoproteasome by the IFN-γ induced LMP2 and LMP7 proteins, both of which are encoded within the HLA complex in the interval between the HLA-DP and the HLA-DQ gene loci (Figure 2). The LMP2 and LMP7 proteins alter the proteolytic specificity of the proteasome, enhancing the production of peptide fragments of appropriate length and charge for binding in the groove of the HLA class I proteins. The addition of another IFN-γ induced protein termed the PA28 proteasome activator also enhances the generation of antigenic peptides that are favorable for presentation in HLA class I molecules.20 After exiting from the immunoproteasome, peptide fragments are transported into the endoplasmic reticulum (ER) by the action of a specific multi-subunit transmembrane transporter. This transporter contains two ATP-binding cassette subunits designated TAP-1 and TAP-2 (Transporters Associated with antigen Presentation) encoded by genes that are located within the MHC gene complex in the same region that encodes LMP2 and LMP7 (Figure 2). Once in the ER, the peptides are loaded into the class I protein binding groove under the direction of the ER protein tapasin with the help of the calcium-binding chaperone protein calreticulin and the oxidoreductase Erp57.21, 22 Prior to its interaction with β2-microglobulin, the class I protein is maintained in a conformation that favors interaction with peptide fragments by association with the chaperone protein calnexin. Interaction with β2-microglobulin stabilizes the complex, causing dissociation of calnexin, and permitting transport of the peptide-loaded class I molecule via the Golgi complex into exocytic vesicles that release the intact complexes onto the cell surface. This pathway is well-adapted to delivering viral peptides produced in a virus infected cell to the cell surface bound to class I HLA molecules in a form that can be recognized by cytotoxic CD8+ T cells. It may also be used to present tumor specific protein fragments that may be useful targets for anti-tumor immunotherapy.
Figure 4. Cellular Pathway for Processing and Presentation of Endogenous Antigens.

Endogenous proteins are digested by the immunoproteasome to small peptide fragments. Production of the immunoproteasome is induced by IFN-γ, which leads to expression of LMP2 and LMP7 (which replace certain components of the conventional cellular proteasome) and the PA28 proteasome activator that modifies the proteasome so that it produces antigenic peptide fragments that are optimal for loading into Class I molecules. Peptides are transferred from the immunoproteasome to the endoplasmic reticulum (ER) via the TAP transporter. There the peptides are loaded, with the help of tapascin, calreticulin and the chaperone Erp57 into a class I heavy chain that associates with a β2m subunit prior to transport to the cell surface where it can be recognized by CD8+ T cells. The association of β2microglobulin with the Class I heavy chain is facilitated by an additional chaperone protein, calnexin. Modified with permission from Huston.114
Studies over the past several years have shown that under certain circumstances exogenous antigens (synthesized outside of the APC) can also be internalized by endocytosis and presented in HLA class I molecules. This uptake of exogenous antigens and display to T cells in HLA class I proteins is known as “cross presentation.”23 Cross presentation is particularly important in antiviral immunity where it helps the host to overcome the ability of some viruses to suppress antigen processing through the endogenous pathway.24
Class II Major MHC Molecules
Like the class I molecules, the class II HLA molecules consist of two polypeptide chains, but in this case both are MHC-encoded transmembrane proteins and are designated α and β. There are three major class II proteins designated HLA-DR, HLA-DQ, and HLA-DP.16 Molecules encoded in this region were initially defined serologically and using cellular immune assays, and consequently their nomenclature does not always reflect the underlying genes encoding the molecules. This is particularly true for HLA-DR, where the genes in the HLA-DR sub-region encode 1 minimally polymorphic (1 common and 2 very rare alleles) α chain (designated DRA) and 2 polymorphic β chains (designated DRB1 and DRB3) (Figure 2). Pairing of the common a chain with the DRB1 chain produces the HLA-DRB1 protein. Over 500 HLA-DRB1 alleles have been defined. Pairing of the common α chain with the DRB3 chain produces molecules designated HLA-DRB2 through HLA-DRB9. There are in total 60 HLA-DRB2 through HLA-DRB9 alleles. The HLA-DQ sub-region encodes 1 polymorphic α chain (25 alleles) and 1 polymorphic β chain (72 alleles). The HLA-DP sub-region encodes 1 polymorphic α chain (16 alleles) and 1 polymorphic β chain (118 alleles). Because both the α chains and the β chains of the HLA-DQ and HLA-DP proteins are polymorphic, each person can express 4 different HLA-DQ and 4 different HLA-DP proteins based on pairing between the gene products of both the maternal and the paternal chromosome. Furthermore, because the minimally polymorphic HLA-DR α chain can pair with an HLA-DRB1 and an HLA-DRB3 chain from both the maternal and the paternal chromosome, each person can express 4 distinct HLA-DR proteins as well. Each of these has the potential to bind a large repertoire of antigenic peptides, making it difficult for a pathogenic microbe to mutate its structure to a form that cannot be recognized by binding in an HLA class II protein.
Each chain of the class II proteins contains a short cytoplasmic anchor, a transmembrane domain, and two extracellular domains designated for the α chain α1 and α2, and for the β chain β1 and β2.16 When the α and β chains pair, the α1 and β1 domains combine to form a peptide-binding groove very similar in structure to that formed by the association of the α1 and α2 domains of the class I proteins. The α2 and β2 domains of the proteins provide a support for this peptide-binding domain and the β2 domain also interacts with the CD4 molecule. This provides a mechanism by which CD4 expressed on helper T cells can enhance the interaction between these T cells and the class II-expressing APC in a fashion similar to the way binding of the HLA class I molecule by CD8 enhances cytotoxic T cell activation.25
The class II proteins are expressed constitutively on B cells, dendritic cells, monocytes and macrophages, all cells that present antigens to CD4+ T cells. Expression of MHC class II proteins can also be induced on many additional cell types, including epithelial and endothelial cells following stimulation with IFN͂γ, permitting these cells to present antigens to CD4+ T cells at sites of inflammation.
Antigens that are presented by class II proteins are loaded into the class II peptide-binding groove via the ‘exogenous’ pathway that starts by endocytosis or phagocytosis of extracellular proteins (Figure 5). The exogenous antigens include antigenic proteins of extracellular pathogens such as most bacteria, parasites, and virus particles that have been released from infected cells and taken up by phagocytosis as well as environmental proteins and glycoproteins such as pollens and venoms, and alloantigens. The ingested antigens are processed to linear peptide fragments by proteolysis after fusion of lysosomes with the phagocytic vacuoles or endosomes to form an acidic compartment.26 The peptide fragments then accumulate in the MHC II loading compartment where they encounter nascent class II proteins. The α and β chains of the class II molecules are synthesized in the ER. In order to protect the class II molecule’s peptide-binding groove so that it can later accommodate an antigenic peptide, the α and β chains associate with the non-polymorphic invariant chain (Ii), assisted by the chaperone protein calnexin. A portion of the Ii chain designated CLIP (class II-associated invariant-chain peptide) lies in the peptide-binding groove of the class II heterodimer, preventing binding of antigenic peptides. Once the class II-Ii complex has formed, it dissociates from calnexin and is transported to the class II loading compartment.27 In the class II loading compartment, the bulk of the invariant chain is degraded by acid proteases such as cathepsins and exchange of the CLIP peptide for an antigenic peptide is catalyzed by the action of the HLA-DM molecule, resulting in the formation of a mature class II protein.28 The class II proteins loaded with antigenic peptide are then delivered to the cell surface by fusion of the class II+ endosome to the plasma membrane.
Figure 5. Cellular Pathway for Processing and Presentation of Exogenous Antigens.

In the endoplasmic reticulum (ER), newly synthesized class II proteins associate with the help of calnexin with an invariant chain protein that protects the antigen-binding groove of the class II molecule until it is transported to the class II+ endosomal protein loading compartment. Exogenous antigens are taken up by phagocytosis or endocytosis, digested by the action of lysosomal enzymes, and transported to the class II+ peptide loading compartment for loading into a class II protein. There the invariant chain is proteolytically degraded and replaced by antigenic peptide with the help of the HLA-DM protein. The assembled class II protein-peptide complex is then delivered to the plasma membrane for recognition by CD4+ T cells. Modified with permission from Huston.114
Association of HLA Types and Disease Susceptibility
Epidemiological studies have demonstrated that over 40 diseases are found more frequently in individuals carrying certain HLA class I or II alleles than in the general population.29 The magnitude of these effects can be quite large, but are probably never absolute. For example, they range from the finding that between 90 and 95% of Caucasian patients with ankylosing spondylitis are HLA-B2730 to the observation that between 30% and 50% of Caucasian patients with type I diabetes mellitus are heterozygous for HLA-DQ2/DQ8.31 Interestingly, HLA-DQ6 appears to provide dominant protection from development of type I diabetes. Most diseases that show linkage of susceptibility to particular HLA genes have a prominent autoimmune character. Although the mechanisms by which HLA genotypes control susceptibility to these diseases remains imprecisely defined, it is likely that the participation of HLA molecules in the establishment of immune tolerance or permitting immune recognition of environmental antigens underlies this phenomenon.32, 33 Protective HLA gene alleles may mediate the elimination in the thymus of potentially pathogenic T cells, whereas susceptibility HLA gene alleles may fail to contribute appropriately to elimination of pathogenic T cells. HLA genotypes can also underlie responsiveness or non-responsiveness to certain vaccines. For example, subjects who are HLA-DR3 have a substantially increased incidence of non-responsiveness to vaccination with hepatitis B surface antigen34 and subjects who are HLA-DRB1*03 or HLA-DQA1*0201 have an increased incidence of seronegativity after measles vaccination.35
HLA-independent Presentation of Antigen
Antigen presentation by class I and class II HLA molecules to CD8+ and CD4+ T lymphocytes is limited to protein antigens. Initially, it was thought that responses to polysaccharide antigens and lipid antigens was restricted to T cell-independent responses that resulted in direct activation of B cells by an antigen with a repeating structure; however, recently it has become clear that there is a class of T cells that recognizes antigens presented by molecules that are not classical HLA class I or class II antigens. One of these classes of T cells uses an antigen receptor composed of α and β chains and recognizes lipid antigens that are presented bound to CD1 molecules.6 CD1 molecules are structurally related to class I HLA molecules, being transmembrane proteins with 3 extracellular domains and associating with β2-microglobulin. There are 5 human CD1 isoforms designated CD1a-CD1e, encoded by linked genes that are not associated with the MHC. X-ray crystallography shows that the α1 and α2 domains of CD1 molecules associate like class I MHC molecules to form a binding groove that can accommodate glycolipid components of microbial pathogens.36 CD1-glycolipid complexes can also serve as targets for recognition by T cells that use the γδ TCR (see below). This presentation of microbial glycolipids by CD1 molecules appears to underlie the MHC-independent recognition of mycobacteria by both αβ and γδ T cells. Glycosphingolipids, a class of carbohydrate-containing lipids that are found in both eukaryotic and prokaryotic cells can also be presented by the CD1d molecule to NK-T cells, leading to their release of large quantities of immunoregulatory cytokines.37 Human γδ T cells can also recognize target cells by virtue of their expression of the stress-inducible MHC class I-related chains A and B (MICA and MICB). MICA and MICB are encoded by genes that lie between the TNF gene cluster in the class III region of the MHC and the HLA-B locus in the class I region (Figure 2). They share structural characteristics with the class I protein heavy chains but appear not to associate with β2microglobulin and not to bind antigenic peptides. Rather, they act as stress-induced molecules that are targets for intestinal γδ T cells, further expanding the repertoire of molecules that can contribute to activation of responding T lymphocytes. In addition to the two functional MICA and MICB genes, there are at least three inactive MIC pseudogenes encoded within the class I region of the MHC (Figure 2).38
T Lymphocytes
The major class of T cells is defined by its surface expression of the αβ TCR. This receptor has evolved primarily to recognize peptide antigens presented in a complex with class I or class II MHC proteins. αβ-T cells differentiate into several different subsets, some of which (CD8+ T cells) act primarily to kill cells infected with intracellular microbes, and others (CD4+ T cells) act primarily to regulate the cellular and humoral immune responses. A small subset of αβ-T cells which expresses the NK1.1 (CD161) NK cell antigen (NK-T cells) are usually CD4 and CD8 double negative, recognize glycolipid antigens presented by the CD1d molecule, and appear to be immunoregulatory based on their ability to release rapidly large quantities of the cytokines IFN-γ, IL-4, granulocyte-macrophage colony stimulating factor (GM-CSF), TNF, and others.39 Details of the mechanisms by which T cells develop, acquiring their antigen specificity, and then are regulated as they encounter antigen in the peripheral tissues are discussed in Chapter 3. An introductory overview is presented here.
