

Abstract
A key challenge in the synthesis of multi-component nanoparticles (NPs) for therapy or diagnosis is obtaining reproducible monodisperse NPs with a minimum number of preparation steps. Here we report the use of microfluidic rapid mixing using hydrodynamic flow focusing in combination with passive mixing structures to realize the self-assembly of monodisperse lipid-polymer and lipid-quantum dot (QD) NPs in a single mixing step. These NPs are composed of a polymeric core for drug encapsulation or a QD core for imaging purposes, a hydrophilic polymeric shell and a lipid monolayer at the interface of the core and the shell. In contrast to slow mixing of lipid and polymeric solutions, rapid mixing directly results in formation of homogeneous NPs with relatively narrow size distribution that obviates the need for subsequent thermal or mechanical agitation for homogenization. We identify rapid mixing conditions that result in formation of homogeneous NPs, and show that that self-assembly of polymeric core occurs independent of the lipid component, which only provides stability against aggregation over time and in the presence of high salt concentrations. Physicochemical properties of the NPs including size (35 to 180 nm) and zeta potential (−10 to +20 mV in PBS) are controlled by simply varying the composition and concentration of precursors. This method for preparation of hybrid NPs in a single mixing step may be useful for combinatorial synthesis of NPs with different properties for imaging and drug delivery applications.
Keywords: microfluidics, PLGA, nanoparticles, lipid, self-assembly
The development of smart multifunctional targeted nanoparticles (NPs) that can deliver drugs at a sustained rate to specific cells and carry nanoscale imaging agents may provide better efficacy, lower toxicity, and enhanced prognosis for treatment of multiple diseases1–4. Two promising families of NPs that can encapsulate and deliver therapeutic agents are polymeric NPs and liposomes.1, 2 Biocompatibility, biodegradability, reduced toxicity, and capacity for size and surface manipulations are benefits that these NPs offer in comparison to other delivery systems. Recently, it has been shown that hybrid lipid-polymeric NPs combine the desirable characteristics of polymeric NPs and liposomes such as high drug encapsulation yield, slow drug release, and high serum stability.3, 4 In addition, temporal controlled release of two different therapeutic agents has been achieved with these hybrid NPs by entrapping one agent in the lipid envelope and the other one in the polymeric core.5
The development of novel nanosystems such as hybrid lipid-polymeric NPs for drug delivery is necessary to advance the frontiers of drug delivery, but the ability to precisely control and predict properties of these systems is critical for their success in clinical translation.6 Furthermore, it may be necessary to screen and select NPs with optimal properties for a certain application, which demands reproducible synthesis of NPs with distinct size, charge, and ligand density.7 Among some of the technologies developed to prepare polymeric and lipid NPs of well-defined properties, continuous-flow microfluidic synthesis offers better control over NP formation compared to conventional synthesis and has the potential to tune NP characteristics in a reproducible manner, which is critical for identifying optimal NP formulations for any given application.8–11 Continuous-flow microfluidics has been used to synthesize polymeric nano and microparticles using controlled emulsification.12 and droplet formation through hydrodynamic flow focusing.13, 14 Recently, hydrodynamic flow focusing was used for the synthesis of liposomes with sizes of less than 200 nm15, 16, and PLGA-PEG NPs by nanoprecipitation (also known as solvent-displacement method).9
However, conventional methods for synthesizing lipid-polymeric NPs are more complex as compared to the preparation of liposomes or polymeric NPs. These methods involve mixing of polymeric NPs with liposomes to form lipid-polymer complexes, in which a lipid bilayer or lipid multilayer fuses on the surface of polymeric NPs.17–19 These complexes usually require a two-step formulation process: (i) development of polymeric NPs, and (ii) encapsulation of polymeric NPs within liposomes, resulting in poor control over the final NP physicochemical structure. Recently, our group developed a single step bulk hybrid lipid NP preparation in which a solution of poly-(lactic-co-glycolic) acid (PLGA) in acetonitrile was added to an aqueous solution of lecithin and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[carboxy(polyethylene glycol)] (DSPE-PEG) resulting in the formation of hybrid lipid-polymeric NPs.3, 4 Although this preparation yields sub-100 nm NPs and simplifies the synthesis, intermediate steps such as heating, vortexing, and long incubation time does not make the process easily amenable to combinatorial synthesis and can introduce variability in the properties of the NPs. A single-step process for reproducible synthesis of hybrid lipid-polymer NPs would enable preparation of libraries of NPs with distinct properties and allow for identification of optimal NP properties for different applications.
In this work, we show that rapid mixing of polymer and lipid solutions using continuous-flow microfluidics results in core-shell lipid-polymeric NPs in a single-step nanoprecipitation process. These NPs are composed of PLGA, lecithin and DSPE-PEG. We investigated the conditions under which stable hybrid NPs were formed and showed that by simply changing the precursors, NPs with tunable size from 35–180 nm and tunable zeta potential from −20 to 10 mV in PBS could be synthesized. Using the same microfluidic platform, we prepared homogenous lipid-QD NPs composed of CdSe/ZnS QDs coated by a lecithin and DSPE-PEG layer for potential imaging applications. Simplicity and reproducibility make this technology suitable for the combinatorial synthesis and screening of NPs with different properties without resorting to labor-intensive processing.
Results and Discussions
Controlled nanoprecipitation of NPs can be obtained by minimizing the mixing time to ensure homogeneous environment for nucleation and growth of the NPs.20 One way to decrease mixing time in microchannels is the use of micromixing structures, such as serpentine, staggered-herringbone, and zig-zag structures.21 Here we used an in-plane micromixing structure called Tesla mixer22 that operates at Reynolds numbers greater than 20. These micromixing structures show contributions from both diffusion and convection at high flow rates.
