

Abstract
Concerns about the possible use of Variola virus, the causative agent of smallpox, as a weapon for bioterrorism have led to renewed efforts to identify new antivirals against orthopoxviruses. We identified a peptide, EB, which inhibited infection by Vaccinia virus with an EC50 of 15 μM. A control peptide, EBX, identical in composition to EB but differing in sequence, was inactive (EC50 > 200 μM), indicating sequence specificity. The inhibition was reversed upon removal of the peptide, and EB treatment had no effect on the physical integrity of virus particles as determined by electron microscopy. Viral adsorption was unaffected by the presence of EB, and the addition of EB post-entry had no effect on viral titers or on early gene expression. The addition of EB post-adsorption resulted in the inhibition of β-galactosidase expression from an early viral promoter with an EC50 of 45 μM. A significant reduction in virus entry was detected in the presence of the peptide when the number of viral cores released into the cytoplasm was quantified. Electron microscopy indicated that 88% of the virions remained on the surface of cells in the presence of EB, compared to 37% in the control (p < 0.001). EB also blocked fusion-from-within, suggesting that virus infection is inhibited at the fusion step. Analysis of EB derivatives suggested that peptide length may be important for the activity of EB. The EB peptide is, to our knowledge, the first known small molecule inhibitor of Vaccinia virus entry.
Introduction
In 1980, the World Health Organization declared that smallpox had been eradicated (WHO, 1980). However, recent concerns about the use of Variola virus as a bioweapon have led to a reevaluation of the methods available to control an outbreak of smallpox or a related orthopoxvirus. Vaccination against smallpox is currently restricted because of the risk of adverse side effects (Baggs et al., 2005; Casey et al., 2005; Fulginiti et al., 2003), due in part to the higher percentage of first-time vaccinees, an increase in the number of individuals with compromised immune systems, and contraindications for those with heart disease (Baggs et al., 2005; Rotz et al., 2001; Schwartz and Lebwohl, 2005). Possible side effects of vaccination range in severity from mild fever and rash to keratitis, systemic infection, encephalitis, myocarditis and death (2003a; 2003b; Baggs et al., 2005; Casey et al., 2005; Garde, Harper, and Fairchok, 2004; Kim et al., 2005; Schwartz and Lebwohl, 2005). The risks associated with vaccination, and the potential for deliberate release of Variola virus, has led to renewed efforts to identify novel compounds with activity against poxviruses for potential therapeutic use.
Recently, several compounds have been identified that inhibit various steps in poxvirus infection, including virion morphogenesis (Byrd et al., 2004; Yang et al., 2005) and DNA synthesis (Baker, Bray, and Huggins, 2003; Buller et al., 2004; Magee, Hostetler, and Evans, 2005; Prichard et al., 2006; Yang and Schneller, 2005). Cidofovir diphosphate (CDV) inhibits poxvirus infection in vitro and in vivo and is approved for those with adverse vaccination reactions (Baker, Bray, and Huggins, 2003; Buller et al., 2004; Kern, 2003; Magee, Hostetler, and Evans, 2005; Neyts and Clercq, 2003; Smee and Sidwell, 2003). However, CDV is highly nephrotoxic, thus there is a need to identify additional compounds with activity against poxviruses.
Here, we describe the antiviral activity of EB, a novel peptide inhibitor of Vaccinia virus (VACV), the prototypic poxvirus. EB consists of the 16 amino acid signal sequence of the human fibroblast growth factor 4 protein (Lin et al., 1995) with an additional 4 amino acid solubility tag (RRKK) at the amino terminal end. EB is one of a class of cell penetrating peptides (CPPs), including those derived from the HIV tat protein (Frankel and Pabo, 1988), poly-arginine peptides (Nakase et al., 2004; Richard et al., 2003) and the Drosophila antennapedia homeobox protein (Joliot et al., 1991), that can facilitate the uptake of covalently attached moieties into cells (Lin et al., 1995). CPPs are increasingly being investigated as tools for delivering proteins, DNA, and other cargoes of interest into the cytoplasm or nuclei of cells both in vitro and in vivo (for a recent review, see (Gupta, Levchenko, and Torchilin, 2005).
Initially, EB was investigated for its ability to facilitate the transport of a covalently-linked inhibitor of Herpes simplex virus type 1 (HSV-1) ribonucleotide reductase into cells; however, EB alone was shown to be a more potent inhibitor than the conjugated peptide (Bultmann, Busse, and Brandt, 2001). The EB peptide exhibited several different activities against HSV-1, including inactivation of HSV-1 virions and inhibition of HSV-1 entry into cells, and was thus designated EB for “entry blocker” (Bultmann, Busse, and Brandt, 2001); unpublished data). A scrambled derivative of EB, EBX, lacked inhibitory activity against HSV-1 (Bultmann, Busse, and Brandt, 2001). EB was not cytotoxic to HeLa or Vero cells at concentrations as high as 100 μM, and displayed no cytotoxic effects when applied at a concentration of 6.1 mM to mouse corneas (Akkarawongsa et al., 2006). Subsequent studies have shown that EB is also effective against influenza virus, including H5 avian strains (Jones et al., 2006). We now show that EB also inhibits infection by VACV via a mechanism or mechanisms that appear to be distinct from its activity against HSV-1 or influenza virus.
Materials and Methods
Cells and viruses
HeLa cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 5% calf serum and 5% fetal bovine serum. Infections using the wild-type WR strain of vaccinia virus or the recombinant vMJ343 strain were performed in DMEM supplemented with 1% calf serum and 1% fetal bovine serum (infection media). The recombinant vaccinia virus vMJ343 expresses lacZ under the control of an artificial early promoter (Davison and Moss, 1989), and was kindly provided by Dr. Bernard Moss (Laboratory of Viral Diseases, NIAID, Bethesda, MD).
To propagate virus, confluent HeLa cells were infected at a multiplicity of infection (m.o.i.) of 0.1. Three days post infection, supernatants and cells were harvested. The cells were frozen and thawed three times and the nuclei were pelleted by centrifugation. The supernatants were combined and layered onto a cushion of 36% sucrose and centrifuged at 20,000 × g for 80 minutes in an SW28 rotor (Beckman, Fullerton, CA). The pelleted virus was then resuspended and stored in 10 mM Tris-HCl, pH 9.0 at −80°C until use. To purify virions for attachment assays, viruses were centrifuged in a continuous sucrose gradient (25%–40% in 10 mM Tris-HCl, pH 9.0) at 19,000 × g for 50 minutes using an SW41 rotor (Beckman, Fullerton, CA). The resulting band of virus was then removed and stored at −80°C until use.
Peptides
Synthesis and analysis of the EB and EBX peptides were done at the Biotechnology Center of the University of Wisconsin-Madison as has previously been described (Bultmann and Brandt, 2002; Bultmann, Busse, and Brandt, 2001); (Bultmann, Teuton, and Brandt, 2007). Briefly, synthesis was carried out at a 25 pmol scale using an automated synthesizer (Applied Biosystems model 432A). The cleaved peptides were precipitated with cold t-butylmethylether, and their relative mass was confirmed by electrospray ionization mass spectroscopy. The purity of the peptides was determined by HPLC, and peptide concentrations in solution were determined by absorbance readings at 215 and 225 nm. The peptide library of EB derivatives was synthesized by EZBiolab Inc. (Westfield, IN). The sequences of the tested peptides are given in Tables 4 and 5.
Table 4.
Effect of truncation on EB activity against Vacciniaa
| Name | Sequence | Activityb | deviation |
|---|---|---|---|
| EB | RRKKAAVALLPAVLLALLAP | −0.27 | 0.4 |
| -1 | RRKKAAVALLPAVLLALLA | −0.10 | 0.2 |
| -2 | RRKKAAVALLPAVLLALL | 0.01 | 0.1 |
| -3 | RRKKAAVALLPAVLLAL | 8.76 | 2.2 |
| -4 | RRKKAAVALLPAVLLA | 3.45 | 2.0 |
| -5 | RRKKAAVALLPAVLL | 5.55 | 0.7 |
| -6 | RRKKAAVALLPAVL | 4.76 | 0.8 |
| -7 | RRKKAAVALLPAV | 4.34 | 0.5 |
| -8 | RRKKAAVALLPA | 5.55 | 0.1 |
| -9 | RRKKAAVALLP | 4.45 | 1.1 |
| -10 | RRKKAAVALL | 4.83 | 0.1 |
| -11 | RRKKAAVAL | 4.30 | 0.1 |
| -12 | RRKKAAVA | 3.02 | 0.3 |
| -13 | RRKKAAV | 3.97 | 0.3 |
| -14 | RRKKAA | 4.10 | 0.5 |
| -16 | RRKKA | 4.46 | 0.4 |
| -17 | RRKK | 2.567 | 2.0c |
| 1- | RRKK AVALLPAVLLALLAP | −0.20 | 0.5 |
| 2- | RRKK VALLPAVLLALLAP | 0.021 | 0.6 |
| 3- | RRKK ALLPAVLLALLAP | 0.46 | 0.4 |
| 4- | RRKK LLPAVLLALLAP | 0.07 | 0.2 |
| 5- | RRKK LPAVLLALLAP | 4.68 | 0.5 |
| 7- | RRKK VLLALLAP | 8.91 | 0.4 |
| 8- | RRKK LLALLAP | 5.67 | 0.5 |
| 9- | RRKK ALLAP | 4.00 | 0.5 |
| 10- | RRKK LLAP | 4.50 | 0.9 |
| 11- | RRKK LAP | 4.58 | 0.9 |
| 12- | RRKK AP | 4.72 | 0.1 |
| 13- | RRKK P | 6.36 | 2.7 |
| EBX | RRKKLAALPLVLAAPLAVLA | 8.98 | 1.3 |
| Mock | 4.46 | 0.1 |
Highlighted derivatives retained levels of antiviral activity not significantly different (p > 0.05) by one-component ANOVA from EB.
