

Abstract
Tumor heterogeneity is one of the most important and challenging problems not only in studying the mechanisms of cancer development but also in developing therapeutics to eradicate cancer cells. Here we report the use of multiplexed quantum dots (QDs) and wavelength-resolved spectral imaging for molecular mapping of tumor heterogeneity on human prostate cancer tissue specimens. By using a panel of just four protein biomarkers (E-cadherin, high-molecular-weight cytokeratin, p63, and α-methylacyl CoA racemase), we show that structurally distinct prostate glands and single cancer cells can be detected and characterized within the complex microenvironments of radical prostatectomy and needle biopsy tissue specimens. The results reveal extensive tumor heterogeneity at the molecular, cellular, and architectural levels, allowing direct visualization of human prostate glands undergoing structural transitions from a double layer of basal and luminal cells to a single layer of malignant cells. For clinical diagnostic applications, multiplexed QD mapping provides correlated molecular and morphological information that is not available from traditional tissue staining and molecular profiling methods.
Keywords: quantum dot, tumor heterogeneity, prostate cancer, multiplexing, spectral imaging, biomarker, immunohistochemistry
Semiconductor quantum dots (QDs) have unique optical and electronic properties such as size-tunable light emission, superior signal brightness, resistance to photobleaching, and simultaneous excitation of multiple fluorescence colors.1 – 4 These properties are most promising for improving the sensitivity and multiplexing capabilities of molecular histopathology and disease diagnosis.5 – 7 In contrast to in vivo imaging applications, where the potential toxicity of cadmium-containing QDs is a major concern,4 immunohistological staining is performed on in vitro clinical patient samples. As a result, the use of multicolor QD probes in immunohistochemistry (IHC) is likely one of the most important and clinically relevant applications in the near term.8 – 16 In particular, the multiplexing capability of QDs is well-suited for investigating tumor heterogeneity and complexity, one of the most important and challenging problems in studying the mechanisms of cancer development and also in developing therapeutics to eradicate cancer cells.16 – 18
Human cancer is especially complex because it evolves over a long time course and shows a multitude of molecular, cellular, and architectural heterogeneity.18 At the molecular level, cancer cells are heterogeneous both in their genetic mutations and in their phenotypic expression profiles. At the cellular level, malignant tumors are characterized by a complicated mix of benign cells, malignant cells, fibroblasts, and other stromal cells, vascular cells, and infiltrating inflammatory cells (such as macrophages and lymphocytes). Also, a small number of stem cells and progenitor cells are believed to be embedded in the perivascular region and could be responsible for tumor growth and recurrence.19 – 21 At the architectural level, normal and cancer cells are often arranged into distinct structures (such as glands in prostate, ducts in breast, and crypts in colon), and multiple lesions of varying grades or malignancy are commonly found within the same tumor. This level of complexity represents an extraordinary challenge to the cancer research community because most experimental data are obtained by ensemble averaging over heterogeneous cell populations.
To address these complexity and heterogeneity problems, Liotta and co-workers22,23 have developed laser capture microdissection (LCM) to capture or “punch out” morphologically distinct cells from tissue specimens, followed by real-time polymerase chain reaction (RT-PCR) or mass spectrometric analysis. Also promising is a technology developed by Rimm and co-workers24,25 that combines immunofluorescence staining and automated quantitative image analysis (called AQUA). In comparison with adsorption-based immunohistochemical methods, fluorescence imaging provides a number of advantages such as higher detection sensitivity, wider signal dynamic ranges, and more linear relationships for biomarker quantification and prediction of therapeutic response. Indeed, recent advances have shown that fluorescence-based analysis of protein biomarkers is strongly correlated with clinical outcome.24 – 27 However, the use of organic dyes for multicolor fluorescence measurement is often limited by photobleaching, low signal intensity (low brightness), spectral overlapping, and the need for multiple light sources to excite different fluorophores.
In this work, we report the use of multiplexed QD – antibody conjugates and wavelength-resolved fluorescence imaging (spectral imaging)28,29 to detect a panel of protein biomarkers directly on human tissue specimens. We show that QD-based spectral imaging can be utilized for high-throughput digital mapping of molecular, cellular, and glandular variations on surgical prostate cancer specimens. Without physically removing any cells from heterogeneous tissue sections, this nanotechnology approach allows the molecular profiles and morphological features to be “digitally” extracted from individual cells, cellular clusters, glands, and complex histopathological loci. By using just four protein biomarkers (E-cadherin, high-molecular-weight cytokeratin, p63, and α-methylacyl CoA racemase), we demonstrate that a single malignant tumor cell can be detected and identified from the complex tissue microenvironment encountered in formalin-fixed paraffin-embedded (FFPE) histologic materials obtained from radical prostatectomy and needle biopsy specimens. The results reveal that complex architectural changes are associated with cancer development and progression, including prostate glands undergoing structural transitions from a double layer of basal and luminal cells to a single layer of malignant cells. As discussed below, multiplexed QD mapping provides new molecular and morphological information that is not available from traditional H&E (hematoxylin and eosin) and immunohistochemical methods, especially at complex and suspicious disease loci.
Results
Multiplexed QD Staining
We have developed and optimized a sequential staining method in which primary and secondary antibodies from two animal species (e.g., primary rabbit and mouse antibodies, and secondary anti-rabbit and anti-mouse antibodies) are repeatedly used to build up the degrees of QD multiplexing (see Figure 1). An important finding is that an excess of QDs conjugated with secondary antibodies can be used to deplete the antigenic sites on primary antibodies when the binding mixture is incubated for an extended period of time (2 – 3 h). Under these conditions, the percentage of “empty” epitopes on the primary antibody is largely determined by the equilibrium constant of primary antibody and QD – secondary antibody binding, thereby avoiding the complications of time-dependent binding kinetics. As a result, during the second round of staining, QD binding takes place mainly at the new primary antibody sites (introduced during the second round). A major advantage of this sequential staining approach is that the use of common antibodies and QD conjugates from just two animal species is well-suited for scale-up applications at low costs and for improving the consistency and reproducibility of repeated staining (discussed in more detail below). A potential problem, however, is that the secondary antibody could dissociate from the primary antibody, and that the primary antibody itself could dissociate from the tissue antigen. Both of these processes could occur during the washing steps as well as during the second round of staining. The release of QD probes from the primary antibody could cause “QD crossover” because the QDs used in the second round of staining can bind to the same primary antibody sites that become available by dissociation. Thus, the second QD probe could “colocalize” with the first QD probe if both of them bind to the same primary antibody sites. A simple strategy to overcome this problem is to cross-link the QD – antibody – antigen complex by using common tissue fixatives such as formaldehyde, glutaraldehyde, or ethyl dimethylaminopropyl carbodiimide (EDC), a procedure that has been used to prevent the loss of microRNA during in situ hybridization.30
Figure 1.
