

Abstract
Background
ERBB receptor tyrosine kinases can mediate proliferation, migration, adhesion, differentiation, and survival in many types of cells and play critical roles in many malignancies. Recent reports suggest a role for EGFR signaling in proliferation and survival of neuroblastoma, a common form of pediatric cancer that often has an extremely poor outcome.
Methods
We examined ERBB family expression in neuroblastoma cell lines and patient samples by flow cytometry, western blot and Q-PCR. Response to ERBB inhibition was assessed in vitro by cell cycle analysis and western blot, and in vivo by serial tumor size measurements.
Results
A panel of neuroblastoma cell lines and primary patient tumors expressed EGFR, HER-3, and HER-4, with Her-2 in some tumors. Her-4 mRNA was expressed predominantly in cleavable isoforms. While EGFR inhibition with erlotinib and pan-ERBB inhibition with CI-1033 inhibited EGF-induced phosphorylation of EGFR, AKT, and ERK1/2, only CI-1033 induced growth inhibition and dose-dependent apoptosis in vitro. Both CI-1033 and erlotinib treatment of neuroblastoma xenograft tumors resulted in decreased tumor growth in vivo, though CI-1033 was more effective. In vivo expression of EGFR was observed predominantly in vascular endothelial cells.
Conclusions
Pan-ERBB inhibition is required for ERBB-related neuroblastoma apoptosis in vitro, though EGFR contributes indirectly to tumor growth in vivo. Inhibition of EGFR in endothelial cells may be an important aspect of erlotinib's impact on neuroblastoma growth in vivo. Our results suggest that non-EGFR ERBB family members contribute directly to neuroblastoma growth and survival, and pan-ERBB inhibition represents a potential therapeutic target for treating neuroblastoma.
Introduction
Neuroblastoma is a severe cancer of childhood, causing more than 7% of malignancies in children under 15 and approximately 15% of all pediatric cancer deaths. Outcome for children with high-risk neuroblastoma remains less than 40% with current treatment regimens 1. The poor survival rates of high-risk patients warrants investigation into novel treatment options.
Dependence on growth factors is one of the hallmarks of cancer 2. The ERBB family, comprised of EGFR, HER-2, HER-3, and HER-4, is a group of receptor tyrosine kinases with common and unique signaling properties 3 that can provide these essential signals. Constitutive activation of EGFR and HER-2 through autocrine ligand production, receptor overexpression, or activating mutation occurs commonly in carcinomas and is correlated with poor prognosis. Less is known about HER-3 and HER-4 in cancer, and few reports correlate HER-3 and HER-4 expression or activation with prognosis 4.
The role of the ERBB family has not been well established in neuroblastoma. EGFR expression and effectiveness of anti-EGFR treatment in vitro has been reported 5, 6, though others found no response to erlotinib in vivo 7. Surprisingly, exposure to EGF induced apoptosis in a dose-dependent manner 8-10. None of these reports evaluated contributions of other ERBB family members, nor tested their importance in vivo.
We report that pan-ERBB inhibition is more effective than EGFR-specific inhibition in neuroblastoma. A pan-ERBB inhibitor caused significant growth inhibition in vitro and in vivo, while EGFR-specific inhibition was ineffective in vitro and produced a modest reduction in tumor growth in vivo. These results suggest that HER-4 signaling may be important in neuroblastoma, and that a pan-ERBB inhibitor may be a novel therapeutic approach for children with neuroblastoma.
Materials and Methods
Chemicals
CI-1033 (Pfizer, New York, NY) was dissolved in water, 5 mM for in vitro use and at 4.5 mg/ml for in vivo use. Erlotinib (OSI Pharmaceuticals, Melville, NY) was dissolved in dimethyl sulfoxide, 10 mM for in vitro use and 7.5 mg/ml for in vivo use.
Cell lines
Neuroblastoma cell lines SK-N-AS, SK-N-SH, SH-EP, SH-SY5Y, IMR-32, SMS-KCNR, LA1-55N, NGP, and CHP-134 were obtained from Dr. Susan Cohn (The University of Chicago Children's Hospital, Chicago, IL) and Dr. John Maris (Children's Hospital of Philadelphia, Philadelphia, PA) and have been previously described 11-18. Cells were incubated at 37°C and 5% CO2 in RPMI 1640 (Mediatech, Manassas, VA) with 10% fetal bovine serum (Hyclone, Logan, UT), 1% L-glutamine (Lonza, Allendale, NJ), and 1% Penicillin/Streptomycin (Gemini, Woodland, CA). A431 and SKOV-3 cells 19, 20 were used as positive controls and grown in DMEM (Invitrogen, Carlsbad, CA) with all other conditions the same. The breast cancer cell line T47D was obtained from Dr. Seth Corey (The Children's Cancer Hospital at M.D. Anderson Cancer Center, Houston, TX) and was grown in DMEM/F12 (Mediatech), with all other conditions as above. For all experiments, cells were harvested when confluence was less than 80%.
Patient-derived Tumor Samples
For primary tumors, approval for specimens was obtained from the Children's Oncology Group (COG) Neuroblastoma committee. Frozen primary tumor samples were then received from the Cooperative Human Tissue Network (CHTN) funded by the National Cancer Institute. Other investigators may have received specimens from the same subjects.
Flow Cytometry
Adherent neuroblastoma cells were washed with PBS and mobilized with enzyme-free cell dissociation buffer (Invitrogen) and washed with cold PBS/1 % bovine serum albumin (Fisher, Pittsburgh, PA). Antibodies to EGFR and HER-2 directly conjugated to phycoerythrin (PE) (Becton Dickinson, Franklin Lakes, NJ), HER-3 and HER-4 (Neomarkers, Fremont, CA) antibodies, or the appropriate IgG isotype control (Becton Dickinson) were then incubated with the cells on ice for 30 minutes. The cells were then washed with PBS/1% BSA three times. For the HER-3 and HER-4, a secondary PE-conjugated anti-mouse (R&D Systems, Minneapolis, MN) was incubated with the cells for 30 minutes on ice. ERBB expression was then analyzed by flow cytometer (FACSCalibur, Becton Dickinson).
Her-4 Isoform Analysis
Sample Preparation
RNA was extracted from neuroblastoma cells grown to 70% confluency with the RNeasy Mini Kit (Qiagen) and treated with DNase I (Qiagen) according to the manufacturer's instructions. RNA was reverse transcribed using the Omniscript Reverse Transcriptase Kit (Qiagen) with oligo-dTs (Invitrogen) according to the manufacturer's instructions.
