

Abstract
Protein stability and ligand-binding affinity measurements are widely required for the formulation of biopharmaceutical proteins, protein engineering and drug screening within life science research. Current techniques either consume too much of often precious biological or compound materials, in large sample volumes, or alternatively require chemical labeling with fluorescent tags to achieve measurements at submicrolitre volumes with less sample. Here we present a quantitative and accurate method for the determination of protein stability and the affinity for small molecules, at only 1.5–20 nL optical sample volumes without the need for fluorescent labeling, and that takes advantage of the intrinsic tryptophan fluorescence of most proteins. Coupled to appropriate microfluidic sample preparation methods, the sample requirements could thus be reduced 85,000-fold to just 108 molecules. The stability of wild-type FKBP-12 and a destabilizing binding-pocket mutant are studied in the presence and absence of rapamycin, to demonstrate the potential of the technique to both drug screening and protein engineering. The results show that 75% of the interaction energy between FKBP-12 and rapamycin originates from residue Phe99 in the binding site.
Keywords: drug screening, FKBP-12, microfluidics, protein fluorescence, Rapamycin
Introduction
There is a significant need for high-throughput techniques that are capable of measuring dissociation constants for combinatorially-derived compound libraries, towards drug susceptible protein targets.1 The need is increasingly pressing because previously containable pathogenic organisms such as Mycobacterium tuberculosis, the seventh leading cause of death worldwide,2 Clostridium difficiles, and Streptococcus aureus continue to develop multidrug resistance. At the same time, new targets are being identified through genome sequencing and proteomics at an increasing pace.3,4 Identifying new drug leads from large chemical libraries requires sensitive high-throughput assays for measuring protein-drug interactions. Existing approaches rely heavily on microplate-based assays, use large quantities of precious protein target and drug compound samples, require high capital investment in automation platforms, have increased errors due to multiple small volume injections, and are prone to evaporation of sample liquids.5 Furthermore, most assays are not generally applicable to measuring a multitude of protein-drug interactions. There is therefore a need for smaller volume assays that conserve samples and can be operated with greater throughput and accuracy.
We have previously established a microplate-based technique for measuring protein stability to denaturants, the effects of mutations, complex denaturation pathways, and ligand affinities, based on the classical measurement of intrinsic protein fluorescence,6,7 removing the need for extrinsic fluorophores such as 1-anilino-8-naphthalene-sulfonate (ANS) used in earlier experiments at the microplate scale,8 but which can alter the solubility and stability of proteins and also their interactions with other molecules. While this technique improved throughput considerably over autotitration methods in fluorometers,9 there is a further need for increased throughput and decreased sample volume to match the increasing size and small-scale syntheses of combinatorial compound libraries in drug discovery,10 for the evolutionary engineering of proteins,11 and for protein bioprocess formulation.12
Towards this aim, microfluidic techniques have been described previously for monitoring the denaturation or refolding of proteins by the measurement of extrinsically added fluorophores,13 or Forster resonant energy transfer (FRET) with fluorescent protein labels.14,15 The binding affinity between two proteins has also been achieved using two-photon excitation and a fluorescent label.16 However, the addition of fluorescent labels to proteins can affect their ligand binding, solubility and stability, and it is also difficult to ensure the attachment of only a single label per protein molecule.17,18 The measurement of intrinsic protein fluorescence has also been demonstrated previously in microchannels for the label-free detection of proteins and their conformational changes, by using lasers19-21 or light-emitting diodes (LED's)22,23 for the excitation of samples. However, these techniques have not yet been sufficiently accurate to obtain thermodynamic parameters such as the free-energy of protein unfolding, as is required for use in drug-discovery, formulation or protein engineering applications. More recently, fluorescence lifetime microscopy has been used to detect the interaction between two label-free proteins at the single molecule level.17 The sensitivity of this technique took advantage of the interaction of two large proteins beta-galactosidase (116 kDa) and an antibody (ca. 150 kDa). However it is not yet able to determine dissociation constants for proteins, measure the conformation and stability of proteins, or detect and quantify the interaction of a protein with small drug-like molecules or ligands. Greater sensitivity at the single molecule level has been achieved recently for the measurement of conformational changes by fluorescence correlation spectroscopy, though this again relies upon the labeling of proteins with a strong fluorophore such as Alexa or AttoOxa11 dyes,24,25 or the use of highly fluorescent proteins such as yellow fluorescent protein (YFP).26
Here we present a microfluidic measurement technique that enables the thermodynamic stability of reversibly unfolding proteins, and the affinity of a small molecule drug compound for a target protein, to be determined using small samples and with high precision. The technique measures the intrinsic fluorescence intensity of proteins under a range of denaturing conditions to determine the stability of the target protein. This is repeated in the presence of a drug compound and the resulting stabilization used to estimate the binding affinity of the compound to the protein. We demonstrate this technique for the first time in a microfluidic system by measuring the binding of rapamycin to the 12 kDa FK506 binding protein (FKBP-12), which is a target for prostate cancer treatment,27 immunosuppression28,29 and most recently for aging in mice.30 We also compare the results with those obtained using an equivalent microplate-based assay.