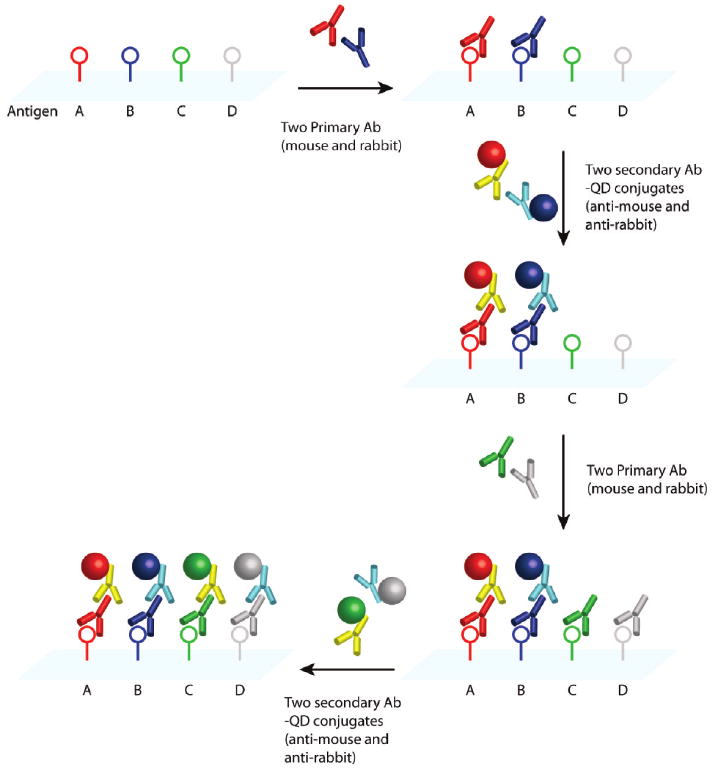
Schematic illustration of sequential QD staining in which two primary antibodies from two animal species are used to recognize two tissue antigens. A mixture of two primary antibodies from two species (e.g., rabbit and mouse) is first used to recognize two antigens (A and B) in the tissue section (first arrow, step 1). After washing, a mixture of two secondary antibody QD conjugates (e.g., goat anti-rabbit QD655 and goat anti-mouse QD605) is applied to stain the two primary antibodies (second arrow, step 2). The same procedure is repeated by using primary antibodies for two additional antigens (third arrow, step 3) followed by the use of two secondary antibody QD conjugates (e.g., goat anti-rabbit QD565 and goat anti-mouse QD705 or QD525) (fourth arrow, step 4).
We have experimentally evaluated the performance of sequential QD staining by using two protein biomarkers (p63 and HMW cytokeratin) that do not overlap in their cellular locations (that is, with distinct distribution patterns).31 – 34 The rationale is that after two rounds of QD – antibody staining, the different QD signals (different colors) should be spatially separated if QD crossing-over and nonspecific binding do not take place during the staining process. Figure 2 shows the color-separated images of single cells that are extracted from human tissue specimens stained sequentially with red and green QD conjugates. High-molecular-weight cytokeratin (CK HMW) is predominantly associated with the cytoplasm and its membrane, while the protein biomarker p63 is exclusively localized in the cell nuclei. Quantitative image analysis indicates that the nonspecific CK signal (detected in the nucleus) is only 5% of its membrane/cytoplasmic signal. Similarly, the nonspecific QD signal for the nuclear antigen p63 at the membrane is about 10% of its specific binding signal in the nucleus, a level low enough for detecting distinct multicolor QD staining patterns on intact tissue specimens. It should be noted, however, that accurate biomarker quantification will require calibration data to normalize the brightness levels of different-colored QDs and the use of housekeeping gene products as internal controls for “on-tissue correction” of QD staining efficiencies.5
Figure 2.
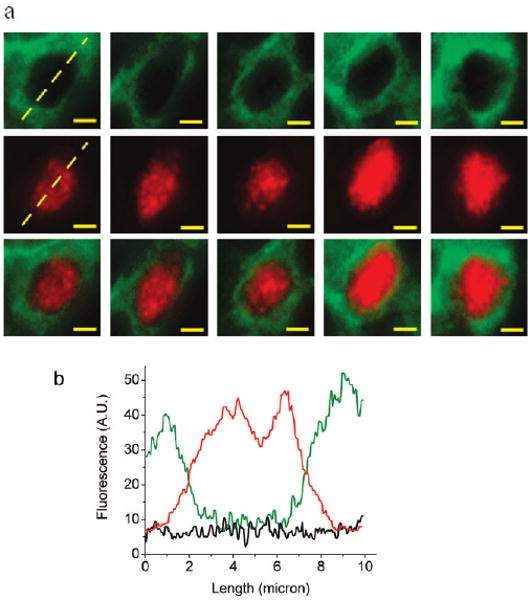
Fluorescence images and quantitative biomarker analysis of single prostate cells extracted from human tissue specimens. The tissue sections were stained by using two primary antibodies and two secondary antibody QD conjugates in a sequential manner for two tissue antigens (p63 and cytokeratin HMW) with distinct cellular localization patterns. The nuclear biomarker p63 (red) was stained first, followed by QD staining of the membrane/cytoplasmic biomarker cytokeratin HMW (green). (a) Deconvolved images of single basal cells in a benign prostate gland showing the membrane/cytoplasmic distribution of cytokeratin HMW (top panel), the nuclear distribution of p63 (middle panel), and their composite images showing minimal spatial overlap (bottom panel). (b) Line plots of QD staining fluorescence intensities for cytokeratin HWM (green), p63 (red), and background (black); see the dashed lines in panel a. Scale bar = 2 μm.
We have used spectral imaging to measure multiple molecular targets based on small but meaningful spectral differences (see Figure 3). We find that there are major differences between cells and tissues including the following: (i) the level of autofluorescence is much higher and more complex with fixed tissues than with cancer cells; (ii) cells in tissue sections are rarely intact, making whole cell quantification difficult; (iii) there is considerable variability in tissue sample source, preservation, and processing. These factors make biomarker quantification more difficult on tissue samples, but they do not alter the QD staining and spectral imaging patterns.
Figure 3.
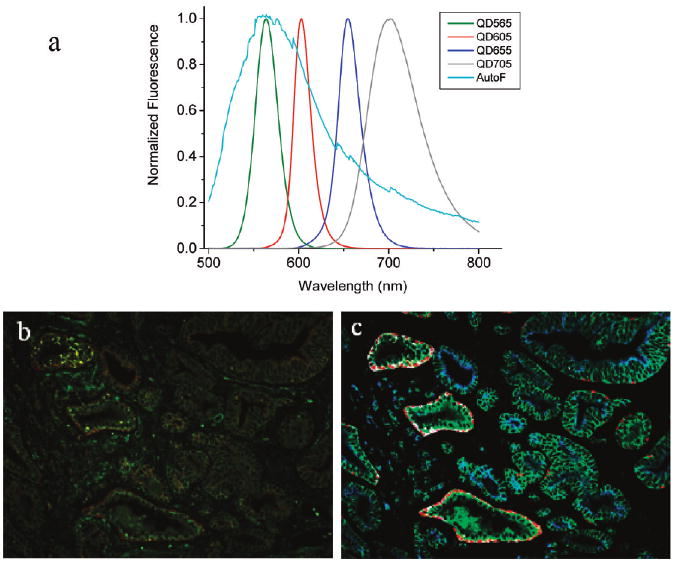
(a) QD emission spectra and tissue autofluorescence data used for color coding and spectral deconvolution. (b) Raw and (c) processed fluorescence images of prostate tissue specimens after multiplexed QD immunostaining for four protein biomarkers. Note that spectral imaging allows color-encoded biomarkers to be extracted and highlighted in the deconvolved image. The protein biomarkers are E-cadherin (green), cytokeratin HMW (white), p63 (red), and AMACR (blue), as shown in encoded pseudocolors in image c.