Primer Design
Primers were designed to anneal uniquely to the four HER-4 JM a-d isoforms according to previously reported sequences 21. Forward primers: HER4 JM-a (5′-CTGCACCCAAGGGTGTAACG-3′), HER4 JM-b (5′-GGCCTGATGGATAGAACTCC-3′), HER4 JM-c (5′-CAAACTGCACCCAAGGAACTC-3′), and HER4 JM-d (5′-CGGCCTGATGGATAGGTGTAAC-3′). A common reverse primer was utilized for all four HER4 JM isoforms (5′-GCAAATGTCAGACCCACAATG-3′).
The HER4 JM isoform data was normalized by comparing the number of isoform copies to the number of GAPDH copies with the forward primer (5′-GCATCCTGGGCTACACTGAG-3′) and the reverse primer (5′-CCACCACCCTGTTGCTGTAG-3′).
Q-PCR
Q-PCR was performed using iQ SYBR Green Supermix (Biorad) according to the manufacturer's instructions. Thermal cycling of HER4 JM a-d consisted of 38 cycles of: 95°C for 30 s; 61, 63, 64, or 64°C for 30 s, respectively; and 72°C for 25, 15, 15, or 20 s, respectively. Expected amplification products for HER4 JM a-d were: 155, 82, 84, and 158 bp, respectively. Thermal cycling of GAPDH consisted of 38 cycles of: 95°C for 30 s, 60°C for 30 s, and 72°C for 15 s with an expected product of 161 bp.
The mRNA transcript copy number was quantified absolutely by creating standard curves of the target message. HER4 JM isoforms standard curves were created by amplifying pcDNA3.1 (Invitrogen) containing HER4 JM isoform cDNA; GAPDH standard curve was created by amplifying pCR2.1 (Invitrogen) containing a 161 bp fragment of GAPDH. Products were evaluated for specificity by electrophoresis on a 1.8% agarose gels.
Western Blotting
For analysis of target inhibition, cells were treated with CI-1033 or erlotinib for 24 hours, followed by 5 minutes of exposure to EGF (100 ng/ml), then washed in cold PBS and incubated on ice with protein lysis buffer (50 mM Hepes, 150 mM NaCl, 1 mM EGTA, 10 mM Sodium Pyrophosphate, 10 mM NaF, 10% glycerol, 1.5 mM MgCl2, 1% Triton X-100) containing phosphatase inhibitor (Sigma-Aldrich, St. Louis, MO) and protease inhibitor (Roche, Basel, Switzerland) for 10 minutes. Lysate was then scraped from the plate and centrifuged at 12,000 rpm for 10 minutes at 4° C. Supernatant was collected and protein concentration determined with the BCA Protein Assay kit (Pierce, Rockford, IL). Frozen patient samples were homogenized using a Dismembrator (Sartorius, Edgewood, NY), then lysed in the same lysis buffer. 30 μg of protein lysate was loaded into an 8% SDS-PAGE gel, then transferred to nitrocellulose using standard methods. Membranes were blocked in 5% milk in TBST (100 mM Tris, 1.5 M NaCl, 0.1% Tween-20, pH 7.9) for 1 hour at room temperature, then washed with TBS-Tween 3 times for 5 minutes. Membranes were then incubated with primary antibody (EGFR, pEGFR, AKT, pAKT, ERK 1/2, pERK 1/2, and PARP at 1:1000 (Cell Signaling, Danvers, MA), HER-3 and HER-4 at 1:50 (Neomarkers), HER-2 at 1:1500 (Epitomics, Burlingame, CA), and actin at 1:1000 (Sigma)) overnight at 4° C. Membranes were washed 3 times for 10 minutes in TBST, then incubated with the appropriate secondary antibody/HRP for 1 hour at room temperature, then washed 3 times for 10 minutes in TBST. Chemiluminescence detection reagents (Pierce) were incubated with the membranes for 1 minute, followed by film development on a Kodak developer.
Growth Analysis
Cells were plated at 100,000 cells per well in a 6 well plate. After cells became adherent, drug was added and refreshed every 24 hours. After 3 or 7 days of triplicate treatments with 0, 0.01, 0.1, 1, 3, or 5 μM CI-1033 or 0, 0.1, 1, 3, 5, or 10 μM erlotinib, media and dead cells were removed by washing with PBS. Intact nuclei were extracted by addition of a 0.01M hepes-0.015 M MgCl2 buffer for 5 minutes, followed by addition of 5% Bretol (ethyl hexadecyldimethylammonium bromide) in water and agitation for 10 minutes. Nuclei were then fixed with a 0.9% NaCl and 0.5% Formalin solution in water and counted using the ViCell Cell Viability Analyzer (Beckman Coulter, Fullerton, CA) as previously described 22. IC50 values were calculated using Microsoft Excel with a best fit trendline.
EGFR and K-Ras mutation analysis
Genomic DNA was isolated from neuroblastoma cell lines using the DNeasy tissue kit (Qiagen, GmbH, Hilden, Germany). The somatic status of the EGFR gene was investigated by PCR using primers specific for exons 18-21, encompassing the tyrosine kinase domain. For the K-RAS gene, PCR was performed using exon 2 specific primers. Subsequently, PCR fragments were sequenced and analyzed by direct sequencing in both sense and antisense direction. For the ease of sequencing, M13 tails were attached to every primer pair. Primer sequences were as follows:
EGFR exon 18: forward primer: CCTGAGGTGACCCTTGTCTCTGTGTTCTT,
reverse primer: GAGGCCTGTGCCAGGGACCTTA,
EGFR exon 19: forward primer: CGCACCATCTCACAATTGCCAGTTA,
reverse primer: AAAGGTGGGCCTGAGGTTCA,
EGFR exon 20, forward primer: CACACTGACGTGCCTCTCC,
reverse primer: TATCTCCCCTCCCCGTATCT,
EGFR exon 21: forward primer: CCCTCACAGCAGGGTCTTCTCTGT,
reverse primer: TCAGGAAAATGCTGGCTGACCTA,
K-RAS exon 2: forward primer: CGTCCTGCACCAGTAATATGC,
reverse primer: GTATTAACCTTATGTGTGACA.
The following PCR program was applied: 5 min 95°C, 30 sec 95°C, 30 sec 62°C, 30 sec 68°C (last three steps were repeated 42 times), 7 min 68°C.