FKBP is a peptidyl-prolyl isomerase enzyme and also a cytosolic immunophilin found in many mammalian cell types. In the latter role, it is known to inhibit T-cell proliferation via the calcineurin-dependent signal transduction pathway, in the presence of FK506 or rapamycin.31 For this purpose, rapamycin and FK506 are both used as immunosuppressants to minimize organ rejection after transplant surgery, or in patients that suffer from autoimmune disorders. The structure of native human FKBP-12 has been obtained both in the presence and absence of rapamycin, by X-ray crystallography32 and NMR.33,34 The native protein contains an amphiphilic five-stranded antiparallel β-sheet packed against an amphiphilic α-helix via a hydrophobic core. The structure forms a large hydrophobic binding pocket to which rapamycin associates with high affinity (Fig. 1). It has been shown previously to denature in a simple two-state transition35 which apparently remains two-state when the native state is stabilized by the association of rapamycin,36 and also for a considerable series of destabilizing and stabilizing mutations.37 Here we have used the new microfluidic technique for the measurement of protein stability and rapamycin dissociation constants for the wild-type FKBP-12, and also for a previously unpublished mutant of FKBP-12 (F99L). Residue Phe99 is in the binding site for rapamycin (Fig. 1) and is also involved in protein stabilization. We use the free energies determined by the capillary-scale technique in a double-mutant cycle type of analysis to determine the intermolecular coupling energy between rapamycin and Phe99.
Figure 1.
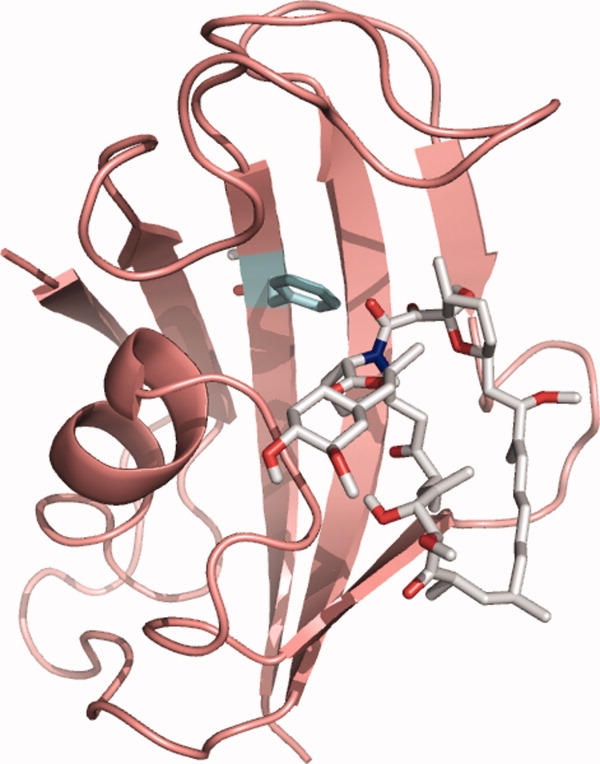
Structure of FKBP-12 bound to rapamycin. Residue F99 (mutated to Leucine in F99L) is highlighted in the binding pocket (cyan sticks). Bound rapamycin is shown in gray sticks. The image was rendered using the PyMOL molecular graphics system38 from the protein databank file 1fkb.
To demonstrate our nanolitre-scale optical measurement advances we have used microlitre-scale equipment for the sample preparation and handling. However, our new technique demonstrates the potential for rapid drug, or protein stability screening by achieving a reduction in sample optical measurement volume from 260 μL in microplates, down to 1.5–20 nL in the microfluidic system, and a limit of detection reduced over 85,000-fold from 1.2 × 1013 in microplates to 1.4 × 108 molecules in the microfluidic system. The technique is therefore suitable for coupling directly to nanolitre volume microfluidic devices for sample preparation and handling.
Results and Discussion
The microcapillary-based fluorometer we designed and built is shown schematically in Figure 2(a,b) and the set up described in detail in the “Methods” section. Figure 2(c) shows the raw PMT/PD ratio throughout a cycle of preparation, sample measurement and cleaning. The signal is stable for the initial buffer measurement, and after cleaning and reverting to buffer, the same signal returns, indicating complete removal of any protein that binds to the capillary surface over prolonged measurement times. The sample measurement time was deliberately extended in the cycle shown in Figure 2(c) to demonstrate both the signal stability and also to show that the cleaning cycle is capable of removing protein from the capillary even for much longer exposure times than are required for accurate measurement. Occasional peaks are observed when the air is passed through the capillary. These are due to the air de-wetting the capillary surface and pushing out the resulting water droplets.