Reproducibility of Multiplexed QD Staining
To experimentally evaluate the consistency and reproducibility of multiplexed QD staining, we have used four QD – antibody conjugates with fluorescence emission at 525 (or 705), 565, 605, and 655 nm to measure four protein biomarkers on adjacent tissue slices of human prostate cancer (removed by prostatectomy). These consecutively cut tissue sections show similar cellular contents and architectural features and are well-suited for a direct comparison of single-color and multiplexed QD staining results from one tissue slice to another. The results demonstrate that the multiplexed QD images are not only highly reproducible but also strongly correlated with both IHC and H&E staining (Figure 4 and Supporting Information Figure S1). Furthermore, the QD staining data provide additional molecular and cellular information for differentiating benign glands, prostatic intraepithelial neoplasm (PIN), and malignant tumor glands, as annotated in Figure 4. More quantitative measurement further reveals that the QD signal variations are limited to about 10% for adjacent tissue sections (Supporting Information Figure S2).
Figure 4.
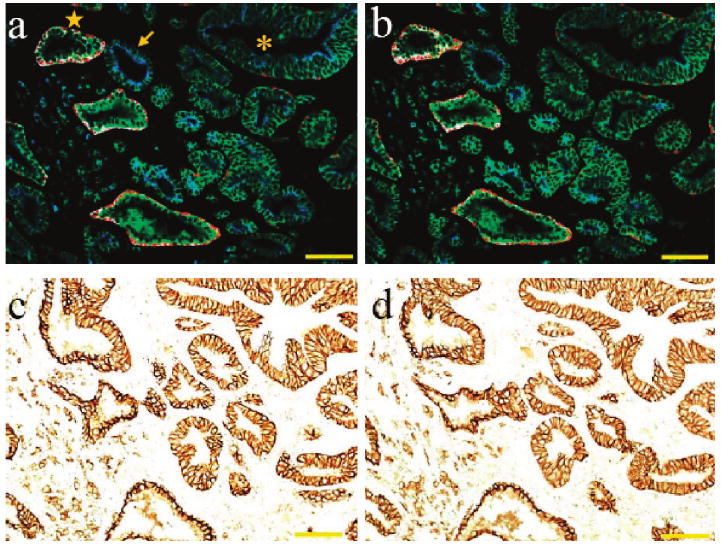
Four biomarker multiplexed QD staining images (a,b) and single biomarker immunohistochemical staining images (c,d) of adjacent prostate cancer tissue sections. The patient was a 68 year old Caucasian male who underwent radical prostatectomy. The remarkable similarities in staining patterns and intensities indicate that the multiplexed QD staining images are highly consistent and reproducible and also are strongly correlated with IHC and H&E stained images (see Supporting Information Figure S1). The protein biomarkers in panels a and b are E-cadherin (green), CK HMW (white), p63 (red), and AMACR (blue). The single biomarker in panels c and d is E-cadherin. In image a, normal prostate glands are annotated with a star symbol; malignant glands with an arrow; glands showing features of prostatic intraepithelial neoplasia (PIN) are annotated with an asterisk. Objective = 20×. Scale bar = 100 μm.
This mapping and visualization of heterogeneous cells and glands on human prostatectomy specimens is achieved by using a panel of just four protein biomarkers, selected based on their cellular localization patterns or their abilities to differentiate the major types of benign and malignant prostate cells (see Table 1). The first biomarker is E-cadherin, a transmembrane protein involved in cell – cell adhesion, tumor invasion, and epithelial-to-mesenchymal transition (EMT) and is present in both normal prostate cells (basal and luminal) and malignant tumor cells.35 The second biomarker is high-molecular-weight cytokeratin (CK HMW), a protein that is present in the cytoplasm and membrane of benign prostate basal cells but is absent in malignant prostatic epithelium.33,34 The third marker is p63, a homologue of the cancer suppressor protein p53 and is present only in the nuclei of normal basal cells.31,32 Finally, we selected the biomarker p504s (α-methylacyl CoA racemase or AMACR), which is an enzyme involved in fatty acid oxidation and is present in the cytoplasm of premalignant and malignant tumor cells but is absent in benign and normal cells.36 Significantly, AMACR is a marker that has been found in many types of human cancer including colorectal, prostatic, ovarian, breast, bladder, lung and renal cell carcinomas, lymphoma, and melanoma.37 It is detectable in up to 90–95% of both prostatic intraepithelial neoplasm36 and invasive prostatic carcinoma but is not found in prostate glands with benign prostatic hyperplasia (BPH).37,38
TABLE 1. Summary of Human Prostate Protein Biomarkers, Their Cellular Localization, and Expression Patterns for QD Mapping of Tumor Heterogeneity in Prostatectomy Tissue Specimens.
| cell type | biomarkers | |||
|---|---|---|---|---|
| P504s (AMACR) | p63 | CK HMW | E-cadherin | |
| normal basal | − | + | + | + |
| normal luminal | − | − | − | + |
| malignant tumor | + | − | − | + |
| cellular location | cytoplasmic | nuclear | membrane and cytoplasmic | membrane |
| coding color | blue | red | white | green |
Molecular Mapping of Benign and Malignant Cells
Figure 5 shows quantitative biomarker data for the three major types of cells found in human prostate cancer specimens. We note that the brightness of QDs is determined by both their fluorescence quantum yields (QE) and their absorption coefficients and shows considerable variations from one color to another, especially when the fluorescence emission color is tuned by changing the QD size.5 However, same-color signal intensities can be compared to estimate the relative expression levels of a given protein biomarker in different cell populations. Indeed, the multiplexed QD mapping data are consistent with that of previous research.31 – 33,36 – 38 showing that malignant tumor cells have greatly overexpressed AMACR levels (by 4 – 5-fold), reduced CK expression, and nearly zero p63 expression. Using three-dimensional (3D) scatter plots, it is clear that the major types of prostate cells can be separated with very high precision (Figure 5b). The E-cadherin is not used in these plots because it is expressed in all three types of cells and is less informative for cellular differentiation.
Figure 5.
