

Abstract
PII constitutes a family of signal transduction proteins that act as nitrogen sensors in microorganisms and plants. Mycobacterium tuberculosis (Mtb) has a single homologue of PII whose precise role has as yet not been explored. We have solved the crystal structures of the Mtb PII protein in its apo and ATP bound forms to 1.4 and 2.4 Å resolutions, respectively. The protein forms a trimeric assembly in the crystal lattice and folds similarly to the other PII family proteins. The Mtb PII:ATP binary complex structure reveals three ATP molecules per trimer, each bound between the base of the T-loop of one subunit and the C-loop of the neighboring subunit. In contrast to the apo structure, at least one subunit of the binary complex structure contains a completely ordered T-loop indicating that ATP binding plays a role in orienting this loop region towards target proteins like the ammonium transporter, AmtB. Arg38 of the T-loop makes direct contact with the γ-phosphate of the ATP molecule replacing the Mg2+ position seen in the Methanococcus jannaschii GlnK1 structure. The C-loop of a neighboring subunit encloses the other side of the ATP molecule, placing the GlnK specific C-terminal 310 helix in the vicinity. Homology modeling studies with the E. coli GlnK:AmtB complex reveal that Mtb PII could form a complex similar to the complex in E. coli. The structural conservation and operon organization suggests that the Mtb PII gene encodes for a GlnK protein and might play a key role in the nitrogen regulatory pathway.
Keywords: nitrogen regulatory protein, PII, GlnK, GlnB, Mycobacterium tuberculosis, binary complex, homology model, glutamine synthetase, ammonium transporter, crystal structure
Introduction
The PII family of small signal transduction proteins plays a pivotal role as nitrogen sensors in both microorganisms and plants.1,2 Nitrogen sensing is achieved by regulating the intracellular nitrogen to carbon ratio represented largely by glutamine and 2-oxoglutarate (2OG), respectively. Intracellular nitrogen regulation is carried out by direct and indirect control of glutamine synthetase (GS) activity through the PII proteins. E. coli has two homologues of the PII protein, GlnB (also referred to as PII), encoded by the glnB gene, and GlnK, encoded by the glnK gene. Under nitrogen limiting conditions, E. coli GlnB is uridylylated at a conserved Tyr51 residue by a uridylyl transferase (GlnD), and then interacts with adenylyl transferase to directly activate GS through deadenylylation.3,4 Conversely, under nitrogen rich conditions, E. coli GlnB is deuridylylated by GlnD, and then interacts through the adenylyl transferase to deactivate GS by adenylylation. GlnB has also been shown to form a direct complex with GS in its activation/deactivation cycle in Archaeal Methanosarcina mazei.5 In the indirect control system, E. coli GlnB interacts with the two component signal transduction system proteins, NtrB and NtrC, to regulate the transcription of the gene glnA which encodes for GS.6,7 When GlnB is deuridylylated it can interact with a histidine kinase, NtrB, which regulates the phosphorylation state of NtrC, controlling the expression of glnA. Thus GlnB is indispensible to the pathway of nitrogen control in E. coli.
GlnK is another PII homolog that plays an essential role in the nitrogen control systems of several organisms including E. coli8,9. In all known instances the glnK gene lies in an operon immediately adjacent to the amtB gene10 which codes for the integral membrane protein, AmtB, that acts as ammonia/ammonium channel.11 Under nitrogen rich conditions in E. coli the GlnK trimer complexes with the AmtB trimer blocking the influx of ammonium into the cellular cytoplasm.9,12 Complex formation between the GlnK protein and AmtB is modulated by the presence of effector molecules like ATP, ADP, and 2OG and is also affected by the post-translational modification of the crucial Tyr51 residue present on the functionally important T-loop of the GlnK protein.12 In the E. coli GlnK, the Tyr51 residue is uridylylated by the GlnD enzyme, but in the Gram-positive soil bacteria Corynebacterium glutamicum and Streptomyces coelicolor GlnD has been shown to act as an adenylyl transferase, adenylylating instead of uridylylating the GlnK protein at this conserved Tyr51 residue.13–15 Under nitrogen limiting conditions, the GlnK protein is uridylylated/adenylylated at the Tyr51 residue, which prevents its interaction with AmtB allowing an unhindered flow of extracellular ammonium into the cell through AmtB.16
The AmtB:GlnK complex is also implicated in the membrane sequestration of enzymes involved in nitrogen fixation, such as the glycohydrolase DraG in Azospirillum brasilense.17,18 A ternary complex of the AmtB:GlnK with the nitrogen stress transcription factor, TnrA, has also been reported in Bacillus subtilis.19 Similarly, in Corynebacterium glutamicum, GlnK interacts with transcriptional repressor AmtR,20 and in Streptomyces coelicolor an OmpR type transcriptional activator, GlnR, has been shown to regulate the expression of glnK, amtB, glnA and glnD.21–23 Thus the AmtB:GlnK complex is capable of functioning with other protein partners like DraG and TnrA to achieve nitrogen regulation and therefore the complex formation is tightly regulated by the presence of effector molecules or post-translational modification at Tyr51 on the T-loop. The PII protein can also complex with N-acetylglutamate kinase (NAGK) to regulate arginine biosynthesis, as seen in the Arabidopsis thaliana24 and Synechococcus elongates25 PII-NAGK complex structures.
Mycobacterium tuberculosis (Mtb) has a single PII homolog, Rv2919c.26 The PII gene was found to be essential for survival in primary murine macrophages by transposon site hybridization (TraSH) in H37Rv.27 Its essentiality, its roles in nitrogen regulation, and its absence in humans makes PII an attractive drug target in Mtb, the causative agent of tuberculosis (TB). The Mtb PII gene was annotated glnB based on its higher sequence homology to E. coli GlnB (www.webTB.org). However, it exists in an operon with glnD-PII-amtB (Rv2918c-Rv2919c-Rv2920c) leading to speculation that it might encode for a GlnK protein on the basis of the operon organization observed in other microorganisms.2 The apparent ambiguity requires clear structural and biochemical data to confirm its identity. As part of the Mtb structural genomics consortium, we have solved the crystal structure of this protein, both in the apo and ATP bound forms, with the aim of understanding its true identity (and hence its role) in Mtb, potentially opening avenues for inhibition studies.28 The crystal structures presented in this report provide important structural and functional insights into the role of Mtb PII as GlnK.
Results and Discussion
Structure of the Mtb apo PII protein
The crystal structure of the Mtb apo PII protein was solved at 1.4 Å resolution by molecular replacement using the E. coli apo GlnK structure (PDB ID: 1GNK) as the search model. The x-ray diffraction data collection and refinement statistics are in Table I. The crystallographic asymmetric unit contains one subunit, however a homotrimer with identical subunits is formed in the crystal lattice [Fig. 1(A)]. Size exclusion chromatography confirmed that the Mtb PII protein exists as a trimer in solution (data not shown).
Table I.
Crystallographic and Refinement Statistics
| Mtb apo PII | Mtb PII:ATP | |
|---|---|---|
| PDB ID | 3BZQ | 3LF0 |
| Data collection | ||
| Space group | R3 | P43 21 2 |
| Wavelength (Å) | 0.96 | 0.98 |
| Temperature (K) | 100 | 100 |
| a (Å) | 77.15 | 69.74 |
| b (Å) | 77.15 | 69.74 |
| c (Å) | 51.84 | 146.68 |
| α (°) | 90 | 90 |
| β (°) | 90 | 90 |
| γ (°) | 120 | 90 |
| Resolution (Å) | 1.40 (1.40–1.44) | 2.40 (2.40–2.46) |
| Unique reflections | 21338 (1595) | 14020 (982) |
| % completeness | 99.10 (100.0) | 99.78 (97.92) |
| R (merge) | 0.06 (0.35) | 0.07 (0.63) |
| 〈I/σI〉 | 23.94 (7.77) | 12.20 (2.02) |
| Refinement | ||
| R value (%) | 20.80 | 21.57 |
| Free R value, random, 5% | 22.60 | 29.21 |
| Molecules per asymmetric unit | 1 | 3 |
| Protein residues | 99 | 312 |
| ATP molecules | 0 | 3 |
| Rmsd bond length (Å)a | 0.006 | 0.017 |
| Rmsd bond angle (Å)a | 1.007 | 1.862 |
| Rmsd between subunits (Å) | – | 0.31, 0.39, 0.45 |
| Residues ϕ-ψ angles | ||
| Most favored (%) | 98.80 | 96.60 |
| Allowed (%) | 1.20 | 3.40 |
| Generously allowed/Disallowed (%) | 0.0 | 0.0 |
Deviations from restraint targets.