Figure 2.
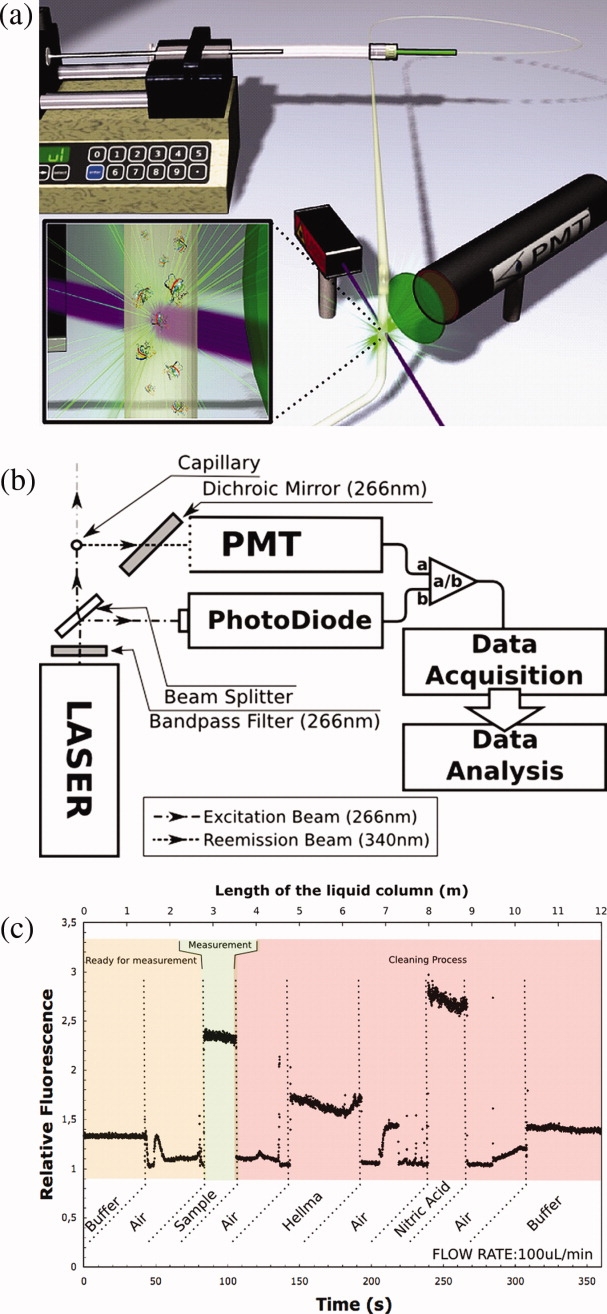
The microcapillary fluorometer system. Schematic configuration of (a) the fluidics, and (b) the optics: components are described in text. (c) Output for one complete cycle of channel preparation, sample fluorescence measurement and channel cleaning. An extended measurement time is shown to demonstrate the signal stability.
The sensitivity, linearity, and dynamic range of the capillary based fluorometer was determined from the fluorescence intensities of BSA solutions at different protein concentrations, as shown in Figure 3. The fluorescence emission was linearly proportional to the protein concentration with an R2 of 0.994, and a standard error for each measurement of 0.015 RFU at above 1.5 μM BSA (from 100 laser pulses). The dynamic range of the linear response was 0.15 μM (0.01 mg/mL) to at least 75 μM (5 mg/mL) of BSA, corresponding to 0.45 to 225 μM tryptophan residues. The limit of detection was 0.15 μM BSA, and the signal-to-noise based on the standard deviation value ranged from 1.27 at 0.15 μM to 1568 at 75 μM BSA. The background fluorescence of the buffer was subtracted from all measurements and had a relative fluorescence intensity of 16.15 with a signal-to-noise of 323. To maintain the dynamic range and sensitivity it was necessary to alter the voltage applied to the PMT from 500 V for measurements between 1.5 μM and 75 μM BSA, to 600 V for less than 1.5 μM. The higher PMT voltage resulted in an increased (electronic) noise-induced measurement error.
Figure 3.
