

Abstract
Quantifying the number of deleterious mutations per diploid human genome is of critical concern to both evolutionary and medical geneticists1–3. Here, we combine genome-wide polymorphism data from PCR-based exon re-sequencing, comparative genomic data across mammalian species, and protein structure predictions to estimate the number of functionally consequential mutations carried by each of 15 African American (AA) and 20 European American (EA) individuals. We find that AAs show significantly higher levels of nucleotide heterozygosity than do EAs for all categories of functional mutations considered including synonymous, nonsynonymous, predicted “benign”, predicted “possibly damaging” and predicted “probably damaging” mutations. This result is wholly consistent with previous work showing higher overall levels of nucleotide variation in African populations as compared to Europeans4. EA individuals, on the other hand, have significantly more genotypes homozygous for the derived allele at synonymous and nonsynonymous SNPs and for the damaging allele at “probably damaging” SNPs than AAs do. Surprisingly, for SNPs segregating only in one population or the other, the proportion of nonsynonymous SNPs is significantly higher in the EA sample (55.4%) than in the AA sample (47.0%; P<2.3 ×10−37). We observe a similar proportional excess of SNPs that are inferred to be “probably damaging” (15.9% EA; 12.1% AA; P<3.3 ×10−11). Using extensive simulations, we show that this excess proportion of segregating damaging alleles in Europeans is likely a consequence of a bottleneck that Europeans experienced around the time of the migration out of Africa.
Current estimates of the number of deleterious mutations per diploid human genome vary by several orders of magnitude. Using a correlation in inbreeding rates within consanguineous marriages and mortality, Morton, Crow, and Muller5 estimated each of us carries 3–5 lethal equivalents (i.e., an allele or combination of alleles that if made homozygous would be lethal) whereas Kondrashov6 has predicted that the number may be as high as 100 lethal equivalents. Comparative genomic methods suggest that approximately 38% of amino-acid changing polymorphisms are deleterious, with 1.6 new deleterious mutations arising per individual per generation7 while studies based on segregating polymorphisms estimate that each person carries between 500 and 1,200 deleterious mutations3,8. It is very difficult to reconcile these estimates since each study used different methods and data. Furthermore, studies that used DNA sequences only included data from several hundred genes. Thus, there is a critical need for an unbiased genome-wide estimate of the number of damaging mutations carried by individuals in different populations.
We quantify the number of damaging mutations per diploid human genome by combining the Applera genome-wide survey of SNPs found by resequencing of 20 European Americans (EAs) and 15 African Americans (AAs)9 with comparative genomic data including the PanTro2 build of the chimpanzee genome and protein structure prediction data. After applying strict quality control criteria, the data set we analyzed contains 39,440 autosomal SNPs free of ascertainment bias comprising 10,150 unique transcripts in the human genome (see Methods). Of these SNPs, 20,893 were synonymous (nucleotide changes that do not change the amino acid) and 18,547 were nonsynonymous (nucleotide changes that change the amino acid).
At each SNP, an individual can be homozygous for the ancestral allele (carry zero copies of the mutant allele), heterozygous (carry one copy of the mutant allele), or homozygous for the derived allele (carry two copies of the mutant allele). We find that an individual is heterozygous, on average, for 1,962.4 nonsynonymous SNPs (SD: 275.1; Fig 1a; Supplementary Table 1). These numbers are an underestimate since only SNPs with good quality sequence and a matching chimp base are considered. Perhaps for these reasons, our estimate is slightly smaller than that by Cargill et al.10, even after decreasing their estimate to account for the current estimated number of genes in the genome. For both synonymous and nonsynonymous SNPs, AA individuals are heterozygous at a greater number of SNPs than are EA individuals (Fig. 1a; P < 6.2 ×10−10, Mann-Whitney U-test (MWU) for synonymous SNPs; P<6.2 × 10−10, MWU for nonsynonymous SNPs), consistent with previous studies finding higher levels of genetic variability in Africa4. Interestingly, for both types of SNPs, we find that EA individuals are homozygous for the derived allele at a greater number of SNPs than AA individuals (Fig. 1b; P < 6.2 ×10−10, MWU). These patterns are largely due to an elevated number of SNPs fixed for the derived allele in the EA sample while segregating for two alleles in the AA sample. Excluding SNPs that are not segregating in the particular subpopulation, we observe that AAs have more homozygous derived genotypes per individual at synonymous SNPs and EAs slightly more homozygous derived genotypes per individual at nonsynonymous SNPs.