Figure 1.
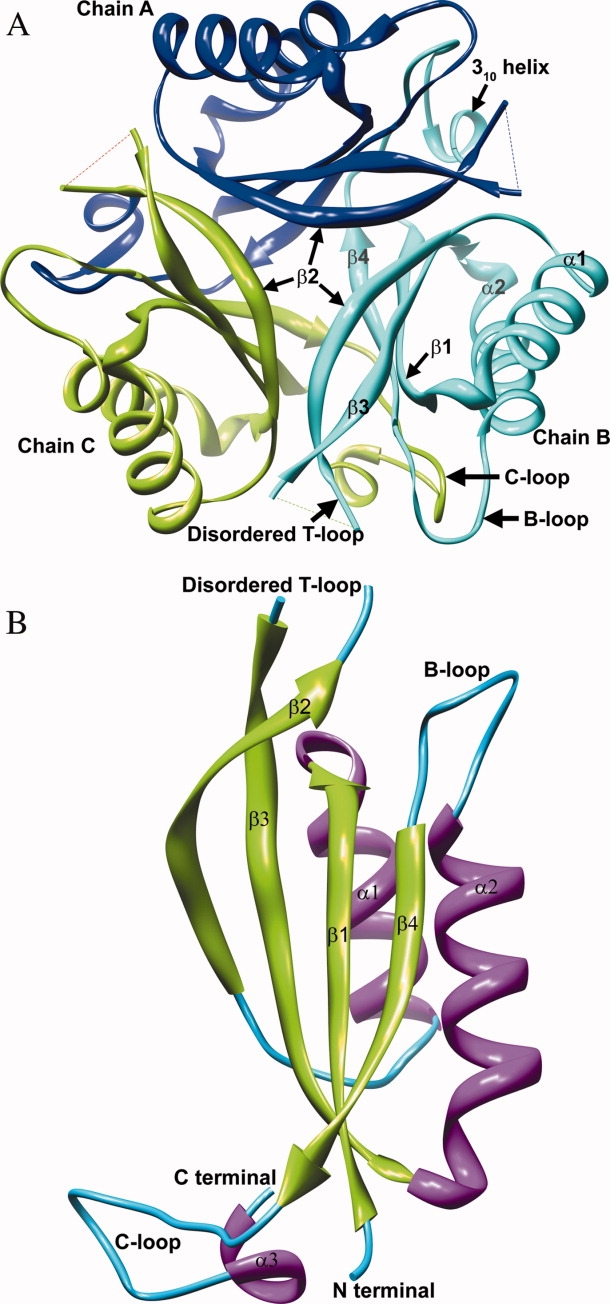
A). A trimer of the Mtb apo PII protein viewed down the crystallographic triad showing the three subunits in ribbon representation–A (blue), B (cyan), and C (green). The T-loop region between Gln39 and Val53 is disordered and missing in the Mtb apo PII structure. The surface of the trimer assembly carrying the T-loop is designated as the top of the triad and is shown facing up. The β2 strands from the individual subunits form the mouth at the top of the β-barrel, while the β4 strand of each subunit bends inward at the bottom to close the base of the concave β-barrel. The secondary structures are labeled on subunit B (cyan). Molecular graphics images were produced using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIH P41 RR-01081).56 (B). A single subunit of the Mtb apo PII protein is shown in ribbon representation and colored by its secondary structures. The secondary structure features are labeled based on the E. coli GlnK structure.30 The functionally important T-loop is disordered in the Mtb apo PII crystal structure.
The structural features of the PII family of proteins from different organisms are well conserved in the Mtb PII structure (Fig. 1). The r.m.s.d. of a subunit of Mtb apo PII with E. coli GlnB (PDB ID: 2PII) is 0.78 Å (for 86 Cα atom pairs–excluding residues 36 to 55 and 109 to 112)29 and the r.m.s.d. with E. coli GlnK (PDB ID: 1GNK–subunit A) is 0.70 Å (for 91 Cα atom pairs–excluding residues 26 and 36 to 55).30 Figure 1(B) shows a subunit of the Mtb apo PII protein with the respective secondary features labeled.30 In each subunit, connecting strands β2 and β3, is a large loop region of 20 residues that extends out from the main body of the molecule, designated the T-loop (Tyr36 to Phe55). The flexible region of this T-loop, encompassing residues Gln39 through Val53, is disordered in the electron density map and is therefore omitted from the final model of the Mtb apo PII structure. The rest of the molecule, including the smaller loop (B-loop) connecting helix α2 and strand β4 is fully ordered. The C-terminal residues extend out from the β4 strand to form a C-loop (Asp97 to Leu112) and has a 310 helix embedded within it between Gly108 and Ala111, consistent with other PII like protein structures.16,24,30–35
Structure of the ATP bound Mtb PII protein
The Mtb PII:ATP binary complex crystals were obtained by co-crystallization. X-ray data was collected to 2.4 Å resolution and the structure was solved by molecular replacement using the Mtb apo PII structure (PDB ID: 3BZQ) as the search model (Data collection and refinement statistics are presented in Table I). Unlike the Mtb apo PII protein and the ATP bound E. coli GlnK, which have a single subunit in the asymmetric unit, the Mtb PII:ATP structure contains a trimer in the asymmetric unit with one ATP molecule bound per subunit [Fig. 2(A) and Supporting information Fig. 1). The crystal structure of Mtb PII:ATP shows that ATP binds in three symmetrically equivalent positions in the trimer in a pocket formed by the base of the T-loop, B-loop, and β4 strand of one subunit, and part of the β2 strand, β3 strand, and the C-loop of a neighboring subunit [Fig. 2(B)]. In this structure the ribose ring of ATP adopts the C-3′-endo ring pucker with the adenine ring in the anti-orientation consistent with its position in the E. coli GlnK:ATP structure30 (Supporting information Fig. 1).
Figure 2.