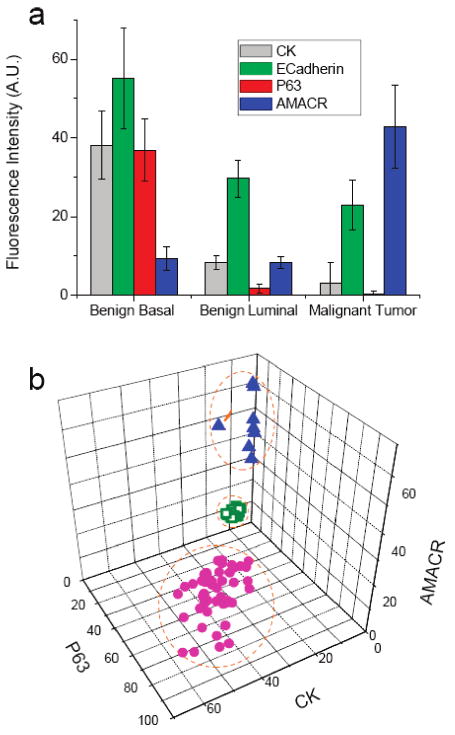
Quantitative biomarker data for three major cell types “digitally” extracted from multiplexed QD staining images of human prostate cancer specimens. (a) Profiles of four biomarkers (CK HMW, E-cadherin, p63, and AMACR) for benign basal cells, benign luminal cells, and malignant tumor cells. (b) Scatter plots of three biomarkers (CK HMW, p63, and AMACR) showing that the major types of prostate cells can be separated with nearly 100% precision. Purple dots, benign basal cells; green dots, benign luminal cells; and blue dots, malignant tumor cells. The protein marker E-cadherin is not used in these plots because it is expressed in all three types of prostate cells.
To examine how a benign prostate gland is transformed into a malignant one, we have observed early events involving only a single cell in the complex environment of surgical tissue specimens. Figure 6 demonstrates the detection of a single cell with a molecular phenotype suggestive of neoplasia, surrounded by an otherwise benign prostate gland. This cell is most likely malignant because of its strong AMACR staining and the concomitant absence of p63 staining in the nucleus. The possibility of this abnormal cell being an artifact can be ruled out because the adjacent tissue slice shows similar staining at exactly the same location (Figure 6d). The AMACR staining signal is not caused by crossover or non-specific QD binding because this antigen was stained in the first step and should not suffer from subsequent crossover or nonspecific binding problems. The blue QDs bound to the AMACR antigen, however, could lose intensity during additional washing and staining steps, but the detection of intense blue signals indicates that this is not a major concern.
Figure 6.
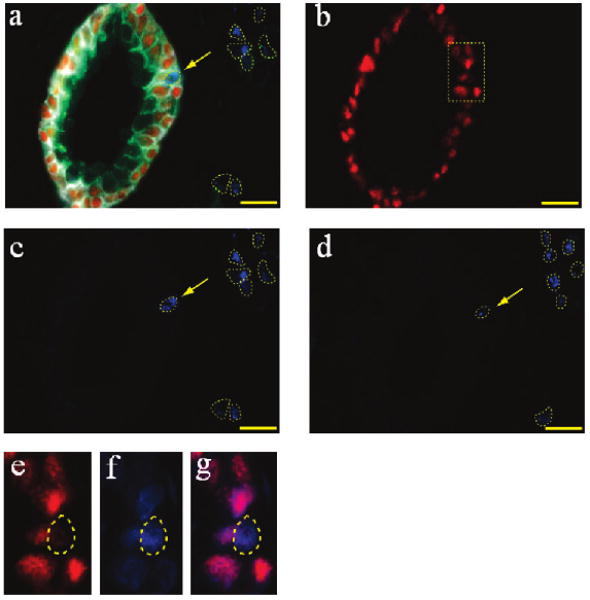
Identification of single malignant tumor cells in a predominantly benign prostate gland by QD multiplexed staining of four protein biomarkers (E-cadherin, green; CK HMW, white; p63, red; and AMACR, blue). (a) Composite image showing the distinct staining patterns of a largely benign gland (central) and surrounding malignant cells (blue signals in dotted circles). A single malignant tumor cell in the basal gland layer is indicated with an arrow. (b) Deconvolved image showing the distribution of p63-positive benign basal cells. (c,d) Deconvolved images showing the distribution of AMACR-positive malignant tumor cells in two adjacent tissues. The staining similarities provide strong evidence that the malignant cells identified in panel a are indeed cancer and are not experimental artifacts. (e–g) Zoomed-in views of the boxed area in panel b showing that the single malignant cells identified by AMACR staining are negative in p63 staining, as expected for truly malignant cells. A cell is, in fact, present at that location, as shown by the DAPI nuclear staining (also in blue) (f), and by the composite p63 and DAPI image (g). Objective = 100×. Scale bar = 20 μm.
A collection of heterogeneous glandular architectures is depicted in Figure 7. These structures range from completely benign (judged by their characteristic basal – luminal cell layers and absent AMACR staining; see Figure 7a) to completely malignant (judged by intense AMACR staining and absent basal cell CK and p63 signals; see Figure 7f). Between there are complex and heterogeneous structures undergoing benign-to-malignant transition at various stages. A surprise finding is that a single prostate gland can contain both a malignant segment (yellow arrows in Figure 7c) and a benign segment (intense red p63 staining), as well as intermediate structures (low AMACR and absent p63 staining). In addition, a benign gland can be situated next to a malignant gland (Figure 7d) (separated by only 5 – 10 μm). In a complex “folded” prostate gland, one portion can have malignant features (Figure 7e, yellow arrows) while the rest is largely benign. These results underscore the pervasive nature of tumor heterogeneity at the cellular and glandular levels.
Figure 7.
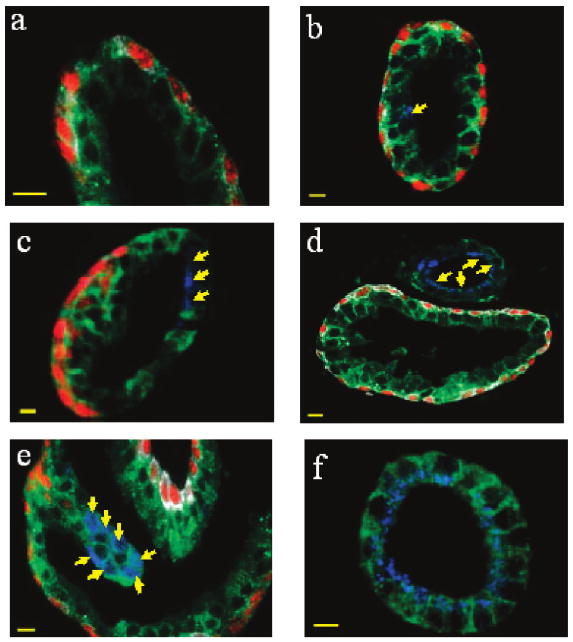
Multiplexed QD staining fluorescence images obtained from human prostatectomy specimens highlighting cellular and glandular heterogeneity. (a) Benign prostate gland, as judged by characteristic basal – luminal cell layers and the absence of AMACR staining. (b) Largely benign prostate gland with a single malignant or premalignant cell in the luminal layer, as judged by positive AMACR staining (see arrow). (c) Prostate gland with a malignant segment (yellow arrows) and a benign segment (intense red p63 staining), as well as intermediate structures (low AMACR and absent p63 staining). (d) Malignant gland (yellow arrows) sitting next to a benign gland (separated by only 5 – 10 μm). (e) Complex “folded” prostate gland in which one portion is malignant (yellow arrows) while the rest is largely benign. (f) Completely malignant gland, as judged by intense AMACR staining and absent basal cell CK and p63 signals. Scale bar = 10 μm.