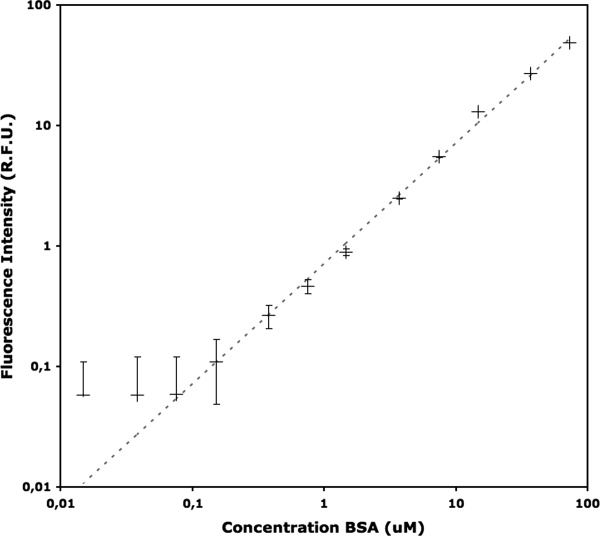
Linearity and sensitivity of fluorescence intensity measurements in the microcapillary. Fluorescence intensities in relative fluorescence units (RFU) measured at a range of bovine serum albumin (BSA) concentrations in sodium phosphate buffer, pH 7.0, 21°C using the capillary technique. Each data point was obtained from an average of 100 laser pulses at 1 kHz. Error bars shown are standard deviations. The dashed curve shows a linear fit to the logarithmic data.
The limit of detection obtained in the capillary technique is compared to that obtained in a microplate in Table I. While the microplate reader was able to detect approximately half the concentration of BSA, the volume required was over 105 times greater than for the capillary technique. This gave a limit of detection of 1.4 × 108 protein molecules in the capillary, which is 85,000-fold lower than for the microplate reader.
Table I.
Limits of Detection for BSA Using Fluorescence at Different Scales
| Technique | Minimum [protein] (μM) | Measurement volume (L) | Number of protein molecules |
|---|---|---|---|
| Microplate | 0.076 | 2.6 × 10−4 | 1.16 × 1013 |
| Micro-capillary | 0.15 | 1.5 × 10−9a | 1.4 × 108 |
Reduction of the actual sample volume needed to the minimum 1.5 nL optical sample volume will require the optical technique to be coupled to an appropriate microfluidic sample preparation and handling device. The dead volume in our standard commercial syringe pumps increased the total sample volume for the present test measurements to 50 μL.
BSA was used initially to optimize the capability of the technique to obtain thermodynamic parameters from the equilibrium denaturation curves of proteins. The effect of urea denaturant concentration upon the measured fluorescence intensity of a 7.14 μM BSA solution at pH 7.2 is shown in Figure 4 for both the capillary-based and microplate methods. The raw data in Figure 4(a) demonstrate the need for continuously monitoring the incident laser intensity with a photodiode (PD) and also illustrate the good signal-to-noise obtained with BSA. Figure 4(b) shows the time-dependence of the processed data as each sample enters the optical measurement zone. These data are then used to obtain the plot, in Figure 4(c), of denaturant versus fluorescence. A sharp sigmoidal transition is observed, as expected for the cooperative two-state unfolding of proteins upon addition of a chemical denaturant.39 The thermodynamic parameters obtained by fitting the data to Eqs. (2) and (3) are shown in Table II. Transition midpoints (C1/2), of 3.7 ± 0.2 M and 3.6 ± 0.2 M were obtained by the capillary and microplate techniques respectively. The associated m-values (mG) are more difficult to determine accurately and the relatively low density of datapoints in this initial test explains the differences between the values obtained of 1.9 ± 0.2 kcal mol−1 M−1 and 1.2 ± 0.2 kcal mol−1 M−1 respectively for the capillary and microplate techniques.
Figure 4.

Fluorescence intensity measurements of BSA denaturation at equilibrium. (a) Typical raw data obtained in the micro-capillary method for a single replicate as fluorescence emission at the photomuliplier tube (PMT) and excitation laser intensity at the photodiode (PD) for a typical denaturation curve acquisition. The final fluorescence measurement (labeled “processed dataset”) was obtained using Eq. (1) as the ratio of the PMT to the PD signals, to account for time-dependent variations of the incident laser source between measurements, with a similar ratio for the blank subtracted. These data are then normalized to unity for high [urea] to produce the curves shown in panel c. (b) Typical time-dependent signal processed as in panel a, at various urea concentrations. The signal jumps initially as the sample enters the optical measurement zone. A signal for buffer only is shown for comparison. (c) Comparison of capillary (open symbols) and 96-well microplate-based (closed symbols) equilibrium denaturation of 7.14 μM BSA by urea as measured by the change in the normalized intrinsic fluorescence intensity of samples in 50 mM Tris.HCl, pH 7.2, 22°C. Error bars shown are standard deviations from triplicate measurements. The best fits to Eq. (2) are also shown for each dataset.
Table II.