Figure 1. Distribution of the number of heterozygous and homozygous genotypes per individual.
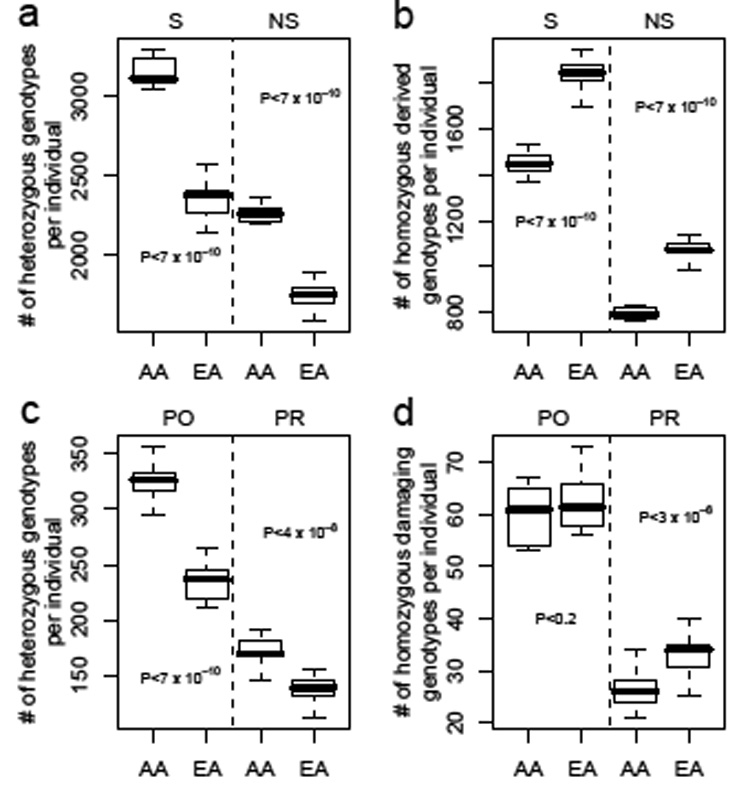
a, Number of heterozygous genotypes per individual at synonymous (S) or nonsynonymous (NS) SNPs. b, Number of genotypes homozygous for the derived allele per individual at synonymous (S) or nonsynonymous (NS) SNPs. c, Number of heterozygous genotypes per individual at possibly damaging (PO) or probably damaging (PR) SNPs. d, Number of genotypes homozygous for the damaging allele at possibly damaging (PO) or probably damaging (PR) SNPs. Dark horizontal lines within boxes indicate medians, and the whiskers indicate the ranges of the distributions. EA: European American; AA: African American.
To estimate the number of damaging alleles carried by each individual in our sample, we used the PolyPhen algorithm8,11 to predict which nonsynonymous SNPs might disrupt protein function. PolyPhen predicts whether a SNP is “benign”, “possibly damaging”, or “probably damaging” based on evolutionary conservation and structural data. In order to assess whether “damaging” SNPs were more likely to be deleterious, we compared the allele frequency distribution of SNPs predicted to be “benign”, “possibly damaging”, and “probably damaging” for each population. We find that the three distributions are significantly different from each other, with more low frequency SNPs in the “probably damaging” category (Table 1, P<5.9 × 10−81 AA, P<2.3 × 10−101 EA, Kruskal-Wallis test), suggesting that the majority of SNPs classified as damaging are also evolutionarily deleterious.
Table 1.
Distribution of Applera SNPs by population and functional class
| Category | Shared | Private AA | Private EA | Mean derived frequency AA1 | Mean derived frequency EA2 |
|---|---|---|---|---|---|
| Synonymous (%) | 8,056 | 8,958 | 3,879 | 0.211 | 0.266 |
| (58.3%) | (53.0%) | (44.6%) | |||
| Nonsynonymous (%) | 5,771 | 7,950 | 4,826 | 0.174 | 0.202 |
| (41.7%) | (47.0%) | (55.4%) | |||
| Benign (%) | 4,448 | 5,260 | 2,928 | 0.200 | 0.238 |
| (78.6%) | (67.7%) | (62.1%) | |||
| Possibly damaging (%) | 795 | 1,572 | 1,035 | 0.113 | 0.119 |
| (14.0%) | (20.2%) | (22.0%) | |||
| Probably damaging (%) | 422 | 942 | 749 | 0.099 | 0.108 |
| (7.4%) | (12.1%) | (15.9%) |
Average frequency using SNPs segregating in the AA sample. No correction for ancestral mis-identification was used.