Complex Histopathological Loci
For clinical diagnostic applications, Figure 8 shows traditional H&E staining and multiplexed QD mapping images of two “suspicious foci”. Each pair of H&E and QD images was obtained from two adjacent tissue sections of human prostate cancer, so that the identical regions of interest can be compared directly. The results indicate that multiplexed QD mapping provides valuable morphological information (similar to H&E) and also yields new molecular and cellular information. For the structurally complex loci in Figure 8, the QD data reveal that both of them contain malignant glands, whereas the first one (Figure 8a) contains more malignant cells (AMACR positive) than the second one (Figure 8b). This level of molecular and cellular mapping is not possible with standard H&E or single-color IHC. Previous studies have shown that many biopsy pathology reports contain a suspicious diagnosis of atypical foci.39,40 Conventional H&E and IHC techniques cannot make a clear conclusion on these specimens. The patients in this category have a risk of approximately 40% carcinoma occurrence.39 The ability of multiplexing by QDs allows a better chance to differentiate these specimens with suspicious foci, but more systematic clinical studies are needed to determine whether the QD mapping data are correlated with cancer behavior and patient outcome.
Figure 8.
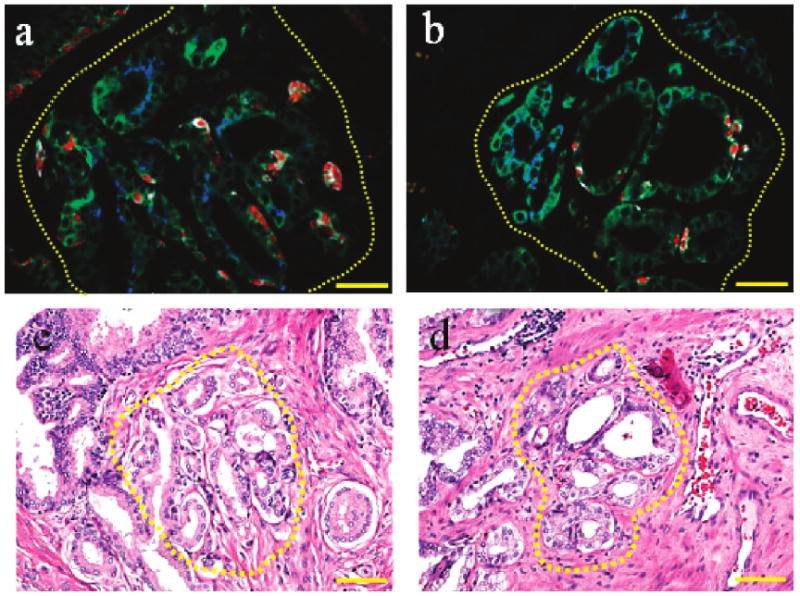
Comparison of multiplexed QD mapping (a,b) and traditional H&E staining images (c,d) for two histopathologically complex foci on adjacent prostate cancer tissue sections. The dashed circles in images a and c correspond to one complex region, and the dashed circles in images b and d correspond to another complex region. In panels a and b, objective = 40×, scale bar = 50 μm. In panels c and d, objective = 20×; scale bar = 100 μm.
Discussion
The glandular architecture of the prostate gland is complex, yet all benign glands share certain essential characteristics, chief among them being a two-cell layer configuration with both basal and luminal epithelial cells.41 A histopathological hallmark of malignant prostate glands is the loss of the basal cell layer. While high-grade prostatic intraepithelial neoplasia (HG-PIN) has been widely accepted as a premalignant lesion,42 it is still unclear how this physical rearrangement of the gland is associated with malignant transformation. One of the obvious difficulties in placing HG-PIN in the transition from normal gland to malignant gland is that HG-PIN is characterized by multiple layers of luminal epithelial cells (see the gland marked with an asterisk in Figure 4a), and that cancer is characterized by a single layer of truly malignant cells (see the gland marked by an arrow in Figure 4a). Little is known about how this dramatic architectural change takes place during the growth of prostate cancer. Our QD mapping data indicate that the architectural changes start with a single malignant cell in the basal or luminal layer (see Figures 6a and 7b), go through many intermediate step (in which a single gland could contain both a malignant region and a benign region), and end with completely malignant structures showing a single luminal cell layer.
Multiplexed QD mapping images represent a “snapshot” of heterogeneous molecular and cellular events that are captured on surgical or needle biopsy tissue specimens at a single time point. This snapshot is likely to reflect (but does not fully represent) the chronology of benign-to-malignant transition that takes place in human patients over several years. It is enticing to use multiplexed QD mapping for “reconstructing” human cancer events by a retrospective deduction process. Also, it will be interesting to use QD mapping to search for rare cell types such as cancer stem cells on human tissue specimens, which are believed to be responsible for tumor reoccurrence after therapies.19 – 21
If cancer could be reliably identified at the earliest possible stage of cellular transition, then novel therapeutics could be designed to arrest the process of malignant transformation. If the histological appearance of these cells in transition could be certain, then the response to such novel therapies could be followed by the changing appearance of prostate histology over time while a patient is enrolled in a clinical trial designed to arrest the process. The accurate identification of cells that under standard light microscopy appear benign but by protein marker analysis are malignant could greatly assist in identifying the earliest molecular alterations accompanying malignant transformation. Current diagnostic and prognostic classifications, based on clinical and pathologic factors, are insufficient to reflect the whole clinical heterogeneity of tumors and are unable to predict successful treatment and outcome at the time of diagnosis.43 Thus, the ability to correlate glandular architectures with molecular biomarkers should provide new diagnostic and prognostic insights that are not available from either morphology or molecular analysis alone.
A deadly step in prostate cancer is the appearance of so-called “lethal phenotypes” that are bone metastatic, hormone independent, and chemotherapy and radiation refractory.44,45 It has been hypothesized that each of these aggressive behaviors or phenotypes could be understood and predicted by a defining set of biomarkers. By critically defining the interrelationships among these biomarkers, it could be possible to diagnose and prognosticate cancer based on a patient's molecular profile, leading to personalized and predictive oncology. That is, a unique molecular profile can be used to predict the tumor's invasive and metastatic potential, its ability to survive and grow under androgen-deprived and hypoxia and metabolic stress conditions, and the potential of certain cancer cells to evade host immune surveillance. In this work, we have carried out multiplexed QD mapping studies on clinical tissue specimens from 16 prostate cancer patients, and the results have revealed extensive tumor heterogeneity in protein biomarkers, in benign and malignant cells, as well as in glandular architectures. The results clearly demonstrate that multiplexed QD staining is sufficiently consistent and reproducible for clinical translation, but the patient number is too small to yield any significant correlation of molecular and architectural features with clinical outcomes. The next step is to conduct large-scale clinical studies in order to establish the protocols and practices for QD-based molecular pathology.