Figure 1a shows a schematic of the microchannel used to synthesize the hybrid lipid-polymeric NPs. An aqueous solution composed of lecithin and DSPE-PEG (lecithin:DSPE-PEG, 8.4:1.6 by mol) was mixed with an organic solution of PLGA dissolved in acetonitrile (1 mg/mL) at a volume ratio of 10:1. At the junction of the streams, the organic stream is hydrodynamically focused and enhanced mixing occurs through the Tesla structures as the focused streams flows along the channel. The hybrid NPs generated have PLGA-lecithin-PEG core-shell structure and properties as those previously synthesized using bulk synthesis methods (Figure 1b)3, 4. TEM imaging allowed qualitative assessment of the product stream including its core-shell structure and monodispersity (Figure 1c). The average NP size is 40 nm and its size distribution by volume indicated that over 85% of the NPs have a size within 30–60 nm (Figure 1d). A fluorescent dye was used to observe the unfolding-folding flow pattern inside the micromixer and used to illustrate the degree of mixing within the channel (Figure 1e). From this image, observation of complete mixing at a total flow rate of 50 µL/min occurs within the fourth turn of the Tesla mixer on a timescale of 10 ms (see supporting information for determination of mixing timescale in microchannel). This flow rate was fast enough to ensure good mixing in the Tesla mixer, yet low enough to limit the pressure drop in the device and hence prevent device failure.
Figure 1.

Nanoprecipitation of lipid-polymeric NPs. (a) A representative schematic of input and output streams within hybrid lipid-polymeric nanoparticle formation in microchannels with Tesla structures (b) Illustrative figure of microfluidic synthesized NP component layers (c) TEM image of uranyl acetate stained hybrid NPs after synthesis which highlights differences in density of the core versus near the surface of the NP potentially illustrating the lipid-PEG layer. Bar is labeled at 100 nm (d) Reproducible average size distribution of hybrid NPs generated through microfluidics. Average size is 40 nm. (e) Solvent mixing in the Tesla micromixing structures using fluorescent dye and water at 5 µL/min and 50 µL/min, respectively, shows complete mixing at the fourth turn in the channel (scale bar: 100 µm).
Characterization of PLGA-lipid NPs
The overall structure of the NPs synthesized was tested by multiple measures to ensure that they were hybrid particles of both lipid and polymeric nature rather than a combination of liposomes and unprotected PLGA NPs. Using different components in the input stream such as polymer alone, lipid/ lipid-PEG alone, or a combination of both illustrated differences in size of particles generated through the Tesla mixer (Figure 2a). When only polymer was present, NPs formed with a size of 40–50 nm and experienced slow aggregation within a few hours of formation, resulting in sizes ranging from 50–100 nm. When only lipid and lipid-PEG were added in the input streams, NPs obtained had a much larger size (~250 nm) and wider size distribution, which is typical for liposomes. However, particles prepared with both polymer and lipid had a size of 40 nm that did not significantly change over a long period of time (see figure S2, supporting information) This difference in size suggests that when both polymer and lipid are present in the input streams, polymeric NPs formed are stabilized by a lipid coating that helps maintain a size of 40 nm. Moreover, NPs generated with varying flow ratios of aqueous to organic stream did not appear to have a significant influence on the NP size (Figure 2a).
Figure 2.

Characterization of Lipid-PLGA structure (a) Comparison of average NP size from the product stream with aqueous : organic flow ratios of 10:1 and 5:1 respectively where the input organic stream is either PLGA, PLGA and lipid, or lipid alone. (b) Determination of lipid coverage of polymeric NPs. Zeta potential and size of NPs as the ratio of lipid to PLGA (w/w) is decreased. (c) Size distributions in water and PBS of NPs as the ratio of lipid to PLGA is changed. Complete lipid coverage of polymeric cores is observed at a ratio of lipid to PLGA ratio of 1:10. Above this ratio, the remaining lipid forms other nanostructures such as liposomes and below this ratio, NPs are not stable in PBS due to inadequate lipid coverage.
To investigate the optimal amount of lipid needed to obtain a stable hybrid NP that do not experience aggregation over time and under biological conditions, we first determined the extent of lipid coverage through measurements of NP zeta potential. A hybrid NP completely covered by lipid and lipid-PEG-COOH had a zeta potential in PBS of approximately −20 mV while a NP without lipid and only PLGA-OCH3 had a zeta potential of −8 mV (Figure 2b). Therefore, by decreasing the amount of lipid with respect to PLGA while keeping other conditions consistent, one may expect to see an increase in zeta potential as the NP goes from complete lipid coverage to no coverage. Variation of the amount of lipid to PLGA from 1:10 to 1:100 lead to an increase in zeta potential without a significant change in size (Figure 2b) which suggests that complete lipid surface coverage is not required to maintain NP size stability at 40 nm in water. In fact, a lipid to polymer ratio of 1:1000 was sufficient to prevent aggregation of the NPs. When lipid was absent, the size of the polymeric NP increased to that of PLGA NPs, similar to the trend observed in Figure 2a. The size increase in the case of 1:1 lipid to PLGA indicates that there is enough lipid to completely cover all PLGA NPs, and the excess lipid present results in an increase in the average NP size either by addition of layers, or by the formation of liposomes.