T Cell Development
Each individual T cell bears antigen receptors of a single specificity. A repertoire of T cells that can protect against the vast universe of microbial pathogens must, therefore, include a very large number of cells encoding a huge array of discrete TCR. These receptors are somatically assembled from variable, diversity, and joining gene elements to generate mature VαJα chains and VβDβJβ chains (see chapter 3). The assembly of these gene elements is initiated by the lymphoid-specific RAG1 and RAG2 proteins which cleave the DNA near the V, D, and J segments and the gene segments are rejoined by a collection of non-lymphoid-specific DNA repair enzymes including DNA-dependent protein kinase (DNA-PK), Ku, XRCC4, XLF, DNA ligase IV, and the Artemis nuclease.40 XRCC4, XLF, and DNA-PK help recruit the enzyme, terminal deoxynucleotidyl transferase (TdT), which adds deoxynucleotides into some of the VDJ junctions providing extra junctional diversity to the recombined gene sequences.41 The action of these recombinase enzymes results in the V, D, and J gene elements assembling in an apparently random process, producing huge diversity of receptor sequences, but also frequently producing non-functional genes. Selection of cells carrying functional TCR genes occurs in the thymus (Figure 6), a complex lymphoid organ located in the anterior mediastinum at the base of the neck.42 The thymus contains 3 compartments. The first, the subcapsular zone, is where bone marrow-derived pro-thymocytes begin to differentiate, proliferate, and rearrange their TCR β chains. The cells then move to the thymic cortex where the α chain gene elements rearrange, potentially forming a functional, mature αβ TCR. In the cortex, cells test whether their receptors have sufficient affinity for self-MHC molecules to permit them ultimately to recognize antigen-MHC complexes. This involves interactions between the developing lymphocyte and the specialized cortical epithelium.43 If the lymphocyte fails this positive selection, then it undergoes apoptosis and is cleared by thymic cortical macrophages. Finally, in the thymic medulla, cells are screened for potential autoreactivity. This screening includes testing for reactivity for an extensive array of tissue-specific proteins that are expressed by a population of thymic medullary epithelial cells under the control of a gene called AIRE (autoimmune regulator). Defective expression of AIRE gives rise to the severe autoimmune syndrome called autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED).44 Cells that recognize self-peptides expressed by these epithelial cells are removed by apoptosis, and cells that have survived this negative selection are exported to the circulation. Fewer than 5% of the developing T cells survive positive and negative selection.
Figure 6. Differentiation and Maturation of T Cells in the Thymus.

Hematopoietic stem cells which do not express CD3, CD4 or CD8 but which are committed to T cell differentiation move from the bone marrow to the thymic subcapsular zone. There they begin rearrangement of the TCR genes. Once a productive TCR β chain has been produced, they move the thymic cortex where TCR α chain rearrangement occurs and surface expression of the CD3, CD4 and CD8 proteins is induced. These CD4+CD8+ (‘double positive’) cells are positively selected on cortical epithelial cells for their ability to recognize self Class I or Class II HLA proteins. If the developing T cell has adequate affinity to recognize a self Class I protein, then it retains expression of CD8 and extinguishes expression of CD4. If the cell recognizes a self Class II protein, then it retains expression of CD4 and extinguishes expression of CD8. Selected CD4/CD8 single positive (SP) cells then move to the thymic medulla where they are negatively selected on medullary epithelial cells to remove cells with excessive affinity for self-antigens presented in HLA molecules. Cells emerge from positive selection SP for CD4 or CD8 expression and then are exported to the periphery. Cells that fail positive or negative selection are removed by apoptosis. A small fraction of cells differentiate from to rearrange their TCR γ and δ chains, rather than their TCR α and β chains. Modified with permission from Huston.114
Approximately 90–95% of circulating T cells use the αβTCR described above. The other 5–10% use an alternate heterodimeric TCR composed of γ and δ chains. The γ and δchains also assemble by RAG1/RAG2-mediated rearrangement of V, D (for the δ chain only), and J elements. A portion of the γδ T cells is generated in the thymus, but a major fraction appears to be generated in an extrathymic compartment, resulting in cells that largely populate the GI tract.45
T Cell Antigen Receptor Complex
The antigen-specific α and β chains of the TCR associate with invariant accessory chains that serve to transduce signals when the TCR binds to antigen-MHC complexes.46 These accessory chains make up the CD3 complex, consisting of the transmembrane CD3γ, CD3δ, and CD3ε chains plus a largely intracytoplasmic homodimer of two CD3ζ chains. Although the stoichiometry of the CD3 complex is not definitively established, it appears that each TCR αβ pair associates with a CD3γε heterodimer, a CD3δε heterodimer, and a CD3ζ homodimer (Figure 7).
Figure 7. The T Cell Receptor Complex and T Cell Activation.

A, the complete TCR complex includes the rearranged TCR α and β chains and also the CD3γ, CD3δ, CD3ε, and CD3ζ chains. The CD3 chains contain ITAMs in their cytoplasmic domains that can be phosphorylated to activate the intracellular signaling cascade for T cell activation. The signaling protein tyrosine kinases Lck and Fyn associate with the intracellular portions of the CD4 and CD3 chains respectively. TCR engagement by MHC plus peptide without the presence of costimulatory proteins fails to activate phosphorylation of the CD3 ITAMs and results in anergy. B, TCR engagement by MHC plus peptide with costimulatory interactions between CD28 on the T cell and CD80 or CD86 (B7.1 or B7.2) on the APC results in Lck- and Fyn-dependent phosphorylation of the CD3 chains, and recruitment of the adapter protein ZAP-70 to the CD3 complex. This leads to phosphorylation of ZAP-70, which induces the downstream program of T cell activation. C, polyclonal activation of T cells can be elicited by superantigens which interact outside the peptide binding groove with the β1 chain of the class II molecule and with all Vβ chains of a particular subclass. This activates CD4-independent, but Fyn-dependent phosphorylation of the CD3 chains, recruitment and phosphorylation of ZAP-70, and cell activation.
Interaction of the TCR/CD3 complex with antigenic peptide presented in an HLA molecule provides only a partial signal for cell activation. Full activation requires the additional participation of a co-stimulatory molecule, such as CD28 on the T cell and CD80 (also designated B7.1) or CD86 (B7.2) on the antigen-presenting cell (Figure 7).47 In fact, interaction of peptide-MHC with the TCR without a co-stimulator can lead to an anergic state of prolonged T cell non-responsiveness.
The cytoplasmic portions of each of the CD3 chains contain sequence motifs designated immunoreceptor tyrosine-based activation motifs (ITAM). When key tyrosines in these ITAMs are phosphorylated by the receptor-associated kinases Lck and Fyn, this initiates an activation cascade involving the proteins ZAP-70, and farther downstream LAT, and SLP-76. Activation of these proteins leads to stimulation of phospholipase C, activation of the G proteins Ras and Rac, and both protein kinase C and the mitogen-associated protein (MAP) kinases. Together, this complex of activation events leads to activation of genes that control lymphocyte proliferation and differentiation.
The pathways that down regulate this activation pathway are becoming increasingly well defined. The membrane molecule CD45 is a key tyrosine phosphatase that occupies a central position in this de-activating process. In addition, a specific receptor-ligand pair, PD-1 (programmed death-1) and PD-L1 (programmed death ligand 1), transduces signals to the activated lymphocyte to inhibit its proliferation and effector functions, thus extinguishing the T cell response.48 Mutations affecting the function of many of the molecules involved in intracellular lymphoid cell signal transduction processes underlie congenital primary immunodeficiency syndromes (chapter 15).
T Cell Subpopulations
During their progress through the thymus, αβ T cells differentiate into discrete subpopulations, each with defined repertoires of effector functions. The major subsets are defined by their selective surface expression of CD4 or CD8. In the thymus, most developing T cells follow a developmental program in which in the cortex they first express neither CD4 nor CD8 (double negative) and then express both CD4 and CD8 (double positive [DP]).49 DP cells are tested by positive selection in the thymic cortex and those that are selected on class I MHC molecules become CD4−CD8+, and those that are selected on class II MHC molecules become CD4+CD8−. The fact that the CD4 molecule contributes to a stable interaction of the developing T cell with class II MHC molecules on the selecting APC and that CD8 contributes to interactions with class I molecules is central to the association of CD4 with class II MHC restricted antigen recognition and of CD8 with class I restricted antigen recognition. Cells that survive positive selection then move to the thymic medulla for negative selection and export to the periphery. In the blood and secondary lymphoid organs, 60–70% of T cells are CD4+CD8− (CD4+) and 30–40% are CD4−CD8+ (CD8+). CD4+ T cells are generally designated ‘helper cells’ and activate both humoral immune responses (B cell help) and cellular responses (delayed type hypersensitivity responses, others). CD8+ cells show a major cytotoxic activity against cells infected with intracellular microbes and against tumor cells, but also contain regulatory cells that down-regulate immune responses (suppressor cells). A portion of the circulating CD4+ T cells play an important regulatory role that acts to down modulate immune responses. These regulatory T (Treg) cells fall into two groups. The first group develops its regulatory function in the thymus and is known as natural Treg cells. These cells are characterized by surface expression of the CD4 and CD25 antigens and by nuclear expression of the forkhead box P3 transcription factor (Foxp3) that is essential for their development. A major portion of this population’s regulatory activity is due to its secretion of the immunomodulatory cytokines TGFβ and IL-10.50 Under some conditions, suppression of effector T cell proliferation by Treg cells requires cell-cell contact. In this situation, it has been reported that TGFβ acts in a membrane-associated form.51 The second group of Treg cells is thought to differentiate in the periphery from naïve CD4+ T cells. Because they appear to develop in response to stimulation with specific antigen, they are called adaptive or induced Treg cells. Their differentiation appears to depend on the presence of IL-10 during their initial activation. Expression of Foxp3 is variable in this subset, and IL-10 is a prominent secreted product with TGFβ also participating.52 The phenotype of these cells can be unstable, with Foxp3 expression disappearing soon after withdrawal of the inductive IL-10 or TGFβ. Recent studies have indicated that epigenetic modification of the Foxp3 locus, in the form of both histone acetylation and altered DNA methylation in the area around the Foxp3 promoter, are essential for establishment of stable expression of Foxp3 and maintenance of the Treg phenotype.53
Approximately 5–10% of T cells in the peripheral blood, lymph nodes, and spleen are CD4−CD8−. Some of these cells use αβ TCR and others use γδTCR. Double negative cells do not recognize antigen in the context of MHC class I or class II. Some of these cells recognize antigen in the class I-related protein CD1 that is adapted to presentation of glycolipid components of mycobacteria and other microbes.36 A subset of double negative γδ T cells recognizes the MHC class I chain-related proteins designated MIC.38
Both CD4+ and CD8+ T cells differentiate into functionally distinct subsets following exposure to antigen. This is best described for the transition of CD4+ T cells from naïve to effector populations. Resting naïve CD4+ T cells (designated T helper cells, Th) release very low levels of cytokines. Early after stimulation by antigen and APC, the Th cells begin to produce IL-2 and are designated Th0. As the Th cells continue to respond to the activating signal, they progress towards polar extremes of differentiation designated Th1, Th2, and Th17 depending on the nature of the cytokines present at the site of activation.54 IL-12 produced by macrophages or NK cells induces differentiation towards Th1, IL-4 produced by NK1.1+ T cells, basophils, or mast cells induces differentiation towards Th2 and TGFβ and IL-6 produced by yet to be defined cells induce differentiation towards Th17. Th1 cells are characterized by their expression of the transcription factor t-bet and by the production of IL-2, IFN-γ, and lymphotoxin. Th2 cells are characterized by their expression of the transcription factor GATA-3 and produce IL-4, IL-5, IL-9, IL-13, and GM-CSF, and Th17 cells express the transcription factor RORC2 and produce the cytokines IL-6 and IL-1755 (see chapter 3). Th17 cells are induced early in the adaptive response to extracellular bacteria and help to recruit the neutrophil response that eliminates these pathogens. They also direct the destructive inflammatory responses that are part of many autoimmune diseases. Th1 and Th2 cells often participate together in immune responses; however, after prolonged immunization, the response can become dominantly Th1-like or Th2-like. Generally, Th1 cells support cell mediated immune responses, and Th2 cells support humoral and allergic responses. CD8+ T cells also can manifest type 1 and type 2 cytokine responses, in which case the cells are designated T cytotoxic cell type 1 (Tc1) and T cytotoxic cell type 2 (Tc2).56 Understanding the factors that govern whether a Th response adopts a predominantly Th1-type, a Th2-type, or a Th17-type response is crucial to the allergist/clinical immunologist. Recent progress using immunization with different types of adjuvants (for example, CpG DNA) demonstrates the feasibility of reprogramming, in atopic patients, allergic Th2-type responses to non-allergic Th1-type responses.57
Superantigens
Conventional antigens bind to a subset of MHC molecules and to a very small fraction of the huge array of TCR. Thus, a conventional peptide antigen activates only a very small fraction of the total pool of T cells. Superantigens, in contrast, are microbial products that bind to large subsets of TCR proteins and MHC molecules, so that a single superantigen can activate up to 20% or more of the total T cells in the body. The superantigen does this by binding without proteolytic processing to the MHC molecule outside of the antigen-binding groove and to TCR proteins outside of their antigen-MHC binding site (Figure 7). For example the Toxic Shock Syndrome Toxin-1 (TSST-1) produced by Staphylococcus aureus can activate all T cells with TCR that use the Vβ2 and Vβ5.1 chains. The activation of large numbers of T cells induced by superantigens results in the massive release of cytokines producing clinical conditions such as toxic shock syndrome.58
B Lymphocytes
B Cell Development and the B Cell Antigen Receptor
B cells constitute approximately 15% of peripheral blood leukocytes. They are defined by their production of Ig. Except as noted below, Ig molecules are composed of two identical 50 kDa heavy chains and two identical 25 kDa κ or λ light chains (see chapter 3). The amino terminal portions of the heavy and light chains vary in amino acid sequence from one antibody molecule to another. These variable portions are designated VH and Vκ or Vλ, respectively. The juxtaposition of one VH segment and one Vκ or Vλ segment creates the antigen-binding portion of the intact Ig molecule. The variable regions of both the heavy and light chains contain three sub-regions that are highly variable between different antibody molecules. These hypervariable sequences are brought together in the Ig protein to form the antigen-binding domain of the molecule. Thus, each Ig has two identical antigen binding sites. The carboxyl terminal portions of the heavy and light chains are constant in each subclass of antibody. The heavy chain constant regions pair to form the Fc domain of the molecule that is responsible for most of the effector functions of the Ig molecule, including binding to Fc receptors and activating the complement system.