Cell cycle analysis
Untreated and cells treated with CI-1033 or erlotinib for 48 hours were evaluated for changes in cell cycle by propidium iodide staining. Adherent and non-adherent cells were harvested using trypsin (Gibco), washed with PBS, and resuspended in a 0.005% propidium iodide and 0.1% Triton solution in PBS. Cells were incubated overnight, and evaluated on the FACS Calibur Flow Cytometer (Becton Dickinson).
In vivo xenografts and administration of ERBB inhibitors
12 week old NOD-SCID-IL2R gamma knockout mice (Stock # 005557, The Jackson Laboratories) were injected subcutaneously in the right flank with 5 million SK-N-SH cells. Mice were randomized into control, CI-1033 treatment, and erlotinib treatment groups at the time of injection. Treatment began when tumors became palpable (approximately 50 mm3). CI-1033 (30 mg/kg) and erlotinib (10 mg/kg) were given by gavage daily. The dose of CI-1033, which is the murine maximum tolerated dose, was chosen to give a serum level exceeding the in vitro IC50 identified in our preliminary data for SH-N-SH 23. For erlotinib, we wished to use the minimum biologically effective dose, as determined by inhibition of the EGFR-driven tumor HN5 24. Note that at a dose of 10 mg/kg, the inhibition of EGFR phosphorylation is identical whether the drug is given ip or po 24. Tumor size was measured in triplicate every other day with electric calipers, and the average measurement was used for analysis. Tumor volume was determined with the formula for the area of an ellipse, 4/3 π × R1 × R22. All mice were sacrificed 18 days after first tumor appearance.
Immunohistochemistry
Mice were euthanized and tumors were excised and flash frozen in OCT (Sakura, Torrance, CA). Frozen tumors were sectioned and fixed with ice cold acetone. H&E staining was performed by standard techniques. Slides were incubated in 4% fish gelatin in PBS for 20 minutes, followed by addition of anti-CD31 antibody (Becton Dickinson) overnight at 4° C. Slides were rinsed with PBS and incubated with Texas Red labeled goat anti-rat secondary antibody (Jackson ImmunoResearch, West Grove, PA) for 1 hour. Slides were then washed in PBS and incubated with anti-EGFR antibody (Santa Cruz) at 4° C overnight. Alexa488 labeled anti-rabbit secondary antibody was incubated for 1 hour. Slides were then rinsed, counter-stained with Hoechst 33342 (Invitrogen) for 2 minutes, and coverslips were placed with anti-fade reagent (Invitrogen).
Results
Expression of ERBB receptors in neuroblastoma cell lines and primary tumors
We evaluated the cell surface expression of EGFR, HER-3, and HER-4 by flow cytometry in nine human neuroblastoma tumor cell lines (Figure 1A). EGFR was variably expressed on the cell surface in six of the cell lines, with highest expression in the SK-N-AS and SH-EP cell lines. Interestingly, SK-N-SH cells had bi-modal expression, which may reflect its dual population of N- type (SH-SY5Y) and S-type (SHEP) cells. HER-2 was not detected (data not shown). Low HER-3 expression was detected in several cell lines by flow cytometry. HER-4 was the most consistently detected ERBB receptor, with expression in seven of the nine cell lines.
Figure 1.
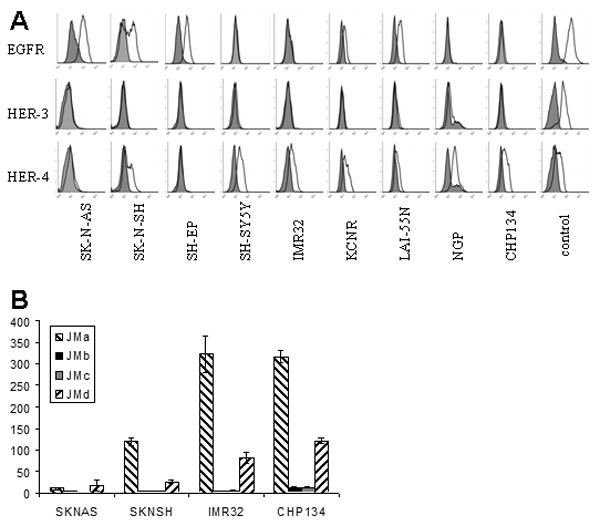
Cell surface expression of ERBB. A: Nine neuroblastoma cell lines were incubated with anti-ERBB antibody either directly conjugated to PE (EGFR) or then incubated with a secondary PE conjugated antibody (HER-3 and HER-4). Positive controls were A431 (EGFR) and T47D (HER-3 and HER-4). Filled histograms represent the isotype control and unfilled histograms represent ERBB-PE expression. B: RNA from cell lines SK-N-AS, SK-N-SH, IMR32 and CHP134 was reverse-transcribed and assessed by Q-PCR for expression of each of the Her-4 juxtramembranous isoforms. Histograms represent copies of each of the four isoforms (JMa, JMb, JMc and JMd) per million copies of GAPDH. Triplicate samples were analyzed; error bars represent standard deviation. Cleavable isoforms of Her-4 (JMa and JMd) were much more abundant than non-cleavable isoforms (JMb and JMc) for all lines tested.
Four cell lines (SK-N-AS, SK-N-SH, IMR-32 and CHP134) were further evaluated by Q-PCR to determine the proportion of mRNA in each of the juxtramembranous isoforms 21 (Figure 1B). Expression of Her-4 mRNA was proportional to cell surface expression, and cleavable isoforms of Her-4 (JMa and JMd) were expressed at much higher levels than non-cleavable isoforms (JMb and JMc).
Western blots evaluated total ERBB expression (Figure 2A). EGFR was detected in all cell lines by western blot, with highest levels in the SK-N-AS and SH-EP cell lines. The detection of EGFR by western blot in surface-negative lines suggests internalization of activated protein in these samples 25. HER-3 was variably expressed in all cell lines, with highest levels in SK-N-SH, SH-SY5Y, and LA1-55N cells. HER-4 was expressed in all nine neuroblastoma cell lines, with highest expression in SH-SY5Y, IMR-32, and KCNR cells. Consistent with the flow cytometry results, HER-2 expression was below the threshold of detection for western blot (data not shown).
Figure 2.