Average frequency using SNPs segregating in the EA sample. No correction for ancestral mis-identification was used.
Fig. 1c–d shows the distribution of the number of SNPs per individual where individuals were heterozygous (Fig. 1c) and homozygous for the damaging allele (Fig. 1d) for SNPs predicted to be “possibly damaging” and “probably damaging”. We find that an individual typically carries 426.1 damaging (here defined as possibly or probably damaging) SNPs in the heterozygous state (SD: 65.4, range: 340–534) and 91.7 in the homozygous state (SD: 8.6, range: 77–113). Since we surveyed just over 10,000 genes, the actual number of damaging mutations in a person’s genome may be as much as twice that given here. Every individual in our sample is heterozygous at fewer “probably damaging” SNPs than synonymous SNPs, consistent with purifying selection eliminating damaging SNPs from the population. AAs have significantly more heterozygous genotypes than do EAs for all three PolyPhen categories (Fig. 1c, P < 6.2 × 10−10, for “possibly damaging” SNPs; P < 3.7 × 10−8, for “probably damaging” SNPs). The two populations differ significantly in the distribution of homozygous genotypes for the damaging allele at “probably damaging SNPs” (Fig. 1d; P < 2.7 × 10−6), with EAs having approximately 26% more homozygous damaging genotypes than AAs. The lack of a statistical difference at “possibly damaging” SNPs (P=0.17) is likely due to a lack of power since, overall, all other categories of SNPs (synonymous, non-synonymous, “benign”, and “probably damaging”) follow the same pattern of excess homozygosity for the derived/damaging allele in EAs relative to AAs.
Classical analyses of human inbreeding suggest that each individual carries 1.44–5 lethal equivalents5,12. However, inbreeding studies cannot determine whether a single lethal equivalent is due to one lethal allele, two alleles each with a 50% chance of lethality, 10 alleles each with a 10% chance of lethality, or other combinations. Since we find that individuals carry hundreds of damaging alleles, it is likely that each lethal equivalent consists of many weakly deleterious alleles. Our finding that each person carries several hundred potentially damaging SNPs suggests that large-scale medical re-sequencing will be useful to find common and rare SNPs of medical consequence2.
We next examined the distribution of synonymous and nonsynonymous SNPs between AA and EA population samples (Table 1). As expected4, there are more of both types of SNPs in the AA sample than in the EA sample. However, when classifying synonymous and nonsynonymous SNPs as being shared, private to AA, or private to EA, we strongly reject homogeneity (Table 2, P < 3.0 × 10−88). We find the proportion of private SNPs that are nonsynonymous (49.9%) is higher than the proportion of shared SNPs that are nonsynonymous (41.7%; P < 4.3 × 10−54), which is not surprising since nonsynonymous SNPs are more likely to be at lower frequency and thus be population specific. However, considering only the private SNPs, we find that the EA sample has a higher proportion of nonsynonymous SNPs (55.4%) than the AA sample (47.0%; P < 2.3 ×10−37). We observed a similar significant proportional excess of private nonsynonymous SNPs in an independent data set collected by the SeattleSNPs project (Supplementary Table 3; Supplementary Note 1). The SeattleSNPs data, additional quality control analyses (Supplementary Note 2 and Supplementary Table 4), and a similar finding reported for the ANGPTL4 locus13 indicate that this pattern is not an artefact of the Applera data. Our further analyses using Yoruba individuals from Nigeria collected by the International HapMap Consortium14, support this result indicating that it is robust to admixture (Supplementary Note 3).
Table 2.