The genes encoding the κ light chain are encoded on chromosome 2, and the genes encoding the λ light chain are on chromosome 22. The complex heavy chain locus is encoded on chromosome 14. The light chain and heavy chain loci are each composed of a series of V (variable) gene elements, followed by several D (diversity) segments (for the heavy chain gene only), some J (joining) segments, and C (constant region) exons. The constant regions of both the κ and the λ light chain genes are encoded as single exons. The heavy chain gene, in contrast, contains exons that encode 9 different constant regions that are used to produce the different classes and subclasses of Ig (Table 1).
Table I.
Structure, Function, and Distribution of Antibody Isotypes
| IgM | IgD | IgG1 | IgG2 | IgG3 | IgG4 | IgA1 | IgA2 | IgE | |
|---|---|---|---|---|---|---|---|---|---|
| Subunit Form1 |
5 | 1 | 1 | 1 | 1 | 1 | 1,2 | 1,2 | 1 |
| Molecular Weight, kDa |
950 | 175 | 150 | 150 | 150 | 150 | 160,400 | 160,400 | 190 |
| Concentration In Serum, mg/ml |
2 | 0.03 | 10 | 4 | 1 | 0.5 | 2 | 0.5 | 0.003 |
| Complement Activating C/A2 |
+/− | −/+ | ++/+ | +/+ | ++/+ | −/+ | −/+ | −/+ | −/− |
| Macrophage FcR Binding |
+ | − | ++ | ++ | ++ | − | ++ | ++ | − |
| Mast Cell Sensitizing |
− | − | − | − | + | − | − | − | +++ |
| Placental Transport |
− | − | ++ | + | ++ | +/− | − | − | − |
| Mucosal Transport3 |
− | − | − | − | − | − | +++ | +++ | − |
5=pentamer, 2=dimer, 1=monomer
C=Classical pathway, A=Alternative pathway
Dimer only
B cells differentiate from hematopoietic stem cells in the bone marrow. It is here that their antigen receptors (surface Ig) are assembled from genetic building blocks in a RAG1/RAG2-mediated process similar to that used for the production of functional TCR.59 The amino terminal portion of each heavy chain is created by somatic joining of genes encoding a variable (VH), diversity (DH), and joining (JH) region. Joining of genes encoding variable and constant light chain gene elements generates the amino terminal portion of the light chain. The VDJ junctions formed by this recombination make up the 3rd hypervariable region that contributes to the antigen-binding site. The amino acid sequence diversity of the 3rd hypervariable region is the result of combinatorial V-D-J joining, and also of non-gene-encoded sequences added into the junction sites by the action of the enzyme TdT that is expressed in developing B cells during the time this gene rearrangement is occurring.
Establishment of the B Cell Repertoire
Differentiation of stem cells to the B lineage depends on bone marrow stromal cells that produce IL-7. The developing B cells follow a program of differential surface antigen expression and sequential heavy and light chain gene rearrangement (Figure 8). First, the recombinase enzyme complex catalyzes the fusion of one of the DH region genes to a JH region gene with the deletion of the intervening DNA sequences. This DHJH recombination occurs on both chromosomes. Next, the recombinase joins one of the VH region genes to the rearranged DHJJ gene. TdT is expressed during this period, resulting in the addition of random nucleotides into the sites of DH-J and VH-DHJH joining, adding to the potential diversity of amino acid sequences encoded by the rearranged VHDHJH gene. The rearranged VHDHJH element forms the most 5’ exon of this rearranged heavy chain gene, and is followed downstream by exons encoding the constant region of the m chain that pairs with a light chain to produce IgM and farther downstream by exons encoding the constant region of the d chain that is used to make IgD. μ chains and δ chains are produced as a result of alternative RNA splicing of the VHDHJH exon to either the μ exons or the δ exons. If the rearrangements of the VH, DH, and JH elements yields a heavy chain transcript that is in-frame and encodes a functional heavy chain proteins, this heavy chain is synthesized and pairs in the cell with two proteins, λ5 and VpreB, which act as a surrogate light chain (Figure 8). Expression of this pre-B cell receptor on the cell surface prevents VH to DHJH rearrangement on the other chromosome, assuring that the developing B cell produces only one antigenic specificity. This process is called allelic exclusion. If the first VHDHJH rearrangement is out of frame and does not produce a functional heavy chain protein, then a VH gene proceeds to rearrange on the other chromosome in a second attempt to generate a successful heavy chain rearrangement. If this second rearrangement is unsuccessful, the cell undergoes apoptosis and is removed.
Figure 8. B Cell Differentiation and Development.

B cells differentiate in the bone marrow from stem cells to become mature surface IgM and IgD expressing cells. This occurs in the absence of antigen. In peripheral lymphoid tissues, the B cell can then mature further under the influence of antigen and T cell help to undergo isotype switching and affinity maturation by somatic mutation. The factors controlling the final differentiation from antibody-secreting B cell to plasma cell are incompletely characterized, but require the participation of the transcription factors Blimp1, Xbp1 and IRF4. Correlations are show between the stage of cell differentiation and the expression of key molecules in the cell (TdT, RAG1/RAG2, cytoplasmic μ) and on the cell surface (class II, CD19, CD21, CD25, CD45, and surface Ig). Modified with permission from Huston.114
Once a functional heavy chain is produced, the cell down regulates its TdT gene and initiates light chain rearrangement. First, a Vκ element rearranges to a Jκ element. If this forms a functional light chain, then the κ light chain pairs with the heavy chain to form a functional Ig protein and further light chain rearrangement stops. If the first κ rearrangement fails, then rearrangement proceeds on the other chromosome. If that fails, then rearrangement of the λ chains proceeds. The RAG1 and RAG2 genes are only expressed during times of heavy and light chain rearrangement, except that some B cells that express autoreactive receptors appear able to re-express their RAG genes and undergo receptor editing by secondary rearrangements of their already rearranged Ig genes.60 These processes result in the assembly of the antigen-binding component of the B cell receptor. Like the TCR, the fully mature B cell receptor also includes additional transmembrane proteins designated Igα and Igβ that activate intracellular signals after receptor binding to antigen.61, 62 B cells also have a co-receptor complex consisting of CD19, CD81, and CD21 (complement receptor 2) that is activated by binding to the activated complement protein C3d.63 Both Igα and Igβ have ITAM domains in their cytoplasmic regions, and utilize similar signal transduction pathways compared to that for T cells. The B cell pathway includes the src-family of kinases Blk, Fyn, and Lyn that phosphorylate the ITAMs on the Igα and Igβ chains. The activation signal is then passed via the tyrosine kinase Syk and the linker protein BLNK to the downstream signaling components phospholipase C and guanine nucleotide exchange factors. Ultimately, as in T cells, activation of protein kinase C, calcium mobilization, and Ras/Rac-dependent activation of MAP kinases leads to activation of new gene transcription that causes cell proliferation and maturation.
Isotype Switching and Affinity Maturation
Naïve B cells express on their cell surfaces IgM and IgD. These two Ig isotypes are expressed by alternative splicing of the same VHDHJH exon to the m and d heavy chain exons. For all heavy chain genes, alternative splicing also permits expression of both membrane-bound (splicing in a transmembrane exon) and secreted (transmembrane exon spliced out) antibody. As B cells mature under the influence of helper T cells, T cell-derived cytokines induce isotype switching. Isotype switching is a process of DNA rearrangement mediated in part by the RNA-editing enzyme activation-induced cytidine deaminase (AID), uracil-DNA glycosylase (UNG), the endonuclease APE1, and the DNA repair enzyme DNA-PK. Switching moves the rearranged VHDHJH exon into a position immediately upstream of alternative heavy chain exons. This permits a functionally rearranged VHDHJH exon to be used to produce antibodies of different isotypes but the same antigenic specificity.64 T cell derived IL-10 causes switching to IgG1 and IgG3. IL-4 and IL-13 cause switching to IgE, and TGFβ causes switching to IgA. IFN-γ or some other undefined product of Th1 cells appears to induce switching to IgG2.