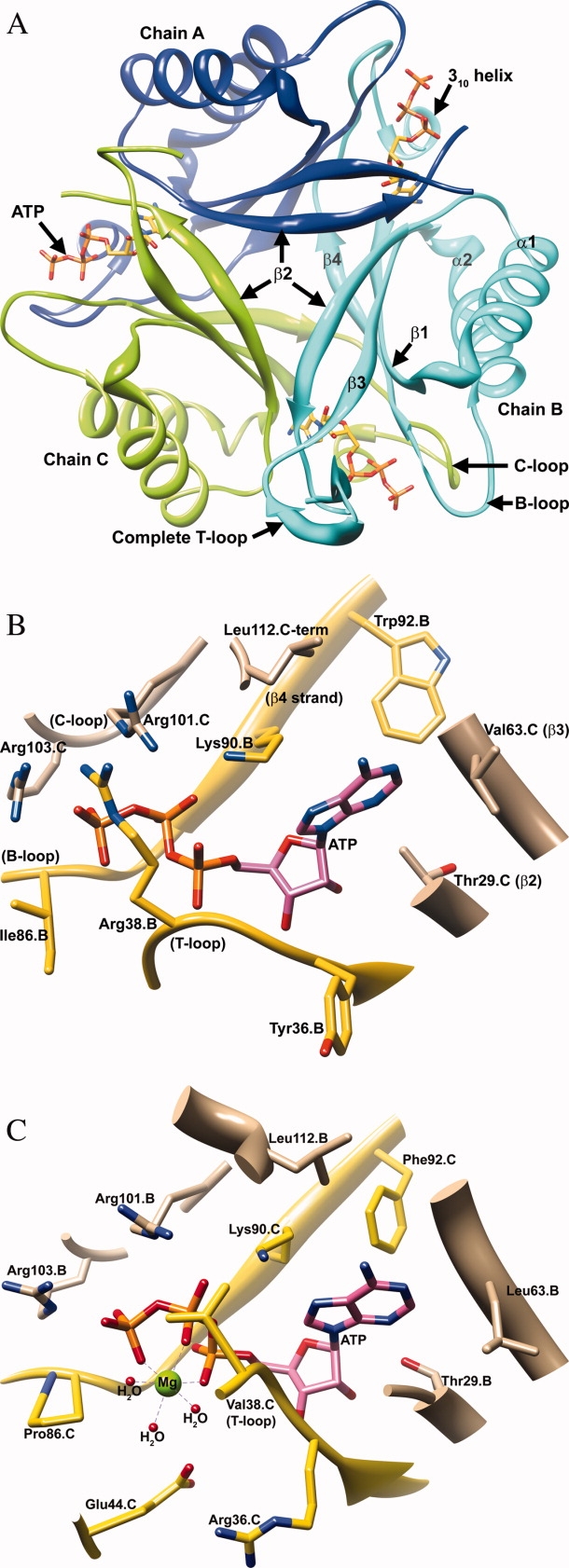
A). A trimer of the Mtb PII:ATP binary complex structure is shown viewing down the crystallographic triad. The orientation and color scheme for the trimer assembly is consistent with Figure 1A. Subunit A (blue), B (cyan), and C (green) are represented as ribbons, whereas the bound ATP molecules are shown in yellow stick representation. A complete T-loop is visible in subunit B. (B) The active site of the Mtb PII protein bound with ATP (pink). Residues contributed by the neighboring subunit are shown in tan. Residues from the main-chain (Subunit B) are shown in yellow. Conserved residues Gly87.B (B-loop), Gly89.B (B-loop), Lys90.B (β4 strand), and Arg103.C (C-loop) hydrogen bond with the polar phosphate groups of ATP. Residues contributing to adenine binding include Ile7.B (β1 strand), Trp92.B (β4 strand), Leu26.C, Gly27.C, Met28.C, and Thr29.C (in the loop connecting the α1 helix and β2 strand), Val64.C (β3 strand), and Leu112.C (C-loop). (Not all the residues in the ATP binding pocket have been displayed in the interest of clarity). (C) A side by side comparison of the ATP binding pocket of Mtb PII:ATP and the M. jannaschii GlnK1:ATP(Mg2+):2OG ternary complex structure (PDB ID: 2J9E) is presented.35 Note that the Mtb PII's Arg38 residue is a Val38 in M. jannaschii GlnK1. The side chain of Arg38 of Mtb PII occupies the Mg2+ position of M. jannaschii GlnK1.
The trimeric Mtb PII:ATP structure is superposed onto the trimeric Mtb apo PII with an r.m.s.d. (between 94 x 3 Cα atom pairs) of 0.69 Å. The r.m.s.d. between the three subunits of the Mtb PII protein is 0.31 Å between subunits A and B, 0.39 Å between subunits B and C, and 0.45 Å between subunits A and C. The differences lie in the positions of the residues in the ATP binding region, whereas the rest of the molecular backbone remains relatively unchanged compared to the Mtb apo PII. The disordered T-loop of the apo PII structure is completely ordered in subunit B of the ATP bound Mtb PII trimer with the exception of Lys40 and the partially ordered Gln39 [Fig. 2(A) and 3]. However, residues Gln39 to Val53 of subunit A and residues Gln39 to Glu50 of subunit C are disordered and were omitted from the final model of the Mtb PII:ATP structure.
The trimer has some distinct structural features that indicate the possible function of the Mtb PII protein. In the trimer, the β-sheet from one subunit tilts toward the neighboring subunit at a sheer angle typical of β-barrel structures.36,37 Three β-sheets, one from each subunit, line the interior of the trimer and enclose a concave β-barrel at the center of the trimer, whereas the α-helices of each subunit lie on the outside of the trimer [Fig. 2(A)]. Glu32 from each β2 strand comes closest to the center at the mouth of the β-barrel, lying symmetrically away from each other at a distance of ∼9.1 Å and creating a negatively charged region at the mouth of the β-barrel [Supporting information Fig. 2(A)]. The inner lining of the β-barrel encloses a concave cavity with hydrophobic residues Leu3, Thr5, and Ile7 from the β1 strand, Thr29 and Ile33 from the β2 strand, Val64 from the β3 strand and Trp92 from the β4 strand. In addition, Arg60 and Glu62 from the β3 strands of each subunit insert the polar ends of their side chains into the center of the cavity forming hydrogen bonds to create a charged center, stabilizing the trimer assembly [Supporting information Fig. 2(B)]. Finally, Pro95 residues from each β4 strand are brought into close proximity at the bottom of this barrel at a distance of ∼4.0 Å [Supporting information Fig. 2(C)].
An important feature that becomes apparent in the trimeric state is how part of the long C-loop of one subunit lies parallel to the β4 strand of the neighboring subunit making several interactions. For instance, there are hydrogen bonding backbone interactions between Thr98 (C-loop of subunit C) and Val93 (β4 strand of subunit B), Val100 (C-loop of subunit C) with Val91 and Trp92 (β4 strand of subunit B), and Val102 (C-loop of subunit C) with Lys90 (β4 strand of subunit B). The extensive inter-subunit hydrogen bonds between the C-loop of one subunit and the β4 strand of another could be the defining factor in the trimerization of Mtb PII. The C-loop of one subunit also approaches the B-loop and the ATP binding site of the neighboring subunit, which may also preserve the structural integrity of the functional trimer. For example, the backbone carbonyl group of Thr104 (C-loop of subunit C) hydrogen bonds with the guanido group of Arg82 (B-loop of subunit B) at a 2.8 Å distance (Subunit B of Mtb PII with a complete T-loop was used to calculate all inter-atomic distances in the ATP binding pocket). The 310 helix at the end of the C-loop contributes to the ATP binding site of the neighboring subunit and is partly stabilized by the hydrogen bonding of the carbonyl group of Ala111 (C-loop) with the amino side chain of Lys90 (β4 strand) (2.8 Å). The importance of the trimeric architecture is that the secondary structural elements involved in the trimerization also form part of the ATP binding pocket indicating that trimerization is critical for its function.
ATP binding site and loop movements–comparison between the apo and ATP bound Mtb PII proteins
There are a number of apo and ATP bound PII crystal structures that have been reported from E. coli16,29–31,34 Herbaspirillum seropedicae,32 Thermotoga maritima,38 Cyanobacteria Synechococcus sp. PCC 7942 and Synechocystis sp. PCC 6803,39 Thermus thermophilus,33 Neisseria meningitidis,40 Methanococcus jannaschii,35 Arabidopsis thaliana,24 and Synechococcus elongatus.25 Similar to the discussion of these structures, the protomer ATP interactions in the Mtb PII protein can be viewed in reference to three important structural elements, namely the C-loop (310 helix), the B-loop, and the T-loop [Fig. 2(B)]. The C-terminus of the neighboring subunit of Mtb PII, aided by a 310 helix, curls back to form part of the ATP binding pocket. The C-terminal residue Leu112 lines a hydrophobic pocket for the adenine ring of ATP, while the terminal carboxylic oxygen atom of Leu112 hydrogen bonds with Arg101 at a distance of 2.8 Å, which in turn hydrogen bonds with the bridge oxygen between the β- and γ-phosphates of ATP (2.9 Å). The 310 helix must play a role in the binding of the ATP because it brings Leu112 into proximity with ATP. Furthermore, in the Mtb apo PII structure, the Cα atom of Leu112 moves away from the ATP binding site by 1.6 Å and loses its hydrogen bonding interaction with Arg101 although the 310 helix is conserved in the apo structure.