In summary, we have developed a high-throughput digital mapping method based on the use of multiplexed quantum dot (QD) – antibody conjugates and wavelength-resolved fluorescence imaging for mapping the molecular, cellular, and glandular heterogeneity of human prostate cancer specimens. This method allows molecular information and morphological features to be “digitally” extracted from individual cells, cellular clusters, glands, and complex histopathological foci. By using just four protein biomarkers, we have shown that a single malignant tumor cell can be detected and identified from the complex tissue microenvironment. A fundamental finding is that the architectural changes of prostate glands start with a single malignant cell in the luminal or basal layer, go through many intermediate steps involving partially benign glands, and finally lead to completely malignant glands with a single luminal cell layer. For clinical diagnostic applications, multiplexed QD mapping can provide new molecular and morphological information that is not available from traditional H&E and IHC, especially at complex and suspicious disease foci. These results have raised exciting possibilities in integrating morphological and molecular biomarker information for cancer diagnosis and treatment selection.
Methods
Human Prostate Cancer Tissue Specimens
Formalin-fixed paraffin-embedded (FFPE) human prostatectomy or needle biopsy specimens were obtained from the Veteran Affairs Medical Center in Atlanta, Georgia. Tissue sections (5 μm thickness) were preheated at 60 – 65 °C for 15 min and were then deparaffinized by immersion in xylene three times for 3, 1, and 1 min, respectively. Tissue hydration was carried out by a series of immersion steps at decreasing ethanol concentrations (100, 95, 80, and 70% ethanol, twice and 2 min each), followed by rinsing in water for 5 min. Antigen retrieval was performed by using a decloaking chamber at 125 °C for 30 s, then at 90 °C for 10 s in standard decloaking buffers (Biocare Medical). After cooling in the decloaking buffer for another 20 min, the tissue slides were washed by water and were stored in 1× PBS plus buffer (containing 0.05% Tween 20) until use.
Multiplexed QD Staining
Multiplexed QD staining was carried out by using a robotic and automated system (Nemesis 7200, Biocare Medical) for high throughput and reproducibility. The use of such an automated staining instrument was found to reduce slide-to-slide variations. The preprogrammed procedure started with blocking the slide surfaces by a mixture of 2% BSA, 5% goat serum, and 1× PBS for 30 min at room temperature. The blocked tissue slides were incubated with a mixture of two primary antibodies at room temperature for 1 h. These two primary antibodies were from two animal species used to recognize two tissue antigens: antigen p504s by a rabbit polyclonal antibody (CP200AK, 1:50 dilution, Biocare Medical) and antigen p63 by a mouse monoclonal antibody (CM163A, 1:50 dilution, Biocare Medical). After washing with 1× PBS plus buffer twice, a mixture of two secondary antibody QD conjugates (goat anti-rabbit QD655 and goat anti-mouse QD605, Invitrogen) was applied to the slides and incubated for 2 h at room temperature. After washing with PBS plus buffer three times (5 min each), the same staining protocols were used for more tissue antigens coupled with more QD colors. Briefly, two additional antigens (E-cadherin and cytokeratin HMW) were stained by using two primary antibodies (E-cadherin by rabbit polyclonal antibody H108, 1:50 dilution from Santa Cruz Biotechnology, and cytokeratin HMW by mouse monoclonal antibody CM127A, 1:50 dilution from Biocare Medical), and two secondary antibody QD conjugates (goat anti-rabbit QD565 and goat anti-mouse QD705 or QD 525, supplied by Invitrogen). The overall staining procedure was finished by DAPI counterstaining, dehydration, and mounting on coverslips for fluorescence imaging.
Multispectral Imaging
Wavelength-resolved fluorescence imaging was achieved by using a multispectral imaging system (Nuance, CRI, Woburn, MA) attached to an inverted fluorescence microscope (Olympus 1×71). With near-UV lamp excitation at 350 – 360 nm and a long-pass dielectric filter (cut-on wavelength 500 nm), a wavelength-resolved stack (λ-stack) of fluorescence images (called an image cube) was acquired across the spectral range of 500 – 800 nm at 10 nm increments, yielding a total of 30 images in the λ-stack. With a data integration time of 50 ms for each wavelength, the complete image stack was finished in less than a few seconds (plus time for data storage and processing). A library of five spectra (four for QDs and one for tissue autofluorescence) was also acquired from standard antibody – QD samples spotted on glass slides and from the control tissue sections (going through the same staining procedure without QDs). This collection of spectral components was used for spectral deconvolution (spectral unmixing).
Image Analysis and Biomarker Quantification
Deconvolution algorithms were applied image each image cube, generating a set of “single-color” images representing each individual QD/biomarker and the tissue autofluorescence. The colors of QDs were reassigned to pseudocolors for graphic visualization or emphasis. A computer-aided threshold was determined from the experimental data and was used for background subtraction. Feature extraction and pattern recognition algorithms were used to identify areas of interest such as single cells, cellular clusters, and whole glands. Biomarker expressions in these identified areas were quantified by simple pixel-based intensity measurement.
Traditional Immunohistochemistry
Consecutive slides from the same tissue block were stained by using standard DAB chromagen immunohistochemistry. One protein biomarker was stained on one tissue section. Briefly, after tissue pretreatment as described above and subsequent endogenous enzyme blocking for 15 min, the slides were loaded into the same autostaining machines. The MACH-4 detection system (Biocare Medical) was used for enhanced sensitivity. The routine counterstaining step with hematoxylin was omitted so that monocolor images from control slides could be used for comparison. Images of IHC-stained tissues were acquired by using a color CCD camera attached to an inverted Olympus microscope.
Supplementary Material
Acknowledgments
This work was supported by grants from the NCI Centers of Cancer Nanotechnology Excellence (CCNE) Program (U54CA119338) and the Bioengineering Research Partnerships Program (BRP) (R01CA108468). We are grateful to Dr. Haiyen Zhau, Dr. Leland Chung, Ms. Yachna Sharma, Mr. Will Hudson, and Mr. Qaiser Chaudry for helpful discussions. M.D.W., B.L.J., and S.M.N. are Distinguished Cancer Scholars of the Georgia Cancer Coalition (GCC).