Thermodynamic Parameters for the Denaturation of BSA, FKBP-12 wt and FKBP-12 F99L
| Experiment | C1/2a (M) | mG (kcal mol−1 M−1) | ΔG〈mG〉b (kcal mol−1) | ΔGH2Oc (kcal mol−1) | ΔΔGX-WTd (kcal mol−1) |
|---|---|---|---|---|---|
| Capillarymethod | |||||
| FKBP-12 wt | 0.74 ± 0.02 | 5.4 ± 0.7 | 3.84 ± 0.3 | 4.03 ± 0.5 | 0 |
| FKBP-12 wt +Rapamycin | 2.28 ± 0.02 | 3.5 ± 0.3 | 7.70 ± 0.5 | 8.08 ± 0.7 | 4.05 ± 0.9 |
| FKBP-12 F99L | 0.63 ± 0.03 | 4.9 ± 0.8 | 3.26 ± 0.3 | 3.11 ± 0.5 | −0.92 ±0.7 |
| FKBP-12 F99L +Rapamycin | 1.31 ± 0.05 | 3.2 ± 0.7 | 4.41 ± 0.3 | 4.20 ± 0.9 | 0.17 ± 1.0 |
| BSA | 3.7 ± 0.2 | 1.9 ± 0.2 | na | 7.0 ± 0.8 | na |
| Microplatemethod | |||||
| FKBP-12 wt | 0.77 ± 0.01 | 6.6 ± 0.4 | 4.72 ± 0.3 | 5.05 ± 0.3 | 0 |
| FKBP-12 wt +Rapamycin | 2.36 ± 0.01 | 3.7 ± 0.2 | 8.26 ± 0.4 | 8.73 ± 0.4 | 3.68 ± 0.5 |
| FKBP-12 F99L | 0.69 ± 0.01 | 5.7 ± 0.4 | 4.23 ± 0.3 | 3.9 ± 0.3 | −1.15 ± 0.4 |
| FKBP-12 F99L +Rapamycin | 1.53 ± 0.01 | 3.3 ± 0.2 | 5.36 ± 0.3 | 5.05 ± 0.3 | 0 ± 0.4 |
| BSA e | 3.6 ± 0.2 | 1.2 ± 0.2 | na | 4.4 ± 0.8 | na |
| Literature | |||||
| FKBP-12 wt f | 0.78 ± 0.005 | 6.6 ± 0.3 | na | 5.13 ± 0.2 | 0 |
| FKBP-12 wt +Rapamycin f | 2.10 ± 0.01 | 3.9 ± 0.3 | na | 8.21± 0.6 | 3.08 ± 0.6 |
All errors quoted on C1/2 and mG are curve fit errors given in Sigmaplot (Systat Software, Hounslow, UK) which indicate the range of values possible without significantly altering R2 for the fit. Wild-type FKBP-12 measurements were at 10 μM protein, and FL99 FKBP-12 at 11.1 μM, 50 mM Tris.HCl, pH 7.5, 1 mM DTT.
Transition mid-points are quoted for GdnHCl as denaturant except for BSA where urea was used.
ΔG〈mG〉 values were obtained using the average mG values for each protein type.
ΔGH2O values were obtained using the mG values from each independent curve fit.
ΔΔGX-WT values are all relative to wild-type FKBP-12 without rapamycin and are obtained from ΔGH2O values.
From Aucamp et al. 2005.6
From Main et al. 1999.40
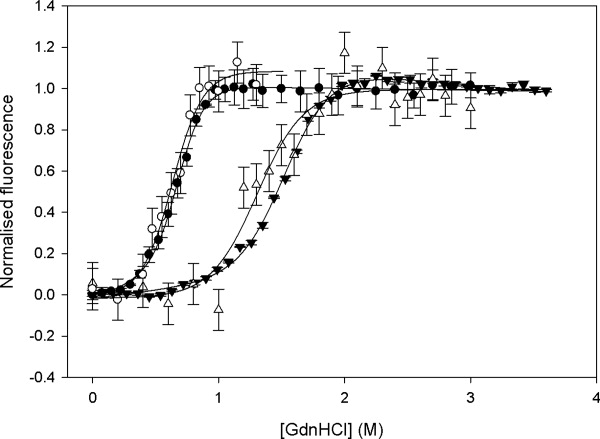
Our main aim was to demonstrate the use of a capillary-based fluorescence technique to obtain dissociation constants for compounds binding to a protein target of interest. The principle for determining dissociation constants from a change in protein stability has been demonstrated previously,41 and is shown in Figure 5 using FKBP-12 as an example. This method is particularly useful for characterizing very tightly binding ligands and also for compounds that equilibrate slowly between bound and unbound forms. Chemical denaturation of wild-type and the mutant FL99 of FKBP-12 by guanidine hydrochloride (GdnHCl) was carried out in a microplate using our previous technique6 and the same samples then measured directly afterwards using the capillary technique. The denaturation curves for wild-type FKBP-12 are compared directly in Figure 6 and show very good agreement between those measured in microplates and those made using the capillary-based technique. The raw data in Figure 6(a) again demonstrate the need for continuously monitoring the incident laser intensity with a PD and also illustrate the good signal-to-noise obtained. The curves for the F99L mutant are also compared in Figure 7. The sharp sigmoidal transition, expected for cooperative two-state unfolding, was observed in all cases. Tables II and III display the thermodynamic parameters obtained from each curve and also compares them to known values from the literature. As expected, the stability of both the wild-type FKBP-12 and the mutant F99L increase in the presence of rapamycin. For wild-type the unfolding free energy increases by between 3 and 4 kcal mol−1, whereas for F99L an increase of only 1.15 kcal mol−1 is observed indicating that the mutation affects the binding affinity of the rapamycin. The stability of the native protein is also decreased by 0.92 kcal mol−1 upon mutation to F99L which is in close agreement with the 1.2 ± 0.11 kcal mol−1destabilization observed previously in a standard fluorometer.42
Figure 5.