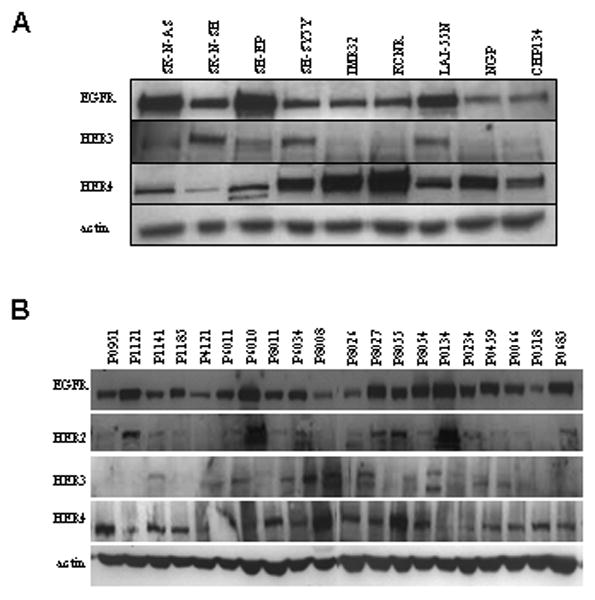
Expression of ERBB in neuroblastoma cell lines and patient samples. (A) Protein expression of EGFR, HER-3, and HER-4 of nine neuroblastoma cell lines was evaluated by western blot. (B) Protein lysates from primary patient tumors were evaluated for expression of EGFR, HER-2, HER-3, and HER-4. Actin was used as the loading control.
Expression of the ERBB receptors in primary neuroblastoma tumor samples was determined by western blot. All four receptors, EGFR, HER-2, HER-3, and HER-4, were variably expressed in twenty patient samples (Figure 2B). EGFR and HER-4 were the predominant ERBB receptors expressed, while HER-2 and HER-3 had variable expression. These results suggest that the role of HER-4 in neuroblastoma should be explored.
Sensitivity of neuroblastoma to EGFR and Pan-ERBB Inhibition
Given the expression of ERBB family members in neuroblastoma cell lines and patient samples, we compared the effects of the selective EGFR inhibitor erlotinib 24, with a pan-ERBB inhibitor, CI-1033 26, on a panel of neuroblastoma cell lines. Cells were treated with inhibitors for three days, and proliferation and survival were assessed (Table 1). CI-1033 treatment caused a dramatic reduction in cell yield for all lines, with IC50 values ranging from 0.94 μM to 2.45 μM. [CI-1033 is specific for the ERBB family of receptor tyrosine kinases in whole cells up to concentrations of 20 micromolar 27.] Expression of EGFR tended to correlate with CI-1033 sensitivity, in that the lines with the highest EGFR expression by either flow cytometry or western blot had the lowest IC50 values with CI-1033. The level of Her-4 expression did not appear to correlate with CI-1033 responsiveness. In contrast, erlotinib induced minimal growth inhibition, achieving an IC50 <10 μM in only two of the nine cell lines (SK-N-AS and LA1-55N). All neuroblastoma cell lines examined were negative for mutations in all investigated EGFR exons and K-RAS exon 2 (data not shown).
Table 1. IC50 Values for CI-1033 and erlotinib Treatment for 72 Hours.
| Cell Line | CI-1033 IC50 (μM) | erlotinib IC50 (μM) |
|---|---|---|
| SK-N-AS | 1.0 | 4 |
| LA1-55N | 1.0 | 10 |
| SK-N-SH | 1.0 | > 10 |
| SH-EP | 1.3 | > 10 |
| CHP-134 | 1.7 | > 10 |
| SH-SY5Y | 1.8 | > 10 |
| IMR-32 | 1.9 | > 10 |
| SMS-KCNR | 2.0 | >10 |
| NGP | 2.5 | >10 |
Differential induction of apoptosis with CI-1033 and erlotinib treatment in vitro
Cell cycle effects of CI-1033 and erlotinib in SK-N-SH cells were determined by propidium iodide staining (Figure 3A). CI-1033 treatment at 3 μM for 48 hours increased subdiploid SK-N-SH cells from 3% to 56% (p<0.005). In contrast, erlotinib, only slightly increased the proportion of sub-diploid cells, even at 10 μM (p<0.05). Similarly, in SK-N-AS cells, significant apoptosis was induced with CI-1033 at 5 μM (p<0.005), but not with erlotinib (data not shown).
Figure 3.
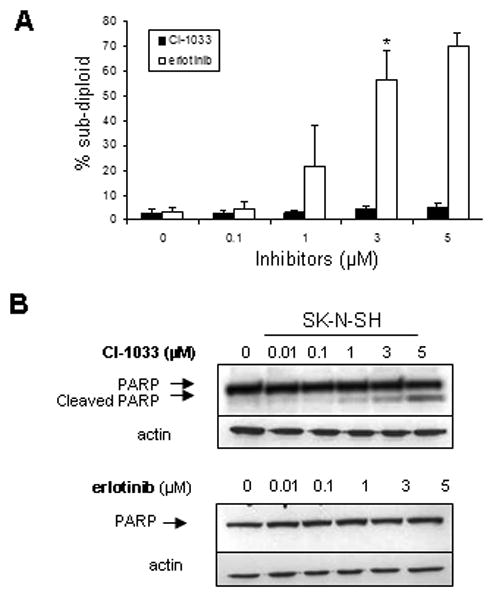
Cell cycle analysis and apoptosis of SK-N-SH cells treated with CI-1033 or erlotinib. (A) SK-N-SH cells were treated with a range of concentrations of CI-1033 or erlotinib for 24 hours and DNA content was measured by flow cytometry. The percent of cells in the sub-diploid population are shown. Error bars represent standard deviation (*= p<0.005). (B) SK-N-SH cells were treated with CI-1033 or erlotinib for 24 hours and analyzed for cleavage of PARP by western blot. Actin was used as the loading control.
To confirm that CI-1033 treatment was inducing apoptosis, we assessed for PARP cleavage by western blot. A dose-dependent increase in PARP cleavage was evident in SK-N-SH cells by 24 hours of CI-1033 treatment. In contrast, erlotinib caused no PARP cleavage (Figure 3B). We obtained similar results with SK-N-AS (data not shown).