Footnotes
Supporting Information Available: Supporting Figures S1 and S2 showing traditional H&E staining data, comparison of single-color and multiplexed QD staining, and biomarker quantification data. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.Alivisatos AP. The Use of Nanocrystals in Biological Detection. Nat Biotechnol. 2004;22:47–52. doi: 10.1038/nbt927. [DOI] [PubMed] [Google Scholar]
- 2.Michalet X, Pinaud FF, Bentolila LA, Tsay JM, Doose S, Li JJ, Sundaresan G, Wu AM, Gambhir SS, Weiss S. Quantum Dots for Live Cells, In Vivo Imaging, and Diagnostics. Science. 2005;307:538–544. doi: 10.1126/science.1104274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gao XH, Yang L, Petras JA, Marshall FF, Simons JW, Nie SM. In Vivo Molecular and Cellular Imaging with Quantum Dots. Curr Opin Biotechnol. 2005;16:63–72. doi: 10.1016/j.copbio.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 4.Smith AM, Duan H, Mohs AM, Nie SM. Bioconjugated Quantum Dots for In Vivo Molecular and Cellular Imaging. Adv Drug Delivery Rev. 2008;60:1226–1240. doi: 10.1016/j.addr.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xing Y, Chaudry Q, Shen C, Kong KY, Zhau HE, Chung LW, Petros JA, O'Regan RM, Yezhelyev MV, Simons JW, et al. Bioconjugated Quantum Dots for Multiplexed and Quantitative Immunohistochemistry. Nat Protoc. 2007;2:1152–1165. doi: 10.1038/nprot.2007.107. [DOI] [PubMed] [Google Scholar]
- 6.Yezhelyev MV, Al-Hajj A, Morris C, Marcus AI, Liu T, Lewis M, Cohen C, Zrazhevskiy P, Simons JW, Rogatko A, et al. In Situ Molecular Profiling of Breast Cancer Biomarkers With Multicolor Quantum Dots. Adv Mater. 2007;19:3146–3151. [Google Scholar]
- 7.Smith AM, Dave S, Nie SM, True L, Gao XH. Multicolor Quantum Dots for Molecular Diagnostics of Cancer. Expert Rev Mol Diagn. 2006;6:231–244. doi: 10.1586/14737159.6.2.231. [DOI] [PubMed] [Google Scholar]
- 8.Ghazani AA, Lee JA, Klostranec J, Xiang Q, Dacosta RS, Wilson BC, Tsao MS, Chan WCW. High Throughput Quantification of Protein Expression of Cancer Antigens in Tissue Microarray Using Quantum Dot Nanocrystals. Nano Lett. 2006;6:2881–1886. doi: 10.1021/nl062111n. [DOI] [PubMed] [Google Scholar]
- 9.Fountaine TJ, Wincovitch SM, Geho DH, Garfield SH, Pittaluga S. Multispectral Imaging of Clinically Relevant Cellular Targets in Tonsil and Lymphoid Tissue Using Semiconductor Quantum Dots. Modern Pathol. 2006;19:1181–1191. doi: 10.1038/modpathol.3800628. [DOI] [PubMed] [Google Scholar]
- 10.Sweeney E, Ward TH, Gray N, Womack C, Jayson G, Hughes A, Dive C, Byers R. Quantitative Multiplexed Quantum Dot Immunohistochemistry. Biochem Biophys Res Commun. 2008;374:181–186. doi: 10.1016/j.bbrc.2008.06.127. [DOI] [PubMed] [Google Scholar]
- 11.Tholouli E, Sweeney E, Barrow E, Clay V, Hoyland JA, Byers RJ. Quantum Dots Light Up Pathology. J Pathol. 2008;216:275–285. doi: 10.1002/path.2421. [DOI] [PubMed] [Google Scholar]
- 12.Nie SM, Xing Y, Kim GJ, Simons JW. Nanotechnology Applications in Cancer. Annu Rev Biomed Eng. 2007;9:257–288. doi: 10.1146/annurev.bioeng.9.060906.152025. [DOI] [PubMed] [Google Scholar]
- 13.Ferrari M. Cancer Nanotechnology: Opportunities and Challenges. Nat Rev Cancer. 2005;5:161–171. doi: 10.1038/nrc1566. [DOI] [PubMed] [Google Scholar]
- 14.Heath JR, Davis ME. Nanotechnology and Cancer. Annu Rev Med. 2008;59:251–265. doi: 10.1146/annurev.med.59.061506.185523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen C, Peng J, Xia HS, Yang GF, Wu QS, Chen LD, Zeng LB, Zhang ZL, Pang DW, Li Y. Quantum Dots-Based Immunofluorescence Technology for the Quantitative Determination of HER2 Expression in Breast Cancer. Biomaterials. 2009;30:2912–2918. doi: 10.1016/j.biomaterials.2009.02.010. [DOI] [PubMed] [Google Scholar]
- 16.Heppner GH. Tumor Heterogeneity. Cancer Res. 1984;44:2259–2265. [PubMed] [Google Scholar]
- 17.Fidler IJ. Tumor Heterogeneity and Biology of Cancer Invasion and Metastasis. Cancer Res. 1978;38:2651–2660. [PubMed] [Google Scholar]
- 18.Heppner GH, Miller BE. Tumor Heterogeneity—Biological Implications and Therapeutic Consequences. Cancer Met Rev. 1983;2:5–23. doi: 10.1007/BF00046903. [DOI] [PubMed] [Google Scholar]
- 19.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem Cells, Cancer, and Cancer Stem Cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 20.Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ. Prospective Identification of Tumorigenic Prostate Cancer Stem Cells. Cancer Res. 2005;65:10946–10951. doi: 10.1158/0008-5472.CAN-05-2018. [DOI] [PubMed] [Google Scholar]
- 21.Wicha MS, Liu SL, Dontu G. Cancer Stem Cells: An Old Idea—A Paradigm Shift. Cancer Res. 2006;66:1883–1890. doi: 10.1158/0008-5472.CAN-05-3153. [DOI] [PubMed] [Google Scholar]
- 22.Emmert-Buck MR, Bonner RF, Smith PD, Chuaqui RF, Zhuang ZP, Goldstein SR, Weiss RA, Liotta LA. Laser Capture Microdissection. Science. 1996;274:998–1001. doi: 10.1126/science.274.5289.998. [DOI] [PubMed] [Google Scholar]
- 23.Simone NL, Bonner RF, Gillespie JW, Emmert-Buck MR, Liotta LA. Laser-Capture Microdissection: Opening the Microscopic Frontier to Molecular Analysis. Trends Genet. 1998;14:272–276. doi: 10.1016/s0168-9525(98)01489-9. [DOI] [PubMed] [Google Scholar]
- 24.Mccabe A, Dolled-Filhart M, Camp RL, Rimm DL. Automated Quantitative Analysis (AQUA) of In Situ Protein Expression, Antibody Concentration, and Prognosis. J Nat Cancer Inst. 2005;97:1808–1815. doi: 10.1093/jnci/dji427. [DOI] [PubMed] [Google Scholar]