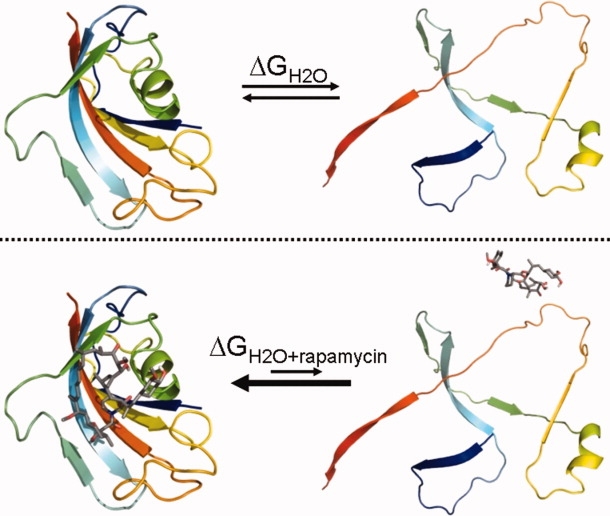
Method used to determine the ligand dissociation-constant of rapamycin with FKBP-12. Top: Equilibrium denaturation of FKBP-12. Bottom: Equilibrium denaturation of FKBP-12 in the presence of rapamycin. The equilibrium is shifted more towards the native state as the ligand stabilizes it. The native FKBP-12 structure shown and the interaction of rapamycin was rendered in the PyMOL molecular graphics system38 from the protein databank file 1fkb. The denatured state is schematic only. The dissociation constant for rapamycin binding can be derived from the change in the free energy of protein unfolding using Eq. (4). [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Figure 6.
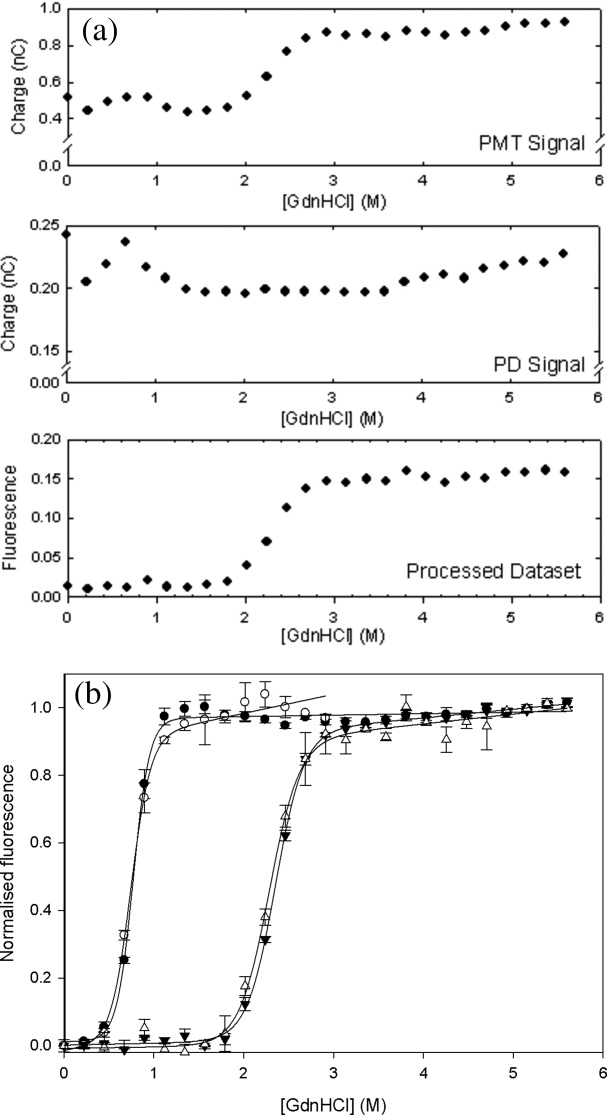
Fluorescence measurements of FKBP-12 denaturation at equilibrium. (a) Typical raw data obtained for a single replicate as fluorescence emission at the photomuliplier tube (PMT) and excitation laser intensity at the photodiode (PD) for a typical denaturation curve acquisition. The final fluorescence measurement (labeled “processed dataset”) was obtained using Eq. (1) as the ratio of the PMT to the PD signals, to account for time-dependent variations of the incident laser source between measurements, with a similar ratio for the blank subtracted. These data are then normalized to unity for high [GdnHCl] to produce the curves shown in panel b. (b) Effect of rapamycin on the equilibrium denaturation of 10 μM wild-type FKBP by guanidine HCl in the presence (triangles) and absence (circles) of 15 μM rapamycin, as measured by the change in the normalized intrinsic fluorescence intensity of samples in 50 mM Tris.HCl, pH 7.5, 24°C in a capillary (open symbols) and in a 96-well microplate (closed symbols). Error bars shown are standard deviations from triplicate measurements. The best fits to Eq. (2) are also shown for each dataset.