Average mOD/min of two separate trials.
High variability between trials prevented the loss of activity from being statistically significant.
Table 5.
Effect of leucine substitution on EB activity against VACVa
| Name | Sequenceb | Activityc | Deviation |
|---|---|---|---|
| EB | RRKKAAVALLPAVLLALLAP | 0.26 | 0.2 |
| EBX | RRKKLAALPLVLAAPLAVLA | 7.28 | 0.4 |
| F2 | RRKKAAVAALPAVLLALLAP | 1.09 | 0.0 |
| F1 | RRKKAAVALAPAVLLALLAP | 1.09 | 1.6 |
| C7 | RRKKAAVALLPAVALALLAP | 3.38 | 0.1 |
| C6 | RRKKAAVALLPAVLAALLAP | 0.80 | 0.6 |
| C4 | RRKKAAVALLKAVLLAALAP | −0.06 | 0.1 |
| C3 | RRKKAAVALLKAVLLALAAP | −0.21 | 0.0 |
| D2 | RRKKAAVAKLPAVLLALLAP | 4.06 | 0.0 |
| D1 | RRKKAAVALKPAVLLALLAP | 1.58 | 0.8 |
| A7 | RRKKAAVALLPAVKLALLAP | −0.03 | 0.4 |
| A6 | RRKKAAVALLPAVLKALLAP | 1.05 | 0.0 |
| A4 | RRKKAAVALLPAVLLAKLAP | 2.46 | 2.9 |
| A3 | RRKKAAVALLKAVLLALKAP | 0.11 | 0.2 |
| E2 | RRKKAAVAELPAVLLALLAP | 5.11 | 1.8 |
| E1 | RRKKAAVALEPAVLLALLAP | 4.70 | 1.0 |
| B7 | RRKKAAVALLPAVELALLAP | 4.32 | 0.5 |
| B6 | RRKKAAVALLPAVLEALLAP | 0.53 | 0.4 |
| B4 | RRKKAAVALLPAVLLAELAP | 6.43 | 0.9 |
| B3 | RRKKAAVALLPAVLLALEAP | 4.87 | 0.2 |
| Mock | 4.06 | 1.7 |
Highlighted derivatives displayed significantly less activity (p < 0.05 by one-component ANOVA) than EB.
Bold residues indicate substitutions.
Average mOD/min of two separate trials.
Comprehensive inhibition assay
To determine the EC50 of the peptides when they were present at all times during the infection, 4 × 104 pfu/mL of virus were incubated with increasing concentrations of peptide ranging from 1.56 μM to 200 μM for 1 hr at 37°C in a final volume of 1.2 mL. Confluent HeLa cells grown in 24 well plates were then infected in triplicate with the virus/peptide mixture at an m.o.i. of 0.1 for 1 hr at 37°C before additional media and peptide were added to bring the final volume of each well to 1 mL. Peptide was present continuously during the infection. Three days post-infection the samples were harvested, frozen and thawed 3 times, and yields were determined by plaque assay on HeLa cells.
To test the dependence of the EC50 on virus concentration, HeLa cells were plated at a density of 1 × 104 cells/well of a 96-well plate and grown to confluency. Peptide (1.56 μM to 200 μM) was incubated with 5 × 104, 5 × 105, 5 × 106 or 5 × 107 pfu/mL of vMJ343 for 1 h at 37°C. Ten microliters of the mixture was then plated in quadruplicate on HeLa cell monolayers and incubated for 1 hr at 37°C before the cells were re-fed with infection media. After an additional 3 hr incubation at 37°C, an equal volume of lysis buffer (0.5% NP-40, 2 mM MgCl2 in PBS) was added to each well, and the samples were incubated at room temperature for 10 min. Substrate was added (CPR-gal; Chlorophenol red-β-D-galactopyranoside, #10884308001, Roche Applied Science, IN), and the initial rate of product formation was measured at 570 nm using an ELX-800 plate reader (Biotek, Winooski, VT) in the linear range.
Post-entry assay
HeLa cells grown to confluency in 24-well plates were infected with virus at an m.o.i. of 2 or an m.o.i. of 5 for 1 hr. One set of wells was then harvested (T = 0), and the supernatants were removed from the remaining wells. One milliliter of infection media alone or infection media with 50 μM EB (EC50 for m.o.i. of 2) or 85 μM EB (EC50 for m.o.i. or 5) was added to the remaining wells, and cells were harvested at 1, 6, 12, 24 and 48 hrs after the addition of peptide. Each sample was titered in triplicate on HeLa cells.
Virus attachment ELISA
The recombinant virus vMJ343 (1 × 106 pfu) was incubated with increasing concentrations of peptide for 1 hr at 37°C and chilled. The virus solution was plated on chilled HeLa cells in a 96-well plate (50 μL per well, m.o.i. = 2) for 1 hr at 4°C, washed with chilled PBS, and fixed at RT for 30 min in 4% paraformalde and stored overnight in PBS at 4°C. The plate was then blocked for 30 min in 3% BSA, probed for 1 hr with a 1:500 dilution of rabbit anti-VACV antibody (#8101, Virostat, Portland, ME) for 1 hr at 37°C in a humid chamber, and rinsed 3 x with PBS. Secondary antibody (HRP goat anti-rabbit, #sc2030, Santa Cruz Biotechnology, Santa Cruz, CA) was added at a 1:1000 dilution for 1 hr at 37°C in a humid chamber. The wells were rinsed 3 x with PBS and developed using the SigmaFAST™ OPD kit (#P9187, Sigma Aldrich, St. Louis, MO) according to the manufacturer’s instructions. The plate was read at 490 nm using an ELX-800 plate reader (Biotek, Winooski, VT).
Detection of early gene expression
Confluent HeLa cells grown in 96-well plates were infected with mock-treated vMJ343 (8.75 × 105 pfu) or virus pre-treated with 25 μM or 100 μM EB in a final volume of 100 μL for 1 hr. After incubation at 37°C for 3 hr, the supernatants were removed and the cells were lysed and developed as for the comprehensive inhibition assay. A control for virus-associated β-galactosidase was run in the presence of 300 μg/mL cycloheximide to inhibit β-galactosidase expression and the signal was subtracted from the cycloheximide-free samples.
Post-attachment assay
HeLa cells were grown to confluency in 96-well plates and infected with vMJ343 at an m.o.i. of 10 in a volume of 50 μL at room temperature for 1 hr. Concentrations of EB ranging from no peptide (mock-treated control) to 200 μM were then added in a final volume of 100 μL per well. The samples were then incubated at 37°C for 3 hrs prior to the measurement of β-galactosidase activity as described above.
Fluorescent microscopy
The entry assay was modified from the protocol described by Law and Smith (Law and Smith, 2004). Briefly, HeLa cells were grown to confluency on glass coverslips. VACV (1 × 104 pfu/mL) was incubated with 50 μM EB or EBX and 300 μg/mL cycloheximide for 1 hr at 37°C in a total volume of 500 μL. The mixture was then plated onto the cells, and the cells were spinoculated at 650 × g for 1 hr at 4°C using a Beckman T6-J centrifuge. The samples were transferred to 37°C for one hour, then fixed with 10% formaldehyde in PBS for 20 minutes and rinsed 3 times with PBS before being stored overnight at 37°C in PBS. The samples were then washed three times for 5 min in 100 mM glycine and then blocked for 30 min with 10% FBS in PBS. The coverslips were probed for 1 hr at 37°C with polyclonal goat anti-VACV antisera (1:250 dilution; # V0500-01, USBiologicals, Swampscott, MA), rinsed 3 times with PBS, and stained with 1:200 IgG Alexa Fluor 488 donkey anti-goat (A11055, Molecular Probes, CA) for 1 hr at 37°C. After rinsing 3 times with PBS, the cells were permeabilized for 5 min with 0.1% Triton-X in PBS, washed 3 times in PBS, and blocked an additional 30 min. Virus cores within the cytoplasm were probed with 1:500 polyclonal rabbit anti-core serum (gift of G. Griffiths, EMBL, Heidelberg, Germany) and stained with 1:200 IgG Alexa Fluor 594 goat anti-rabbit (#A11012, Molecular Probes, CA). The cells were rinsed once in PBS, incubated for 5 minutes in PBS + 1 ng/mL Hoechst 33342 (#H1399, Molecular Probes, CA), and rinsed a final time before the coverslips were mounted on slides with ImmunoMount (ThermoShandon, Pittsburgh, PA). Coverslips were viewed with an Axioplan 2 fluorescent microscope (Zeiss, Gottingen, Germany), photographed, and the number of attached virus and viral cores was counted for 50 cells. All images using the same filter were taken using the same exposure.