To confirm this hypothesis, we obtained the size distributions by volume of NPs in water as the amount of lipid introduced in the input stream was decreased. In addition, since PLGA NPs covered by lipid are stable in PBS as opposed to those without lipid coverage, we obtained the size distribution of the NPs in PBS as way to assay their stability (Figure 2c). Our studies indicate that two populations of particles are formed at a lipid to PLGA ratio of 1:1 with sizes at 40 nm and 250 nm. The smaller particles correspond to hybrid NPs with full lipid coverage while the larger particles correspond to liposomes similar to ones observed in Figure 2a. For a lipid to PLGA ratio of 1:10, a homogenous size distribution was observed in water and PBS. It must be noted that a small shift of the average NP size peak is observed when the samples were measured in water versus PBS. This peak shift could be explained by an aggregation of 2 or 3 NPs after immersion in PBS and it can be controlled by tuning the ratio of lipid to lipid-PEG in the formulation (Figure S3, supporting information). For lipid to PLGA ratio of 1:100 and 1:1000, there is enough lipid present to keep the NP at size of 40 nm in water, yet it is not enough to avoid NPs aggregation in PBS. Our studies suggest that the optimal lipid coverage is obtained at a lipid to PLGA ratio of approximately 1:10. In fact, NPs prepared at this ratio were stable for a period of 24 hours in 10% BSA and 10% serum, which are surrogates for in vivo protein adsorption and biofouling (Supporting Information, Figure S2).3 Considering the thickness and hydrodynamic radius of a lecithin layer with DSPE-PEG, estimation of the amount of lecithin and DSPE-PEG needed to completely cover NP core of 40 nm lead to a calculated ratio of 1.4:10 of lipid to PLGA. Although this calculation relies on several assumptions (e.g. all NPs are spheres and monodisperse, constant ratio of lecithin to DSPE-PEG, full lipid coverage on surface etc), it still offers an adequate approximation of the amount of lipid necessary to form a stable hybrid lipid-polymeric NP.
At the same lipid to PLGA ratio, we found that there was no significant difference in the size of NPs prepared at aqueous to organic flow ratios of 10:1 and 5:1 (see figure S1 supporting information). Larger flow ratios (e.g. 20:1, aqueous to organic) resulted in pulsing of the flow due to the syringe pumps, and required higher PLGA concentrations that made the device more susceptible to fouling. On the contrary, lower flow ratios (e.g. 1:1, aqueous to organic) would result in an inadequate environment for NP formation since for optimal nanoprecipitation a flow ratio of aqueous to acetonitrile streams at least 3:1 is desired.9 Therefore, all the NPs prepared in this work were obtained at a flow ratio of 10:1 or 5:1.
Control of NP physicochemical properties: Size and surface charge
After confirming the core-shell structure of the lipid-PLGA NPs, and knowing their range of optimal lipid coverage, we investigated the possibility of controlling the NP’s physicochemical properties, mainly size and surface charge. Figure 3a illustrates a change in zeta potential of the lipid-polymeric NPs when different end-functional groups of DSPE-PEG were introduced in the input streams. The zeta potential of the NPs could be controlled from positive to neutral to negative charge by utilizing -COOH, -CH3 and -NH2 respectively, while the size remained essentially unchanged. Specific values for zeta potentials of different modified end groups agree with those previously published.23 These results not only show that the surface charge of the NP can be finely tuned but they also confirm that lipid-PEG is on the surface of the polymeric core. Finally, NP size was controlled by varying the inherent viscosity of the polymer and the PLGA concentration while keeping other conditions such as organic and aqueous flow rates and flow ratio the same. Decrease in inherent viscosity and increase in initial polymer concentration lead to the generation of larger NPs as reported similarly in bulk synthesis of these particles (Figure 3b)4. This illustrates that hybrid NPs made from microfluidics are similar in nature to those made in bulk and their physicochemical properties such as size and charge can be controlled.
Figure 3.
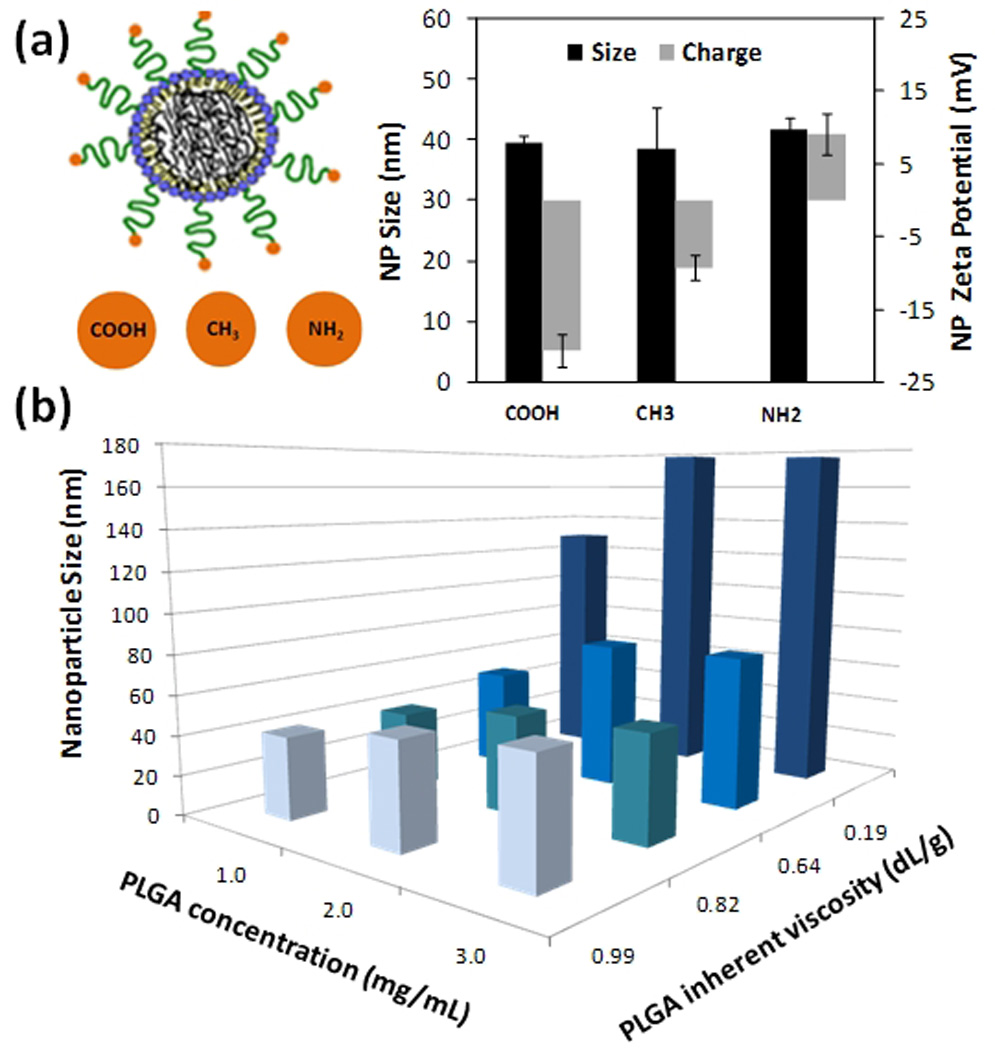
Control of NP’s physicochemical properties. (a) Control of surface charge and lipid coverage of the hybrid NPs is elucidated by changes in zeta potential of the NPs in PBS using DSPE-PEG with modified functional groups of carboxyl, methyl, and amine (b) Control of NP size by varying PLGA viscosity and concentration in the organic stream. Flow ratios of aqueous to organic streams and rate were kept constant at 10 : 1 at a total flow rate of 55 µL/min.