Both CI-1033 and erlotinib inhibit EGFR and downstream signaling
We wished to know whether the ERBB inhibitors were effectively blocking signaling through EGFR in neuroblastoma. Both inhibitors decreased EGF-induced phosphorylation of EGFR at a concentration of 0.01μM (Figure 4). Near complete inhibition of EGFR phosphorylation occurred by 1 μM CI-1033, which correlated with a decrease in ERK 1/2 and AKT phosphorylation, with no changes in corresponding total protein levels (Figure 4A). However, despite dramatic inhibition of EGFR and ERK1/2 phosphorylation by 1 μM erlotinib, the phosphorylation of AKT only modestly decreased, even at high concentrations of erlotinib, suggesting a role for other ERBB receptors in AKT activation (Figure 4B).
Figure 4.
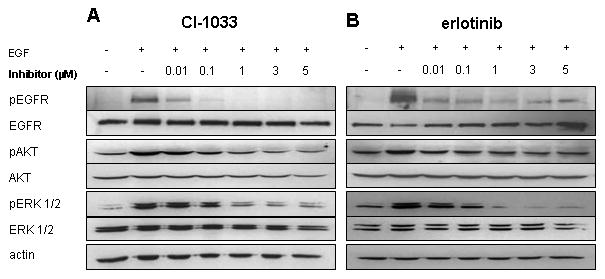
Inhibition of targets and downstream proteins with CI-1033 and erlotinib treatment in SK-N-SH cells. SK-N-SH cells were treated with multiple concentrations of CI-1033 (A) or erlotinib (B) for 24 hours, followed by the addition of EGF (100 ng/ml) for five minutes prior to collection of protein lysate. The phosphorylation of EGFR, AKT, and ERK 1/2 was analyzed by western blot. Blots were then stripped and reprobed with total EGFR, AKT, and ERK 1/2. Actin was used as the loading control.
In vivo tumor growth with CI-1033 and erlotinib treatment
An established neuroblastoma xenograft model using SK-N-SH cells in immunodeficient NOD-SCID-IL2R gamma knockout mice was used to test the effects of EGFR and pan-ERBB inhibition in vivo. Tumor bearing mice were treated by daily gavage with erlotinib, CI-1033, or vehicle control. Treatment with CI-1033 for 18 days resulted in a significant reduction in tumor growth (p<0.0001). Surprisingly, erlotinib treatment also reduced tumor growth after 18 days of treatment, but to a lesser degree than CI-1033 (p<0.005) (Figure 5A). Similarly, treatment with CI-1033 reduced final tumor weight by an average of 56% (p<0.005), compared with a 34% reduction with erlotinib treatment (Figure 5B).
Figure 5.
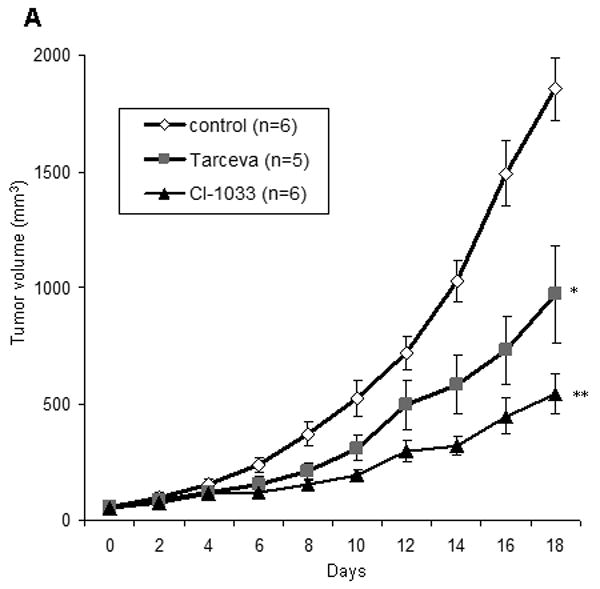
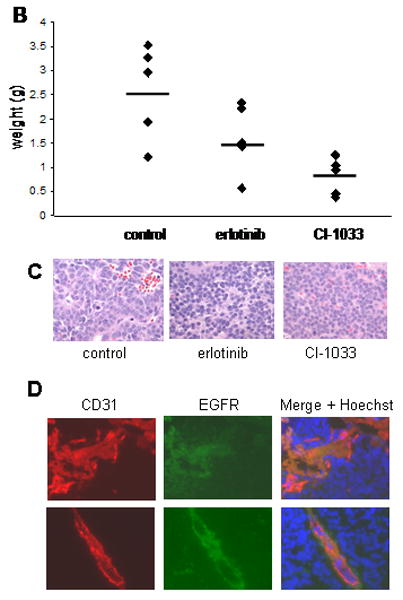
In vivo effects of CI-1033 and erlotinib. (A) Subcutaneous SK-N-SH xenografts were either untreated, treated with 10 mg/kg/day of erlotinib, or treated with 30 mg/kg/day of CI-1033. Tumor volume was measured every other day and all mice were sacrificed after 18 days of treatment. Error bars represent the standard error of the mean (* = p< 0.005, ** = p< 0.001). (B) The final weight of each of the subcutaneous tumors are shown (dots), along with the average for each group (bars) (CI-1033 p<0.005, erlotinib p>0.05). (C) Representative H&E sections from tumors of mice treated with vehicle control, erlotinib or CI-1033. (D) Neuroblastoma tumor sections were stained with anti-EGFR (green) and anti-CD31 (red). Hoechst 33342 dye (blue) identifies the nuclei. Images are from a control (top) and erlotinib treated (bottom) tumor.
To evaluate whether there was a role for ERBB signaling in neuroblastoma tumor angiogenesis, we measured CD31 staining in tumor sections. Mean vessel density after treatment with CI-1033 or erlotinib treatment showed a trend toward decreased vascularity in the erlotinib group (data not shown). Interestingly, anti-CD31 staining co-localized with EGFR staining, whereas EGFR staining was also seen in surrounding tumor tissue (Figure 5D). This suggests that EGFR inhibition on tumor vasculature contributes to ERBB inhibitors' in vivo effect, as has been described in other tumor models 28-31, but further studies are required.
Discussion
Children with high-risk neuroblastoma have a survival rate of less than 40%, even with aggressive treatment 1. Neuroblastoma survivors often have serious long-term adverse effects following treatment 32. Children with high-risk neuroblastoma therefore need novel therapies to improve overall outcomes and reduce the incidence and severity of late effects.