The B-loop carries the conserved mononucleotide binding motif ‘Gly(84)-X-X-Gly(87)-X-Gly(89)-Lys(90)’ and thus acts as the primary binding site for ATP.41 Compared to the Mtb apo PII structure, the B-loop in Mtb PII:ATP moves away from the ATP binding region, making space for the ATP to bind. The Cα atom of Lys85 at the tip of the B-loop moves away by 1.5 Å. The backbone nitrogen atom of Gly89 hydrogen bonds with the oxygen atom of the β-phosphate at a 2.9 Å distance. While the Cα atom of ATP bound and unbound Lys90 are in identical positions, the side chain moves in the ATP bound structure so that the N-Z atom of Lys90 is 5.3 Å away from its position in the apo structure and hydrogen bonds with the β-phosphate of ATP 2.5 Å away.
Arg38 extends out from the base of the T-loop and hydrogen bonds with the oxygen atom of the γ-phosphate at a 3.0 Å distance [Fig. 2(B) and 3]. The binding of ATP is involved in stabilizing the side chain conformation of Arg38, and therefore should partly stabilize the T-loop conformation. In the Mtb apo PII structure, the side chain of Arg38 is disordered (along with the T-loop) and is represented as alanine, which could result from a lack of stabilizing interactions in the absence of ATP [Fig. 3]. Indeed, the Cα atom of Arg38 in the Mtb apo PII moves away from the active site by 8.3 Å compared to the ATP bound structure, opening this cavity to solvent. This effectively changes the flexibility of the T-loop conformation in Mtb apo PII. Comparing the apo and ATP bound structures of the Mtb PII protein, it seems apparent that Arg38's interaction with the γ-phosphate is the reason the base of the T-loop moves back into the active site [Fig. 3]. Interestingly, in contrast to the Mtb PII and E. coli GlnK/B structures, the crystal structure of the M. jannaschii GlnK1 shows a Mg2+ ion bound next to ATP which coordinates with all three phosphate oxygen atoms35 [Fig. 2(B,C) for a comparison of the Mtb PII and M. jannaschii GlnK1 ATP binding sites]. Yildiz et al.35 have argued that Mg2+ and ATP are necessary to change the T-loop conformation in the M. janaschii GlnK1 protein allowing the creation of the 2OG binding site. In the Mtb PII:ATP protein, Arg38 of the T-loop occupies the Mg2+ binding site [Fig. 2(B)] to form a direct hydrogen bond with the γ-phosphate oxygen atom of the ATP (3.0 Å). Arg38 of Mtb PII is a Val38 in M. jannaschii GlnK1 suggesting that a Mg2+ is critical for M. jannaschii as it is to many other ATP binding proteins. Binding of the Mg2+ ion pushes the base of T-loop away from the ATP phosphates in M. jannaschii GlnK1, whereas in the Mtb PII:ATP structure the base of the T-loop approaches ATP (the main chain amide group of Arg38 hydrogen bonds with the ATP α-phosphate at a 3.1 Å distance). The same amide group of Val38 in M. jannaschii GlnK1 is 4.7 Å away from the ATP α-phosphate,35 therefore, it may be that a Mg2+ ion is not required by Mtb PII for ATP or 2OG binding. In fact, recent in vitro studies have shown that the Mtb PII binds to both ATP (Kd = 1.93 μM) and 2OG, in the absence of Mg2+.42 Our attempts to crystallize Mtb GlnK in the presence of Mg2+ ions were unsuccessful.
Figure 3.
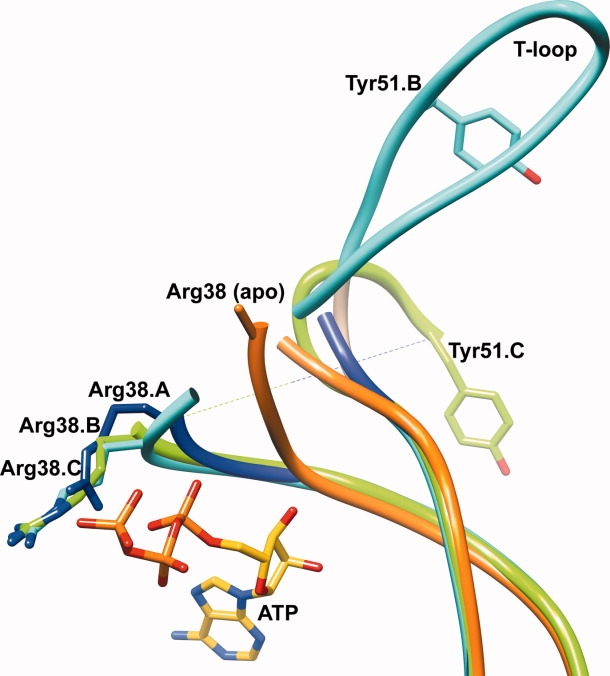
Overlay of the T-loop regions of subunits A (blue), B (cyan), C (green) of the Mtb PII:ATP and Mtb apo PII (purple) structures, shown in a worm representation. The bound ATP molecule is shown in yellow stick representation. The relative positions of the Tyr51 residues on the T-loop of subunit B (cyan) and subunit C (green) of Mtb PII highlight the different orientations of the T-loop in both subunits. Arg38 of the Mtb apo PII protein (orange) is modeled as an Ala. Upon binding ATP, Arg38 of all three subunits of the Mtb PII:ATP structure (blue, cyan, and green) are stabilized and move relative to their position in the Mtb apo PII structure.
While Arg38 forms one part of the enclosure for the phosphates of ATP, Arg103 from the C-loop of the neighboring molecule forms another part of this enclosure forming a hydrogen bond with an oxygen on the γ-phosphate (2.8 Å). Similarly, the C-loop also contributes Arg101, which further stabilizes the ATP binding site; in subunits A, B, and C Arg101 hydrogen bonds with the bridge oxygen between the β- and γ-phosphates of ATP (at distances of 2.8, 2.9, and 3.2 Å, respectively). While Arg38 moves in closer to the active site upon ATP binding, Tyr36 moves away (the Cα atom of Tyr36 moves by 2.9 Å) compared to its original position in the Mtb apo PII structure. In the ATP bound structure, the backbone nitrogen atom of the Tyr36 forms a hydrogen bond with the 3′ hydroxyl group of ATP (3.2 Å). Therefore, in the ATP bound PII structure, the Tyr36 orients its side chain more closely to the side chain of Phe55 (3.5 Å). As Tyr36 is the first residue and Phe55 is the last residue of the functionally important T-loop, ATP binding can be said to partly influence the orientation of the T-loop itself. Taken together, the binding of ATP along with the positions of Tyr36, Arg38, and Phe55 might contribute to the stability of at least the base of the T-loop of Mtb PII and point it in the right orientation to form a complex with PII's target proteins, such as AmtB or GlnD.
Although the residues at the base of the T-loop, including Arg38, are fully ordered in all subunits of Mtb PII:ATP (Arg38 is disordered in the apo structure), only subunit B contained the fully ordered T-loop. Upon analyzing the crystal packing effect from the residues of symmetry related molecules on the stability and the conformation of the visible T-loop of subunit B in the Mtb PII:ATP structure, we observed that part of the T-loop of subunit B packs inside a groove created by two symmetry related molecules (Mtb PII′ and PII″) [Fig. 4(A)]. Tyr51 of the T-loop makes hydrophobic interactions with Val53′ (3.8 Å) of the T-loop from the symmetry related molecule, Tyr46 makes hydrophobic interactions with Ser52′ (3.7 Å), and Glu50 makes a hydrogen bond interaction with Thr98″ (2.5 Å). The backbone amide group of Arg47 hydrogen bonds with the carboxylic acid group of Asp71″ (3.0 Å), and the Cα atom of Arg47 makes a van der Waal's interaction with the side chain of Val70″ (3.8 Å). Overlaying subunit B on subunit A reveals that the T-loop of subunit A is exposed to solvent without any stabilizing interactions, and overlaying subunit B on subunit C of Mtb PII revealed clashes of the T-loop with the T-loop from a neighboring symmetry molecule, requiring drastic changes in the T-loop conformation of subunit C to avoid these clashes. Because of this the T-loops in subunits A and C of the ATP bound Mtb PII were only partially ordered. It is clear from the positions of the residues leading up to the missing section of the T-loop in subunit C that the loop has a very different conformation compared to subunits A and B (Fig. 3). Collectively, the interactions of the T-loop of subunit B with the symmetry molecules help stabilize the flexible part of the T-loop, which would otherwise need to be stabilized by forming a complex with a target protein or an effector molecule such as 2OG.12 Perhaps, the crystal packing effect on the stabilization of the T-loop of subunit B of the Mtb PII protein mimics the formation of a complex with a target protein [Fig. 4(A)]. Thus, the T-loop requires some additional stabilizing interactions mediated through the binding of a target protein to Mtb PII, in addition to the ATP binding. However, the present conformation of the T-loop is subjective and may adopt different conformations in the presence of a physiological binding partner like AmtB.