Investigating the role of rapid mixing and mechanism of self-assembly of the hybrid NPs
To gain more insight into the role of rapid mixing in self-assembly of the NPs, we compared the formation of NPs under rapid mixing conditions versus slow mixing conditions obtained by pipetting the same volume of polymer solution used in the microfluidic device into a lipid solution without sonication or heating. Figure 4 shows the size distributions in water and in PBS of NPs prepared under rapid mixing with the microfluidic chip and slow mixing conditions. Under slow mixing conditions at a 1:1 ratio of lipid to PLGA, the formation of liposomes is noted by the peak around 400 nm. A peak around 10 µm in PBS, characteristic of aggregates of polymeric NPs, indicates the formation of polymeric NPs. Finally some hybrid lipid-polymeric NPs are formed since there is a stable peak around 100 nm in water and PBS. The hybrid NPs formed under slow mixing are not homogenous, noted from the irregularity in the peak around 100 nm. This observation suggests uneven distribution of lipid-PEG among the polymeric NPs since lipid-PEG confers stability to the hybrid NPs through formation of the PEG corona (Figure S3, supporting information). Formation of polymeric NPs, lipid-polymeric NPs and liposomes, was also observed for a lipid to a PLGA ratio of 1:10. However, the larger peaks in water and PBS around 100–1000 nm are not as prominent as for the 1:1 case since the percentage by volume of polymeric NPs formed is much higher than that of liposomes. Under rapid mixing within the microfluidic channel, homogenous and stable hybrid NPs and liposomes are obtained at a lipid to polymer ratio of 1:1 and only stable homogenous hybrid NPs are obtained at a lipid to polymer ratio of 1:10, as inferred by the absence of larger aggregates upon addition of PBS. A size distribution by intensity is shown in the supporting information where the formation of distinct populations of NPs is clearly evident in the case of slow mixing of lipid and PLGA solutions, but only a single population is seen in the case of rapid mixing of lipid and PLGA solutions (Figure S4, supporting information).
Figure 4.

Slow versus rapid mixing. Comparison of NP size distribution in water and PBS for rapid and slow mixing of lipid and PLGA solutions with lipid : PLGA ratios of 1:1 and 1:10. Under slow mixing conditions without the input of any form of energy, aggregation upon addition of PBS indicates the presence of heterogeneous NPs (i.e. polymeric, lipid, and lipid-polymeric). Under rapid mixing conditions, absence of aggregation upon addition of PBS indicates that only homogenous hybrid lipid-polymeric NPs are formed, except for the 1:1 ratio that results in homogeneous hybrid NPs and liposomes.
These results give some insight into the role of rapid mixing in the self-assembly of hybrid lipid NPs. Under rapid mixing, there is uniform lipid and lipid-PEG coverage around polymeric cores resulting in the formation of homogenous hybrid lipid-polymeric NPs. In contrast, under slow mixing, some lipid and lipid-PEG is deposited onto polymeric NPs while the rest forms lipid structures leaving polymeric NPs with uneven or no coverage. The result is the formation of a combination of liposomes, polymeric NPs, and hybrid-lipid NPs (Figure S4, supporting information). An input of energy to the system in the form sonication and/or raise in the temperature as provided in the bulk methods of synthesis may assist in the disassembly of lipid structures and their reassembly around the polymeric cores forming homogenous lipid-polymeric NPs. Integration of rapid mixing using microfluidics bypasses the intermediate steps needed in slow mixing conditions in the preparation of homogenous lipid-polymeric NPs.
While the above experiment suggests that rapid mixing plays an important role in ensuring uniform lipid coverage on the NPs, it does not explain the invariance of NP size with lipid concentration. Interestingly, even a 1:1000 lipid : PLGA ratio was sufficient to maintain the hybrid NP at a size of 40 nm as opposed to NPs made with only PLGA, which increased in size over a period of few hours. In addition, subsequent increase of the lipid : PLGA ratio by three orders of magnitude did not affect the hybrid NP size (although some liposomes were formed in the 1:1 case). To investigate the role of lipid in self-assembly of the NPs, we designed experiments to elucidate to what extent the self assembly of PLGA was affected by the presence of the lipid component.
In these experiments, we prepared lipid-PLGA NPs using a ‘two-stage’ manner, where a PLGA polymeric core of a specific size was formed first followed by deposition of the lipid onto PLGA core by flowing the solutions through the microfluidic device for a second time. We then compared the size and size distributions of these NPs with those prepared using the conventional one-step method. First, a PLGA solution in acetonitrile and water were injected into the device. These unprotected PLGA NPs (NP1) were washed and suspended in water at a concentration of 1 mg/mL. Next, NP1 particles in water were immediately reintroduced into the center inlet of the channel and the lipid solutions were introduced through the side channels. The size and size distribution of the lipid-covered NPs (NP2) obtained at the outlet were measured. NP2 particles were placed in PBS to test their stability in comparison to those NPs synthesized through conventional one-step microfluidic method (i.e. mixing PLGA solution with the lipid solution) (Figure 5). NP2 particles prepared using the two-stage manner had similar size distribution and stability properties to the hybrid NPs prepared in the conventional one-step microfluidic method. In other words, NPs made from a one-step method and NP2 particles had a uniform distribution with an average size of 40 nm and remained stable when placed in PBS.