Results of G-tests of homogeneity for Table 1.
| Nonsynonymous vs. | Benign vs. Possibly vs. | |||||
|---|---|---|---|---|---|---|
| Synonymous | Probably damaging | |||||
| G | df | P-value | G | df | P-value | |
| Shared vs. private AA vs. | 403.1 | 2 | 3.0 × 10−88 | 377.8 | 4 | 1.8 × 10−80 |
| private EA | ||||||
| Shared vs. Private | 239.9 | 1 | 4.3 × 10−54 | 329.5 | 2 | 2.9 × 10−72 |
| Private AA vs. Private EA | 163.2 | 1 | 2.3 × 10−37 | 48.3 | 2 | 3.3 × 10−11 |
We hypothesized that the proportional excess of nonsynonymous polymorphism in the EA sample could be due to varying efficacy of purifying selection due to differences in demographic histories between the two populations. Our hypothesis has two testable predictions: 1) if this proportional excess of nonsynonymous polymorphisms in EAs is due to an excess of damaging alleles, we would also expect to find a proportional increase of “probably damaging” SNPs as predicted by PolyPhen in the EA sample, and 2) we should be able to recapitulate this pattern using simulations with reasonable demographic parameters. When dividing nonsynonymous SNPs into the three PolyPhen categories, we find a significant excess of “probably damaging” SNPs in private SNPs compared to shared SNPs (Table 1 and Table 2). When considering only the private SNPs, we find a significantly higher proportion of “probably damaging” SNPs in the EA sample relative to the AA sample (P<3.3 × 10−11, Table 1 and Table 2), supporting our hypothesis that the excess proportion of nonsynonymous SNPs in the EA sample is due to a higher proportion of damaging SNPs.
In order to assess whether these observations are consistent with plausible demographic histories of the two populations, we developed a large-scale forward simulation program that includes non-stationary demography and a negative log-normal distribution of selective effects for deleterious mutations. Our program used demographic parameters estimated from the data and the literature15 for each population (Supplementary Table 2). For example, for the simulations in Fig. 2a,b, we used a population expansion model for the AAs and a bottleneck model for the EAs (Supplementary Fig. 1). We sampled from these simulated populations and found that the proportion of nonsynonymous SNPs is greater in the bottlenecked population than in a population that has expanded (Fig 2a; Supplementary Table 2; Supplementary Fig. 2a). Furthermore, as shown in Fig. 2a, the simulated proportions agree with the observed proportions for the Applera dataset (here the proportion includes all SNPs, not just private ones). For all demographic models considered, we observed a higher proportion of nonsynonymous SNPs in the population that underwent a bottleneck as compared to a population of constant size, or that has expanded; the degree to which these other models fit the observed data is variable, however (Supplementary Table 2; Supplementary Fig. 2a). For all models tested, we find that a higher proportion of SNPs in the simulated EA sample are weakly or strongly deleterious (−0.001< s < −0.5) than in the simulated AA sample (Fig 2b; Supplementary Table 2, Supplementary Fig. 2b), which supports our hypothesis that a higher proportion of deleterious alleles have accumulated in the bottlenecked population. Our analysis illustrates that plausible models of human demography and purifying selection are sufficient to account for the observed increase in the proportion of nonsynonymous SNPs in the EA sample relative to the AA sample.
Figure 2. Demography and selection can cause a proportional excess of nonsynonymous SNPs in Europeans.
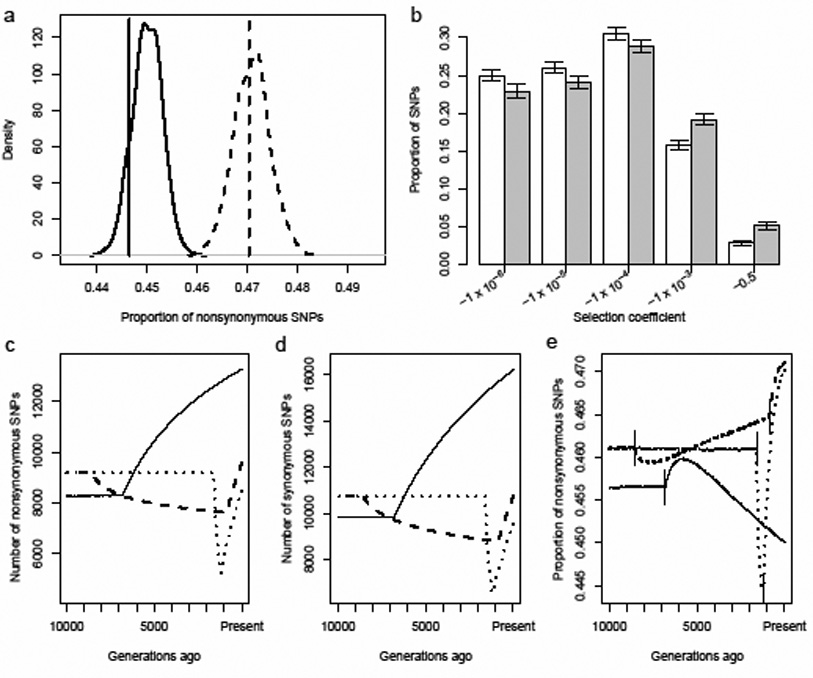
a,b Results of forward-simulations of a population that expanded (AA 2 in Supplementary Table 2), to represent the African American (AA) population and a population that experienced a bottleneck to represent the European (EA) population (EA 1 in Supplementary Table 2).