At the same time as B cells undergo isotype switching, an active process produces mutations, apparently randomly, in the antigen-binding portions of the heavy and light chains. This process, designated somatic mutation, also appears to require AID, UNG, APE1, and DNA repair enzymes.65 If these mutations result in loss of affinity for the antigen, the cell loses important receptor-mediated growth signals and dies. If, however, the mutations resulted in increased affinity for the antigen, then the cell producing that antibody has a proliferative advantage in response to antigen and grows to dominate the pool of responding cells. Somatic mutation and clonal expansion of mutated cells occurs in the germinal centers of secondary lymphoid tissues.66
T Cell-Dependent B Cell Responses
Antigens that activate T cells as well as B cells establish Ig responses in which T cells provide ‘help’ for the B cells to mature. This maturation includes both induction of isotype switching, in which the T cell cytokines control the isotype of Ig produced, and activation of somatic mutation. The cellular interactions underlying T cell help are driven by the specific antigen and take advantage of the ability of B cells to serve as APC. B cells that capture their cognate antigen via their membrane Ig can internalize the antigen and process it intracellularly for presentation on the cell surface in the B cell’s class II HLA proteins. Uptake of antigen induces increased class II expression and expression of CD80 and CD86. T cells activated by this combination of co-stimulator and antigen-class II complex on the B cell then signal reciprocally to the B cell by the interaction of the T cell CD40 ligand (CD40L) with B cell CD40. Signaling through CD40 is essential for induction of isotype switching, and human patients with defects in the X chromosome encoded CD40L gene manifest X-linked hyper-IgM syndrome and patients with mutant CD40 show autosomal recessive hyper-IgM syndrome.67
Isotype switching and somatic mutations are strongly associated with the development of B cell memory. Memory responses, defined as rapid induction of high levels of high affinity antibody after secondary antigen challenge, are characterized by production of IgG, IgA, and IgE antibodies, and by somatic mutations in the antigen-binding domains of the heavy and light chains of these antibodies.68 The development of B cell memory is critical to the success of vaccination against pathogens and also perpetuates the pathology of many autoimmune and allergic syndromes. Understanding how to enhance or reduce memory responses will provide important new therapeutic opportunities to the clinical immunologist.
T Cell Independent B Cell Responses
B cells can also be activated successfully without T cell help. T cell independent B cell activation occurs without the assistance of T cell co-stimulatory proteins. In the absence of co-stimulators, monomeric antigens are unable to activate B cells. Polymeric antigens with a repeating structure, in contrast, are able to activate B cells, probably because they can cross-link and cluster Ig molecules on the B cell surface. T cell independent antigens include bacterial lipopolysaccharide (LPS), certain other polymeric polysaccharides, and certain polymeric proteins. Somatic mutation does not occur in most T cell independent antibody responses. Consequently, immune memory to T cell independent antigens is generally weak. This is why it is difficult to create fully protective vaccines directed against polysaccharide components of microbes. Covalent attachment of the polysaccharide component to a carrier protein, in order to recruit T cell help to the response, can induce a beneficial memory response. The value of coupling a polysaccharide antigen to a carrier protein was observed in the Haemophilus influenzae type B vaccine. The original polysaccharide vaccine provided low antibody titers, and no protection for children less than 18 months of age. The current conjugate vaccine generally provides protection beginning at 2–4 months of age.
Lymphoid Tissues
Cellular interactions are essential for a normally regulated, protective immune response. In particular, T cell help is needed to generate high affinity antibody with memory against most protein antigens. A major challenge for the immune system of a naïve subject is to bring rare antigen-specific B cells together with rare antigen-specific T cells and antigen-charged APC. The primary role of the secondary lymphoid tissues is to facilitate these interactions. Generally, the secondary lymphoid organs contain zones enriched for B cells (follicles) and other zones enriched for T cells.69 The B cell zones contain clusters of follicular dendritic cells (FDC) that bind antigen-antibody complexes and provide sites adapted to efficient B cell maturation, somatic mutation and selection of high affinity B cells. The T cell zones contain large numbers of dendritic cells that are potent APC for T cell activation. The tissues also contain specialized vascular structures for recruitment of cells into the tissue. High endothelial venules in lymph nodes, Peyer’s patches, and mucosal associated lymphoid tissues are vascular sites that efficiently extract naïve T and B cells from the circulation into the lymphoid organ. The marginal sinus probably serves a similar function in the spleen. Afferent lymphatic vessels provide efficient entry of antigen-charged antigen-transporting cells (such as epidermal Langerhans cells) from peripheral tissues into lymph nodes. Efferent lymphatic vessels permit efficient export of antigen-experienced cells back into the circulation. Programmed release of distinct chemokines within the lymphoid tissues orchestrate the coming together of antigen-responsive B cells and T cells and then migration of the activated B cells and selected T cells to the FDC clusters where they can form a germinal center.70 In addition to chemokines signals that control leukocyte entry into and migration within secondary lymphoid tissues, it is now understood that specific signals, especially provided by the lysophospholipid sphingosine 1-phosphate, regulate the egress of cells out of the lymphoid tissues and into the circulation.71
Although potent adjuvants can induce some degree of affinity maturation in the setting of congenital absence of lymph nodes and Peyer’s patches, these secondary lymphoid organs are generally essential for the induction of an efficient, protective immune response. Ectopic lymph node-like structure designated tertiary lymphoid tissues can form at sites of chronic inflammation such as the synovial membrane of a joint affected by rheumatoid arthritis. Immune reactions ongoing in these tertiary lymphoid tissues can contribute importantly to the pathogenesis of the inflammatory disease.
Signaling by Cytokines
Cytokines act on cells via transmembrane cell surface receptors. Binding of the cytokine to the receptor elicits its cellular response by activating an intracellular signal transduction pathway that ultimately leads to induction of new gene transcription and synthesis of new cellular proteins. Most cytokine receptors signal using one of the Janus kinase (Jak) family of molecules that then acts on the signal transducers and activators of transcription (STAT) family of proteins. Specific Jak proteins associate with the cytoplasmic domains of cytokine receptor. When the receptor is activated by binding the cytokine, the Jak phosphorylates its respective STAT protein, causing the STAT to dimerize, and translocate into the nucleus where it then initiates new gene transcription. The essential role of Jak and STAT proteins in immune regulation is seen in individuals with inherited deficiency of these molecules (see chapter 12). Jak3 interacts with the γc protein, a subunit of several cytokine receptors including the receptors for IL-2, IL-4, IL-7, IL-9, IL-15 and IL-21. Deficiency of the autosomally encoded Jak3 protein causes autosomal recessive severe combined immune deficiency (SCID).72 Deficiency of the X chromosome encoded γc protein causes X-linked SCID.73 Mutation of STAT1 causes susceptibility to infection with mycobacteria and a variable increase in susceptibility to virus infections because of impaired ability to respond to signals from either type I or type II interferons.74 Homozygous deficiency of STAT3 in mice is embryonic lethal, but heterozygous deficiency of STAT3 in humans causes autosomal dominant hyper-IgE syndrome associated with deficiency of Th17 cell differentiation.75 Deficiency of STAT4 blocks IL-12 signal transduction resulting in impaired development of Th1 cells. And STAT6 deficient mice showed impaired signaling through the IL-4 receptor and inability to generate Th2 cell-dependent responses.76
Effectors of Innate Immunity
While the adaptive T and B cell immune responses provide important protection for the host and permit the development of immune memory, mutations in elements of the innate immune response demonstrate that innate immune effectors are critical for effective host defense. Initially, the innate and adaptive immune responses were thought to act independently, with the innate response providing the first line of defense against invading microbes, and the adaptive response being activated later to sterilize the infection. It is now apparent that the adaptive response has co-opted many of the innate effector mechanisms to enhance its effectiveness. Additionally, the adaptive immune system requires innate signals for its activation. By using innate signals to help initiate its responses, the adaptive immune system takes advantage of the innate system’s ability to discriminate between contact with dangerous pathogens and innocuous or even beneficial microbes and environmental factors. This ability of the innate immune system to sense danger is essential for well-regulated immune responses. Thus, the innate and adaptive arms of the immune response should be viewed as complementary and cooperating.
Toll-Like Receptors
Toll was first identified in Drosophila where Toll was found to control polarity of the developing embryo and later was recognized to participate in the fly’s anti-fungal immunity. Cloning of Drosophila Toll showed that it encoded a transmembrane receptor whose extracellular domain contained leucine-rich repeating units, while its cytoplasmic domain had homology to the cytoplasmic domain of the IL-1 receptor of mammals (designated the Toll/IL-1 Receptor domain, TIR). This suggested that there might be Toll homologues in mammals. Indeed, 10 human Toll-like receptors (TLR) have now been defined. The TLR appear largely to recognize pathogen-associated molecular patterns (PAMP).77 These include LPS from Gram-negative bacteria, peptidoglycan, lipoteichoic acid, lipoarabinomannan, bacterial flagellar proteins, viral double stranded RNA, and unmethylated DNA with CpG motifs characteristic of microbial DNA. TLR are particularly found on macrophages and dendritic cells, but also are expressed on neutrophils, eosinophils, epithelial cells, and keratinocytes. Although activation of some TLR can activate or potentiate an allergic, Th2 type response, activation of most TLR elicits mediators that program CD4 T cells towards a non-atopic Th1 response. TLR9, activated by interaction with CpG DNA, provides the molecular basis for efforts to divert Th2-driven atopic responses to non-atopic Th1 dominated responses.78 Downstream signal transduction through most TLR is dependent on MyD88, a cytoplasmic protein encoded by the myeloid differentiation primary response gene 88. MyD88 also mediates signaling through the IL-1 receptor. MyD88-defficiency leads to life threatening, recurrent pyogenic infections.79
Nucleotide-Binding Domain, Leucine-Rich Repeat (NLR) Proteins and the Inflammasome
All of the TLR proteins are transmembrane molecules, some expressed on the plasma membrane of the cell where they can interact with extracellular triggering molecules, and some expressed on intracellular membranes where they can interact with structures on intracellular microbes and viruses. Another set of pattern recognition molecules, designated NLR, has also been identified. These molecules are cytosolic and appear to interact with soluble intracellular ligands. Like the TLR, the NLR are characterized by the presence of leucine-rich repeat structures that are thought to contribute to their ability to bind to conserved microbial structures. The NLR can also recognize endogenous signals of cellular damage, such as uric acid crystals. Over 20 NLR-encoding genes have been identified in the human genome. Most are characterized by the presence of a C-terminal leucine-rich repeat domain that is thought to interact with microbial structures, a central nucleotide-binding oligomerization domain that is used to form multimeric complexes of the NLR, and an N-terminal effector domains that allow the NLR to recruit a class of intracellular cysteine proteinases (caspases) that activate the cellular apoptosis pathways or that activate the NF-kB transcription factor to induce a broad pro-inflammatory response.80 One of the NLR proteins, NALP3, has a special function in the innate immune response. Activation of NALP3 leads to its association with the intracellular adapter protein that is designated apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC), which combines with and activates caspase-1, leading to an active enzyme complex termed the inflammasome. The inflammasome functions to activate the potent proinflammatory molecules IL-1, IL-18, and IL-33.81 Recent studies have shown that alum, the most common adjuvant in vaccines administered to humans, is taken up by phagocytic cells where it activates NALP3, activating the inflammasome. This is crucial for its adjuvant activity. If any one of NALP3, ASC, or caspase-1 is absent or defective, then alum can no longer serve to augment the antibody response.82
Dectin-1, Collectins, Pentraxins and Ficolins
Additional pattern recognition proteins that contribute to the innate response to microbes include dectin-1, the collectins, certain of the pentraxins, and the ficolins. Dectin-1 is a transmembrane receptor that is activated when it binds b-glucans that are major components of the cell walls of yeast.83 The three major collectins in humans, mannan-binding lectin, and surfactant proteins A and D are all expressed at substantial levels in the human airway and recognize microbial carbohydrates through their carbohydrate recognition domains (CRD). Activation of the collectins opsonizes the microbe for phagocytosis, and activates the expression of proinflammatory cytokines and the production of anti-microbial reactive oxygen free radicals.84 The pentraxins are a group of homopentameric proteins that also recognize microbial molecular patterns. The best known are the short pentraxins, C-reactive protein (CRP) and serum amyloid P-component (SAP). CRP binds to bacterial low-density lipoproteins, a variety of bacterial polysaccharides, apoptotic host cells and nuclear material and induces activation of the complement system (see below) and phagocytosis. SAP recognizes microbial carbohydrates, nuclear substances, and amyloid fibrils and thus contributes to the host response to clear infections, autoimmunity and amyloidosis.85 The ficolins contain CRD domains that share structure with fibrinogen.2 After binding to carbohydrates on a microbe, they activate complement through the lectin pathway (see below) and thus contribute importantly to clearance of the microbe.