This study reports for the first time characterization of the ERBB family in neuroblastoma by flow cytometry. We report cell surface expression of EGFR, HER-3, and HER-4 in a panel of cell lines. Interestingly, comparison with western blot results suggests that much of the expressed EGFR is not displayed at the cell surface, pointing toward regulation of EGFR signaling through receptor recycling. Future studies of ERBB's role in neuroblastoma should include a more detailed evaluation of the intracellular location of each family member, to better define the roles that each molecule plays and how small molecule inhibitors might affect them. The ERBB expression we observe is similar to prior reports of ERBB expression by RT-PCR 5. Primary tumor samples also expressed abundant EGFR and Her-4, with varied Her-2 and Her-3, suggesting that signaling through more than one ERBB receptor, particularly HER-4, may play a role in neuroblastoma. It is not clear why Her-2 was detected in some primary samples but no established cell lines. It is possible that the conditioned used to establish neuroblastoma cell cultures select against Her-2 expression, or that a broader panel of cell lines might identify samples with Her-2 expression. It is clear, however, that Her-2 expression is not essential in neuroblastoma pathogenesis, to the extent that many cell lines and at least some patient samples had no Her-2 expression.
Supporting a role for ERBB signaling in neuroblastoma, we observed different effects from the pan-ERBB inhibitor CI-1033 26, 27 compared to selective EGFR inhibition by erlotinib 33, 34. While both inhibitors blocked EGF-induced EGFR and ERK1/2 phosphorylation at similar concentrations, CI-1033 was more effective at inhibiting proliferation, inducing PARP-cleavage and apoptosis and inhibiting AKT phosphorylation. This effect correlated with more potent in vivo growth inhibition with CI-1033 than erlotinib. These results suggest that ERBB receptors other than EGFR may contribute to neuroblastoma growth, and that differential effects on downstream targets, such as AKT are an essential feature. Given its expression in nearly all neuroblastoma cell lines and patient samples, and particularly its consistent surface expression by flow cytometry, HER-4 is the most likely ERBB receptor to explore in neuroblastoma. As mentioned before, little is known about the role of HER-4 in tumorigenesis, however its multiple isoforms, gamma-secretase-mediated cleavage and the transcriptional effects of its liberated intracellular domain, provide for multiple mechanisms of dysregulation to be explored 35-39.
Activating mutations in EGFR of NSCLC can predict for a response to erlotinib treatment 40, 41. An analysis of 36 neuroblastoma cell lines revealed no EGFR mutations. Further, no mutations of K-Ras exon 2, which can cause resistance to cetuximab in colorectal cancer 42, were found in 36 neuroblastoma cell lines. Therefore, the lack of neuroblastoma response to erlotininb was not due to the presence of any of the known resistance-causing mutations.
Since there was no effect on neuroblastoma cell growth in vitro with erlotinib treatment, we did not expect suppression of tumor cell growth in vivo with erlotinib. We were surprised, therefore, to find that erlotinib-treated xenograft tumors grew more slowly than the untreated tumors. Given the limited effects of erlotinib in vitro, the in vivo growth inhibition likely arose due to modulation of the tumor microenvironment. We saw strong co-localization of CD31 and EGFR by immunohistochemistry, and with erlotinib, found a trend toward reduced mean vessel density. Given that erlotinib-treated tumors were smaller than the untreated tumors and also tended to have fewer vessels per high-powered field, it seems likely that the total vessel content of erlotinib-treated tumors was truly less than that of untreated tumors. However, our in vivo experiments were not powered or designed to assess this difference. Although several reports have shown EGFR expression in endothelial cells of tumor models and anti-EGFR treatment reducing endothelial cell number 28-31, others have not confirmed these findings. For example, Amin et al have reported a decreased in vivo tumor volume in a melanoma xenograft with gefitinib treatment, presumably through targeting of blood vessels, but they did not find a decrease in vessel density 31. Further analysis is required to determine the contribution of erlotinib and CI-1033 treatment to angiogenesis in vivo. Given that interactions between tumor cells and stromal elements appear to contribute to ERBB's impact on neuroblastoma growth, orthotopic models in which neuroblastoma cells are placed under the adrenal capsule may be the best models for addressing these questions in vivo.
CI-1033, through its pan-ERBB inhibition, is likely having an anti-angiogenic effect through blockade of EGFR, and also a direct anti-tumor effect as seen in vitro. This would account for smaller final tumor volume with CI-1033 treatment, compared to erlotinib treatment. However, the different targeting profiles, the irreversible binding of CI-1033, and the bio-availability of erlotinib and CI-1033 may also contribute to differences observed in vivo. CI-1033 has been reported to reach a plasma level of 1-3 μM in a mouse model with an oral dose of 20 mg/kg 23, so the 30 mg/kg/day that we administered was expected to reach concentrations where effects were observed in vitro. It also is still expected to be specific for all ERBB kinases at this concentration 27. The dose given of erlotinib, 10 mg/kg/day, is equivalent to the dose currently given to lung cancer patients (150 mg). This dose was also shown to have little toxicity in mice, and to reach a peak plasma level of about 13 μM after 0.5 hours 43, higher than the levels we found to be ineffective in vitro. We chose this dose also because we wished to evaluate the minimum biologically effective dose 24, and to be certain that we were within the dose range in which the drug remains specific for EGFR, since erlotinib can inhibit Her-2 signaling at higher concentrations 44. At a dose of 10 mg/kg/day, serum concentrations of erlotinib are identical with either the iv or po routes 24, and po administration at this dose has been shown to inhibit EGFR autophosphorylation and EGFR-mediated growth in xenograft models.
In summary, we report the cell surface protein expression of multiple ERBB receptors in neuroblastoma, notably EGFR and HER-4. We demonstrate multiple significant differences in the effects of the EGFR selective inhibitor erlotinib when compared to the pan-ERBB inhibitor CI-1033, particularly pro-apoptotic and anti-tumor effects. Though further work to explore the direct effects of non-EGFR ERBB signaling in neuroblastoma is needed, we present data that support the use of pan-ERBB inhibitors such as the successor compound to CI-1033, PF-00299804, in neuroblastoma over the use of EGFR-specific agents.
Acknowledgments
This work was supported by the Lorrie Olivier Family Neuroblastoma Research Fund and by NIH grant 5K08CA118730 to DPMH.
Footnotes
The authors have no other financial disclosures.