Electron microscopy (EM)
A total of 1 × 107 pfu/mL of gradient-purified vMJ343 were incubated at 37°C in media with or without 50 μM EB and supplemented with 300 μg/mL cycloheximide. HeLa cells grown to confluency on 10 mm glass coverslips were then infected at a multiplicity of infection of 100 for 1 hr at 37°C. Samples were fixed in 2.5% glutaraldehyde, 2.0% paraformaldehyde in 0.1M sodium cacodylate buffer, pH 7.4 for 1 hr at room temperature. The cells were then post fixed in 1% osmium tetroxide (OsO4) in the same buffer for 1 hr at room temperature. Following OsO4 post-fixation, the samples were dehydrated in a graded ethanol series, further dehydrated in propylene oxide and embedded in Epon. The samples were then etched with 52% hydrofluoric acid to remove the glass cover slips and sectioned using a Reichert-Jung Ultracut-E Ultramicrotome. After staining with Reynolds lead citrate and 8% uranyl acetate in 50% ethanol, ultrathin sections were viewed with a Philips CM120 electron microscope and images were captured with a MegaView III side-mounted digital camera. Virus particles from ~30 fields of view per condition were scored based on whether they were adsorbed to the cell surface (adsorbed), whether the virus cores had been released into the cytoplasm (entered), or whether the virus particles were associated with vesicles within the cytoplasm (vesicles).
Inactivation of virions
To determine if EB inactivated virions, 1 × 106 pfu/mL of virus were incubated with increasing concentrations of peptide ranging from 1.56 μM to 200 μM for 1 hr at 37°C. The virus mixture was then dialyzed overnight at 4°C against 10 mM Tris with 0.1 M NaCl, pH 7.4 (Slide-A-Lyzer dialysis cassette, #66425, Pierce, Rockford, IL) and titered by plaque assay on HeLa cells.
Cell pretreatment assay
HeLa cells grown to confluency in 96-well plates were pretreated in triplicate with 50 μL of peptide (1.5625 μM to 200 μM) in infection media for 1 hr at 37°C. The medium was then removed, and 500 pfu of vMJ343 were added per well (m.o.i. = 0.2). The amount of virus used fell within the linear range of β-galactosidase signal detected from cells infected by vMJ343 as determined by standard curve (data not shown). Infected cells were incubated for 3 hr at 37° C, when the medium was removed. Lysis buffer (50 μL) was then added, and β-galactosidase activity was measured as described above.
Determining the contribution of peptide-cell interactions to EB activity
HeLa cells grown in 96-well plates were chilled to 4°C and exposed to EB, infected with vMJ343 (m.o.i.=1), or treated with media for 1 hr. The liquid was then decanted and replaced with vMJ343, EB, or EB-treated vMJ343 (m.o.i.=1), respectively, for 1 hr at 4°C. The temperature was then raised to 37°C for 4 hr. The medium was removed, lysis buffer was added, and the β-galactosidase activity measured as described above. Data were normalized to endogenous β-galactosidase levels in uninfected cells. The EC50 of EB under all three conditions was determined relative to the β-galactosidase activity measured in cells infected with mock-treated vMJ343.
Fusion from within
Confluent HeLa cells grown in 24-well plates were infected at a m.o.i. of 5 for 24 hrs. The medium was then removed, and the wells incubated for 3 min at room temperature with PBS adjusted to a pH of 7.0 or 5.0. The wells were then washed twice with media and incubated at 37°C for 3 hrs in media with or without 150 μM EB. The cells were then fixed in 4% paraformaldehyde in PBS for 30 min and stored in PBS until viewed with an Axiovert 200M inverted microscope (Carl Zeiss, Thornwood, NY). Pictures are representative of two independent experiments per condition.
To verify that EB was not toxic to cells when added immediately after the low pH wash, HeLa cells grown to confluency in a 96-well plate were treated with PBS adjusted to a pH of 7 or 5 as above, rinsed twice with DMEM + 2% FBS, and then incubated for 1 hr at 37°C in a 1:5 dilution of CellTiter 96® AQueous One Solution (#G3580, Promega Corporation, Madison, WI) in DMEM + 2% FBS with or without the addition of 150 μM EB (total volume, 100 μL). The reaction was then halted by the addition of 25 μL of 2.5% NP-40 in DMEM, and product formation measured at 490 nm using an ELX-800 plate reader (Biotek, Winooski, VT). Data represent 6 independent samples per condition, and were normalized to the signal from wells containing CellTiter 96® AQueous One Solution alone.
Screening of the peptide library
Confluent HeLa cells in 96-well plates were infected for 3 hrs at 37°C with 5 × 104 pfu/well of vMJ343 that had been pretreated for 1 hr with 100 μM of peptide. The supernatants were removed and the cells were lysed and developed as above. To control for virion-associated β-galactosidase, one set of samples was run in the presence of 300 μg/mL cycloheximide and the signal was subtracted from the cycloheximide-free samples.
Statistical analysis
Student’s paired t-tests were performed using Microsoft Excel. One-way ANOVA tests were performed using Sigmaplot 11 (Systat Software Inc., San Jose, CA).
Results
The antiviral activities of EB and the control peptide EBX (Table 1) against VACV were initially tested using a comprehensive yield reduction assay where the peptides were present at all times. Virus (4 × 104 pfu/mL) was incubated with increasing concentrations of peptide for 1 hr. The viral inoculum was then added to HeLa cells, and the virus yield 3 days p.i. was determined by plaque assay. There was a dose dependent decrease in virus yield in the presence of EB, with an EC50 value of 15 μM (Table 1). In contrast, the EC50 for the scrambled peptide was 150 μM (Table 1), indicating that the antiviral effect was sequence specific.
Table 1.
Antiviral properties of peptides used in this study.
| EC50 (μM) | |||||
|---|---|---|---|---|---|
| Peptide | Sequence | Comprehensivea | Pretreated cellsb | Post-adsorptionc | CC50 (μM)d |
| EB | RRKKAAVALLPAVLLALLAP | 15 | 63 | 45 | >100 |
| EBX | RRKKLAALPLVLAAPLAVLA | 150 | n.d. | n.d. | >100 |
n.d. = not done
HeLa cells were infected with peptide-treated virus for 3 days prior to harvesting. Peptide was present continuously during the infection. Viral titers were determined by plaque assay.
HeLa cells were treated with peptide for 1 hr prior to infection with the recombinant virus vMJ343. After 3 hrs incubation at 37°C, the infected cells were lysed and the β-galactosidase activity was measured.
HeLa cells were infected with the recombinant virus vMJ343 at an m.o.i. of 2 for 1 hr at room temperature. Peptide was then added and the samples were shifted to 37°C for 3 hrs, lysed, and the β-galactosidase activity was measured.
50% cytotoxic concentration, as reported in (Akkarawongsa et al., 2006)
We previously observed that high concentrations of HSV-1 in solution could overwhelm the antiviral activity of EB (unpublished data). To determine whether EC50 was dependent on virus concentration, increasing concentrations of the vMJ343 strain of virus, which expresses β-galactosidase from an early promoter (Davison and Moss, 1989), were incubated with up to 200 μM EB and then added to HeLa cells. Four hours later, β-galactosidase activity was measured. As the concentration of virus increased, the EC50 of EB increased (Fig. 1), indicating that the activity of the peptide could be overwhelmed by excessive amounts of virus.
Figure 1. High concentrations of virus can overwhelm EB activity.

HeLa cells grown in 96-well plates were infected with increasing concentrations of vMJ343 that had been pretreated with EB (1.56 μM to 200 μM). Four hours post-infection, the cells were lysed and β-galactosidase activity was measured. The EC50 value of EB against each concentration of virus was determined from a plot of peptide concentration versus β-galactosidase activity (data not shown). The data represent the mean of four replicates.
The EB peptide can penetrate cell membranes when added exogenously (Lin et al., 1995), suggesting that EB may inhibit an intracellular step in VACV infection. To test whether EB reduced infection when added after virus infection had been established, HeLa cells were infected at a m.o.i. of 2 or 5, and 50 μM EB or 85 μM EB, respectively, was added at 1 hr p.i. The titers of virus from EB-treated cells were the same as those from untreated cells at all time points measured, indicating that EB was not able to block an established VACV infection (Fig. 2). These data suggested that EB was not acting intracellularly.