- 25.Camp RL, Chung GG, Rimm DL. Automated Subcellular Localization and Quantification of Protein Expression in Tissue Microarrays. Nat Med. 2002;8:1323–1327. doi: 10.1038/nm791. [DOI] [PubMed] [Google Scholar]
- 26.Camp RL, Dolled-Filhart M, King BL, Rimm DL. Quantitative Analysis of Breast Cancer Tissue Microarrays Shows That Both High and Normal Levels of HER2 Expression Are Associated with Poor Outcome. Cancer Res. 2003;63:1445–1448. [PubMed] [Google Scholar]
- 27.Rubin MA, Zerkowski MP, Camp RL, Kuefer R, Hofer MD, Chinnaiyan AM, Rimm DL. Quantitative Determination of Expression of the Prostate Cancer Protein α-Methylacyl-CoA Racemase Using Automated Quantitative Analysis (AQUA)—A Novel Paradigm for Automated and Continuous Biomarker Measurements. Am J Pathol. 2004;164:831–840. doi: 10.1016/s0002-9440(10)63171-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Levenson RM. Spectral Imaging and Pathology: Seeing More. Lab Med. 2004;35:244–252. [Google Scholar]
- 29.Cheng L, Nagabhushan M, Pretlow TP, Amini SB, Pretlow TG. Expression of E-Cadherin in Primary and Metastatic Prostate Cancer. Am J Pathol. 1996;148:1375–1380. [PMC free article] [PubMed] [Google Scholar]
- 30.Pena JTG, Sohn-Lee C, Rouhanifard SH, Ludwig J, Hafner M, Mihailovic A, Lim C, Holoch D, Berninger P, Zavolan M, et al. MiRNA In Situ Hybridization in Formaldehyde and EDC-Fixed Tissues. Nat Methods. 2009;6:139–141. doi: 10.1038/nmeth.1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weinstein MH, Signoretti S, Loda M. Diagnostic Utility of Immunohistochemical Staining for p63, a Sensitive Marker of Prostatic Basal Cells. Mod Pathol. 2002;15:1302–1308. doi: 10.1097/01.MP.0000038460.95912.6E. [DOI] [PubMed] [Google Scholar]
- 32.Reiner T, De Las Pozas A, Parrando R, Perez-Stable C. Progression of Prostate Cancer from a Subset of p63-Positive Basal Epithelial Cells in FG/Tag Transgenic Mice. Mol Cancer Res. 2007;5:1171–1179. doi: 10.1158/1541-7786.MCR-07-0024. [DOI] [PubMed] [Google Scholar]
- 33.Mirtti T, Alanen K, Kallajoki M, Rinne A, Söderström KO. Expression of Cystatins, High Molecular Weight Cytokeratin, and Proliferation Markers in Prostatic Adenocarcinoma and Hyperplasia. Prostate. 2003;54:290–298. doi: 10.1002/pros.10196. [DOI] [PubMed] [Google Scholar]
- 34.Rhodes DR, Sanda MG, Otte AP, Chinnaiyan AM, Rubin M. Multiplex Biomarker Approach for Determining Risk of Prostate-Specific Antigen-Defined Recurrence of Prostate Cancer. J Nat Cancer Inst. 2003;95:661–668. doi: 10.1093/jnci/95.9.661. [DOI] [PubMed] [Google Scholar]
- 35.Mansfield JR, Gossage KW, Hoyt CC, Levenson RM. Autofluorescence Removal, Multiplexing, and Automated Analysis Methods for In Vivo Fluorescence Imaging. J Biomed Optics. 2005;10:41207. doi: 10.1117/1.2032458. [DOI] [PubMed] [Google Scholar]
- 36.Rubin MA, Zhou M, Dhanasekaran SM, Varambally S, Barrette TR, Sanda MG, Pienta KJ, Ghosh D, Chinnaiyan AM. α-Methylacyl Coenzyme A Racemase as a Tissue Biomarker for Prostate Cancer. J Am Med Assoc. 2002;287:1662–1670. doi: 10.1001/jama.287.13.1662. [DOI] [PubMed] [Google Scholar]
- 37.Zhou M, Chinnaiyan AM, Kleer CG, Lucas PC, Rubin MA. α-Methylacyl-CoA Racemase: A Novel Tumor Marker Over-Expressed in Several Human Cancers and Their Precursor Lesions. Am J Surg Pathol. 2002;26:926–931. doi: 10.1097/00000478-200207000-00012. [DOI] [PubMed] [Google Scholar]
- 38.Leav I, Mcneal JE, Ho SM, Jiang Z. α-Methylacyl-CoA Racemase (P504S) Expression in Evolving Carcinomas within Benign Prostatic Hyperplasia and in Cancers of the Transition Zone. Human Pathol. 2003;34:228–233. doi: 10.1053/hupa.2003.42. [DOI] [PubMed] [Google Scholar]
- 39.Epstein JI, Herawi M. Prostate Needle Biopsies Containing Prostatic Intraepithelial Neoplasia or Atypical Foci Suspicious for Carcinoma: Implications for Patient Care. J Urol. 2006;175:820–834. doi: 10.1016/S0022-5347(05)00337-X. [DOI] [PubMed] [Google Scholar]
- 40.Montironi R, Scattoni V, Mazzucchelli R, Lopez-Beltran A, Bostwick DG, Montorsi F. Atypical Foci, Suspicious but Not Diagnostic of Malignancy in Prostate Needle Biopsies. Eur Urol. 2006;50:666–674. doi: 10.1016/j.eururo.2006.07.048. [DOI] [PubMed] [Google Scholar]
- 41.Helpap B, Bonkhoff H, Cockett A, Montironi R, Troncoso P, Waters D, Bostwick D. Relationship between Atypical Adenomatous Hyperplasia (AAH), Prostatic Intraepithelial Neoplasia (PIN) and Prostatic Adenocarcinoma. Pathologica. 1997;89:288–300. [PubMed] [Google Scholar]
- 42.McNeal JE. In: Prostate in Histology for Pathologists. 3rd. Mills SE, editor. Lippincott Williams & Wilkins; Philadelphia, PA: 2007. [Google Scholar]
- 43.Epstein JI. What's New In Prostate Cancer Disease Assessment In 2006? Curr Opin Urol. 2006;16:146–151. doi: 10.1097/01.mou.0000193389.31727.9b. [DOI] [PubMed] [Google Scholar]
- 44.Holmberg L, Bill-Axelson A, Helgesen F, Salo JO, Folmerz P, Häggman M, Andersson SO, Spangberg A, Busch C, Nordling S, et al. A Randomized Trial Comparing Radical Prostatectomy with Watchful Waiting in Early Prostate Cancer. N Engl J Med. 2002;347:781–789. doi: 10.1056/NEJMoa012794. [DOI] [PubMed] [Google Scholar]
- 45.Loberg RD, Bradley DA, Tomlins SA, Chinnaiyan AM, Pienta KJ. The Lethal Phenotype of Cancer: The Molecular Basis of Death Due to Malignancy. CA Cancer J Clin. 2007;57:225–241. doi: 10.3322/canjclin.57.4.225. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.