a, Distribution of the proportion of nonsynonymous SNPs segregating in samples simulated under European (dashed curve) and African (solid curve) demographic models. Vertical lines show the observed proportions in the Applera dataset. b, Distribution of selection coefficients for simulated SNPs in the AA (white bars) and the EA (shaded bars) samples. The labels on the x-axis are the more negative limits of the bins. Error bars denote 95% intervals on the proportion of SNPs in each group.
c–e, Expected distribution of SNPs over time during a population expansion (AA 2, solid lines), a long, mild bottleneck (EA 1, dashed lines), and a short, severe bottleneck (EA 6, dotted lines). Time moves forward in the figures from left to right. Solid vertical lines indicate when the populations changed size. Further details are given in Supplementary Table 2. c, The number of nonsynonymous SNPs, d, the number of synonymous SNPs and e, the proportion of nonsynonymous SNPs.
To determine how the bottleneck contributed to the increased proportion of nonsynonymous SNPs in the EA sample, we recorded the number of SNPs at different time points throughout our forward simulations (see Supplementary Methods). Fig. 2 c–e show how the number of synonymous SNPs, nonsynonymous SNPs, and the proportion of nonsynonymous SNPs change over time for the EA and AA models described above as well as for a second bottleneck model, having a shorter, but more severe reduction in population size. At the start of the bottleneck, the proportion of nonsynonymous SNPs drops below the pre-bottleneck value (due to the preferential loss of low frequency nonsynonymous SNPs). Then, the proportion increases during the bottleneck due to the accumulation of slightly deleterious SNPs that almost behave neutrally in the small population but are eliminated efficiently from larger populations16. Once the population expands, the proportion of nonsynonymous SNPs increases dramatically since the increase in population size results in many more mutations (most of which are nonsynonymous, due to the genetic code) entering the population (Fig. 2c, 2d). Since growth was recent, purifying selection has not had sufficient time to decrease the proportion of nonsynonymous SNPs to the equilibrium value for the larger population. A related effect has been noted in spatial expansion models, where deleterious mutations can “surf” to high frequency on the edge of the expansion17. Our simulations for African demography suggest that once the African population expanded, the proportion of nonsynonymous SNPs also increased initially. But, since the African expansion occurred further back in time than the most recent European expansion, the proportion of nonsynonymous SNPs has had more time to decrease closer to the equilibrium value in the AA sample. At the present time, the absolute numbers of SNPs are higher in the non-bottleneck model (AA 2) than in the bottleneck models (EA 1 and EA 6). The bottleneck dynamics were robust to the distribution of selective effects used in our simulations (Supplementary Fig. 3).
Thus, both the PolyPhen analysis and the forward simulations suggest that given the lower levels of genetic diversity compared to Africans, EAs have a higher proportion of deleterious alleles which can be explained by the Out-of-Africa bottleneck and subsequent expansion that outbred European populations endured. This result is important for two reasons. First, while previous work has highlighted examples of European-specific positive selection14,18–21, the importance of adaptations for the evolution of European populations needs to be tempered by our finding that negative selection is less effective at removing slightly deleterious alleles from European populations. Second, the idea that bottlenecks and founder effects could lead to an increase of damaging alleles in human populations was historically reserved for isolated populations that experienced severe founder effects (e.g. Ashkenazi Jews22 and Finns23). Our work suggests that the interaction of demographic processes and purifying selection can have an important impact on the distribution of deleterious variation, even in populations that did not undergo a severe founder effect.
Methods summary
We used an improved bioinformatics pipeline to analyze SNPs described in ref. 9. We mapped the SNPs to the RefSeq v18 gene model to determine whether they were synonymous or nonsynonymous. Ancestral and derived states for each SNP were determined using the syntenic net alignments between hg18 and panTro2 (refs. 24, 25). When counting the number of genotypes per individual, we added a correction for misidentification of the ancestral allele26. SNPs were dropped from the analysis if they failed to meet our bioinformatics quality controls, but we did not filter SNPs based upon frequency.