References
- 1.Maris JM, Hogarty MD, Bagatell R, Cohn SL. Neuroblastoma. Lancet. 2007 Jun 23;369(9579):2106–2120. doi: 10.1016/S0140-6736(07)60983-0. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000 January 7;100(1):57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 3.Schulze WX, Deng L, Mann M. Phosphotyrosine interactome of the ErbB-receptor kinase family. Mol Syst Biol. 2005 January 1;1 doi: 10.1038/msb4100012. 2005.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Holbro T, Hynes NE. ErbB receptors: directing key signaling networks throughout life. Annu Rev Pharmacol Toxicol. 2004 January 1;44:195–217. doi: 10.1146/annurev.pharmtox.44.101802.121440. [DOI] [PubMed] [Google Scholar]
- 5.Ho R, Minturn JE, Hishiki T, et al. Proliferation of Human Neuroblastomas Mediated by the Epidermal Growth Factor Receptor. Cancer Res. 2005 November 1;65(21):9868–9875. doi: 10.1158/0008-5472.CAN-04-2426. [DOI] [PubMed] [Google Scholar]
- 6.Tamura S, Hosoi H, Kuwahara Y, et al. Induction of apoptosis by an inhibitor of EGFR in neuroblastoma cells. Biochem Biophys Res Commun. 2007 June 22;358(1):226–232. doi: 10.1016/j.bbrc.2007.04.124. [DOI] [PubMed] [Google Scholar]
- 7.Beaudry P, Nilsson M, Rioth M, et al. Potent antitumor effects of ZD6474 on neuroblastoma via dual targeting of tumor cells and tumor endothelium. Mol Cancer Ther. 2008 February 1;:1535–7163. doi: 10.1158/1535-7163.MCT-07-0568. MCT-1507-0568. [DOI] [PubMed] [Google Scholar]
- 8.Chiu B, Mirkin B, Madonna MB. Epidermal growth factor can induce apoptosis in neuroblastoma. J Pediatr Surg. 2007 Mar;42(3):482–488. doi: 10.1016/j.jpedsurg.2006.10.055. [DOI] [PubMed] [Google Scholar]
- 9.Chiu B, Mirkin B, Madonna MB. Mitogenic and apoptotic actions of epidermal growth factor on neuroblastoma cells are concentration-dependent. J Surg Res. 2006 Oct;135(2):209–212. doi: 10.1016/j.jss.2006.04.018. [DOI] [PubMed] [Google Scholar]
- 10.Chiu B, Mirkin B, Madonna MB. Novel action of epidermal growth factor on caspase 3 and its potential as a chemotherapeutic adjunct for neuroblastoma. J Pediatr Surg. 2007 Aug;42(8):1389–1395. doi: 10.1016/j.jpedsurg.2007.03.047. [DOI] [PubMed] [Google Scholar]
- 11.Reynolds CP, Tomayko MM, Donner L, et al. Biological classification of cell lines derived from human extra-cranial neural tumors. Prog Clin Biol Res. 1988;271:291–306. [PubMed] [Google Scholar]
- 12.Tumilowicz JJ, Nichols WW, Cholon JJ, Greene AE. Definition of a continuous human cell line derived from neuroblastoma. Cancer Res. 1970 Aug;30(8):2110–2118. [PubMed] [Google Scholar]
- 13.Biedler JL, Helson L, Spengler BA. Morphology and growth, tumorigenicity, and cytogenetics of human neuroblastoma cells in continuous culture. Cancer Res. 1973 Nov;33(11):2643–2652. [PubMed] [Google Scholar]
- 14.Reynolds CP, Biedler JL, Spengler BA, et al. Characterization of human neuroblastoma cell lines established before and after therapy. J Natl Cancer Inst. 1986 Mar;76(3):375–387. [PubMed] [Google Scholar]
- 15.Brodeur GM, Sekhon G, Goldstein MN. Chromosomal aberrations in human neuroblastomas. Cancer. 1977 November 1;40(5):2256–2263. doi: 10.1002/1097-0142(197711)40:5<2256::aid-cncr2820400536>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 16.Brodeur GM, Green AA, Hayes FA, Williams KJ, Williams DL, Tsiatis AA. Cytogenetic Features of Human Neuroblastomas and Cell Lines. Cancer Res. 1981 November 1;41(11_Part_1):4678–4686. [PubMed] [Google Scholar]
- 17.Ciccarone V, Spengler BA, Meyers MB, Biedler JL, Ross RA. Phenotypic Diversification in Human Neuroblastoma Cells: Expression of Distinct Neural Crest Lineages. Cancer Res. 1989 January 1;49(1):219–225. [PubMed] [Google Scholar]
- 18.Foley J, Cohn S, Salwen H, et al. Differential expression of N-myc in phenotypically distinct subclones of a human neuroblastoma cell line. Cancer Research. 1991;51(23):6338–6345. [PubMed] [Google Scholar]
- 19.Merlino GT, Xu YH, Ishii S, et al. Amplification and enhanced expression of the epidermal growth factor receptor gene in A431 human carcinoma cells. 1984;224:417–419. doi: 10.1126/science.6200934. [DOI] [PubMed] [Google Scholar]
- 20.King BL, Carter D, Foellmer HG, Kacinski BM. Neu proto-oncogene amplification and expression in ovarian adenocarcinoma cell lines. 1992;140:23–31. [PMC free article] [PubMed] [Google Scholar]
- 21.Gilbertson R, Hernan R, Pietsch T, et al. Novel ERBB4 juxtamembrane splice variants are frequently expressed in childhood medulloblastoma. Genes Chromosomes Cancer. 2001 July 1;31(3):288–294. doi: 10.1002/gcc.1146. [DOI] [PubMed] [Google Scholar]
- 22.Hughes DPM, Thomas DG, Giordano TJ, McDonagh KT, Baker LH. Essential erbB family phosphorylation in osteosarcoma as a target for CI-1033 Inhibition. Pediatric Blood and Cancer. 2006;46(5):614–623. doi: 10.1002/pbc.20454. [DOI] [PubMed] [Google Scholar]
- 23.Nyati MK, Maheshwari D, Hanasoge S, et al. Radiosensitization by Pan ErbB Inhibitor CI-1033 in Vitro and in Vivo. Clin Cancer Res. 2004 January 15;10(2):691–700. doi: 10.1158/1078-0432.ccr-1041-03. [DOI] [PubMed] [Google Scholar]
- 24.Pollack VA, Savage DM, Baker DA, et al. Inhibition of Epidermal Growth Factor Receptor-Associated Tyrosine Phosphorylation in Human Carcinomas with CP-358,774: Dynamics of Receptor Inhibition In Situ and Antitumor Effects in Athymic Mice. J Pharmacol Exp Ther. 1999 November 1;291(2):739–748. [PubMed] [Google Scholar]