Figure 7.
Effect of rapamycin on the equilibrium denaturation of FKBP F99L by guanidine hydrochloride. Denaturation curves are shown in the presence (triangles) and absence (circles) of 20 (M rapamycin, as measured by the change in normalized intrinsic fluorescence intensity of samples in 50 mM Tris.HCl, pH 7.5, 22 °C in a capillary at 2 (M FKBP F99L (open symbols) and in a 96-well microplate at 10 (M FKBP F99L (closed symbols). Best fits to Eq. (2) are shown for each dataset.
Table III.
Rapamycin Dissociation Constants for FKBP-12 wt and FKBP-12 F99L
| ΔΔGbinding (kcal mol−1)a |
Kd (nM) |
||||
|---|---|---|---|---|---|
| Protein | Capillary b | Microplate b | Capillary | Microplate | Literature |
| FKBP-12 wt | 3.86 ± 0.6 | 3.54 ± 0.5 | 7.4 ± 4.7 | 12.8 ± 6.8 | 0.2c, 0.27d, 0.35e, 99.6f |
| FKBP-12 F99L | 1.15 ± 0.4 | 1.13 ± 0.4 | 1760 ± 810 | 1850 ± 780 | n.a. |
n.a., not available.
ΔΔGbinding and errors were determined from ΔG〈mG〉 and associated error values in Table II.
Capillary and microplate measurements were obtained with 10 μM FKBP-12 wt or 11.1 μM FL99 FKBP-12, in 15 or 20 μM rapamycin, 50 mM Tris. HCl at pH 7.5, 1 mM DTT, 25°C. Literature values were obtained by a range of techniques and conditions:
Radiolabeled competition assay with 1 nM FKBP, 0.3–10 nM rapamycin in 100 mM NaCl, 20 mM phosphate, 1 mM EDTA, pH 7.3, 0.015% Triton X-100.43
Surface plasmon resonance (SPR) with GST-FKBP fusion in PBS pH 7.4, 0.02% Tween-20, 50 nM rapamycin.44
Fluorescence polarization competition assay with a fluorescein-labeled synthetic ligand, 5 nM FKBP, 0.05–100 nM rapamycin, PBS pH 7.4, 0.011% Triton X-100, 0.1 mg/mL bovine γ-globulin.44
Calculated from available intrinsic protein fluorescence equilibrium denaturation data using a 900 μL sample of 2 μM FKBP-12, 20 μM rapamycin, 50 mM Tris.HCl at pH 7.5, 1 mM DTT, 25°C.40
The transition midpoints show very good agreement at all scales of measurement and show only a slight decrease in precision from 0.005 to 0.01 and 0.02 M GdnHCl for wild type at the respectively smaller scales. The m-values for equilibrium denaturation curves typically have the greatest errors which are the dominant contribution to errors in the calculated unfolding free energy. The m-values at different scales agree well within error, although for wild-type FKBP-12 in the absence of rapamycin the value in the capillary method is slightly lower than expected and this is reflected also in the lower free energy of unfolding obtained. The precision of the m-values obtained can in all cases typically be improved by either more repeats at each concentration or by a greater number of data points obtained for each curve, whereas the fluorescence measurements themselves have similar standard deviations for each method. However, a small systematic error associated with the capillary method itself cannot be ruled out, perhaps due to interaction with the capillary which has a greater surface-to-volume ratio than microplates or cuvettes. Photobleaching in the capillary does not occur at the flowrates used as the sample can only be excited once given the 200 μm laser spot and 1 kHz laser pulsing. Photobleaching was only observed at flowrates up to 1 μL min−1 (see Supporting Information Fig. S1).
Ligand dissociation constants can be obtained by measuring the free-energy of denaturation for native proteins in the presence and absence of the ligand.45 The dissociation constants for rapamycin binding to both wild-type and F99L FKBP-12, derived using the capillary technique, are compared to values obtained by other methods in Table III. For wild-type FKBP-12, literature values vary in the range 0.2–99.6 nM from various techniques, samples and solution conditions.40,43,44 The microplate-based fluorescence technique gave a value of 12.8 nM, and the capillary technique gave 7.4 nM, both consistent with the existing literature data. This demonstrates that the capillary technique can obtain useful dissociation constant measurements using significantly reduced quantities of sample.