To predict whether a nonsynonymous SNP will damage protein function, we used an updated version of PolyPhen which has false-positive and false-negative rates below ~15% (Supplementary Methods). When counting the number of damaging genotypes per individual, we used the subset of SNPs where the predicted damaging allele was the derived allele.
An additional four AA individuals were sequenced, but we did not include them (or SNPs private to them) in further analyses since we determined that they had substantially more European admixture than the other AAs (Supplementary Methods, Supplementary Table 5, and Supplementary Fig. 4). If our estimates of admixture are not perfect, this should not drastically affect the comparisons of different classes of SNPs, making our analysis robust to this problem (Supplementary Note 3). The Coriell sample numbers for the individuals used in our study are given in Supplementary Table 1.
To test whether the higher proportion of nonsynonymous SNPs in EAs compared to AAs could be due to the different demographic histories of the two populations, we used forward simulations which allowed us to model demography and purifying selection. We considered a range of demographic models for both populations (Supplementary Table 2) and a distribution of selective effects for nonsynonymous SNPs.
Online Methods
Bioinformatic pipeline
SNPs were mapped onto RefSeq v18 gene model in a two step process. First we aligned the Celera gene models to hg18 using Blat v33.2 (ref. 27, filtering out any hits that had less than 98.5% sequence identity or less than 90% coverage. We then aligned RefSeq v18 CDS sequences28 to hg18 using the same filtering conditions. Having coordinates of both our SNPs and RefSeq gene models relative to the assembly, we converted our SNP positions onto the RefSeq CDS position to determine reading frame. If a SNP mapped to multiple RefSeqs, we chose the longest transcript for analysis. Any sequences in RefSeq that were not covered by PCR amplicons were excluded from analysis. SNPs that mapped to multiple RefSeqs that were out-of-frame were discarded. SNPs were polarized by the chimpanzee genome using the syntenic net alignments between hg18 and panTro2 (refs. 24, 25). SNPs were dropped from the analysis if they aligned to a non-syntenic region in panTro2, neither human allele matched the panTro2 allele, fewer than nine individuals in either population had a successfully called genotypes, or if we detected a departure from Hardy-Weinberg equilibrium (defined as P<0.01) using the exact test of Wigginton et al.29. SNPs mapping to multiple transcripts were only counted once. We used all SNPs passing bioinformatics quality controls, without filtering for frequency. Certain analyses were also done excluding singletons and are described in Supplementary Note 2.
Correction for ancestral mis-identification
Misidentifying the ancestral state of a SNP can lead to miscalculating the proportion of homozygous derived SNPs carried by each individual. We accounted for the probability of ancestral misidentification by adapting the method of ref. 26 to model the number of homozygous SNPs carried by each individual. In this model, the number of homozygous SNPs carried by each individual is considered to be a mixture of sites whose ancestral states were correctly identified using the chimpanzee outgroup and those that were not (two unknown quantities). The corrected number of homozygous derived mutations carried by each individual can then be reconstituted by solving for this unknown quantity as a function of the mixture proportions and observed data. Here, the mixture proportions account for the divergence time between human and chimpanzee using a context-dependent mutation model inferred along the human lineage30.
PolyPhen analysis
We predicted the functional consequences of SNPs using a newer version of PolyPhen that differs slightly from that described in ref. 8, 11. For SNPs mapping to multiple transcripts, we ran PolyPhen on the SNP in each transcript. If a SNP had different PolyPhen predictions in different transcripts, it was excluded from any further PolyPhen analyses. 340 SNPs had multiple PolyPhen predictions and 56 did not have a prediction. For our data, PolyPhen used an average of 18.2 (SD: 28.0) sequences across covered SNPs. SNPs used for analyses, along with their frequencies and PolyPhen predictions are available (Supplementary Data). For approximately 83.9% of the “benign”, 98.2% of the “possibly damaging” and 98.8% of “probably damaging” SNPs, the damaging allele (the allele with the lower PSIC score) is the derived allele, indicating that PolyPhen has a greater ability to distinguish which allele is damaging for “probably damaging” SNPs than for “benign” or “possibly damaging” SNPs. As explained in the Supplementary Methods, PolyPhen classified 85.5% of 3,604 disease mutations annotated in the UniProt database as either probably or possibly damaging, while predicting 86.1% of 12,237 amino acid differences between humans and another mammalian ortholog as benign. These results suggest that the false positive and false negative rates of the algorithm are each below ~15%.