Figure 4.

A) Part of the T-loop of subunit B of the Mtb PII protein is stabilized by its interaction with the symmetry related molecules Mtb PII′ and Mtb PII”. Subunit B of Mtb PII is shown in purple ribbon representation along with the bound ATP in stick representation. The symmetry related molecules Mtb PII′ and Mtb PII” are shown in mesh representation with 50% transparency.(B) Homology model of Mtb AmtB and Mtb PII:ATP based on the crystal structure of the E. coli GlnK:AmtB complex.16,34 AmtB is shown in mesh representation with 50% transparency and PII is shown in green ribbon representation. Subunit B of the crystallized Mtb PII:ATP structure is superposed on to the homology modeled Mtb PII structure and shown in purple ribbon representation along with the bound ATP in stick representation. The superposition highlights the ∼45° movement required by the T-loop of the crystallized Mtb PII protein to interact with the T-loop binding site of the Mtb AmtB protein.
Comparison with the E. coli Glnk:AmtB complex
Mtb encodes a probable amtB gene (Rv2920c), which is next to the glnB gene.26 Comparison between the 428 amino acid E. coli AmtB protein and the 477 amino acid Mtb AmtB protein shows a 41% sequence identity. All essential residues involved in the interaction of AmtB with the T-loop of PII, specifically Phe107, His168, Phe215, Leu259, Ser263, Val299, Asp313, and His318 of the E. coli AmtB, were found to be conserved in Mtb AmtB.34 We constructed a homology model of Mtb AmtB and the T-loop section of the Mtb PII protein based on the crystal structure of the E. coli GlnK:AmtB complex (PDB ID: 2NS1) [Fig. 4(B)] using the SWISS-MODEL program.43 Comparing the T-loops of subunit B of the Mtb PII protein with the homology modeled Mtb PII, we noticed that the region of the T-loop that sticks out from the main body of the PII protein swings away from the T-loop binding pocket of AmtB by ∼45°. The position of the T-loop observed in the crystal structure of Mtb PII:ATP could be due to crystal packing effects (vide ante). Thus, in order to bind Mtb AmtB, the T-loop would have to swing by ∼45° with a maximum movement of ∼11.0 Å (observed for the Cα atom of Arg47) to bring the T-loop inside the groove of AmtB [Fig. 4(B)]. However, the base of the T-loop that forms and interacts with the ATP binding site does not move. This suggests that ATP binding stabilizes the base of the T-loop (residues 36–39), but that the rest of the T-loop must be stabilized by a target protein. Given that PII proteins can bind a multitude of target proteins (particularly, AmtB, and GlnD of the same operon13), it is logical to think that the T-loop exists in different conformations depending on the target protein it complexes with. For example, in the Arabidopsis thaliana PII:NAGK complex structure, the T-loop adopts a very different conformation compared to the E. coli PII:AmtB complex structure.44 The crystal structure of PII:GlnD has not been solved for any organism to date despite strong biochemical evidence for this protein-protein complex formation.45 Obviously other parameters (like 2OG binding) would influence the conformational changes of the T-loop.
Looking into the T-loop binding pocket of the Mtb AmtB homology model, there are few differences when compared with the E. coli AmtB structure. The Mtb PII protein's Tyr51 terminal hydroxyl group makes favorable hydrogen bonding interactions with the backbone amide of Phe224 of Mtb AmtB (2.6 Å). The phenyl ring of Tyr51 makes additional hydrophobic contacts with the side chain of Leu223 (3.3 Å), which does not exist in E. coli AmtB, where it is an Ala192. However some hydrogen bonding interactions observed in E. coli are missing in Mtb, such as the interaction of the backbone carbonyl of Arg47 with Arg253 of E. coli AmtB, in Mtb it is an Asp284 and is 5.9 Å away. There is also a 36 residue insertion at the C-terminus of the Mtb AmtB that hangs on the cytoplasmic side of the membrane that was not included in the homology model, but will be involved in some form of interaction with the PII trimer. Thus the Mtb PII protein would be expected to interact and form a complex with the Mtb AmtB.
Is the Mtb PII protein a GlnB or GlnK?
The gene encoding Mtb PII has been annotated glnB in the Tuberculosis Structural Genomics Consortium (TBSGC). Comparing the sequences of other PII proteins from across different species, the Mtb PII protein shows a 2 to 7% greater sequence identity with known GlnBs than with their respective GlnKs. (See Table II for comparison of percentage sequence identities with PII proteins from other organisms.) Thus, on the basis of sequence identity alone it could be termed Mtb GlnB. However, while the glnB gene in most organisms is found in an operon with the glnA gene encoding glutamine synthetase, the glnK gene in most organisms is linked to an amtB gene forming an amtB-glnK or glnK-amtB operon (and may also contain a third gene, glnD). C. glutamicum and S. coelicolor are two other actinobacteria where the nitrogen regulation pathway has been studied in detail.46,47 Like Mtb they too encode a single homolog of the PII gene that lies between the amtB and glnD genes and the gene product has been annotated as GlnK in both microorganisms based on operon organization. As Mtb follows the same operon organization as C. glutamicum and S. coelicolor it is logical that the gene would also translate into a GlnK protein. Furthermore, it has been suggested that to be annonated glnB, the protein product of these genes should consist of a conserved Lys3 and an Asp5 or Glu5.2 Whereas the E. coli GlnB observes this rule, the Mtb PII protein has a Leu3 and Thr5, both consistent with E. coli GlnK. The Lys3 and Asp5/Glu5 residues among GlnB proteins and the Leu3 and Thr5 residues among GlnK proteins are fairly conserved across species, suggesting that the Mtb PII gene is in fact glnK.2
Table II.
Percentage Identity of the Mtb PII Protein with GlnB and GlnK of Other organisms
| GlnB (% identity with Mtb PII protein) | GlnK (% identity with Mtb PII protein) | |
|---|---|---|
| α Proteobacteria | ||
| Azospirillum brasilense | 60 | 56 |
| Rhodospirillum rubrum | 57 | 60 |
| Azorhizobium caulinodans | 59 | 57 |
| β Proteobacteria | ||
| Herbaspirillum seropedicae | 64 | 61 |
| γ Proteobacteria | ||
| Escherichia coli | 61 | 54 |
| Klebsiella pneumoniae | 61 | 54 |
| Firmibacteria | ||
| Bacillus subtilis | – | 34 |
| Actinobacteria | ||
| Corynebacterium glutamicum | – | 68 |
| Streptomyces coelicolor | – | 68 |
| Archaebacteria | ||
| Methanococcus jannaschii | – | 55 |
| Cyanobacteria | ||
| Synechococcus | 61 | – |
| Plants | ||
| Arabidopsis thaliana | 46 | – |
– a GlnK or GlnB gene does not exist or has not yet been identified.