Figure 2. EB is not active when added post-infection.

Cells were infected at an m.o.i. of 2 (circles) or 5 (triangles) for 1 h with WR, rinsed once with PBS, and then media only (filled symbols) or media containing 50 or 85 μM EB (EC50 for m.o.i. = 2 and m.o.i. = 5, respectively; open symbols) was added. Infected cells were harvested at the indicated times post-addition of EB. All samples were titered in triplicate, with the data representing the means and standard deviations of the means.
To determine whether EB treatment inhibited VACV attachment, virus binding was measured by ELISA. Pretreated vMJ343 was added to HeLA cells (m.o.i. = 2) for 1 h at 4°C. The cells were fixed and probed for attached virus using polyclonal anti-VACV antisera. No significant decrease in virus attachment in the presence of high concentrations of EB was detected (p>0.1, Fig. 3A), indicating that VACV attachment was not inhibited by EB treatment.
Figure 3. EB does not block VACV adsorption.
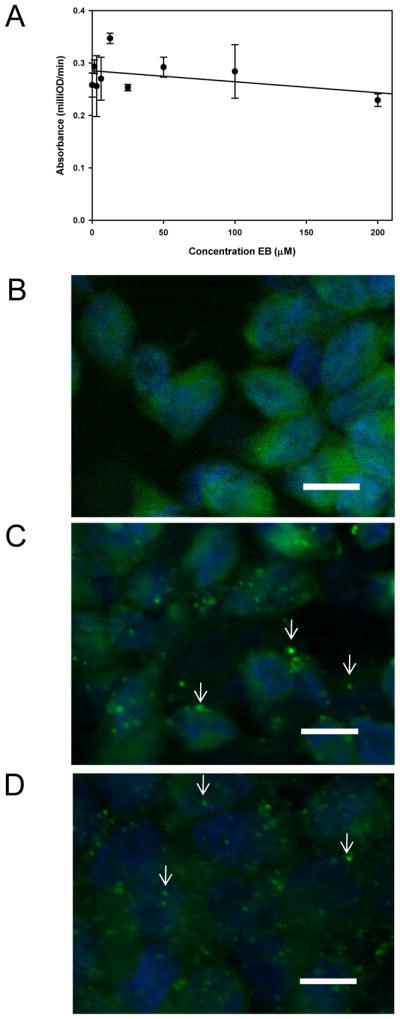
(A) EB does not block VACV adsorption as measured by ELISA. Chilled HeLa cells were exposed to EB-treated vMJ343 at an m.o.i. of 2 for 1 hr at 4°C, fixed, and probed for attached virus using polyclonal anti-VACV antisera and an HRP-conjugated secondary. The plates were developed using the SigmaFASTtm OPD kit and read at 490 nm. Data are the means and standard deviations of three replicates and are representative of two independent runs. (B–D) EB does not block VACV adsorption as measured by immunofluorescence microscopy. HeLa cells grown on glass coverslips were mock infected (B) or infected with mock-treated WR (C) or virus treated with 50 μM EB (D) for 1 hr. Cells were then fixed, and stained for adsorbed virus with polyclonal antiserum (green). Nuclei were stained with Hoechst 33348. Fifty cells per condition were scored for the number of adsorbed virions detected (see Table 2). Arrows denote attached virions. [scale bars = 20 μm]
These results were also confirmed by a standard immunofluorescence assay for virus adsorption (Law and Smith, 2004). Mock- or EB-treated virus was spinoculated onto cells and adsorbed for 1 hr. The cells were then fixed and stained for adsorbed virus. The number of adsorbed virions detected per cell in mock- versus EB-treated samples was not significantly different (p=0.75; Table 2; Fig. 3B–D), confirming the ELISA results that virus adsorption was not quantitatively affected by EB.
Table 2.
Effect of EB on VACV adsorption and entry as determined by IFA
Average number of virions detected on the cell surface
Average number of cores detected in the cytoplasm
p = 0.75 compared to control
p < 0.0001 compared to control
The data indicated that EB blocked VACV infection post-adsorption but prior to the establishment of a productive infection. To determine if EB acted upstream of early gene expression, HeLa cells were infected for 3 hr with vMJ343 that had been mock-treated or incubated with 25 μM or 100 μM EB and then assayed for β-galactosidase activity. Samples treated with 25 μM EB exhibited 80% less β-galactosidase activity than mock-treated samples, while β-galactosidase activity in samples treated with 100 μM EB were indistinguishable from background (Fig. 4). These data are consistent with the hypothesis that EB blocked VACV infection at a step prior to early gene expression.
Figure 4. EB blocks infection before early viral protein synthesis.

The vMJ343 virus was mock treated or treated with EB at the indicated concentrations for 1 hr at 37°C and then used to infect HeLA cells. The cells were lysed 3 hrs p.i., and β-galactosidase activity was measured by ELISA at 570 nm. Cycloheximide (300 μg/mL) was added to one set of samples as a control for background β-galactosidase activity, and the signal was subtracted from the test samples. Data represent the mean ± standard deviation from three samples.
To confirm that EB inhibited VACV entry, the number of VACV cores released into the cytoplasm were quantitated by immunofluorescence. Mock-treated or EB-treated VACV (1 × 104 pfu) was spinoculated onto HeLa cells (Carter et al., 2005) and incubated for 1 hr at 37°C before being fixed and stained for both adsorbed virus and viral cores. Cycloheximide was present during the infection to prevent dissolution of any cores released into the cytoplasm. In the absence of EB there was an average of 2.74 cores visible per cell. When EB was present, the number of cores detected dropped significantly to 0.1 cores per cell (p < 0.0001; Table 2; Fig. 5). Similar results were obtained when virus was allowed to adsorb passively to cells (data not shown), thus spinoculation did not affect the ability of EB to inhibit VACV infection. These results are consistent with EB inhibiting VACV entry.
Figure 5. EB blocks core release into the cytoplasm.

HeLA cells grown on coverslips were mock infected (A) or infected with mock-treated WR (B) or virus treated with 50 μM EB (C) for 1 hr in media containing 300 μg/mL cycloheximide. Cells were then fixed, permeabilized with 0.1% Triton-X and stained for virus cores using polyclonal antisera (red). Nuclei were stained with Hoechst 33348. Fifty cells per condition were scored for the number of viral cores detected (see Table 2). Arrows denote representative viral cores. [scale bars = 20 μm]
As further confirmation that EB inhibited VACV entry, cells were exposed to VACV alone or to VACV with 50 μM EB for 1 hr at 37°C, thin sectioned, and the location of virions determined by TEM. Horizontal and vertical cross-sections were scored for the number of virus particles attached to cell surfaces (attached, Fig. 6A), the number of cores visible in the cytoplasm (entered, Fig. 6B), and the number of virus particles that appeared to be associated with vesicles in the cells (vesicles, Fig. 6C). The quantitative data are shown in Table 3. The percentage of virus particles that were attached to cell surfaces was significantly higher in EB-treated samples than in mock treated samples (88% vs. 37%, p < 0.001), while the percentage of cores visible in the cytoplasm of the EB-treated samples was significantly lower than in mock treated samples (0% vs. 42%, p < 0.001). The percentage of virus particles that appeared to be associated with vesicles was not significantly different between the two treatments (12% in EB treated vs. 21% in untreated, p > 0.05). These data confirm that EB blocked VACV entry.
Figure 6. EB blocks VACV entry into cells.

HeLa cells grown on glass coverslips were infected with mock treated vMJ343 (m.o.i. 100) or virus pretreated with 50 μM EB for 1 hr at 37°C. Horizontal and vertical sections of the cells were viewed by TEM, and an equal number of virus particles from mock-and EB-treated samples were counted and scored for location. (A) Representative sample of virus adsorbed to the surface of the cells (Adsorbed). (B) Representative sample of a virus core visible in the cytoplasm (Entered). (C) Representative sample of a virion contained within a vesicle (Vesicles). Arrows indicate virus cores; arrowheads indicate vesicle membranes.
Table 3.
Location of Vaccinia virus particles as determined by EM
Virions on cell surface
Cores in cytoplasm
Cores in vesicles
p<0.001 compared to control
p<0.001 compared to control
p>0.05 compared to control
To determine whether EB inhibits VACV entry primarily through interactions with the virus or with the cell surface, the EC50s of EB when added prior to VACV adsorption, after VACV adsorption, and simultaneous with VACV adsorption were directly compared. HeLa cells were chilled to 4°C and exposed first to peptide and then VACV, to VACV and then peptide, or to media and then EB-treated VACV. The infected cells were then shifted to 37°C for 4 hrs before β-galactosidase activity was measured. The β-galactosidase activity was compared to that observed in cells infected with untreated VACV. The EC50 for EB when added post-VACV adsorption and when used to pretreat the virus was approximately 45 μM,, while the EC50 for EB when used to pretreat the cells was approximately 170 μM (Fig. 7A). These data indicate that the EB inhibits VACV entry primarily via interactions between the peptide and the virus at low concentrations of peptide, while interactions between EB and the cell surface contribute to the inhibition of VACV at higher concentrations of peptide.