Counting the number of genotypes per individual
To determine whether AA individuals were heterozygous at more SNPs than EA individuals, we used a two-sided Mann-Whitney U test (MWU) to compare the distribution of the number of heterozygous genotypes per individual in AA individuals to the distribution of the number of heterozygous genotypes per individuals in the EA individuals. This comparison was done separately for synonymous, nonsynonymous, “benign”, “possibly” and “probably damaging” SNPs. A similar test was used to test whether EA individuals were homozygous for the derived allele at a greater number of SNPs than EA individuals. When counting the number of SNPs per individual, we wanted to ensure that our counts were not biased because some samples had more complete sequencing than others. We divided the number of genotypes in an individual of each particular category (e.g. number of heterozygous genotypes for synonymous sites in a particular individual) by the total number of genotypes in that category (e.g. total number of genotypes at synonymous sites) in the individual. We then tested if the distribution of these proportions was different between the AA and EA sample. In all cases, we observed the same pattern as shown in Fig. 1 (data not shown), indicating that this result was not due to inconsistent sequencing of different individuals.
Forward simulations
A detailed description of the methods used for forward simulations is given in Supplementary Methods. Briefly, we wanted to test whether the observation of a higher ratio of nonsynonymous to synonymous SNPs in EAs than in Africans could have been due to the different demographic histories of the two populations. We simulated one population forward in time with a demographic history consistent with that of Africans and another population forward in time with demographic history consistent with that of Western Europe. We considered a variety of plausible demographic models for each population15, and simulated the African and European populations independently of each other. In addition to simulating populations where all SNPs were neutral, we also independently simulated a second set of populations for each set of demographic parameters where the selection coefficients were from a distribution of selective effects (Supplementary Methods) to mimic nonsynonymous sites. At the end of the simulation, we sampled 15 individuals from the population that expanded and 20 individuals from the population that underwent a bottleneck. We examined whether one population had a higher proportion of damaging (nonsynonymous) SNPs and whether segregating SNPs in one population had a different distribution of selection coefficient than SNPs segregating in the other population.
Acknowledgments
We thank the Celera Genomics sequencing center, International HapMap Consortium, and SeattleSNPs for generation of these datasets. This work was supported by NIH 1R01HG003229 to AGC, CDB, RN, and Tara Matisse, NSF0516310 to CDB, and an NSF Graduate Research Fellowship to KEL.
Footnotes
Supplementary Information accompanies the paper on www.nature.com/nature.
References
- 1.Muller HJ. Our load of mutations. Am. J. Hum. Genet. 1950;2:111–176. [PMC free article] [PubMed] [Google Scholar]
- 2.Cohen JC, et al. Multiple rare alleles contribute to low plasma levels of HDL cholesterol. Science. 2004;305:869–872. doi: 10.1126/science.1099870. [DOI] [PubMed] [Google Scholar]
- 3.Fay JC, Wyckoff GJ, Wu CI. Positive and negative selection on the human genome. Genetics. 2001;158:1227–1234. doi: 10.1093/genetics/158.3.1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tishkoff SA, Williams SM. Genetic analysis of African populations: human evolution and complex disease. Nat. Rev. Genet. 2002;3:611–621. doi: 10.1038/nrg865. [DOI] [PubMed] [Google Scholar]
- 5.Morton NE, Crow JF, Muller HJ. An estimate of the mutations damage in man from data on consanguineous marriages. Proc. Natl. Acad. Sci. U. S. A. 1956;42:855–863. doi: 10.1073/pnas.42.11.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kondrashov AS. Contamination of the genome by very slightly deleterious mutations: why have we not died 100 times over? J. Theor. Biol. 1995;175:583–594. doi: 10.1006/jtbi.1995.0167. [DOI] [PubMed] [Google Scholar]
- 7.Eyre-Walker A, Keightley PD. High genomic deleterious mutation rates in hominids. Nature. 1999;397:344–347. doi: 10.1038/16915. [DOI] [PubMed] [Google Scholar]