Figure 5.

NP formation to elucidate stepwise formation of hybrid lipid-polymer NPs within microchannel (a) NP size distribution in water and PBS of particles formed in two-stage manner. PLGA NPs were prepared in the microfluidic mixer, then washed and placed as an input along with lipid aqueous stream resulting in the generation of hybrid lipid-PLGA NPs. (b) NP size distribution in water and PBS formed through the current one-step microfluidic method.
These results suggest that self-assembly of the polymeric core of lipid-PLGA NPs in the device was unaffected by the presence of the lipid component. It can be then rationalized that the one-step method indeed involves the above two distinct stages in a very small timescale. The Péclet number (Pe=V·w/D) in this case is over 1000, which indicates that convective transport can enhance mixing of particles as long as the streams move laterally.24, 25 The Tesla structures in our mixing channel enforce such lateral movement of particles at their junctions. PLGA core formation, the first assembly stage, requires that PLGA chains in the focused acetonitrile stream encounter the anti-solvent, (i.e. water molecules), which results in conditions under which PLGA can precipitate9. Our microfluidic mixer ensures that complete solvent displacement of acetonitrile by water (0.2–2 ms, see supporting information) occurs on a timescale that is shorter than that of formation of PLGA cores. Furthermore, solvent displacement is almost complete before a substantial amount of lipid approaches the PLGA cores. Lipid shell formation, the second stage, then follows as soon as the lipid molecules are transported to the vicinity of already-formed PLGA cores. Although the diffusion of lipid molecules is slower than that of water molecules at least by an order of magnitude, embedded Tesla structures considerably enhance particle mixing due to their convective effects. Such laterally-dispersive transport (combination of diffusion and convection) occurs on a mixing timescale of ~10 ms (see supporting information). It is worth noting that the minimal lipid coverage required to stabilize the NPs in water occurs at a lipid : PLGA ratio of 1:1000 and is obtained on a sub-millisecond timescale after sufficient amount of lipid has been transported near the core. In other words, sufficient lipid coverage to prevent long-term aggregation is achieved on a sub-millisecond timescale after complete mixing of solvent and anti-solvent. Thus, the formation of the lipid shell is a transport-limited process since the timescale of coverage is limited by the timescale of mixing. In cases of ratios of lipid : PLGA higher than 1:1000, partial mixing of lipid molecules with acetonitrile would be sufficient for minimum lipid coverage of the PLGA cores. The lipid shell forms on the timescale of 1 ms in the case of 1:1 ratio of lipid : PLGA. Therefore, the timescales for self-assembly of the PLGA cores is on the same order of magnitude as the timescale for minimum lipid coverage, at least for high lipid : PLGA ratios (see supporting information). In contrast to this minimum coverage, complete coverage of the NP with lipid requires complete mixing, which occurs on the timescales of >10 ms. The fact that hybrid NPs could be prepared in two steps (i.e. by first forming PLGA NPs followed by mixing with lipid) indicates that the size of the NPs formed in the device is independent of the presence of the lipid component on short timescales, and the differences in size of PLGA versus hybrid lipid-PLGA NPs shown in Figure 2a occur over a longer timescale before measurement of the NP size. Therefore, these results, along with the invariance of NP size with lipid ratio, show that the lipid component does not play a significant role in the self-assembly of the polymeric NPs but rather it stabilizes the NPs and prevents their aggregation on longer timescales. These results are further supported by measuring the size and size distribution of PLGA NPs immediately upon synthesis, which are in a similar range to hybrid NPs (Figure S2 and S4, supporting information).
Preparation of lipid-QD NPs
To demonstrate the versatility of our platform design, we examined the ability of rapid mixing to synthesize hybrid quantum dot (QD) lipid NPs for imaging applications. QDs are semiconducting nanocrystals that possess excellent optical properties that make them suitable to be used as imaging probes.26 However, the hydrophobicity and poor colloidal stability at physiological conditions frequently renders them inappropriate for clinical use.27 In the same fashion as for hybrid lipid-polymeric NPs, it has been proposed and shown that lipid-coated QD provide enhanced NP hydrophilicity, stability in plasma, and an overall improvement of in their biocompatibility.28, 29 Others have encapsulated other type of particles such as magnetic NPs30 and gold NPs31 inside a lipid and polymeric envelope, respectively.
As a proof of concept to show that a similar microfluidic platform can be used to prepare other NPs, we prepared hybrid NPs composed of QDs encapsulated by lecithin and DSPE-PEG layer (Figure 6a). Lipophilic QDs where dissolved in THF (0.5 mg/mL) and introduced in the middle input stream in place of the polymeric stream from the prior study. The lipid-QD NPs in the product stream showed a homogenous size distribution with an average size of 60 nm and did not need further processing for in vitro and/or in vivo experiments except for the removal of a small fraction of THF through filtration or evaporation (Figure 6b). At an initial concentration of 0.5 mg/mL, TEM images show an average of four quantum dots encapsulated per NP (Figure 6c). The number of QDs could be controlled by varying the lipid to QD ratio. To ensure that the images obtained from TEM are QDs encapsulated on lipid NPs and not QDs adhered to the surface of a lipid matrix, images were taken of the operating channels to study their formation. It was observed that before encapsulation of QDs, some aggregation of QDs was visible inside the channel and after encapsulation, the channel remained free of QD aggregates (Figure S6, supporting information). These results clearly show that one can use the same microfluidic platform to synthesize distinct types of hybrid NPs. The results suggest that by using this continuous-flow microfluidic technology one can entrap other imaging agents such as gold NPs and/or magnetic NPs inside a lipid or polymeric envelope to form multifunctional particles for use of various imaging modalities. Through selection of appropriate solvents and concentration, therapeutic and imaging agents can be introduced into the input stream to form a theranostic system.