Chitinases
Chitin is a biopolymer of N-acetyl-β-D-glucosamine that is the major constituent of the cell walls of fungi and the exoskeletons of helminths, insects, and crustaceans. It is thought to be the second most abundant glycopolymer in the world. Chitinase designates a group of enzymes that digest chitin, both for the purpose of cellular and tissue remodeling during homeostasis in these organisms and for digestion of these organisms by the mammalian innate immune response. Because infestation with helminths and some of the chitin-expressing insects leads to the induction of high levels of IgE antibodies and eosinophil-predominant inflammation, scientists have investigated chitinase in human allergic disease. These studies have shown that chitin is a potent inducer of the production of Th2 cytokines and leads to the accumulation of eosinophils and basophils into chitin-challenged tissues. Although mammals do not synthesize chitin, they do express both enzymatically active chitinases and enzymatically inactive chitinase like proteins (CLP).86 The acidic mammalian chitinase (AMC) rapidly degrades chitin, contributing importantly to host defense against chitin-expressing organisms, dramatically reducing the allergic inflammatory response these organisms induce.87 AMC, which is expressed in epithelial cells as well as tissue leukocytes, and the related chitin digesting enzyme chitotriosidase are anti-inflammatory in settings of chitin challenge, and thus form part of the innate host defense mechanisms. The biologic functions of the enzymatically inactive CLP are not known; however, the fact that many of them, including the human YKL-40 protein, avidly bind and sequester chitin and its degradation products suggests that they may have immunoregulatory functions.87 AMC and CLP are both dramatically increased in the lungs of asthmatic subjects, suggesting that these molecules may contribute to the immunopathology of these disorders and might be appropriate targets for new drug therapy of this important clinical disorder.88, 89
Phagocytic Cells
The major phagocytic cells are neutrophils, macrophages, and monocytes. These cells engulf pathogenic microbes and localize them in intracellular vacuoles were they are exposed to toxic effector molecules such as nitric oxide, superoxide, and degradative enzymes in an effort to destroy the organism. Phagocytic cells use a variety of Fc receptors and complement receptors to enhance uptake of particles that have been marked by the adaptive and innate immune systems for destruction.
Natural Killer (NK) Cells
NK cells are thought to represent a third lineage of lymphoid cells. When activated, they have the morphology of a large granular lymphocyte. They develop in the bone marrow under the influence of IL-2, IL-15, and bone marrow stromal cells. They represent only a small fraction of peripheral blood cells and a small fraction of lymphoid cells in the spleen and other secondary lymphoid tissues. NK cells have no antigen-specific receptors. Their cytotoxic activity is inhibited by encounter with self-MHC molecules via inhibitory receptors on their surface that recognize class I HLA molecules. They thus kill self cells that have down-regulated class I molecule expression. This is important in host defense since several viruses have developed mechanisms to down regulate class I expression in infected cells as a strategy to avoid CD8+ cell killing. NK cells, however, also possess activating receptors. The nature of the ligands for these receptors and the mechanisms by which they contribute to identifying proper targets for NK cell cytotoxicity are currently under investigation. NK cells can destroy targets cells by antibody dependent cell-mediated cytotoxicity. They have prominent anti-tumor effects and are potent killers of virally infected cells.90
Complement
The complement system is a very important effector component of both adaptive and innate immunity. The complement system is composed of over 25 plasma and cell surface proteins that include three activation pathways and soluble and membrane bound down-modulating regulatory pathways.91, 92 Many of the proteins of the activation pathway are proteinases, and activation occurs in a cascade by proteolytic activation of one zymogen that then activates the next zymogen in the pathway. The main goal of the activation pathway is to mark targets permanently for destruction, to recruit other proteins and cells that facilitate target destruction, and in the case of some bacteria and viruses to participate directly in the destructive process by osmotic lysis. Antigen-antibody complexes provide the activating signal for the Classical Pathway of Complement Activation. Sequential activation of complement components C1, C4, and C2 produces the key enzyme in the pathway, the C3 convertase, which acts to cleave and activate C3. The cleavage results in release of the small C3a fragment, a potent anaphylatoxin that induces mast cell degranulation, creates edema and recruits phagocytic cells, and the larger C3b fragment which covalently attaches to the activating antigen, marking it for destruction. C3b serves both as a site for attack of the complement membrane attack complex (MAC), a self-assembling pore forming complex of serum proteins that kills targets by osmotic lysis, and as an opsonin, enhancing phagocytosis by its binding to complement receptors on the surfaces of neutrophils and macrophages.93
The second activation pathway, the Alternative Pathway of Complement Activation, is activated without antibody by microbial structures that neutralize inhibitors of spontaneous complement activation. This activation pathway can deposit >105 molecules of C3b on a single bacterium in less than 5 minutes. C3b deposited in this way then triggers the MAC and also enhances phagocytosis and killing.94
The third activation pathway is triggered by microbial cell wall components containing mannans and is called the Lectin Pathway of Complement Activation.95 The interaction of mannan-containing microbes with plasma mannan binding lectin (MBL) activates the zymogenic plasma proteases MBL-associated serine proteases 1 and 2 (MASP-1, MASP-2). These form a protease analogous to the activated C1 of the classical pathway that then goes on to activate C4, C2 and the remainder of the pathway. The lectin pathway can also be activated by complexes of microbes and host pentraxins and ficolins. Together, these three activation pathways permit complement to participate in the destruction and clearance of a wide variety of pathogens and macromolecules.
The effector mechanism of complement is potent and recruits intense local inflammation. There are several plasma proteins (factor H, C4 binding protein) and membrane proteins (complement receptors 1–4, decay accelerating factor, membrane co-factor protein) that inhibit the complement activation pathways to prevent unwanted damage to host tissues.95
The importance of the activation and regulatory pathways of complement are underscored by the dramatic phenotype of inherited deficiencies of individual components.91 Deficiencies of components of the MAC lead to increased susceptibility to infection with Neisseria. Deficiency of C3 results in life threatening susceptibility to pyogenic infections, often fatal during childhood. Deficiency of C4 or C2 causes a lupus-like immune complex disease, indicating that one of the roles of the classical pathway is to participate in the host response to and clearance of immune complexes. Deficiency of the serum inhibitor of C1 (an inhibitor of spontaneous activation of C1 and also of several components of the fibrinolytic pathway) leads to episodic mast cell-independent episodes of angioedema. Clonal hematopoietic lineage deficiency of the regulatory protein decay-accelerating factor (expressed on erythrocytes, leukocytes, and endothelial cells) causes Paroxysmal Nocturnal Hemoglobinuria.96
Leukocyte Adhesion and Tissue Inflammation
Recruitment of leukocytes both to secondary lymphoid tissues and to peripheral tissue sites of microbial invasion is essential for intact host defense. Cellular adhesion molecules and chemotactic proteins both contribute importantly to this process.97 There are three main families of cell adhesion proteins: selectins, integrins, and Ig domain cell adhesion molecules. In addition to mediating recruitment to tissues, these molecules contribute to cell-cell interactions between leukocyte subsets and can contribute to intercellular and intracellular signaling.98
There are three selectin glycoproteins, designated L-selectin, E-selectin, and P-selectin. Selectins are present on the surfaces of all leukocytes and on endothelial cells. Leukocytes also express ligands for selectins. The interactions between selectin ligands on leukocytes and selectins on vascular endothelial are low affinity and lead to rolling of cells along the vessel wall.97
Rolling cells can then be induced to arrest and adhere firmly to the endothelium by interactions between integrins on the leukocyte surface and Ig domain cell adhesion molecules on the endothelial cells. Integrins are heterodimers of one α and one β chain. Key integrins for leukocyte adhesion are LFA1 (CD11a/CD18, αLβ2), VLA4 (CD49d/CD29, α4β1), and Mac-1 (CD11b/CD18, αMβ2) that bind to the Ig domain cell adhesion molecules ICAM-1, VCAM-1, and ICAM-1/C3b respectively. Binding of leukocytes to endothelial cells is enhanced by the expression of chemokines by the endothelial cells or by underlying damaged cells and tissues (see chapter 5).
Cellular Homeostasis
After an immune response is completed, the majority of antigen responsive cells must be removed in order to prepare for the next immune challenge faced by the organism. Removal of effector cells without causing inflammation and tissue damage is best achieved by inducing the unwanted cells to undergo apoptosis. Molecules of the TNF family provide strong signals for the apoptotic programmed cell death pathway. TNF, signaling through the type I TNF receptor, induces death in tumor cells and at sites of ongoing inflammation. An alternative apoptosis-inducing receptor, Fas, is more specifically involved in regulatory apoptotic events. Fas, for example, transmits important apoptotic signals during thymic T cell selection.50, 99 It also contributes to the regulation of autoreactive cells in the periphery.100 Defects in Fas or in its ligand, FasL, result in autoimmune disorders with prominent lymphoproliferation.101 Thus, deregulated Fas or its ligand may contribute importantly to autoimmune diseases.
Tolerance, Immunopathology and Atopy
The goal of a properly regulated immune response is to protect the host from pathogens and other environmental challenges without causing damage to self-tissues. In the case of infection with viruses or intracellular bacteria and parasites, it is often impossible to eradicate the pathogen without destroying the infected cells. In cases like this, the use of cellular apoptosis as a mechanism for removing infected cells provides an elegant way to reduce damage to nearby uninfected cells. Infected cells that undergo apoptosis are generally fragmented into membrane-enclosed vesicles that can be taken up by healthy phagocytic cells and digested so as to eliminate both the potentially inflammatory contents of the infected cell and also the microbe that was multiplying inside the cell.
Some degree of local inflammation is, however, often an important part of an effective host immune response. The key elements of inflammation are part and parcel of the host’s mobilization of its defense and repair responses. When inflammation is modest and controlled, normal tissue architecture and function can be restored after the pathogen or toxin has been eliminated. If the inflammatory response is excessively severe, however, there is danger of lasting tissue damage, and the development of fibrosis during the resolution of the inflammatory state.102 Mild fibrosis is physiological and generally does not interfere with normal tissue function; however, when inflammation is either very severe or becomes chronic the resulting fibrosis may lead to profound organ dysfunction. There has been important progress over the last several years in understanding the mechanisms that control the transition from physiologically appropriate inflammation and tissue repair to damaging fibrosis. A common theme underlying the fibrotic process is the local production of activated fibroblasts via the action of selected cytokines and other mediators on tissue epithelial cells. Through the process of epithelial to mesenchymal transition, epithelial cells are thought to be converted to activated fibroblasts and myofibroblasts that are then responsible for the tissue changes that lead to fibrosis.103 Development of therapeutics that target the mediators of tissue fibrosis may prevent many of the long-term complications of chronic inflammation.