Figure 7. EB interacts reversibly with virions to inhibit entry.

(A) EB interactions with virions, not cells, are responsible for VACV inhibition at low concentrations of peptide. Confluent cells were exposed in triplicate to either EB (●), vMJ343 (m.o.i. = 1; ○), or media (▼) for 1 hr at 4°C. Virus (m.o.i.= 1; ○,▼) or peptide (●) was then added for 1 hr at 4°C. Cultures were shifted to 37°C for 4 hrs, lysed, and β-galactosidase activity was measured and compared to the signal present in cells infected with virus in the absence of EB. Data are the means and standard deviations of triplicate infections; experiment is representative of three independent trials. Dashed line indicates 50% of the virus-only control. (B) EB inhibition is reversible by dialysis. VACV (WR; 1 × 106 pfu/mL) was incubated with increasing concentrations of EB for 1 hr at 37°C, then dialyzed overnight at 4°C using Slide-a-lyzer dialysis cassettes. Samples were then titered in triplicate and compared to the titer of mock-treated, dialyzed virus (0 μM EB). The bars represent the standard deviations of the means.
EB irreversibly inactivates HSV-1 (Bultmann, Busse, and Brandt, 2001). To determine whether the antiviral activity of EB against VACV was due to the peptide irreversibly inactivating VACV, 1 × 106 pfu of virus were incubated for 1 hr with increasing concentrations of EB in a final volume of 1 mL. The samples were dialyzed and the recovered virus was titered by plaque assay on HeLa cells. The infectivity of EB-treated samples was fully restored regardless of the concentration of EB used (Fig. 7B). Electron microscopy of virus particles treated with EB showed they were morphologically identical to mock-treated particles (data not shown). These data indicate that the inhibition of VACV by EB was reversible and suggested that EB does not grossly disrupt the integrity of VACV particles.
Successful fusion of the viral envelope and the host cell membrane has frequently been associated with the ability of VACV to induce cell-cell fusion from within and from without upon treatment with low pH (Brown, Senkevich, and Moss, 2006; Gong, Lai, and Esteban, 1990; Izmailyan et al., 2006; Moss, 2006; Ojeda, Domi, and Moss, 2006; Ojeda, Senkevich, and Moss, 2006; Senkevich and Moss, 2005; Senkevich, Ward, and Moss, 2004; Townsley, Senkevich, and Moss, 2005a; Townsley, Senkevich, and Moss, 2005b). The very high m.o.i. used for the fusion-from-without assay required concentrations of EB approaching toxicity and therefore was not done. To determine if EB could inhibit fusion-from-within, HeLa cells infected at a m.o.i. of 5 for 24 hrs were briefly treated with PBS with a pH of 7 or 5. The solution was then neutralized, and the cells were refed either with normal media or media supplemented with 150 μM EB. Three hours later, the cells were fixed in paraformaldehyde and visualized. Infected cells treated with low pH showed considerable syncytia formation compared to infected cells treated at a neutral pH (Fig. 8A). Infected cells treated with EB, however, showed little sign of syncytia formation after either pH treatment, indicating that EB inhibited cell-cell fusion (Fig. 8A). The combination of the low pH treatment and subsequent EB exposure was not toxic to the cells (Fig. 8B). These data suggest that EB blocks VACV entry by inhibiting fusion between the virus envelope and the cell membrane.
Figure 8. EB blocks fusion-from-within.

(A) Confluent HeLa cells in 24-well plates were infected at a m.o.i. of 5 with WR and incubated for 24 hrs. The cells were then exposed to PBS adjusted to the indicated pH for 3 min, washed twice with infection media, and incubated 3 hrs with infection media with or without 150 μM EB. The samples were fixed with paraformaldehyde and examined for syncytia formation. Arrows denote syncytia. Bars = 100 μm. (B) EB is not toxic to cells when added immediately after low-pH treatments. Confluent HeLa cells were treated with PBS (pH 7 or 5), rinsed twice with DMEM + 2% FBS, and then incubated for 1 hr at 37°C in a 1:5 dilution of CellTiter 96® AQueous One Solution (#G3580, Promega Corporation, Madison, WI) in DMEM + 2% FBS with or without the addition of 150 μM EB. The reaction was halted by the addition of 25 μL of 2.5% NP-40 in DMEM. Product formation was measured at 490 nm using an ELX-800 plate reader (Biotek, Winooski, VT). Data represent 6 independent samples per condition, and were normalized to the signal from wells containing CellTiter 96® AQueous One Solution alone.
Structural analysis of EB
To map the regions of EB crucial to its activity, we screened a library of sequentially-truncated EB derivatives for the loss of anti-VACV activity (Table 4). The vMJ343 virus was incubated with 1 peptide for 1 hr at a concentration of 100 μM, and then 5 × 104 pfu of treated virus were added to confluent HeLa cells for 3 hr at 37°C. The supernatants were then removed, the monolayers were lysed, and the substrate was added. Beginning after the RRKK solubility tag, up to 3 residues could be removed without significantly reducing activity (compare EB4- to EB5-, Table 4). Up to 3 residues could be deleted from the carboxy terminus without reducing the activity (compare EB-2 and EB-3, Table 4). Note that when deleting from either direction, antiviral activity was lost when the first leucine encountered was removed. These results suggested that the length of the peptide and perhaps the presence of the leucine pairs at positions 9–10 and 17–18 may be important for the activity of EB.
To further characterize the role of the various residues in the antiviral activity of EB, peptide derivatives with alanine, lysine or glutamic acid substitutions for the individual leucines were screened (Table 5). Substituting alanine for any of the six leucines had no significant effect on the activity of the peptide, suggesting that multiple leucine residues may contribute to the antiviral effect. Substituting lysine for the leucine at position 9 significantly reduced activity (D2; Table 5), but single lysine substitutions for the leucines at the other positions had no effect on antiviral activity, indicating that single positive charges can be tolerated at positions 10, 14, 15, 17 and 18 but not at position 9. Single glutamic acid substitutions at positions 9, 10, 14, 17 and 18 significantly reduced peptide activity (E2, E1, B7, B4, B3; Table 5), indicating that negative charges at these positions adversely affect the ability of the peptide to inhibit VACV activity.
Discussion
Concerns about the deliberate release of Variola virus have renewed interest in the development of antiviral drugs effective against orthopoxviruses. Here we describe the anti-VACV activity of a novel antiviral peptide, denoted EB. The EB peptide blocked VACV infection with an EC50 of 15 μM in a sequence-specific manner. The EB peptide was not virucidal and did not interfere with VACV attachment; rather, EB blocked the entry of VACV into cells. Addition of EB after low-pH treatment of infected cells blocked fusion-from-within, supporting the conclusion that EB inhibited virus fusion. Pre-treatment of cells with EB also resulted in resistance to infection but with a higher EC50. Thus the antiviral effect could be due to EB binding to the virus, the cell or both. Further studies will be needed to determine the target or targets of EB. To the best of our knowledge, the EB peptide is the first known non-antibody inhibitor of poxvirus entry.
Several lines of evidence support the claim that EB inhibits VACV entry. First, quantification of the number of VACV cores released into the cytoplasm of infected cells by immunofluorescence microscopy showed that treatment of the virus with 50 μM EB prior to infection reduced the number of cores detected by 85% (Fig. 5). Second, the TEM data showed that significantly fewer virus cores were detected in the cytoplasm of cells when EB was present than in samples with mock-treated virus (Fig. 6D). Third, treatment with EB led to a reduction in expression from an artificial early promoter when the virus was pretreated with the peptide (Fig. 4) or when the peptide was added post-adsorption but prior to initiation of entry (Fig. 7A). Additionally, EB was able to inhibit fusion-from-within in HeLa cells (Fig. 8), although fusion-from-without was not tested as the concentration of virus necessary for the assay (~108 pfu/mL) would overwhelm the ability of the peptide to inhibit infection and the concentration for peptide needed for an effect would be toxic to the cells (Fig. 1). When considered together with the observation that EB did not inhibit adsorption (Fig. 3), these results indicate that the EB peptide blocks VACV entry into cells, likely by inhibiting fusion between the virus envelope and the cell membrane.