Figure 6.

Preparation of hybrid lipid-QD NPs. (a) Schematic of liposome formation in the Tesla mixer with quantum dots encapsulated within the core (b) NP distribution of quantum dot encapsulated liposomes formed through the Tesla mixer (c) TEM image of hybrid lipid-QD NPs stained with 1% phosphotungstic acid aqueous solution showing monodisperse particles with a Z-average size of 60nm. Bar is labeled at 100 nm
In conclusion, we demonstrated that hybrid lipid-PLGA NPs can be prepared by rapid mixing of a polymeric solution with a lipid solution in a microfluidic device. We identified an optimal ratio of lipid : PLGA that resulted in stable and homogeneous NPs. The size and charge of the NPs could be controlled by using PLGA of different viscosities (molecular weights) and by using lipid molecules with different end groups, respectively. The experiments suggest that the self-assembly of PLGA core occurs independent of the lipid component, but the lipid component provides stability to the NP against aggregation over time and in the presence of high salt concentrations. Furthermore, rapid mixing ensures formation of homogeneous NPs; in contrast, slow mixing results in different populations of NPs that are not uniform in composition and size. We also demonstrated that hybrid lipid-QD NPs could be formed in the same system. Reproducible manufacture of monodisperse, stable NPs with the ability to control properties by varying concentrations of different precursors in a simple mixing step could greatly facilitate combinatorial synthesis and prove to be useful in the emerging field of nanomedicine.
Experimental Methods
Device fabrication and experimental setup
Microfluidic devices were fabricated with poly(dimethylsiloxane) (PDMS) using a standard micromolding process. PDMS (Sylgard 184, Dow Corning) monomer and curing agent were mixed in a ratio of 10:1 by weight, pored over the silicon wafer mold, and degassed. After curing, PDMS was peeled off and inlet/outlet holes were drilled using a 300 µm diameter drill bit. The PDMS component was then bonded to a 1” × 2” glass slide using air plasma. The resulting device had one inlet each for water and solvent streams, and one outlet. The water stream was split into two in order to achieve two water streams at the flow focusing junction. The mixing channel was 50 µm wide, 60 µm high and 2.5 mm long. One 500 µL syringe was mounted on a syringe pump (SP230IW, World Precision Instruments) while the other syringe was mounted on another syringe pump (PHD 22/2000, Harvard Apparatus) to control flow through the device. Water flow rate was maintained at 50 µL/min, while the solvent flow rate was varied from 5 µL/min to 10 µL/min.
Preparation of PLGA-lecithin-PEG NPs
A solution of PLGA (intrinsic viscosity 0.82 dL/g; Lactel, Pelham, AL) was dissolved in acetonitrile (1 mg/mL) and lipid solution composed of 4% ethanol aqueous solution of lecithin (soybean, refined, molecular weight: ~330 Da; Alfa Aesar, Ward Hill, MA) and DSPE-PEG (molecular weight: ~2850 Da; Avanti Polar Lipids, Alabaster, AL) (lecithin/DPSE PEG, 8.4/1.6) were prepared independently for separate inlets and mixed within the chip at fixed flow rates using syringe pumps. Lipid solution flow rate was fixed at 50 µL/min while polymer solution flow rate was set at 10 µL/min for some experiments and 5µL/min for others. Lipid concentration was varied from 102 to 0.1 µg/mL at a constant flow rate of 50 µL/min. To prepare NPs at slow mixing conditions, 100 µL of the lipid solution described in the previous paragraph was mixed with 10 µL of PLGA solution in acetonitrile using pipette tips. For the NP stability studies, 20 µL of 10x PBS were added to 60 µL of the NP solution. Size and zeta potential of NPs were immediately measured upon addition of PBS. For hybrid lipid-PLGA NPs prepared with the microchannel, up to four consecutive additions of 20 µL of 10x PBS were pipetted to 60 µL of the sample and its size was monitored for each addition. No change in size was observed after the first addition of the buffer.
Preparation of QD-lecithin-PEG NPs
A solution of lipophilic quantum dots (TOP-coated CdSe/ZnS QDs, Invitrogen, CA, USA) was dissolved in tetrahydrofuran (THF) (0.5 mg/mL) and a lipid solution of 4% ethanol aqueous solution of lecithin and DSPE-PEG (lecithin/DPSE PEG, 8.4/1.6) at a concentration of 0.02 mg/mL were prepared independently for separate inlets and mixed within the chip. Lipid solution flow rate was set at 50 µL/min while QD solution flow rate was set at 5 µL/min. Both flow rates were controlled with syringe pumps.
Particle sizing and zeta potential measurements
Particle sizing was performed using dynamic light scattering with Zetasizer Nano ZS (Malvern Instruments Ltd., U.K.). For each measurement, 100 µL or more volume of the sample was loaded in a disposable low-volume cuvette. Three measurements were performed on each sample. We observed that the presence of acetonitrile changed the NP size by less than 3 % when water-acetonitrile mixtures containing up to 5 % acetonitrile were further diluted in water. All measurements were performed at acetonitrile concentrations of less than 10 % acetonitrile to ensure that any observed variation in particle size was not due to the solvent. The NP surface zeta potential was measured by using ZetaPALS (Brookhaven Instrument., U.S.A). For each measurement, particles were washed with water three times and reconstituted in 1mL of PBS (0.5mg/mL). The zeta potential was recorded as the average of three measurements.
Transmission electron microscopy (TEM)
Lipid-polymeric NPs
TEM experiments were carried out on a JEOL JEM-200CX instrument at an acceleration voltage of 200 kV. The TEM sample was prepared by depositing 10 µL of the NP suspension (1.0 mg/mL) onto a 200-mesh carbon-coated copper grid. Samples were blotted away after 30 min incubation and grids were negatively stained for 20 min at room temperature with freshly prepared and sterile-filtered 2% (w/v) uranyl acetate aqueous solution. The grids were then washed twice with distilled water and air dried prior to imaging.