Perhaps more puzzling are conditions in which tissue inflammation appears to develop without any underlying infectious or noxious stimulus. Prominent in these are autoimmune diseases and atopic illnesses. These disorders appear to represent a fundamental misdirection of the immune response, resulting in tissue damage when no real danger was present. The growing spectrum of autoimmune diseases appears to represent a breakdown in self-tolerance. This leads to the induction of both cellular and humoral immune responses against components of self-tissues. Usually, both the cellular and humoral aspects of these pathological responses have features of a Th1-type or Th17-type CD4 T cell response, suggesting that defective regulation of either T cell differentiation or activation underlies the response.104 Atopic diseases rarely manifest autoimmune character (although some forms of chronic urticaria are thought to have an autoimmune etiology; see chapters 12 and 17). Rather, they appear to represent an overly aggressive Th2 type response leading to hypersensitivity to a broad spectrum of normally encountered environmental antigens. Epidemiological studies have demonstrated that there is an inherited component to both the autoimmune and the atopic diseases.105 There also appears to be a strong interplay with environmental factors, perhaps including unrecognized infectious microbes or toxic agents in the environment. The central role of Treg cells in controlling all aspects of the CD4 T cell response, and the observation that congenital absence of Treg cells (as in the immune dysregulation, polyendocrinopathy, enteropathy, X-linked [IPEX] syndrome) leads to development of an aggressive autoimmune state106 suggest that disturbed Treg function may underlie all autoimmune and atopic diseases. While disordered Th1, Th17 and Th2 responsiveness is a major manifestation of these illnesses, the disorders do not simply represent a predisposition to over-polarization of the CD4+ T cell response. Epidemiological studies have shown that the presence of atopy shows little protection against development of the Th1/Th17-predominant illness rheumatoid arthritis.107 In fact, other studies have suggested that patients with an autoimmune illness are more likely to have an atopic disorder, suggesting that they have a common underlying etiology.108 Development of a thorough understanding of the mechanisms underlying these two types of T cell-mediated inflammation will lead to important new therapeutic options for successful treatment of these common diseases.109
A special situation in which tolerance is modulated in a physiological way concerns the suppression of the maternal immune response to permit the maintenance of the semi-allogeneic fetus and placenta in the setting of normal pregnancy. Recent studies have demonstrated that in mid-gestation human fetuses, 20–25% of the CD4 T cells in lymph nodes and spleen have a Treg phenotype and levels of TGFβ were remarkably high in these lymphoid organs.110 Additionally, lymphocytes in these secondary lymphoid tissues were poorly activated when exposed to an allogeneic stimulus.111 These high numbers of Treg cells returned to normal shortly after delivery. Interestingly, spontaneous abortion has been associated with loss of normal pregnancy-associated immune suppression.110 Alterations in Treg function do not constitute the entire mechanism underlying the tolerance of pregnancy. Other studies have shown very high levels of expression of galectin-1, an immunoregulatory glycan-binding protein, in fetal tissues, and loss of galectin-1 in failing pregnancies.112 Additionally, levels of thymic stromal lymphopoietin (TSLP) are increased in pregnancy, and this induces placental DC to drive the differentiation of Th cells that produce abundant levels of IL-10, a cytokine well adapted to help maintain the fetal allograft.113 Understanding the mechanisms that control tolerance to the fetal allograft may provide new insights into the regulatory systems that have failed in autoimmunity and atopy.
Conclusion
The immune system uses many mechanisms to combat infection by microbes. These mechanisms work together, and the fully integrated immune response draws elements from many effector systems in order to tailor a response to the specific invading pathogen. Abnormal regulation of the various effector mechanisms can lead to chronic or acute tissue damage. Understanding the relationships between the different immune effector pathways will permit improved immunomodulatory therapeutics, development of improved vaccines, and avoidance of unintended tissue injury.
Figure 9. The Activation Pathways of Complement.

Three pathways lead to activation of complement. The Classical Pathway is initiated by complexes of IgM, IgG1, or IgG3 with antigens. This activates proteolysis of C1 that cleaves C4 and C2 to form the classical pathway C3 convertase. The Mannose Lectin Pathway is activated by interaction of mannan-containing microbes with MBL, which activates MASP-1 and MASP-2 to cleave C4 and C2, again forming a C3 convertase. The Alternative Pathway is initiated by interactions between microbial antigens and inhibitory complement regulatory proteins. This permits autoactivation of the pathway in which C3 interacts with factor B and factor D to generate the alternative pathway C3 convertase. These convertases all cleave C3 to generate the anaphylatoxic C3a fragment and depositing C3b on the activating microbial particle or immune complex. This opsonizes the particle for phagocytosis and initiates the activation of the membrane attack complex. The C5a fragment that is proteolytically released from C5 also is a highly anaphylatoxic molecule that induces intense local inflammation.
Abbreviations used
- AID
Activation-induced cytidine deaminase
- AIRE
Autoimmune regulator
- AMC
Acidic mammalian chitinase
- APC
Antigen presenting cell
- APECED
Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy
- ACS
Apoptosis-associated speck-like protein containing a caspase recruitment domain
- Bf
Complement factor B
- CD
Cluster of differentiation
- CD40L
CD40 ligand
- CFU
Colony forming unit
- CLIP
Class II-associated invariant-chain peptide
- CLP
Chitinase-like protein
- CRD
Carbohydrate recognition domain
- CRP
C-reactive protein
- DNA-PK
DNA-dependent protein kinase
- DP
Double positive
- ER
Endoplasmic reticulum
- FcεRI
High affinity receptor for IgE
- FDC
Follicular dendritic cell
- Flt3
Fms-like tyrosine kinase receptor-3
- Foxp3
Forkhead box P3
- GM-CSF
Granulocyte-macrophage colony stimulating factor
- HLA
Human leukocyte-associated
- HLDA
Human leukocyte differentiation antigen
- IFN
Interferon
- Ig
Immunoglobulin
- Ii
Invariant chain
- IL
Interleukin
- IPEX
Immune dysregulation, polyendocrinopathy, enteropathy, X-linked
- ITAM
Immunoreceptor tyrosine-based activation motif
- Jak
Janus kinase
- LPS
Lipopolysaccharide
- MAC
Membrane attack complex
- MAP
Mitogen-associated protein
- MASP
MBL-associated serine protease
- MBL
Mannan binding lectin
- MHC
Major histocompatibility complex
- MIC
MHC class I chain-related
- MyD88
Myeloid differentiation primary response gene 88
- NK
Natural killer
- NLR
Nucleotide-binging domain leucine rich repeat
- PAMP
Pathogen-associated molecular pattern
- PD-1
Programmed death-1
- PD-L1
Programmed death-ligand 1
- RAG
Recombinase-activating gene
- SCID
Severe combined immune deficiency
- STAT
Signal transducers and activators of transcription
- TAP
Transporter associated with antigen presentation
- Tc
T cytotoxic
- TCR
T cell receptor
- TdT
Terminal deoxynucleotidyl transferase
- TGFβ
Transforming growth factor β
- Th
T helper
- TIR
Toll/IL-1 receptor
- TLR
Toll-like receptor
- TNF
Tumor necrosis factor
- Treg
T regulatory
- TSLP
Thymic stromal lymphopoietin
- TSST-1
Toxic shock syndrome toxin-1
- UNG
Uracil DNA glycoslyase
References
- 1.Hiemstra PS. The role of epithelial beta-defensins and cathelicidins in host defense of the lung. Exp Lung Res. 2007;33:537–542. doi: 10.1080/01902140701756687. [DOI] [PubMed] [Google Scholar]
- 2.Holmskov U, Thiel S, Jensenius JC. Collectins and ficolins: humoral lectins of the innate immune defense. Annu Rev Immunol. 2003;21:547–578. doi: 10.1146/annurev.immunol.21.120601.140954. [DOI] [PubMed] [Google Scholar]
- 3.Sjoberg AP, Trouw LA, Blom AM. Complement activation and inhibition: a delicate balance. Trends Immunol. 2009;30:83–90. doi: 10.1016/j.it.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 4.Robin C, Bollerot K, Mendes S, Haak E, Crisan M, Cerisoli F, et al. Human placenta is a potent hematopoietic niche containing hematopoietic stem and progenitor cells throughout development. Cell Stem Cell. 2009;5:385–395. doi: 10.1016/j.stem.2009.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jonsson AH, Yokoyama WM. Natural killer cell tolerance licensing and other mechanisms. Adv Immunol. 2009;101:27–79. doi: 10.1016/S0065-2776(08)01002-X. [DOI] [PubMed] [Google Scholar]
- 6.Balato A, Unutmaz D, Gaspari AA. Natural killer T cells: an unconventional T-cell subset with diverse effector and regulatory functions. J Invest Dermatol. 2009;129:1628–1642. doi: 10.1038/jid.2009.30. [DOI] [PubMed] [Google Scholar]
- 7.Kennedy AD, DeLeo FR. Neutrophil apoptosis and the resolution of infection. Immunol Res. 2009;43:25–61. doi: 10.1007/s12026-008-8049-6. [DOI] [PubMed] [Google Scholar]
- 8.Benoit M, Desnues B, Mege JL. Macrophage polarization in bacterial infections. J Immunol. 2008;181:3733–3739. doi: 10.4049/jimmunol.181.6.3733. [DOI] [PubMed] [Google Scholar]
- 9.Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 10.Minai-Fleminger Y, Levi-Schaffer F. Mast cells and eosinophils: the two key effector cells in allergic inflammation. Inflamm Res. 2009;58:631–638. doi: 10.1007/s00011-009-0042-6. [DOI] [PubMed] [Google Scholar]
- 11.Schroeder JT. Basophils: beyond effector cells of allergic inflammation. Adv Immunol. 2009;101:123–161. doi: 10.1016/S0065-2776(08)01004-3. [DOI] [PubMed] [Google Scholar]
- 12.Lambrecht BN, Hammad H. Biology of lung dendritic cells at the origin of asthma. Immunity. 2009;31:412–424. doi: 10.1016/j.immuni.2009.08.008. [DOI] [PubMed] [Google Scholar]
- 13.Gilliet M, Cao W, Liu YJ. Plasmacytoid dendritic cells: sensing nucleic acids in viral infection and autoimmune diseases. Nat Rev Immunol. 2008;8:594–606. doi: 10.1038/nri2358. [DOI] [PubMed] [Google Scholar]
- 14.Colonna M, Pulendran B, Iwasaki A. Dendritic cells at the host-pathogen interface. Nat Immunol. 2006;7:117–120. doi: 10.1038/ni0206-117. [DOI] [PubMed] [Google Scholar]
- 15.Naik SH, Sathe P, Park HY, Metcalf D, Proietto AI, Dakic A, et al. Development of plasmacytoid and conventional dendritic cell subtypes from single precursor cells derived in vitro and in vivo. Nat Immunol. 2007;8:1217–1226. doi: 10.1038/ni1522. [DOI] [PubMed] [Google Scholar]
- 16.Bjorkman PJ. MHC restriction in three dimensions: a view of T cell receptor/ligand interactions. Cell. 1997;89:167–170. doi: 10.1016/s0092-8674(00)80195-6. [DOI] [PubMed] [Google Scholar]