Analysis of VACV entry both directly, through EM analysis, and indirectly, via the analysis of the effects of endosomal inhibitors and weak bases on infection, currently suggests that VACV is capable of entering cells by fusion at the surface of the cell (Armstrong, Metz, and Young, 1973; Carter et al., 2005; Chang and Metz, 1976; Doms, Blumenthal, and Moss, 1990; Janeczko, Rodriguez, and Esteban, 1987; Law et al., 2006; Vanderplasschen, Hollinshead, and Smith, 1998), macropinocytosis (Mercer and Helenius, 2008), or via an endosomal pathway (Dales, 1963; Dales and Kajioka, 1964; Payne and Norrby, 1978; Vanderplasschen, Hollinshead, and Smith, 1998). Entry by the WR strain of virus is inhibited by endosomal inhibitors and thus appears to enter mostly via endocytosis (Townsley et al., 2006; Vanderplasschen, Hollinshead, and Smith, 1998). Therefore, it was possible that EB could have been disrupting VACV entry by blocking the escape of virus from endocytic vesicles. However, this is unlikely as we observed no significant difference between the percentage of virus particles that appeared to be associated with vesicles in cells infected with EB-treated or mock-treated virus by TEM (Fig. 6C). Determining whether EB blocks VACV entry at the cell surface, prevents the release of virus from endosomes, or both will require further study.
Vaccinia virus infection results in the production of two immunologically distinct infectious particles: the single-enveloped mature virus (MV) and the double-enveloped extracellular virus (EV) (Moss, 2006). The EV particle is composed of an MV particle surrounded by an additional lipid bilayer envelope. EM analysis of EV entry has suggested that the outer envelope of the particle is shed at the surface of the cell in a nonfusogenic, ligand-dependent manner, leaving the interior MV particle free to enter the cell (Law et al., 2006). EV particles are released in low quantity by VACV strain WR grown in HeLa cells (Payne, 1979) and are disrupted by repeated freezing; thus, the majority of the virus particles used in our study were MVs. Further studies will be needed to determine if EB blocks infection by EVs.
Most of these assays were performed using virus that was pre-treated with EB. Previous studies have shown that exposure of cells to EB protects them from infection with HSV-1 (manuscript in preparation), so it was possible that the antiviral activity we observed could have been the result of interactions between EB and the cells. The EB peptide was able to reduce infection by VACV when used to pretreat cells prior to infection, however, at low concentrations of EB interactions between the peptide and the virus were more important to the inhibition of VACV infection than were interactions between the peptide and the cells (Fig. 7A). In terms of therapeutic uses, at drug concentrations that would likely be used, both activities would be occurring.
Our data indicate that removing 4 residues from the N- terminus or 3 residues from the C- terminus of EB had little effect on peptide activity (compare Table 4, EB-2 to EB-3 and EB4- to EB5-) but larger deletions reduced activity. These data suggest peptide length may be important. Derivatives of EB containing single amino acid substitutions for the leucine residues lost activity when the substituting residue was negatively charged (Table 5; E2, E1, B7, B4, B3), suggesting that a negative charge at these positions are detrimental. In contrast, single positive charges, with the exception of position 9, did not significantly reduce activity. The presence of 3 leucine pairs is a distinctive feature of the EB peptide and was potentially important for the antiviral activity. However, derivatives of EB containing alanine substitutions fo single leucines did not affect activity (Table 5; F2, F1, C7, C6, C4, C3), thus leucine pairs are not required. Previously it was shown that the leucine content of signal peptides was critical for interactions with the signal recognition particle as increasing the alanine: leucine ratio negatively affected function (Doud, Chou, and Kendall, 1993; Hatsuzawa, Tagaya, and Mizushima, 1997; Zanen et al., 2005). This raises the possibility that total leucine content is important for the activity of EB, However, EBX has the same leucine content and is inactive. Our observation that the EBX peptide was inactive indicates that the sequence is important and raises the possibility that the spacing of the leucines may play an important role in the antiviral activity of EB. Further studies will be needed to test these alternatives. The effects of the single leucine substitutions cannot be directly compared to the effects of truncation, as the total length of the peptide remains constant in the substituted derivatives. Overall, it seems likely that the activity of EB is dependent on the concurrent action of several amino acids.
The EB peptide is derived from the signal sequence of the human FGF4 protein (Lin et al., 1995). Signal sequences are highly variable (von Heijne, 1985), and are known to bind the SRP in a methionine-rich hydrophobic pocket (Lutcke et al., 1992). The hydrophobicity of this pocket is believed to be crucial for the ability of the SRP to recognize a broad range of signal sequences independent of their specific sequences (Keenan et al., 2001). Similarly, sorting of some transmembrane proteins to the endosomal-lysosomal system has been shown to depend on the interaction between dileucine sorting-signals and hydrophobic pockets (Misra et al., 2002; Shiba et al., 2002). It is possible that EB may be interacting with a similar pocket in one or more of the VACV entry-fusion complex proteins, thereby disrupting complex function. Eleven proteins, A16, A21, A28, F9, G3, G9, H2, I2, J5, L1, and L5, have been implicated in VACV entry, (Bisht, Weisberg, and Moss, 2008; Brown, Senkevich, and Moss, 2006; Izmailyan et al., 2006; Nichols et al., 2008; Ojeda, Domi, and Moss, 2006; Ojeda, Senkevich, and Moss, 2006; Senkevich and Moss, 2005; Senkevich et al., 2005; Senkevich, Ward, and Moss, 2004; Townsley, Senkevich, and Moss, 2005a; Townsley, Senkevich, and Moss, 2005b), and A17 and A27 have been implicated in cell-cell fusion (Kochan et al., 2008). Identification of the viral protein or proteins that bind to EB will be important in describing the mechanism of inhibition and such studies are underway.
The EB peptide has previously been shown to have antiviral activity against both HSV-1 and influenza virus (Bultmann, Busse, and Brandt, 2001; Jones et al., 2006), and we can now add VACV to the list of viruses inhibited by EB. However, the mechanism by which EB inhibits these viruses appears to be different for each virus. The EB peptide was shown to block HSV-1 entry by irreversibly inactivating virus particles (Bultmann, Busse, and Brandt, 2001), whereas EB did not irreversibly inactivate VACV. EB did not irreversibly inactivate influenza; instead, EB blocked virus adsorption by binding specifically to the viral hemagglutinin protein (Jones et al., 2006), which is in contrast to VACV where adsorption was not blocked.
In summary, we have identified a novel antiviral peptide, EB, which inhibits VACV entry with an EC50 of 15 μM. The activity was sequence-specific and reversible. The EB peptide was active when used to pre-treat virus, when added after virus adsorption but prior to the initiation of entry, or when used to pretreat cells, and inhibited fusion from within. These results indicate that EB inhibits virus-cell fusion. The EB peptide is, to our knowledge, the first identified non-antibody inhibitor of VACV entry, and has potential for development in clinical applications and as a reagent to probe VACV entry.
Acknowledgments
The authors would like to thank Dr. Bernard Moss (Laboratory of Viral Diseases, NIAID, Bethesda, MD) for the vMJ343 virus, Dr. Alan Townsley (Laboratory of Viral Diseases, NIAID, Bethesday, MD) for assistance with the fusion-from-within protocol, and Dr. Garreth Griffiths (EMBL Heidelberg, Germany) for the anti-core antibody. The authors would also like to thank Dr. Gary Case and Dr. Ben August for peptide synthesis and EM assistance, respectively. Additionally, Elizabeth Froelich provided administrative assistance and Dr. Donna Peters, Dr. Hermann Bultmann, Dr. Monica Sauter, Dr. Radeekorn Akkarawongsapat, Aaron Kolb and Jeremy Teuton provided valuable comments on the manuscript. This work was funded by grants from the NIH (EY07336 and AI52049 to C.R.B.), DARPA (MDS 972-97-1-005 to C.R.B.), a Core Grant for Vision Research (P30 EY 016665), a Sigma Xi Grant-in-Aid of Research to J.C.J, training grant AI 55397 (to S.E.A), a Research to Prevent Blindness Senior Scientist Award to C.R.B., and an unrestricted grant from Research to Prevent Blindness, Inc. to the Department of Ophthalmology and Visual Sciences.