- 8.Sunyaev S, et al. Prediction of deleterious human alleles. Hum. Mol. Genet. 2001;10:591–597. doi: 10.1093/hmg/10.6.591. [DOI] [PubMed] [Google Scholar]
- 9.Bustamante CD, et al. Natural selection on protein-coding genes in the human genome. Nature. 2005;437:1153–1157. doi: 10.1038/nature04240. [DOI] [PubMed] [Google Scholar]
- 10.Cargill M, et al. Characterization of single-nucleotide polymorphisms in coding regions of human genes. Nat. Genet. 1999;22:231–238. doi: 10.1038/10290. [DOI] [PubMed] [Google Scholar]
- 11.Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 2002;30:3894–3900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bittles AH, Neel JV. The costs of human inbreeding and their implications for variations at the DNA level. Nat. Genet. 1994;8:117–121. doi: 10.1038/ng1094-117. [DOI] [PubMed] [Google Scholar]
- 13.Romeo S, et al. Population-based resequencing of ANGPTL4 uncovers variations that reduce triglycerides and increase HDL. Nat Genet. 2007;39:513–516. doi: 10.1038/ng1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.International HapMap Consortium. A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Voight BF, et al. Interrogating multiple aspects of variation in a full resequencing data set to infer human population size changes. Proc. Natl. Acad. Sci. U. S. A. 2005;102:18508–18513. doi: 10.1073/pnas.0507325102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ohta T. Slightly deleterious mutant substitutions in evolution. Nature. 1973;246:96–98. doi: 10.1038/246096a0. [DOI] [PubMed] [Google Scholar]
- 17.Travis MJ, et al. Deleterious mutations can surf to high densities on the wave front of an expanding population. Mol. Biol. Evol. 2007;24:2334–2343. doi: 10.1093/molbev/msm167. [DOI] [PubMed] [Google Scholar]
- 18.Voight BF, Kudaravalli S, Wen X, Pritchard JK. A map of recent positive selection in the human genome. PLoS Biol. 2006;4:e72. doi: 10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mekel-Bobrov N, et al. Ongoing adaptive evolution of ASPM, a brain size determinant in Homo sapiens. Science. 2005;309:1720–1722. doi: 10.1126/science.1116815. [DOI] [PubMed] [Google Scholar]
- 20.Evans PD, et al. Micrsocephalin, a gene regulating brain size, continues to evolve adaptively in humans. Science. 2005;309:1717–1720. doi: 10.1126/science.1113722. [DOI] [PubMed] [Google Scholar]
- 21.Akey JM, et al. Population history and natural selection shape patterns of genetic variation in 132 genes. PLoS Biol. 2004;2:e286. doi: 10.1371/journal.pbio.0020286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Slatkin M. A population-genetic test of founder effects and implications for Ashkenazi Jewish diseases. Am. J. Hum. Genet. 2004;75:282–293. doi: 10.1086/423146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kere J. Human population genetics: Lessons from Finland. Annu. Rev. Genomics Hum. Genet. 2001;2:103–128. doi: 10.1146/annurev.genom.2.1.103. [DOI] [PubMed] [Google Scholar]
- 24.Kent WJ, Baertsch R, Hinrichs A, Miller W, Haussler D. Evolution's cauldron: duplication, deletion, and rearrangement in the mouse and human genomes. Proc. Natl. Acad. Sci. U. S. A. 2003;100:11484–11489. doi: 10.1073/pnas.1932072100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karolchik D, et al. The UCSC Genome Browser Database. Nucleic Acids Res. 2003;31:51–54. doi: 10.1093/nar/gkg129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hernandez RD, Williamson SH, Bustamante CD. Context Dependence, Ancestral Misidentification, and Spurious Signatures of Natural Selection. Mol. Biol. Evol. 2007;24:1792–1800. doi: 10.1093/molbev/msm108. [DOI] [PubMed] [Google Scholar]
- 27.Kent WJ. BLAT--the BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pruitt KD, Tatusova T, Maglott DR. NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2005;33:D501–D504. doi: 10.1093/nar/gki025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wigginton JE, Cutler DJ, Abecasis GR. A note on exact tests of Hardy-Weinberg equilibrium. Am. J. Hum. Genet. 2005;76:887–893. doi: 10.1086/429864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hwang DG, Green P. Bayesian Markov chain Monte Carlo sequence analysis reveals varying neutral substitution patterns in mammalian evolution. Proc. Natl. Acad. Sci. U. S. A. 2004;101:13994–14001. doi: 10.1073/pnas.0404142101. [DOI] [PMC free article] [PubMed] [Google Scholar]