Lipid-QD NPs
TEM experiments were carried out on a JEOL JEM-2011 instrument at an acceleration voltage of 200 kV. The TEM sample was prepared by depositing 10 µL of the NP suspension (1.0 mg/mL) onto a 300-mesh formvar-coated copper grid. Samples were blotted away after 30 min incubation and grids were negatively stained for 20 min at room temperature with sterile-filtered 1% (w/v) phosphotungstic acid aqueous solution. The grids were then washed twice with distilled water and air dried prior to imaging.
Supplementary Material
Acknowledgments
We thank MIT’s Microsystems Technology Laboratory and staff for their help with device fabrication. We also thank Sangeeta Bhatia (MIT) for allowing the use of particle sizing equipment in her laboratory. Electron microscopy image acquisition was performed in the Center for Material Science and Engineering (CMSE) imaging facility. This research was supported by the Koch-Prostate Cancer Foundation Award in Nanotherapeutics (R.L. and O.C.F.), by the Concept Development Grant 5P50CA090381-09 from the Dana Farber Cancer Institute Prostate SPORE (O.C.F.), and by NIH Grants CA119349 (R.L. and O.C.F.) and EB003647 (O.C.F.). P.M.V. and P.A.B. are supported by the NSF graduate research fellowship. Finally, we thank Dr. Qiaobing Xu and Dr. Christian Kastrup for helpful discussions and Maria I. Rodriguez for her assistance in some of the experiments.
Footnotes
Support Information available: Comparison of lipid-PLGA NPs prepare under rapid mixing conditions versus NPs prepared with previously published method. Change in size over time of NPs made with different precursors and suspended in 10% BSA and 10% serum. Investigation of the effect of DSPE-PEG on lipid-PLGA NP stability. Further characterization of NPs made under slow mixing conditions. Determination of mixing time in channel. Further characterization of self-assembly of lipid-QDs NPs inside microfluidic channel. Estimation of mixing timescales. Estimation of self-assembly timescales of PLGA cores. Estimation of the timescale for minimal lipid coverage on polymeric core for NP stabilization. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an Emerging Platform for Cancer Therapy. Nat Nanotechnol. 2007;2(12):751–760. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 2.Wagner V, Dullaart A, Bock AK, Zweck A. The Emerging Nanomedicine Landscape. Nat Biotechnol. 2006;24(10):1211–1217. doi: 10.1038/nbt1006-1211. [DOI] [PubMed] [Google Scholar]
- 3.Zhang L, Chan JM, Gu FX, Rhee JW, Wang AZ, Radovic-Moreno AF, Alexis F, Langer R, Farokhzad OC. Self-Assembled Lipid--Polymer Hybrid Nanoparticles: A Robust Drug Delivery Platform. ACS Nano. 2008;2(8):1696–1702. doi: 10.1021/nn800275r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chan JM, Zhang L, Yuet KP, Liao G, Rhee JW, Langer R, Farokhzad OC. Plga-Lecithin-Peg Core-Shell Nanoparticles for Controlled Drug Delivery. Biomaterials. 2009;30(8):1627–1634. doi: 10.1016/j.biomaterials.2008.12.013. [DOI] [PubMed] [Google Scholar]
- 5.Sengupta S, Eavarone D, Capila I, Zhao G, Watson N, Kiziltepe T, Sasisekharan R. Temporal Targeting of Tumour Cells and Neovasculature with a Nanoscale Delivery System. Nature. 2005;436(7050):568–572. doi: 10.1038/nature03794. [DOI] [PubMed] [Google Scholar]
- 6.Hall JB, Dobrovolskaia MA, Patri AK, McNeil SE. Characterization of Nanoparticles for Therapeutics. Nanomed. 2007;2(6):789–803. doi: 10.2217/17435889.2.6.789. [DOI] [PubMed] [Google Scholar]
- 7.Gu F, Zhang L, Teply BA, Mann N, Wang A, Radovic-Moreno AF, Langer R, Farokhzad OC. Precise Engineering of Targeted Nanoparticles by Using Self-Assembled Biointegrated Block Copolymers. Proc Natl Acad Sci U S A. 2008;105(7):2586–2591. doi: 10.1073/pnas.0711714105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jahn A, Reiner JE, Vreeland WN, DeVoe DL, Locascio LE, Gaitan M. Preparation of Nanoparticles by Continuous-Flow Microfluidics. J. Nanopart Res. 2008;10:925–934. [Google Scholar]
- 9.Karnik R, Gu F, Basto P, Cannizzaro C, Dean L, Kyei-Manu W, Langer R, Farokhzad OC. Microfluidic Platform for Controlled Synthesis of Polymeric Nanoparticles. Nano Lett. 2008;8(9):2906–2912. doi: 10.1021/nl801736q. [DOI] [PubMed] [Google Scholar]
- 10.Gu FX, Karnik R, Wang AZ, Alexis F, Levy-Nissenbaum E, Hong S, Langer R, Farokhzad OC. Targeted Nanoparticles for Cancer Therapy nanotoday. 2007;2(3):14–21. [Google Scholar]
- 11.Yen BK, Gunther A, Schmidt MA, Jensen KF, Bawendi MG. A Microfabricated Gas-Liquid Segmented Flow Reactor for High-Temperature Synthesis: The Case of Cdse Quantum Dots. Angew Chem Int Ed Engl. 2005;44(34):5447–5451. doi: 10.1002/anie.200500792. [DOI] [PubMed] [Google Scholar]