The Mtb PII protein projects the Leu3 and Thr5 residues from each subunit into the central cavity of the β-barrel formed by the trimer, while E. coli GlnB has Lys3 and Asp5 residues in these positions. The significance of these hydrophilic versus hydrophobic residues at the center of the β-barrel in relation to the function of GlnB and GlnK is unknown and calls for further investigation. Apart from operon organization and sequence specific characterization, the crystal structure of the Mtb PII protein gives a clear indication of the nomenclature of the protein. The presence of a 310 helix at the C-terminus of the Mtb PII protein is consistent with other known GlnK structures, while in GlnB this region is a loop that is positioned away from the ATP binding pocket. It has been proposed earlier that GlnK structures may differ from GlnB due to the presence of a 310 helix in the C-loop.30,39,48 For these reasons we suggest that the gene for the PII protein in Mtb is glnK.
In summary, the operon organization of Mtb PII gene, the existence of GlnK specific structural features in both the apo and ATP bound Mtb PII crystal structures, and the homology model of the Mtb PII:AmtB complex, suggest that the PII homolog of Mtb may play a key role as GlnK in the nitrogen regulatory pathway.
Materials and Methods
A 339 bp DNA fragment containing the PII gene (Rv2919c) was amplified by PCR with Mtb H37Rv genomic DNA as a template, using the following oligonucleotide primers:
5′-GGG AAT TCC ATA TGA AGC TGA TCA CTG CGA TCG TGA AGC-3′
3′-CCC AAG CTT TCA TAA CGC GTC GTG TCC GCG TTC AC–5′.
The amplified DNA fragment was digested with NdeI and HindIII restriction enzymes and subcloned into the corresponding restriction sites in a pET28b vector containing an N-terminal His tag (Novagen). Following sequence confirmation, the plasmid was transformed into E. coli BL21(DE3) cells. Cells were induced with 1 mM isopropyl-1-thio-β-D-galactopyranoside at A600 of 0.8, and grown for 15 h at 25°C. The cells were harvested by centrifugation and lysed by French-press in a buffer containing 20 mM tris-HCl, 500 mM NaCl, 5 mM β-mercaptoethanol (BME), pH 7.5 (buffer A), and a protease inhibitor (Roche Applied Science). The lysate was spun down and the supernatant loaded onto a nickel HiTrap column (GE healthcare). The 12.32 kDa Mtb PII protein was eluted with a 100 mL linear gradient of 10 to 500 mM imidazole in buffer A. The peak fractions were collected and dialyzed in the presence of 20 mM tris-HCl (pH 7.5), 50 mM NaCl, 5% glycerol, and 5 mM BME and concentrated to 15 mg/mL using an amicon filter (Amicon) as measured by the Bradford protein assay (Bio-Rad).
Crystallization
Initial crystallization conditions were obtained at 289 K in 96-well plates by sitting-drop vapor diffusion. Crystals of the Mtb apo PII were obtained by mixing equal volumes of 15 mg/mL protein with a crystallization solution from Crystal Screen™ (Hampton Research) containing 0.1 M sodium cacodylate pH 6.5 and 1.4 M sodium acetate as precipitant. The Mtb PII:ATP binary complex was prepared by mixing the purified 15 mg/mL protein solution with ATP (Sigma) at a final concentration of 5 mM and incubating on ice for 30 min. Crystals of Mtb PII:ATP were obtained in condition containing 0.1 M imidazole pH 6.0 and 1.0 M sodium acetate, also from Crystal Screen™.
Data collection, structure determination, and refinement
X-ray diffraction data for Mtb PII and Mtb PII:ATP crystals were collected on APS beamline 19-ID and 23-ID, respectively. For data collection, a single crystal was cryoprotected by brief soaking in N-paratone and then flash-frozen in a liquid N2 stream (100 K). The HKL-2000 suite49 of programs was used for integration and scaling of the PII and PII:ATP complex structures. Crystals of the Mtb apo PII protein belong to the space group R3 with unit cell parameters a = b = 77.15 Å, c = 51.84 Å with one molecule per asymmetric unit and an estimated solvent content of 48.46%. Crystals of the Mtb PII:ATP belong to the space group P43212 with unit cell parameters a = b = 69.74 Å, c = 146.68 Å with three molecules per asymmetric unit and an estimated solvent content of 48.1%. Data collection details are summarized in Table I.
The structure of Mtb apo PII was solved by molecular replacement using Phaser.50 The truncated E. coli PII structure (PDB ID: 1GNK) lacking the T-loop residues 38–54, was used as search model to solve the Mtb apo PII crystal structure.30 The Mtb apo PII structure was used to solve the three positions of Mtb PII:ATP structure. After rigid body and restrained refinement using CCP4–REFMAC551,52 the model building was carried out in COOT53 and XTALVIEW54 for both the structures, solvent molecules were subsequently added. Bias-minimized electron density maps were obtained using the Shake&wARP (SNW) protocol.55 Each subunit of the Mtb PII:ATP structure contained one molecule of ATP with clear electron density (Supporting information Fig. 1).55,57 and were included in the Refmac refinement. The final R-factor for the Mtb apo PII crystal structure was 20.80% (Rfree = 22.60%) at 1.4 Å resolution and for the Mtb PII:ATP structure it was 21.57% (Rfree = 29.21%) at 2.4 Å resolution (Table I).
Acknowledgments
We appreciate the support of staff scientists at beamlines, 19-ID and 23-ID of the Advanced Photon Source, Argonne National Laboratory for their help in data collection. We also thank Misty D. Watson for her excellent technical assistance and Tracey Musa for her comments on the manuscript.
Glossary
Abbreviations:
- Mtb
Mycobacterium tuberculosis
- M. jannaschii
Methanococcus jannaschii
- E. coli
Escherichia coli
- M. smegmatis
Mycobacterium smegmatis
- 2OG
2-oxoglutarate
- GS
glutamine synthetase
- glnD
uridylyl transferase
- glnE/ATase
adenylyl transferase
- AmtB
ammonia/ammonium transporter
- AmtR
transcriptional regulator/repressor
- GlnR
transcriptional regulator/activator
- ATP
adenosine triphosphate
- ADP
adenosine diphosphate
- BME
β-mercaptoethanol.