References
- Update: adverse events following civilian smallpox vaccination--United States, 2003. MMWR Morb Mortal Wkly Rep. 2003a;52(20):475–7. [PubMed] [Google Scholar]
- Update: cardiac-related events during the civilian smallpox vaccination program--United States, 2003. MMWR Morb Mortal Wkly Rep. 2003b;52(21):492–6. [PubMed] [Google Scholar]
- Akkarawongsa R, Cullinan AE, Zinkel A, Clarin J, Brandt CR. Corneal toxicity of cell-penetrating peptides that inhibit Herpes simplex virus entry. J Ocul Pharmacol Ther. 2006;22(4):279–89. doi: 10.1089/jop.2006.22.279. [DOI] [PubMed] [Google Scholar]
- Armstrong J, Metz D, Young M. The mode of entry of vaccinia virus into L cells. J Gen Virol. 1973;21(3):533–537. doi: 10.1099/0022-1317-21-3-533. [DOI] [PubMed] [Google Scholar]
- Baggs J, Chen RT, Damon IK, Rotz L, Allen C, Fullerton KE, Casey C, Nordenberg D, Mootrey G. Safety profile of smallpox vaccine: insights from the laboratory worker smallpox vaccination program. Clin Infect Dis. 2005;40(8):1133–40. doi: 10.1086/428731. [DOI] [PubMed] [Google Scholar]
- Baker RO, Bray M, Huggins JW. Potential antiviral therapeutics for smallpox, monkeypox and other orthopoxvirus infections. Antiviral Res. 2003;57(1–2):13–23. doi: 10.1016/S0166-3542(02)00196-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisht H, Weisberg AS, Moss B. Vaccinia Virus L1 Protein Is Required for Cell Entry and Membrane Fusion. J Virol. 2008;82(17):8687–8694. doi: 10.1128/JVI.00852-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown E, Senkevich TG, Moss B. Vaccinia Virus F9 Virion Membrane Protein Is Required for Entry but Not Virus Assembly, in Contrast to the Related L1 Protein. J Virol. 2006;80(19):9455–9464. doi: 10.1128/JVI.01149-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buller RM, Owens G, Schriewer J, Melman L, Beadle JR, Hostetler KY. Efficacy of oral active ether lipid analogs of cidofovir in a lethal mousepox model. Virology. 2004;318(2):474–81. doi: 10.1016/j.virol.2003.11.015. [DOI] [PubMed] [Google Scholar]
- Bultmann H, Brandt CR. Peptides containing membrane-transiting motifs inhibit virus entry. J Biol Chem. 2002;277(39):36018–23. doi: 10.1074/jbc.M204849200. [DOI] [PubMed] [Google Scholar]
- Bultmann H, Busse JS, Brandt CR. Modified FGF4 signal peptide inhibits entry of herpes simplex virus type 1. J Virol. 2001;75(6):2634–45. doi: 10.1128/JVI.75.6.2634-2645.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bultmann H, Teuton J, Brandt CR. Addition of a C-terminal cysteine improves the anti-herpes simplex virus activity of a peptide containing the human immunodeficiency virus type 1 TAT protein transduction domain. Antimicrob Agents Chemother. 2007;51(5):1596–607. doi: 10.1128/AAC.01009-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrd CM, Bolken TC, Mjalli AM, Arimilli MN, Andrews RC, Rothlein R, Andrea T, Rao M, Owens KL, Hruby DE. New class of orthopoxvirus antiviral drugs that block viral maturation. J Virol. 2004;78(22):12147–56. doi: 10.1128/JVI.78.22.12147-12156.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter GC, Law M, Hollinshead M, Smith GL. Entry of the vaccinia virus intracellular mature virion and its interactions with glycosaminoglycans. J Gen Virol. 2005;86(5):1279–1290. doi: 10.1099/vir.0.80831-0. [DOI] [PubMed] [Google Scholar]
- Casey CG, Iskander JK, Roper MH, Mast EE, Wen XJ, Torok TJ, Chapman LE, Swerdlow DL, Morgan J, Heffelfinger JD, Vitek C, Reef SE, Hasbrouck LM, Damon I, Neff L, Vellozzi C, McCauley M, Strikas RA, Mootrey G. Adverse Events Associated With Smallpox Vaccination in the United States, January-October 2003. JAMA. 2005;294(21):2734–2743. doi: 10.1001/jama.294.21.2734. [DOI] [PubMed] [Google Scholar]
- Chang A, Metz D. Further investigations on the mode of entry of vaccinia virus into cells. J Gen Virol. 1976;32(2):275–282. doi: 10.1099/0022-1317-32-2-275. [DOI] [PubMed] [Google Scholar]
- Dales S. The uptake and development of vaccinia virus in strain L cells followed with labeled viral deoxyribonucleic acid. J Cell Biol. 1963;18(1):51–72. doi: 10.1083/jcb.18.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dales S, Kajioka R. The cycle of multiplication of vaccinia virus in Earle’s strain L cells I. Uptake and penetration. Virology. 1964;24(3):278–294. doi: 10.1016/0042-6822(64)90167-9. [DOI] [PubMed] [Google Scholar]
- Davison AJ, Moss B. Structure of vaccinia virus early promoters. Journal of Molecular Biology. 1989;210(4):749–769. doi: 10.1016/0022-2836(89)90107-1. [DOI] [PubMed] [Google Scholar]
- Doms RW, Blumenthal R, Moss B. Fusion of intra- and extracellular forms of vaccinia virus with the cell membrane. J Virol. 1990;64(10):4884–92. doi: 10.1128/jvi.64.10.4884-4892.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doud SK, Chou MM, Kendall DA. Titration of protein transport activity by incremental changes in signal peptide hydrophobicity. Biochemistry. 1993;32(5):1251–1256. doi: 10.1021/bi00056a008. [DOI] [PubMed] [Google Scholar]
- Frankel AD, Pabo CO. Cellular uptake of the tat protein from human immunodeficiency virus. Cell. 1988;55(6):1189–93. doi: 10.1016/0092-8674(88)90263-2. [DOI] [PubMed] [Google Scholar]
- Fulginiti VA, Papier A, Lane JM, Neff JM, Henderson DA. Smallpox vaccination: a review, part II. Adverse events. Clin Infect Dis. 2003;37(2):251–71. doi: 10.1086/375825. [DOI] [PubMed] [Google Scholar]
- Garde V, Harper D, Fairchok MP. Tertiary contact vaccinia in a breastfeeding infant. Jama. 2004;291(6):725–7. doi: 10.1001/jama.291.6.725. [DOI] [PubMed] [Google Scholar]
- Gong SC, Lai CF, Esteban M. Vaccinia virus induces cell fusion at acid pH and this activity is mediated by the N-terminus of the 14-kDa virus envelope protein. Virology. 1990;178(1):81–91. doi: 10.1016/0042-6822(90)90381-z. [DOI] [PubMed] [Google Scholar]
- Gupta B, Levchenko TS, Torchilin VP. Intracellular delivery of large molecules and small particles by cell-penetrating proteins and peptides. Adv Drug Deliv Rev. 2005;57(4):637–51. doi: 10.1016/j.addr.2004.10.007. [DOI] [PubMed] [Google Scholar]
- Hatsuzawa K, Tagaya M, Mizushima S. The Hydrophobic Region of Signal Peptides Is a Determinant for SRP Recognition and Protein Translocation across the ER Membrane. J Biochem. 1997;121(2):270–277. doi: 10.1093/oxfordjournals.jbchem.a021583. [DOI] [PubMed] [Google Scholar]
- Izmailyan RA, Huang CY, Mohammad S, Isaacs SN, Chang W. The envelope G3L protein is essential for entry of vaccinia virus into host cells. J Virol. 2006;80(17):8402–10. doi: 10.1128/JVI.00624-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janeczko RA, Rodriguez JF, Esteban M. Studies on the mechanism of entry of vaccinia virus in animal cells. Arch Virol. 1987;92(1–2):135–50. doi: 10.1007/BF01310068. [DOI] [PubMed] [Google Scholar]
- Joliot A, Pernelle C, Deagostini-Bazin H, Prochiantz A. Antennapedia homeobox peptide regulates neural morphogenesis. Proc Natl Acad Sci U S A. 1991;88(5):1864–8. doi: 10.1073/pnas.88.5.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones JC, Turpin EA, Bultmann H, Brandt CR, Schultz-Cherry S. Inhibition of influenza virus infection by a novel antiviral peptide that targets viral attachment to cells. J Virol. 2006;80(24):11960–11967. doi: 10.1128/JVI.01678-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keenan RJ, Freymann DM, Stroud RM, Walter P. The signal recognition particle. Annu Rev Biochem. 2001;70:755–75. doi: 10.1146/annurev.biochem.70.1.755. [DOI] [PubMed] [Google Scholar]
- Kern ER. In vitro activity of potential anti-poxvirus agents. Antiviral Res. 2003;57(1–2):35–40. doi: 10.1016/S0166-3542(02)00198-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Yeo SG, Jang HC, Park WB, Lee CS, Lee KD, Kim HB, Kim NJ, Kim YT, Jee Y, Cho H, Oh M-d, Choe KW. Clinical Responses to Smallpox Vaccine in Vaccinia-Naive and Previously Vaccinated Populations: Undiluted and Diluted Lancy-Vaxina Vaccine in a Single-Blind, Randomized, Prospective Trial. J Infect Dis. 2005;192(6):1066–1070. doi: 10.1086/432765. [DOI] [PubMed] [Google Scholar]
- Kochan G, Escors D, Gonzalez JM, Casasnovas JM, Esteban M. Membrane cell fusion activity of the vaccinia virus A17–A27 protein complex. Cellular Microbiology. 2008;10(1):149–164. doi: 10.1111/j.1462-5822.2007.01026.x. [DOI] [PubMed] [Google Scholar]