- 12.Xu Q, Hashimoto M, Dang TT, Hoare T, Kohane DS, Whitesides GM, Langer R, Anderson DG. Preparation of Monodisperse Biodegradable Polymer Microparticles Using a Microfluidic Flow-Focusing Device for Controlled Drug Delivery. Small. 2009;5(13):1575–1581. doi: 10.1002/smll.200801855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martin-Banderas L, Flores-Mosquera M, Riesco-Chueca P, Rodriguez-Gil A, Cebolla A, Chavez S, Ganan-Calvo AM. Flow Focusing: A Versatile Technology to Produce Size-Controlled and Specific-Morphology Microparticles. Small. 2005;1(7):688–692. doi: 10.1002/smll.200500087. [DOI] [PubMed] [Google Scholar]
- 14.Rondeau E, Cooper-White JJ. Biopolymer Microparticle and Nanoparticle Formation within a Microfluidic Device. Langmuir. 2008;24(13):6937–6945. doi: 10.1021/la703339u. [DOI] [PubMed] [Google Scholar]
- 15.Jahn A, Vreeland WN, DeVoe DL, Locascio LE, Gaitan M. Microfluidic Directed Formation of Liposomes of Controlled Size. Langmuir. 2007;23(11):6289–6293. doi: 10.1021/la070051a. [DOI] [PubMed] [Google Scholar]
- 16.Jahn A, Vreeland WN, Gaitan M, Locascio LE. Controlled Vesicle Self-Assembly in Microfluidic Channels with Hydrodynamic Focusing. J Am Chem Soc. 2004;126(9):2674–2675. doi: 10.1021/ja0318030. [DOI] [PubMed] [Google Scholar]
- 17.De Miguel I, Imbertie L, Rieumajou V, Major M, Kravtzoff R, Betbeder D. Proofs of the Structure of Lipid Coated Nanoparticles (Smbv) Used as Drug Carriers. Pharm Res. 2000;17(7):817–824. doi: 10.1023/a:1007504124603. [DOI] [PubMed] [Google Scholar]
- 18.Thevenot J, Troutier AL, David L, Delair T, Ladaviere C. Steric Stabilization of Lipid/Polymer Particle Assemblies by Poly(Ethylene Glycol)-Lipids. Biomacromolecules. 2007;8(11):3651–3660. doi: 10.1021/bm700753q. [DOI] [PubMed] [Google Scholar]
- 19.Wong HL, Rauth AM, Bendayan R, Manias JL, Ramaswamy M, Liu Z, Erhan SZ, Wu XY. A New Polymer-Lipid Hybrid Nanoparticle System Increases Cytotoxicity of Doxorubicin against Multidrug-Resistant Human Breast Cancer Cells. Pharm Res. 2006;23(7):1574–1585. doi: 10.1007/s11095-006-0282-x. [DOI] [PubMed] [Google Scholar]
- 20.Johnson BK, Prud’homme RK. Mechanism for Rapid Self-Assembly of Block Copolymer Nanoparticles. Phys Rev Lett. 2003;91(11):118302. doi: 10.1103/PhysRevLett.91.118302. [DOI] [PubMed] [Google Scholar]
- 21.DeMello AJ. Control and Detection of Chemical Reactions in Microfluidic Systems. Nature. 2006;442(7101):394–402. doi: 10.1038/nature05062. [DOI] [PubMed] [Google Scholar]
- 22.Hong CC, Choi JW, Ahn CH. A Novel in-Plane Passive Microfluidic Mixer with Modified Tesla Structures. Lab Chip. 2004;4(2):109–113. doi: 10.1039/b305892a. [DOI] [PubMed] [Google Scholar]
- 23.Salvador-Morales C, Zhang L, Langer R, Farokhzad OC. Immunocompatibility Properties of Lipid-Polymer Hybrid Nanoparticles with Heterogeneous Surface Functional Groups. Biomaterials. 2009;30(12):2231–2240. doi: 10.1016/j.biomaterials.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Asgar A, Bhagat S, Papautsky I. Enhancing Particle Dispersion in a Passive Planar Micromixer Using Rectangular Obstacles. Journal of Micromechanics and Microengineering. 2008;18(8) [Google Scholar]
- 25.Rhee M, Burns MA. Drop Mixing in a MicroChannel for Lab-on-a-Chip Platforms. Langmuir. 2008;24(2):590–601. doi: 10.1021/la702575j. [DOI] [PubMed] [Google Scholar]
- 26.Michalet X, Pinaud FF, Bentolila LA, Tsay JM, Doose S, Li JJ, Sundaresan G, Wu AM, Gambhir SS, Weiss S. Quantum Dots for Live Cells in Vivo Imaging and Diagnostics. Science. 2005;307(5709):538–544. doi: 10.1126/science.1104274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klostranec JM, Chan WCW. Quantum Dots in Biological and Biomedical Research: Recent Progress and Present Challenges. Adv. Mater. 2006;18:1953–1964. [Google Scholar]
- 28.Carion O, Mahler B, Pons T, Dubertret B, Synthesis Encapsulation. Purification and Coupling of Single Quantum Dots in Phospholipid Micelles for Their Use in Cellular and in Vivo Imaging. Nat Protoc. 2007;2(10):2383–2390. doi: 10.1038/nprot.2007.351. [DOI] [PubMed] [Google Scholar]
- 29.Dubertret B, Skourides P, Norris DJ, Noireaux V, Brivanlou AH, Libchaber A. In Vivo Imaging of Quantum Dots Encapsulated in Phospholipid Micelles. Science. 2002;298(5599):1759–1762. doi: 10.1126/science.1077194. [DOI] [PubMed] [Google Scholar]
- 30.Park JH, von Maltzahn G, Ruoslahti E, Bhatia SN, Sailor MJ. Micellar Hybrid Nanoparticles for Simultaneous Magnetofluorescent Imaging and Drug Delivery. Angew Chem Int Ed Engl. 2008;47(38):7284–7288. doi: 10.1002/anie.200801810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gindy ME, Panagiotopoulos AZ, Prud'homme RK. Composite Block Copolymer Stabilized Nanoparticles: Simultaneous Encapsulation of Organic Actives and Inorganic Nanostructures. Langmuir. 2008;24(1):83–90. doi: 10.1021/la702902b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.