- 17.Zinkernagel RM, Doherty PC. The discovery of MHC restriction. Immunol Today. 1997;18:14–17. doi: 10.1016/s0167-5699(97)80008-4. [DOI] [PubMed] [Google Scholar]
- 18.Joshi NS, Kaech SM. Effector CD8 T cell development: a balancing act between memory cell potential and terminal differentiation. J Immunol. 2008;180:1309–1315. doi: 10.4049/jimmunol.180.3.1309. [DOI] [PubMed] [Google Scholar]
- 19.Niedermann G. Immunological functions of the proteasome. Curr Top Microbiol Immunol. 2002;268:91–136. doi: 10.1007/978-3-642-59414-4_5. [DOI] [PubMed] [Google Scholar]
- 20.Tanahashi N, Yokota K, Ahn JY, Chung CH, Fujiwara T, Takahashi E, et al. Molecular properties of the proteasome activator PA28 family proteins and gamma-interferon regulation. Genes Cells. 1997;2:195–211. doi: 10.1046/j.1365-2443.1997.d01-308.x. [DOI] [PubMed] [Google Scholar]
- 21.Garbi N, Tanaka S, Momburg F, Hammerling GJ. Impaired assembly of the major histocompatibility complex class I peptide-loading complex in mice deficient in the oxidoreductase ERp57. Nat Immunol. 2006;7:93–102. doi: 10.1038/ni1288. [DOI] [PubMed] [Google Scholar]
- 22.Momburg F, Tan P. Tapasin-the keystone of the loading complex optimizing peptide binding by MHC class I molecules in the endoplasmic reticulum. Mol Immunol. 2002;39:217–233. doi: 10.1016/s0161-5890(02)00103-7. [DOI] [PubMed] [Google Scholar]
- 23.Melief CJ. Mini-review: Regulation of cytotoxic T lymphocyte responses by dendritic cells: peaceful coexistence of cross-priming and direct priming? Eur J Immunol. 2003;33:2645–2654. doi: 10.1002/eji.200324341. [DOI] [PubMed] [Google Scholar]
- 24.Sigal LJ, Crotty S, Andino R, Rock KL. Cytotoxic T-cell immunity to virus-infected non-haematopoietic cells requires presentation of exogenous antigen. Nature. 1999;398:77–80. doi: 10.1038/18038. [DOI] [PubMed] [Google Scholar]
- 25.Konig R, Fleury S, Germain RN. The structural basis of CD4-MHC class II interactions: coreceptor contributions to T cell receptor antigen recognition and oligomerization-dependent signal transduction. Curr Top Microbiol Immunol. 1996;205:19–46. doi: 10.1007/978-3-642-79798-9_2. [DOI] [PubMed] [Google Scholar]
- 26.Turley SJ, Inaba K, Garrett WS, Ebersold M, Unternaehrer J, Steinman RM, et al. Transport of peptide-MHC class II complexes in developing dendritic cells. Science. 2000;288:522–527. doi: 10.1126/science.288.5465.522. [DOI] [PubMed] [Google Scholar]
- 27.Van Kaer L. Accessory proteins that control the assembly of MHC molecules with peptides. Immunol Res. 2001;23:205–214. doi: 10.1385/IR:23:2-3:205. [DOI] [PubMed] [Google Scholar]
- 28.Sadegh-Nasseri S, Chen M, Narayan K, Bouvier M. The convergent roles of tapasin and HLA-DM in antigen presentation. Trends Immunol. 2008;29:141–147. doi: 10.1016/j.it.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ghodke Y, Joshi K, Chopra A, Patwardhan B. HLA and disease. Eur J Epidemiol. 2005;20:475–488. doi: 10.1007/s10654-005-5081-x. [DOI] [PubMed] [Google Scholar]
- 30.Reveille JD. Recent studies on the genetic basis of ankylosing spondylitis. Curr Rheumatol Rep. 2009;11:340–348. doi: 10.1007/s11926-009-0049-6. [DOI] [PubMed] [Google Scholar]
- 31.Pociot F, McDermott MF. Genetics of type 1 diabetes mellitus. Genes Immun. 2002;3:235–249. doi: 10.1038/sj.gene.6363875. [DOI] [PubMed] [Google Scholar]
- 32.Caillat-Zucman S. Molecular mechanisms of HLA association with autoimmune diseases. Tissue Antigens. 2009;73:1–8. doi: 10.1111/j.1399-0039.2008.01167.x. [DOI] [PubMed] [Google Scholar]
- 33.Sonderstrup G, McDevitt HO. DR, DQ, and you: MHC alleles and autoimmunity. J Clin Invest. 2001;107:795–796. doi: 10.1172/JCI12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alper CA, Kruskall MS, Marcus-Bagley D, Craven DE, Katz AJ, Brink SJ, et al. Genetic prediction of nonresponse to hepatitis B vaccine. N Engl J Med. 1989;321:708–712. doi: 10.1056/NEJM198909143211103. [DOI] [PubMed] [Google Scholar]
- 35.Poland GA, Ovsyannikova IG, Jacobson RM, Vierkant RA, Jacobsen SJ, Pankratz VS, et al. Identification of an association between HLA class II alleles and low antibody levels after measles immunization. Vaccine. 2001;20:430–438. doi: 10.1016/s0264-410x(01)00346-2. [DOI] [PubMed] [Google Scholar]
- 36.Grant EP, Beckman EM, Behar SM, Degano M, Frederique D, Besra GS, et al. Fine specificity of TCR complementarity-determining region residues and lipid antigen hydrophilic moieties in the recognition of a CD1-lipid complex. J Immunol. 2002;168:3933–3940. doi: 10.4049/jimmunol.168.8.3933. [DOI] [PubMed] [Google Scholar]
- 37.Prigozy TI, Naidenko O, Qasba P, Elewaut D, Brossay L, Khurana A, et al. Glycolipid antigen processing for presentation by CD1d molecules. Science. 2001;291:664–667. doi: 10.1126/science.291.5504.664. [DOI] [PubMed] [Google Scholar]
- 38.Wu J, Groh V, Spies T. T cell antigen receptor engagement and specificity in the recognition of stress-inducible MHC class I-related chains by human epithelial gamma delta T cells. J Immunol. 2002;169:1236–1240. doi: 10.4049/jimmunol.169.3.1236. [DOI] [PubMed] [Google Scholar]
- 39.Godfrey DI, MacDonald HR, Kronenberg M, Smyth MJ, Van Kaer L. NKT cells: what's in a name? Nat Rev Immunol. 2004;4:231–237. doi: 10.1038/nri1309. [DOI] [PubMed] [Google Scholar]
- 40.Gellert M. V(D)J recombination: RAG proteins, repair factors, and regulation. Annu Rev Biochem. 2002;71:101–132. doi: 10.1146/annurev.biochem.71.090501.150203. [DOI] [PubMed] [Google Scholar]
- 41.Nguyen HH, Zemlin M, Ivanov II, Andrasi J, Zemlin C, Vu HL, et al. Heterosubtypic immunity to influenza A virus infection requires a properly diversified antibody repertoire. J Virol. 2007;81:9331–9338. doi: 10.1128/JVI.00751-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Miller JF. The discovery of thymus function and of thymus-derived lymphocytes. Immunol Rev. 2002;185:7–14. doi: 10.1034/j.1600-065x.2002.18502.x. [DOI] [PubMed] [Google Scholar]
- 43.Nitta T, Murata S, Ueno T, Tanaka K, Takahama Y. Thymic microenvironments for T-cell repertoire formation. Adv Immunol. 2008;99:59–94. doi: 10.1016/S0065-2776(08)00603-2. [DOI] [PubMed] [Google Scholar]
- 44.Mathis D, Benoist C. Aire. Annu Rev Immunol. 2009;27:287–312. doi: 10.1146/annurev.immunol.25.022106.141532. [DOI] [PubMed] [Google Scholar]
- 45.Ishikawa H, Naito T, Iwanaga T, Takahashi-Iwanaga H, Suematsu M, Hibi T, et al. Curriculum vitae of intestinal intraepithelial T cells: their developmental and behavioral characteristics. Immunol Rev. 2007;215:154–165. doi: 10.1111/j.1600-065X.2006.00473.x. [DOI] [PubMed] [Google Scholar]
- 46.Salmond RJ, Filby A, Qureshi I, Caserta S, Zamoyska R. T-cell receptor proximal signaling via the Src-family kinases, Lck and Fyn, influences T-cell activation, differentiation, and tolerance. Immunol Rev. 2009;228:9–22. doi: 10.1111/j.1600-065X.2008.00745.x. [DOI] [PubMed] [Google Scholar]
- 47.Nurieva RI, Liu X, Dong C. Yin-Yang of costimulation: crucial controls of immune tolerance and function. Immunol Rev. 2009;229:88–100. doi: 10.1111/j.1600-065X.2009.00769.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Riley JL. PD-1 signaling in primary T cells. Immunol Rev. 2009;229:114–125. doi: 10.1111/j.1600-065X.2009.00767.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.von Boehmer H, Kisielow P, Kishi H, Scott B, Borgulya P, Teh HS. The expression of CD4 and CD8 accessory molecules on mature T cells is not random but correlates with the specificity of the alpha beta receptor for antigen. Immunol Rev. 1989;109:143–151. doi: 10.1111/j.1600-065x.1989.tb00023.x. [DOI] [PubMed] [Google Scholar]
- 50.Sakaguchi S, Ono M, Setoguchi R, Yagi H, Hori S, Fehervari Z, et al. Foxp3+ CD25+ CD4+ natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol Rev. 2006;212:8–27. doi: 10.1111/j.0105-2896.2006.00427.x. [DOI] [PubMed] [Google Scholar]
- 51.Nakamura K, Kitani A, Strober W. Cell contact-dependent immunosuppression by CD4(+)CD25(+) regulatory T cells is mediated by cell surface-bound transforming growth factor beta. J Exp Med. 2001;194:629–644. doi: 10.1084/jem.194.5.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Curotto de Lafaille MA, Lafaille JJ. Natural and adaptive foxp3+ regulatory T cells: more of the same or a division of labor? Immunity. 2009;30:626–635. doi: 10.1016/j.immuni.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 53.Huehn J, Polansky JK, Hamann A. Epigenetic control of FOXP3 expression: the key to a stable regulatory T-cell lineage? Nat Rev Immunol. 2009;9:83–89. doi: 10.1038/nri2474. [DOI] [PubMed] [Google Scholar]
- 54.Sallusto F, Lanzavecchia A. Heterogeneity of CD4+ memory T cells: functional modules for tailored immunity. Eur J Immunol. 2009;39:2076–2082. doi: 10.1002/eji.200939722. [DOI] [PubMed] [Google Scholar]
- 55.Unutmaz D. RORC2: the master of human Th17 cell programming. Eur J Immunol. 2009;39:1452–1455. doi: 10.1002/eji.200939540. [DOI] [PubMed] [Google Scholar]
- 56.Thomas MJ, MacAry PA, Noble A, Askenase PW, Kemeny DM. T cytotoxic 1 and T cytotoxic 2 CD8 T cells both inhibit IgE responses. Int Arch Allergy Immunol. 2001;124:187–189. doi: 10.1159/000053706. [DOI] [PubMed] [Google Scholar]
- 57.Racila DM, Kline JN. Perspectives in asthma: molecular use of microbial products in asthma prevention and treatment. J Allergy Clin Immunol. 2005;116:1202–1205. doi: 10.1016/j.jaci.2005.08.050. [DOI] [PubMed] [Google Scholar]
- 58.Larkin EA, Carman RJ, Krakauer T, Stiles BG. Staphylococcus aureus: The Toxic Presence of a Pathogen Extraordinaire. Curr Med Chem. 2009;16:4003–4019. doi: 10.2174/092986709789352321. [DOI] [PubMed] [Google Scholar]
- 59.Thomas LR, Cobb RM, Oltz EM. Dynamic regulation of antigen receptor gene assembly. Adv Exp Med Biol. 2009;650:103–115. doi: 10.1007/978-1-4419-0296-2_9. [DOI] [PubMed] [Google Scholar]
- 60.Jankovic M, Casellas R, Yannoutsos N, Wardemann H, Nussenzweig MC. RAGs and regulation of autoantibodies. Annu Rev Immunol. 2004;22:485–501. doi: 10.1146/annurev.immunol.22.012703.104707. [DOI] [PubMed] [Google Scholar]
- 61.Khan WN. B cell receptor and BAFF receptor signaling regulation of B cell homeostasis. J Immunol. 2009;183:3561–3567. doi: 10.4049/jimmunol.0800933. [DOI] [PubMed] [Google Scholar]
- 62.Kurosaki T, Hikida M. Tyrosine kinases and their substrates in B lymphocytes. Immunol Rev. 2009;228:132–148. doi: 10.1111/j.1600-065X.2008.00748.x. [DOI] [PubMed] [Google Scholar]
- 63.Carroll MC. Complement and humoral immunity. Vaccine. 2008;26 Suppl 8:I28–I33. doi: 10.1016/j.vaccine.2008.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Malisan F, Briere F, Bridon JM, Harindranath N, Mills FC, Max EE, et al. Interleukin-10 induces immunoglobulin G isotype switch recombination in human CD40-activated naive B lymphocytes. J Exp Med. 1996;183:937–947. doi: 10.1084/jem.183.3.937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chaudhuri J, Basu U, Zarrin A, Yan C, Franco S, Perlot T, et al. Evolution of the immunoglobulin heavy chain class switch recombination mechanism. Adv Immunol. 2007;94:157–214. doi: 10.1016/S0065-2776(06)94006-1. [DOI] [PubMed] [Google Scholar]