- 25.Wiley H. Trafficking of the ErbB receptors and its influence on signaling. Exp Cell Res. 2003 March 10;284(1):78–88. doi: 10.1016/s0014-4827(03)00002-8. [DOI] [PubMed] [Google Scholar]
- 26.Slichenmyer W, Elliott W, Fry D. CI-1033, a pan-erbB tyrosine kinase inhibitor. Semin Oncol. 2001 October 1;28(5 Suppl 16):80–85. doi: 10.1016/s0093-7754(01)90285-4. [DOI] [PubMed] [Google Scholar]
- 27.Citri A, Alroy I, Lavi S, et al. Drug-induced ubiquitylation and degradation of ErbB receptor tyrosine kinases: implications for cancer therapy. EMBO J. 2002 May 15;21(10):2407–2417. doi: 10.1093/emboj/21.10.2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuwai T, Nakamura T, Sasaki T, et al. Phosphorylated epidermal growth factor receptor on tumor-associated endothelial cells is a primary target for therapy with tyrosine kinase inhibitors. Neoplasia. 2008 May;10(5):489–500. doi: 10.1593/neo.08200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baker CH, Pino MS, Fidler IJ. Phosphorylated epidermal growth factor receptor on tumor-associated endothelial cells in human renal cell carcinoma is a primary target for therapy by tyrosine kinase inhibitors. Neoplasia. 2006 Jun;8(6):470–476. doi: 10.1593/neo.06172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Amin DN, Hida K, Bielenberg DR, Klagsbrun M. Tumor Endothelial Cells Express Epidermal Growth Factor Receptor (EGFR) but not ErbB3 and Are Responsive to EGF and to EGFR Kinase Inhibitors. Cancer Res. 2006 February 15;66(4):2173–2180. doi: 10.1158/0008-5472.CAN-05-3387. [DOI] [PubMed] [Google Scholar]
- 31.Amin DN, Bielenberg DR, Lifshits E, Heymach JV, Klagsbrun M. Targeting EGFR activity in blood vessels is sufficient to inhibit tumor growth and is accompanied by an increase in VEGFR-2 dependence in tumor endothelial cells. Microvasc Res. 2008 Mar 18; doi: 10.1016/j.mvr.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 32.Gurney JG. Neuroblastoma, childhood cancer survivorship, and reducing the consequences of cure. Bone Marrow Transplant. 2007 Oct;40(8):721–722. doi: 10.1038/sj.bmt.1705815. [DOI] [PubMed] [Google Scholar]
- 33.Higgins B, Kolinsky K, Smith M, et al. Antitumor activity of erlotinib (OSI-774, Tarceva) alone or in combination in human non-small cell lung cancer tumor xenograft models. Anticancer Drugs. 2004 June 1;15(5):503–512. doi: 10.1097/01.cad.0000127664.66472.60. [DOI] [PubMed] [Google Scholar]
- 34.Ling YH, Li T, Yuan Z, Haigentz M, Jr, Weber TK, Perez-Soler R. Erlotinib, an effective epidermal growth factor receptor tyrosine kinase inhibitor, induces p27KIP1 up-regulation and nuclear translocation in association with cell growth inhibition and G1/S phase arrest in human non-small-cell lung cancer cell lines. Mol Pharmacol. 2007 Aug;72(2):248–258. doi: 10.1124/mol.107.034827. [DOI] [PubMed] [Google Scholar]
- 35.Ni CY, Murphy MP, Golde TE, Carpenter G. gamma -Secretase cleavage and nuclear localization of ErbB-4 receptor tyrosine kinase. Science. 2001 Dec 7;294(5549):2179–2181. doi: 10.1126/science.1065412. [DOI] [PubMed] [Google Scholar]
- 36.Rio C, Buxbaum JD, Peschon JJ, Corfas G. Tumor Necrosis Factor-alpha -converting Enzyme Is Required for Cleavage of erbB4/HER4. J Biol Chem. 2000 March 31;275(14):10379–10387. doi: 10.1074/jbc.275.14.10379. [DOI] [PubMed] [Google Scholar]
- 37.Junttila T, Sundvall M, Maatta J, Elenius K. Erbb4 and its isoforms: selective regulation of growth factor responses by naturally occurring receptor variants. Trends Cardiovasc Med. 2000 October 1;10(7):304–310. doi: 10.1016/s1050-1738(01)00065-2. [DOI] [PubMed] [Google Scholar]
- 38.Williams CC, Allison JG, Vidal GA, et al. The ERBB4/HER4 receptor tyrosine kinase regulates gene expression by functioning as a STAT5A nuclear chaperone. J Cell Biol. 2004 Nov 8;167(3):469–478. doi: 10.1083/jcb.200403155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Komuro A, Nagai M, Navin NE, Sudol M. WW Domain-containing Protein YAP Associates with ErbB-4 and Acts as a Co-transcriptional Activator for the Carboxyl-terminal Fragment of ErbB-4 That Translocates to the Nucleus. J Biol Chem. 2003 August 29;278(35):33334–33341. doi: 10.1074/jbc.M305597200. [DOI] [PubMed] [Google Scholar]
- 40.Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004 May 20;350(21):2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 41.Paez JG, Janne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004 Jun 4;304(5676):1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 42.Lievre A, Bachet JB, Le Corre D, et al. KRAS Mutation Status Is Predictive of Response to Cetuximab Therapy in Colorectal Cancer. Cancer Res. 2006 April 15;66(8):3992–3995. doi: 10.1158/0008-5472.CAN-06-0191. [DOI] [PubMed] [Google Scholar]
- 43.Zerbe LK, Dwyer-Nield LD, Fritz JM, et al. Inhibition by erlotinib of primary lung adenocarcinoma at an early stage in male mice. Cancer Chemother Pharmacol. 2007 Nov 21; doi: 10.1007/s00280-007-0644-z. [DOI] [PubMed] [Google Scholar]
- 44.Schaefer G, Shao L, Totpal K, Akita RW. Erlotinib Directly Inhibits HER2 Kinase Activation and Downstream Signaling Events in Intact Cells Lacking Epidermal Growth Factor Receptor Expression. Cancer Res. 2007 February 1;67(3):1228–1238. doi: 10.1158/0008-5472.CAN-06-3493. [DOI] [PubMed] [Google Scholar]