- Law M, Carter GC, Roberts KL, Hollinshead M, Smith GL. Ligand-induced and nonfusogenic dissolution of a viral membrane. PNAS. 2006:0601025103. doi: 10.1073/pnas.0601025103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law M, Smith GL. Studying the binding and entry of the intracellular and extracellular enveloped forms of vaccinia virus. Methods Mol Biol. 2004;269:187–204. doi: 10.1385/1-59259-789-0:187. [DOI] [PubMed] [Google Scholar]
- Lin YZ, Yao S, Veach RA, Torgerson TR, Hawiger J. Inhibition of nuclear translocation of transcription factor NF-κB by a synthetic peptide containing a cell membrane-permeable motif and nuclear localization sequence. J Biol Chem. 1995;270(24):14255–14258. doi: 10.1074/jbc.270.24.14255. [DOI] [PubMed] [Google Scholar]
- Lutcke H, High S, Romisch K, Ashford AJ, Dobberstein B. The methionine-rich domain of the 54 kDa subunit of signal recognition particle is sufficient for the interaction with signal sequences. Embo J. 1992;11(4):1543–51. doi: 10.1002/j.1460-2075.1992.tb05199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee WC, Hostetler KY, Evans DH. Mechanism of Inhibition of Vaccinia Virus DNA Polymerase by Cidofovir Diphosphate. Antimicrob Agents Chemother. 2005;49(8):3153–3162. doi: 10.1128/AAC.49.8.3153-3162.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer J, Helenius A. Vaccinia Virus Uses Macropinocytosis and Apoptotic Mimicry to Enter Host Cells. Science. 2008;320(5875):531–535. doi: 10.1126/science.1155164. [DOI] [PubMed] [Google Scholar]
- Misra S, Puertollano R, Kato Y, Bonifacino JS, Hurley JH. Structural basis for acidic-cluster-dileucine sorting-signal recognition by VHS domains. Nature. 2002;415(6874):933–7. doi: 10.1038/415933a. [DOI] [PubMed] [Google Scholar]
- Moss B. Poxvirus entry and membrane fusion. Virology. 2006;344(1):48–54. doi: 10.1016/j.virol.2005.09.037. [DOI] [PubMed] [Google Scholar]
- Nakase I, Niwa M, Takeuchi T, Sonomura K, Kawabata N, Koike Y, Takehashi M, Tanaka S, Ueda K, Simpson JC, Jones AT, Sugiura Y, Futaki S. Cellular uptake of arginine-rich peptides: roles for macropinocytosis and actin rearrangement. Mol Ther. 2004;10(6):1011–22. doi: 10.1016/j.ymthe.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Neyts J, Clercq ED. Therapy and short-term prophylaxis of poxvirus infections: historical background and perspectives. Antiviral Res. 2003;57(1–2):25–33. doi: 10.1016/s0166-3542(02)00197-3. [DOI] [PubMed] [Google Scholar]
- Nichols RJ, Stanitsa E, Unger B, Traktman P. The Vaccinia Virus Gene I2L Encodes a Membrane Protein with an Essential Role in Virion Entry. J Virol. 2008;82(20):10247–10261. doi: 10.1128/JVI.01035-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojeda S, Domi A, Moss B. Vaccinia Virus G9 Protein Is an Essential Component of the Poxvirus Entry-Fusion Complex. J Virol. 2006;80(19):9822–9830. doi: 10.1128/JVI.00987-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojeda S, Senkevich TG, Moss B. Entry of Vaccinia Virus and Cell-Cell Fusion Require a Highly Conserved Cysteine-Rich Membrane Protein Encoded by the A16L Gene. J Virol. 2006;80(1):51–61. doi: 10.1128/JVI.80.1.51-61.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne LG. Identification of the vaccinia hemagglutinin polypeptide from a cell system yielding large amounts of extracellular enveloped virus. J Virol. 1979;31(1):147–55. doi: 10.1128/jvi.31.1.147-155.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne LG, Norrby E. Adsorption and penetration of enveloped and naked vaccinia virus particles. J Virol. 1978;27(1):19–27. doi: 10.1128/jvi.27.1.19-27.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prichard MN, Keith KA, Quenelle DC, Kern ER. Activity and Mechanism of Action of N-Methanocarbathymidine against Herpesvirus and Orthopoxvirus Infections. Antimicrob Agents Chemother. 2006;50(4):1336–41. doi: 10.1128/AAC.50.4.1336-1341.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard JP, Melikov K, Vives E, Ramos C, Verbeure B, Gait MJ, Chernomordik LV, Lebleu B. Cell-penetrating peptides. A reevaluation of the mechanism of cellular uptake. J Biol Chem. 2003;278(1):585–90. doi: 10.1074/jbc.M209548200. [DOI] [PubMed] [Google Scholar]
- Rotz LD, Dotson DA, Damon IK, Becher JA. Vaccinia (smallpox) vaccine: recommendations of the Advisory Committee on Immunization Practices (ACIP), 2001. MMWR Recomm Rep. 2001;50(RR-10):1–25. quiz CE1–7. [PubMed] [Google Scholar]
- Schwartz B, Lebwohl M. Complications of the smallpox vaccine. Int J Dermatol. 2005;44(4):289–292. doi: 10.1111/j.1365-4632.2004.02568.x. [DOI] [PubMed] [Google Scholar]
- Senkevich TG, Moss B. Vaccinia Virus H2 Protein Is an Essential Component of a Complex Involved in Virus Entry and Cell-Cell Fusion. J Virol. 2005;79(8):4744–4754. doi: 10.1128/JVI.79.8.4744-4754.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senkevich TG, Ojeda S, Townsley A, Nelson GE, Moss B. Poxvirus multiprotein entry-fusion complex. PNAS. 2005;102(51):18572–18577. doi: 10.1073/pnas.0509239102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senkevich TG, Ward BM, Moss B. Vaccinia virus entry into cells is dependent on a virion surface protein encoded by the A28L gene. J Virol. 2004;78(5):2357–66. doi: 10.1128/JVI.78.5.2357-2366.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiba T, Takatsu H, Nogi T, Matsugaki N, Kawasaki M, Igarashi N, Suzuki M, Kato R, Earnest T, Nakayama K, Wakatsuki S. Structural basis for recognition of acidic-cluster dileucine sequence by GGA1. Nature. 2002;415(6874):937–41. doi: 10.1038/415937a. [DOI] [PubMed] [Google Scholar]
- Smee DF, Sidwell RW. A review of compounds exhibiting anti-orthopoxvirus activity in animal models. Antiviral Res. 2003;57(1–2):41–52. doi: 10.1016/S0166-3542(02)00199-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsley AC, Senkevich TG, Moss B. The Product of the Vaccinia Virus L5R Gene Is a Fourth Membrane Protein Encoded by All Poxviruses That Is Required for Cell Entry and Cell-Cell Fusion. J Virol. 2005a;79(17):10988–10998. doi: 10.1128/JVI.79.17.10988-10998.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsley AC, Senkevich TG, Moss B. Vaccinia Virus A21 Virion Membrane Protein Is Required for Cell Entry and Fusion. J Virol. 2005b;79(15):9458–9469. doi: 10.1128/JVI.79.15.9458-9469.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsley AC, Weisberg AS, Wagenaar TR, Moss B. Vaccinia Virus Entry into Cells via a Low-pH-Dependent Endosomal Pathway. J Virol. 2006;80(18):8899–8908. doi: 10.1128/JVI.01053-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderplasschen A, Hollinshead M, Smith GL. Intracellular and extracellular vaccinia virions enter cells by different mechanisms. J Gen Virol. 1998;79(4):877–87. doi: 10.1099/0022-1317-79-4-877. [DOI] [PubMed] [Google Scholar]
- von Heijne G. Signal sequences: The limits of variation. Journal of Molecular Biology. 1985;184(1):99–105. doi: 10.1016/0022-2836(85)90046-4. [DOI] [PubMed] [Google Scholar]
- WHO. Declaration of global eradication of smallpox. Weekly Epidemiol Rec. 1980;55(20):148. [Google Scholar]
- Yang G, Pevear DC, Davies MH, Collett MS, Bailey T, Rippen S, Barone L, Burns C, Rhodes G, Tohan S, Huggins JW, Baker RO, Buller RLM, Touchette E, Waller K, Schriewer J, Neyts J, DeClercq E, Jones K, Hruby D, Jordan R. An orally bioavailable antipoxvirus compound (ST-246) inhibits extracellular virus formation and protects mice from lethal orthopoxvirus challenge. J Virol. 2005;79(20):13139–13149. doi: 10.1128/JVI.79.20.13139-13149.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Schneller SW. 5′-Homoaristeromycin. Synthesis and antiviral activity against orthopox viruses. Bioorganic & Medicinal Chemistry Letters. 2005;15(1):149–151. doi: 10.1016/j.bmcl.2004.10.019. [DOI] [PubMed] [Google Scholar]
- Zanen G, Houben ENG, Meima R, Tjalsma H, Jongloed JDH, Westers H, Oudega B, Luirink J, van Dijl JM, Quax WJ. Signal peptide hydrophobicity is critical for early stages in protein export by Bacillus subtilis. FEBS Journal. 2005;272(18):4617–4630. doi: 10.1111/j.1742-4658.2005.04777.x. [DOI] [PubMed] [Google Scholar]