References
- 1.Ninfa AJ, Atkinson MR. PII signal transduction proteins. Trends Microbiol. 2000;8:172–9. doi: 10.1016/s0966-842x(00)01709-1. [DOI] [PubMed] [Google Scholar]
- 2.Arcondeguy T, Jack R, Merrick M. P(II) signal transduction proteins, pivotal players in microbial nitrogen control. Microbiol Mol Biol Rev. 2001;65:80–105. doi: 10.1128/MMBR.65.1.80-105.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jiang P, Peliska JA, Ninfa AJ. The regulation of Escherichia coli glutamine synthetase revisited: role of 2-ketoglutarate in the regulation of glutamine synthetase adenylylation state. Biochemistry. 1998;37:12802–10. doi: 10.1021/bi980666u. [DOI] [PubMed] [Google Scholar]
- 4.Engleman EG, Francis SH. Cascade control of E. coli glutamine synthetase. II. Metabolite regulation of the enzymes in the cascade. Arch Biochem Biophys. 1978;191:602–12. doi: 10.1016/0003-9861(78)90398-3. [DOI] [PubMed] [Google Scholar]
- 5.Ehlers C, Weidenbach K, Veit K, Forchhammer K, Schmitz RA. Unique mechanistic features of post-translational regulation of glutamine synthetase activity in Methanosarcina mazei strain Go1 in response to nitrogen availability. Mol Microbiol. 2005;55:1841–54. doi: 10.1111/j.1365-2958.2005.04511.x. [DOI] [PubMed] [Google Scholar]
- 6.Ninfa AJ, Magasanik B. Covalent modification of the glnG product, NRI, by the glnL product, NRII, regulates the transcription of the glnALG operon in Escherichia coli. Proc Natl Acad Sci U S A. 1986;83:5909–13. doi: 10.1073/pnas.83.16.5909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang Y, Pohlmann EL, Roberts GP. GlnD is essential for NifA activation, NtrB/NtrC-regulated gene expression, and posttranslational regulation of nitrogenase activity in the photosynthetic, nitrogen-fixing bacterium Rhodospirillum rubrum. J Bacteriol. 2005;187:1254–65. doi: 10.1128/JB.187.4.1254-1265.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Heeswijk WC, Hoving S, Molenaar D, Stegeman B, Kahn D, Westerhoff HV. An alternative PII protein in the regulation of glutamine synthetase in Escherichia coli. Mol Microbiol. 1996;21:133–46. doi: 10.1046/j.1365-2958.1996.6281349.x. [DOI] [PubMed] [Google Scholar]
- 9.Javelle A, Merrick M. Complex formation between AmtB and GlnK: an ancestral role in prokaryotic nitrogen control. Biochem Soc Trans. 2005;33:170–2. doi: 10.1042/BST0330170. [DOI] [PubMed] [Google Scholar]
- 10.Thomas G, Coutts G, Merrick M. The glnKamtB operon. A conserved gene pair in prokaryotes. Trends Genet. 2000;16:11–4. doi: 10.1016/s0168-9525(99)01887-9. [DOI] [PubMed] [Google Scholar]
- 11.Khademi S, O'Connell J, 3rd, Remis J, Robles-Colmenares Y, Miercke LJ, Stroud RM. Mechanism of ammonia transport by Amt/MEP/Rh: structure of AmtB at 1.35 Å. Science. 2004;305:1587–94. doi: 10.1126/science.1101952. [DOI] [PubMed] [Google Scholar]
- 12.Durand A, Merrick M. In vitro analysis of the Escherichia coli AmtB-GlnK complex reveals a stoichiometric interaction and sensitivity to ATP and 2-oxoglutarate. J Biol Chem. 2006;281:29558–67. doi: 10.1074/jbc.M602477200. [DOI] [PubMed] [Google Scholar]
- 13.Strosser J, Ludke A, Schaffer S, Kramer R, Burkovski A. Regulation of GlnK activity: modification, membrane sequestration and proteolysis as regulatory principles in the network of nitrogen control in Corynebacterium glutamicum. Mol Microbiol. 2004;54:132–47. doi: 10.1111/j.1365-2958.2004.04247.x. [DOI] [PubMed] [Google Scholar]
- 14.Read R, Pashley CA, Smith D, Parish T. The role of GlnD in ammonia assimilation in Mycobacterium tuberculosis. Tuberculosis (Edinb) 2007;87:384–90. doi: 10.1016/j.tube.2006.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hesketh A, Fink D, Gust B, Rexer HU, Scheel B, Chater K, Wohlleben W, Engels A. The GlnD and GlnK homologues of Streptomyces coelicolor A3(2) are functionally dissimilar to their nitrogen regulatory system counterparts from enteric bacteria. Mol Microbiol. 2002;46:319–30. doi: 10.1046/j.1365-2958.2002.03149.x. [DOI] [PubMed] [Google Scholar]
- 16.Conroy MJ, Durand A, Lupo D, Li XD, Bullough PA, Winkler FK, Merrick M. The crystal structure of the Escherichia coli AmtB-GlnK complex reveals how GlnK regulates the ammonia channel. Proc Natl Acad Sci U S A. 2007;104:1213–8. doi: 10.1073/pnas.0610348104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huergo LF, Merrick M, Pedrosa FO, Chubatsu LS, Araujo LM, Souza EM. Ternary complex formation between AmtB, GlnZ and the nitrogenase regulatory enzyme DraG reveals a novel facet of nitrogen regulation in bacteria. Mol Microbiol. 2007;66:1523–35. doi: 10.1111/j.1365-2958.2007.06016.x. [DOI] [PubMed] [Google Scholar]
- 18.Huergo LF, Chubatsu LS, Souza EM, Pedrosa FO, Steffens MB, Merrick M. Interactions between PII proteins and the nitrogenase regulatory enzymes DraT and DraG in Azospirillum brasilense. FEBS Lett. 2006;580:5232–6. doi: 10.1016/j.febslet.2006.08.054. [DOI] [PubMed] [Google Scholar]
- 19.Heinrich A, Woyda K, Brauburger K, Meiss G, Detsch C, Stulke J, Forchhammer K. Interaction of the membrane-bound GlnK-AmtB complex with the master regulator of nitrogen metabolism TnrA in Bacillus subtilis. J Biol Chem. 2006;281:34909–17. doi: 10.1074/jbc.M607582200. [DOI] [PubMed] [Google Scholar]
- 20.Jakoby M, Nolden L, Meier-Wagner J, Kramer R, Burkovski A. AmtR, a global repressor in the nitrogen regulation system of Corynebacterium glutamicum. Mol Microbiol. 2000;37:964–77. doi: 10.1046/j.1365-2958.2000.02073.x. [DOI] [PubMed] [Google Scholar]
- 21.Wray LV, Jr., Atkinson MR, Fisher SH. Identification and cloning of the glnR locus, which is required for transcription of the glnA gene in Streptomyces coelicolor A3(2) J Bacteriol. 1991;173:7351–60. doi: 10.1128/jb.173.22.7351-7360.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fink D, Weissschuh N, Reuther J, Wohlleben W, Engels A. Two transcriptional regulators GlnR and GlnRII are involved in regulation of nitrogen metabolism in Streptomyces coelicolor A3(2) Mol Microbiol. 2002;46:331–47. doi: 10.1046/j.1365-2958.2002.03150.x. [DOI] [PubMed] [Google Scholar]
- 23.Tiffert Y, Supra P, Wurm R, Wohlleben W, Wagner R, Reuther J. The Streptomyces coelicolor GlnR regulon: identification of new GlnR targets and evidence for a central role of GlnR in nitrogen metabolism in actinomycetes. Mol Microbiol. 2008;67:861–80. doi: 10.1111/j.1365-2958.2007.06092.x. [DOI] [PubMed] [Google Scholar]
- 24.Mizuno Y, Moorhead GB, Ng KK. Structural basis for the regulation of N-acetylglutamate kinase by PII in Arabidopsis thaliana. J Biol Chem. 2007;282:35733–40. doi: 10.1074/jbc.M707127200. [DOI] [PubMed] [Google Scholar]
- 25.Llacer JL, Contreras A, Forchhammer K, Marco-Marin C, Gil-Ortiz F, Maldonado R, Fita I, Rubio V. The crystal structure of the complex of PII and acetylglutamate kinase reveals how PII controls the storage of nitrogen as arginine. Proc Natl Acad Sci U S A. 2007;104:17644–9. doi: 10.1073/pnas.0705987104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE, 3rd, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Krogh A, McLean J, Moule S, Murphy L, Oliver K, Osborne J, Quail MA, Rajandream MA, Rogers J, Rutter S, Seeger K, Skelton J, Squares R, Squares S, Sulston JE, Taylor K, Whitehead S, Barrell BG. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–44. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 27.Rengarajan J, Bloom BR, Rubin EJ. Genome-wide requirements for Mycobacterium tuberculosis adaptation and survival in macrophages. Proc Natl Acad Sci U S A. 2005;102:8327–32. doi: 10.1073/pnas.0503272102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goulding CW, Apostol M, Anderson DH, Gill HS, Smith CV, Kuo MR, Yang JK, Waldo GS, Suh SW, Chauhan R, Kale A, Bachhawat N, Mande SC, Johnston JM, Lott JS, Baker EN, Arcus VL, Leys D, McLean KJ, Munro AW, Berendzen J, Sharma V, Park MS, Eisenberg D, Sacchettini J, Alber T, Rupp B, Jacobs W, Jr., Terwilliger TC. The TB structural genomics consortium: providing a structural foundation for drug discovery. Curr Drug Targets Infect Disord. 2002;2:121–41. doi: 10.2174/1568005023342551. [DOI] [PubMed] [Google Scholar]