- 66.Schmidlin H, Diehl SA, Blom B. New insights into the regulation of human B-cell differentiation. Trends Immunol. 2009;30:277–285. doi: 10.1016/j.it.2009.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Durandy A, Taubenheim N, Peron S, Fischer A. Pathophysiology of B-cell intrinsic immunoglobulin class switch recombination deficiencies. Adv Immunol. 2007;94:275–306. doi: 10.1016/S0065-2776(06)94009-7. [DOI] [PubMed] [Google Scholar]
- 68.Tangye SG, Tarlinton DM. Memory B cells: effectors of long-lived immune responses. Eur J Immunol. 2009;39:2065–2075. doi: 10.1002/eji.200939531. [DOI] [PubMed] [Google Scholar]
- 69.Fu YX, Chaplin DD. Development and maturation of secondary lymphoid tissues. Annu Rev Immunol. 1999;17:399–433. doi: 10.1146/annurev.immunol.17.1.399. [DOI] [PubMed] [Google Scholar]
- 70.Allen CD, Okada T, Cyster JG. Germinal-center organization and cellular dynamics. Immunity. 2007;27:190–202. doi: 10.1016/j.immuni.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schwab SR, Cyster JG. Finding a way out: lymphocyte egress from lymphoid organs. Nat Immunol. 2007;8:1295–1301. doi: 10.1038/ni1545. [DOI] [PubMed] [Google Scholar]
- 72.Pesu M, Candotti F, Husa M, Hofmann SR, Notarangelo LD, O'Shea JJ. Jak3, severe combined immunodeficiency, and a new class of immunosuppressive drugs. Immunol Rev. 2005;203:127–142. doi: 10.1111/j.0105-2896.2005.00220.x. [DOI] [PubMed] [Google Scholar]
- 73.Kovanen PE, Leonard WJ. Cytokines and immunodeficiency diseases: critical roles of the gamma(c)-dependent cytokines interleukins 2, 4, 7, 9, 15, and 21, and their signaling pathways. Immunol Rev. 2004;202:67–83. doi: 10.1111/j.0105-2896.2004.00203.x. [DOI] [PubMed] [Google Scholar]
- 74.Chapgier A, Boisson-Dupuis S, Jouanguy E, Vogt G, Feinberg J, Prochnicka-Chalufour A, et al. Novel STAT1 alleles in otherwise healthy patients with mycobacterial disease. PLoS Genet. 2006;2:e131. doi: 10.1371/journal.pgen.0020131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tangye SG, Cook MC, Fulcher DA. Insights into the role of STAT3 in human lymphocyte differentiation as revealed by the hyper-IgE syndrome. J Immunol. 2009;182:21–28. doi: 10.4049/jimmunol.182.1.21. [DOI] [PubMed] [Google Scholar]
- 76.Kaplan MH, Sun YL, Hoey T, Grusby MJ. Impaired IL-12 responses and enhanced development of Th2 cells in Stat4-deficient mice. Nature. 1996;382:174–177. doi: 10.1038/382174a0. [DOI] [PubMed] [Google Scholar]
- 77.Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature. 2007;449:819–826. doi: 10.1038/nature06246. [DOI] [PubMed] [Google Scholar]
- 78.Spiegelberg HL, Raz E. DNA-based approaches to the treatment of allergies. Curr Opin Mol Ther. 2002;4:64–71. [PubMed] [Google Scholar]
- 79.von Bernuth H, Picard C, Jin Z, Pankla R, Xiao H, Ku CL, et al. Pyogenic bacterial infections in humans with MyD88 deficiency. Science. 2008;321:691–696. doi: 10.1126/science.1158298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Istomin AY, Godzik A. Understanding diversity of human innate immunity receptors: analysis of surface features of leucine-rich repeat domains in NLRs and TLRs. BMC Immunol. 2009;10:48. doi: 10.1186/1471-2172-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sutterwala FS, Ogura Y, Szczepanik M, Lara-Tejero M, Lichtenberger GS, Grant EP, et al. Critical role for NALP3/CIAS1/Cryopyrin in innate and adaptive immunity through its regulation of caspase-1. Immunity. 2006;24:317–327. doi: 10.1016/j.immuni.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 82.Eisenbarth SC, Colegio OR, O'Connor W, Sutterwala FS, Flavell RA. Crucial role for the Nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature. 2008;453:1122–1126. doi: 10.1038/nature06939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gantner BN, Simmons RM, Underhill DM. Dectin-1 mediates macrophage recognition of Candida albicans yeast but not filaments. EMBO J. 2005;24:1277–1286. doi: 10.1038/sj.emboj.7600594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Waters P, Vaid M, Kishore U, Madan T. Lung surfactant proteins A and D as pattern recognition proteins. Adv Exp Med Biol. 2009;653:74–97. doi: 10.1007/978-1-4419-0901-5_6. [DOI] [PubMed] [Google Scholar]
- 85.Agrawal A, Singh PP, Bottazzi B, Garlanda C, Mantovani A. Pattern recognition by pentraxins. Adv Exp Med Biol. 2009;653:98–116. doi: 10.1007/978-1-4419-0901-5_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sutherland TE, Maizels RM, Allen JE. Chitinases and chitinase-like proteins: potential therapeutic targets for the treatment of T-helper type 2 allergies. Clin Exp Allergy. 2009;39:943–955. doi: 10.1111/j.1365-2222.2009.03243.x. [DOI] [PubMed] [Google Scholar]
- 87.Reese TA, Liang HE, Tager AM, Luster AD, Van Rooijen N, Voehringer D, et al. Chitin induces accumulation in tissue of innate immune cells associated with allergy. Nature. 2007;447:92–96. doi: 10.1038/nature05746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kazakova MH, Sarafian VS. YKL-40--a novel biomarker in clinical practice? Folia Med (Plovdiv) 2009;51:5–14. [PubMed] [Google Scholar]
- 89.Ober C, Chupp GL. The chitinase and chitinase-like proteins: a review of genetic and functional studies in asthma and immune-mediated diseases. Curr Opin Allergy Clin Immunol. 2009;9:401–408. doi: 10.1097/ACI.0b013e3283306533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cooper MA, Colonna M, Yokoyama WM. Hidden talents of natural killers: NK cells in innate and adaptive immunity. EMBO Rep. 2009;10:1103–1110. doi: 10.1038/embor.2009.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Carroll MC. The complement system in regulation of adaptive immunity. Nat Immunol. 2004;5:981–986. doi: 10.1038/ni1113. [DOI] [PubMed] [Google Scholar]
- 92.Longhi MP, Harris CL, Morgan BP, Gallimore A. Holding T cells in check--a new role for complement regulators? Trends Immunol. 2006;27:102–108. doi: 10.1016/j.it.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 93.Kim J, Hematti P. Mesenchymal stem cell-educated macrophages: a novel type of alternatively activated macrophages. Exp Hematol. 2009;37:1445–1453. doi: 10.1016/j.exphem.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Harboe M, Mollnes TE. The alternative complement pathway revisited. J Cell Mol Med. 2008;12:1074–1084. doi: 10.1111/j.1582-4934.2008.00350.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Botto M, Kirschfink M, Macor P, Pickering MC, Wurzner R, Tedesco F. Complement in human diseases: Lessons from complement deficiencies. Mol Immunol. 2009;46:2774–2783. doi: 10.1016/j.molimm.2009.04.029. [DOI] [PubMed] [Google Scholar]
- 96.Brodsky RA. Narrative review: paroxysmal nocturnal hemoglobinuria: the physiology of complement-related hemolytic anemia. Ann Intern Med. 2008;148:587–595. doi: 10.7326/0003-4819-148-8-200804150-00003. [DOI] [PubMed] [Google Scholar]
- 97.Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7:678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- 98.Abram CL, Lowell CA. The ins and outs of leukocyte integrin signaling. Annu Rev Immunol. 2009;27:339–362. doi: 10.1146/annurev.immunol.021908.132554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Grant EP, Degano M, Rosat JP, Stenger S, Modlin RL, Wilson IA, et al. Molecular recognition of lipid antigens by T cell receptors. J Exp Med. 1999;189:195–205. doi: 10.1084/jem.189.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fortner KA, Budd RC. The death receptor Fas (CD95/APO-1) mediates the deletion of T lymphocytes undergoing homeostatic proliferation. J Immunol. 2005;175:4374–4382. doi: 10.4049/jimmunol.175.7.4374. [DOI] [PubMed] [Google Scholar]
- 101.Strasser A, Jost PJ, Nagata S. The many roles of FAS receptor signaling in the immune system. Immunity. 2009;30:180–192. doi: 10.1016/j.immuni.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hardie WD, Glasser SW, Hagood JS. Emerging concepts in the pathogenesis of lung fibrosis. Am J Pathol. 2009;175:3–16. doi: 10.2353/ajpath.2009.081170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Guarino M, Tosoni A, Nebuloni M. Direct contribution of epithelium to organ fibrosis: epithelial-mesenchymal transition. Hum Pathol. 2009;40:1365–1376. doi: 10.1016/j.humpath.2009.02.020. [DOI] [PubMed] [Google Scholar]
- 104.Annunziato F, Cosmi L, Liotta F, Maggi E, Romagnani S. Type 17 T helper cells-origins, features and possible roles in rheumatic disease. Nat Rev Rheumatol. 2009;5:325–331. doi: 10.1038/nrrheum.2009.80. [DOI] [PubMed] [Google Scholar]
- 105.Ono SJ. Molecular genetics of allergic diseases. Annu Rev Immunol. 2000;18:347–366. doi: 10.1146/annurev.immunol.18.1.347. [DOI] [PubMed] [Google Scholar]
- 106.Zhou Z, Song X, Li B, Greene MI. FOXP3 and its partners: structural and biochemical insights into the regulation of FOXP3 activity. Immunol Res. 2008;42:19–28. doi: 10.1007/s12026-008-8029-x. [DOI] [PubMed] [Google Scholar]
- 107.Rudwaleit M, Andermann B, Alten R, Sorensen H, Listing J, Zink A, et al. Atopic disorders in ankylosing spondylitis and rheumatoid arthritis. Ann Rheum Dis. 2002;61:968–974. doi: 10.1136/ard.61.11.968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Simpson CR, Anderson WJ, Helms PJ, Taylor MW, Watson L, Prescott GJ, et al. Coincidence of immune-mediated diseases driven by Th1 and Th2 subsets suggests a common aetiology. A population-based study using computerized general practice data. Clin Exp Allergy. 2002;32:37–42. doi: 10.1046/j.0022-0477.2001.01250.x. [DOI] [PubMed] [Google Scholar]
- 109.Rabin RL, Levinson AI. The nexus between atopic disease and autoimmunity: a review of the epidemiological and mechanistic literature. Clin Exp Immunol. 2008;153:19–30. doi: 10.1111/j.1365-2249.2008.03679.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Burlingham WJ. A lesson in tolerance--maternal instruction to fetal cells. N Engl J Med. 2009;360:1355–1357. doi: 10.1056/NEJMcibr0810752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mold JE, Michaelsson J, Burt TD, Muench MO, Beckerman KP, Busch MP, et al. Maternal alloantigens promote the development of tolerogenic fetal regulatory T cells in utero. Science. 2008;322:1562–1565. doi: 10.1126/science.1164511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Blois SM, Ilarregui JM, Tometten M, Garcia M, Orsal AS, Cordo-Russo R, et al. A pivotal role for galectin-1 in fetomaternal tolerance. Nat Med. 2007;1:1450–1457. doi: 10.1038/nm1680. [DOI] [PubMed] [Google Scholar]
- 113.Li DJ, Guo PF. The regulatory role of thymic stromal lymphopoietin (TSLP) in maternal-fetal immune tolerance during early human pregnancy. J Reprod Immunol. 2009;83:106–108. doi: 10.1016/j.jri.2009.07.015. [DOI] [PubMed] [Google Scholar]
- 114.Huston DP. The biology of the immune system. JAMA. 1997;278:1804–1814. [PubMed] [Google Scholar]