- 29.Carr PD, Cheah E, Suffolk PM, Vasudevan SG, Dixon NE, Ollis DL. X-ray structure of the signal transduction protein from Escherichia coli at 1.9 Å. Acta Crystallogr D Biol Crystallogr. 1996;52:93–104. doi: 10.1107/S0907444995007293. [DOI] [PubMed] [Google Scholar]
- 30.Xu Y, Cheah E, Carr PD, van Heeswijk WC, Westerhoff HV, Vasudevan SG, Ollis DL. GlnK, a PII-homologue: structure reveals ATP binding site and indicates how the T-loops may be involved in molecular recognition. J Mol Biol. 1998;282:149–65. doi: 10.1006/jmbi.1998.1979. [DOI] [PubMed] [Google Scholar]
- 31.Cheah E, Carr PD, Suffolk PM, Vasudevan SG, Dixon NE, Ollis DL. Structure of the Escherichia coli signal transducing protein PII. Structure. 1994;2:981–90. doi: 10.1016/s0969-2126(94)00100-6. [DOI] [PubMed] [Google Scholar]
- 32.Machado Benelli E, Buck M, Polikarpov I, Maltempi de Souza E, Cruz LM, Pedrosa FO. Herbaspirillum seropedicae signal transduction protein PII is structurally similar to the enteric GlnK. Eur J Biochem. 2002;269:3296–303. doi: 10.1046/j.1432-1033.2002.03011.x. [DOI] [PubMed] [Google Scholar]
- 33.Sakai H, Wang H, Takemoto-Hori C, Kaminishi T, Yamaguchi H, Kamewari Y, Terada T, Kuramitsu S, Shirouzu M, Yokoyama S. Crystal structures of the signal transducing protein GlnK from Thermus thermophilus HB8. J Struct Biol. 2005;149:99–110. doi: 10.1016/j.jsb.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 34.Gruswitz F, O'Connell J, 3rd, Stroud RM. Inhibitory complex of the transmembrane ammonia channel, AmtB, and the cytosolic regulatory protein, GlnK, at 1.96 Å. Proc Natl Acad Sci U S A. 2007;104:42–7. doi: 10.1073/pnas.0609796104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yildiz O, Kalthoff C, Raunser S, Kuhlbrandt W. Structure of GlnK1 with bound effectors indicates regulatory mechanism for ammonia uptake. Embo J. 2007;26:589–99. doi: 10.1038/sj.emboj.7601492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murzin AG, Lesk AM, Chothia C. Principles determining the structure of beta-sheet barrels in proteins. I. A theoretical analysis. J Mol Biol. 1994;236:1369–81. doi: 10.1016/0022-2836(94)90064-7. [DOI] [PubMed] [Google Scholar]
- 37.Murzin AG, Lesk AM, Chothia C. Principles determining the structure of beta-sheet barrels in proteins. II. The observed structures. J Mol Biol. 1994;236:1382–400. doi: 10.1016/0022-2836(94)90065-5. [DOI] [PubMed] [Google Scholar]
- 38.Schwarzenbacher R, von Delft F, Abdubek P, Ambing E, Biorac T, Brinen LS, Canaves JM, Cambell J, Chiu HJ, Dai X, Deacon AM, DiDonato M, Elsliger MA, Eshagi S, Floyd R, Godzik A, Grittini C, Grzechnik SK, Hampton E, Jaroszewski L, Karlak C, Klock HE, Koesema E, Kovarik JS, Kreusch A, Kuhn P, Lesley SA, Levin I, McMullan D, McPhillips TM, Miller MD, Morse A, Moy K, Ouyang J, Page R, Quijano K, Robb A, Spraggon G, Stevens RC, van den Bedem H, Velasquez J, Vincent J, Wang X, West B, Wolf G, Xu Q, Hodgson KO, Wooley J, Wilson IA. Crystal structure of a putative PII-like signaling protein (TM0021) from Thermotoga maritima at 2.5 Å resolution. Proteins. 2004;54:810–3. doi: 10.1002/prot.10647. [DOI] [PubMed] [Google Scholar]
- 39.Xu Y, Carr PD, Clancy P, Garcia-Dominguez M, Forchhammer K, Florencio F, Vasudevan SG, Tandeau de Marsac N, Ollis DL. The structures of the PII proteins from the cyanobacteria Synechococcus sp. PCC 7942 and Synechocystis sp. PCC 6803. Acta Crystallogr D Biol Crystallogr. 2003;59:2183–90. doi: 10.1107/s0907444903019589. [DOI] [PubMed] [Google Scholar]
- 40.Nichols CE, Sainsbury S, Berrow NS, Alderton D, Saunders NJ, Stammers DK, Owens RJ. Structure of the PII signal transduction protein of Neisseria meningitidis at 1.85 Å resolution. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2006;62:494–7. doi: 10.1107/S1744309106015430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Walker JE, Saraste M, Runswick MJ, Gay NJ. Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. Embo J. 1982;1:945–51. doi: 10.1002/j.1460-2075.1982.tb01276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bandyopadhyay A, Arora A, Jain S, Laskar A, Mandal C, Ivanisenko VA, Fomin ES, Pintus SS, Kolchanov NA, Maiti S, Ramachandran S. Expression and molecular characterization of the Mycobacterium tuberculosis PII protein. J Biochem. 2010;147:279–89. doi: 10.1093/jb/mvp174. [DOI] [PubMed] [Google Scholar]
- 43.Schwede T, Kopp J, Guex N, Peitsch MC. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 2003;31:3381–5. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mizuno Y, Berenger B, Moorhead GB, Ng KK. Crystal structure of Arabidopsis PII reveals novel structural elements unique to plants. Biochemistry. 2007;46:1477–83. doi: 10.1021/bi062149e. [DOI] [PubMed] [Google Scholar]
- 45.Bueno R, Pahel G, Magasanik B. Role of glnB and glnD gene products in regulation of the glnALG operon of Escherichia coli. J Bacteriol. 1985;164:816–22. doi: 10.1128/jb.164.2.816-822.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Burkovski A. Nitrogen control in Corynebacterium glutamicum: proteins, mechanisms, signals. J Microbiol Biotechnol. 2007;17:187–94. [PubMed] [Google Scholar]
- 47.Reuther J, Wohlleben W. Nitrogen metabolism in Streptomyces coelicolor: transcriptional and post-translational regulation. J Mol Microbiol Biotechnol. 2007;12:139–46. doi: 10.1159/000096469. [DOI] [PubMed] [Google Scholar]
- 48.Xu Y, Carr PD, Huber T, Vasudevan SG, Ollis DL. The structure of the PII-ATP complex. Eur J Biochem. 2001;268:2028–37. doi: 10.1046/j.1432-1327.2001.02074.x. [DOI] [PubMed] [Google Scholar]
- 49.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–26. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 50.McCoy AJ. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr D Biol Crystallogr. 2007;63:32–41. doi: 10.1107/S0907444906045975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vagin AA, Steiner RA, Lebedev AA, Potterton L, McNicholas S, Long F, Murshudov GN. REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr D Biol Crystallogr. 2004;60:2184–95. doi: 10.1107/S0907444904023510. [DOI] [PubMed] [Google Scholar]
- 52.Collaborative Computational Project Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–3. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 53.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 54.McRee DE. XtalView/Xfit–A versatile program for manipulating atomic coordinates and electron density. J Struct Biol. 1999;125:156–65. doi: 10.1006/jsbi.1999.4094. [DOI] [PubMed] [Google Scholar]
- 55.Reddy V, Swanson SM, Segelke B, Kantardjieff KA, Sacchettini JC, Rupp B. Effective electron-density map improvement and structure validation on a Linux multi-CPU web cluster: The TB Structural Genomics Consortium Bias Removal Web Service. Acta Crystallogr D Biol Crystallogr. 2003;59:2200–10. doi: 10.1107/s0907444903020316. [DOI] [PubMed] [Google Scholar]
- 56.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 57.Merritt EA, Murphy ME. Raster3D Version 2.0. A program for photorealistic molecular graphics. Acta Crystallogr D Biol Crystallogr. 1994;50:869–73. doi: 10.1107/S0907444994006396. [DOI] [PubMed] [Google Scholar]