The dissociation constant obtained for rapamycin and the F99L mutant of FKBP-12 was determined here for the first time to be 1.8 μM using the capillary technique, showing a 240-fold loss of affinity relative to wild-type. The intermolecular coupling energy between rapamycin and the mutated phenylalanine residue in FKBP-12 could also be calculated from the free-energies determined in the capillary by using a double-mutant cycle analysis46 where the interaction energy, ΔGint, is given by ΔGint = ΔGFL99, rap − ΔGFL99 − ΔGrap + ΔGwt. The interaction energy thus determined does not include the loss of protein stability resulting from the FL99 mutation, but reports only on the free energy for the interaction of rapamycin with the atoms removed by mutation. This interaction energy was found to be 3.0 ± 1.3 kcal mol−1, which is a significant proportion of the total interaction of 4.05 kcal mol−1 between FKBP-12 and rapamycin. It is remarkable that at least 50% of the interaction energy should be lost upon mutation of the Phe99 residue to leucine given that it contributes to only 3% of the FKBP-12 protein surface area that becomes solvent excluded upon binding to rapamycin.
Materials and Methods
Rapamycin was from LC Laboratories (Woburn, MA). All other reagents were from Sigma-Aldrich. Wild-type FKBP-12 and the F99L mutant were expressed and purified from a GST-FKBP fusion gene of the pGST-FKBP-12 plasmid in E. coli BL21 cells as described previously.36
Capillary-based fluorescence measurement
A 266 nm, 5 mW, 1 kHz pulsed QUV266-02 UV laser (Crystalaser, Reno, NV) emitted a 200 μm diameter band-pass filtered (BG3, Newport) ray perpendicular to a fused-silica glass micro-capillary (FS-110, Upchurch Scientific, ID=100 μm, OD = 300 μm) containing sample. Plastic coating on the capillary was removed with a blue flame and the capillary placed on a 561 UltrAlign precision linear stage (Newport Corp., Stratford, CT). Fluorescence emission filtered by a custom dichroic mirror (Andover, Salem, NH) to 320–400 nm, was detected by photomultiplier tube (R1166 PMT, Hamamatsu, Japan). The incident laser was split with a quartz slice (UQS Optics, Cambridge, UK), detected by PD (OSD5.8-7Q, RS components), and used to remove time-dependent source variations from the fluorescence emission. Both signals were processed via a PXI-5124 digitizer and Labview (National Instruments, Austin, TX). Samples were pumped through the capillary at 10 μL min−1 by syringe pump (KD Scientific Inc., Holliston, MA) and a 500 μL Hamilton gas-tight syringe, and samples measured and averaged for 100 laser pulses (corresponding to 16.7 nL of sample). Cleaning was applied between measurements using 1% Hellma (5 min, 10 μL min−1), 2% nitric acid (5 min, 10 μL min−1), an air flush, then sample buffer (5 min, 40 μL min−1). Fluorescence was determined immediately after the sample meniscus had passed. Baselines without protein were obtained and the final output signal (S) obtained from Eq. (1), where PMT and PD are the photomultiplier and PD signals:
| (1) |
Linearity and sensitivity to protein concentration for fluorescence intensity measured in the capillary
Twelve solutions of BSA at 0.015–75 μM were prepared in 60 mM sodium phosphate buffer, pH 7.0, and fluorescence intensities measured as described above.
Equilibrium denaturation of BSA measured using capillary fluorescence
To 2 mL each of a range of urea solutions from 0 to 9 M in 50 mM Tris.HCl, pH 7.2 was added 100 μL 0.15 mM BSA stock (250 mg BSA in 25 mL 50 mM Tris.HCl, pH 7.2), giving final BSA concentrations of 7.14 μM. Samples were equilibrated for 17 hours at 22°C, consistent with the FKBP-12 experiments below. Fluorescence intensities were measured in the capillary as above, and then in a Fluostar Optima plate-reader (BMG Labtechnologies, Aylesbury, UK) with 280 nm excitation and 340 ± 10 nm emission as previously described.6,7
Equilibrium denaturation of wild-type FKBP-12
We added 50 μL FKBP-12 stock (50 μM FKBP-12, 5 mM DTT, 50 mM Tris.HCl, pH 7.5) to each well of a UV transparent Costar (Corning, Lowell, MA) 96-well plate. Twenty-five concentrations of guanidine hydrochloride (GdnHCl) from 0 to 5.6 M, were created by varying the volumes of 0 M and 7 M GdnHCl stock solutions in 50 mM Tris.HCl pH 7.5, autotitrated in each well (Fluostar Optima) to a total of 200 μL. For rapamycin binding, the 0 M and 7 M GdnHCl stock solutions both contained 18.8 μM rapamycin (from a 54 mM stock in EtOH), giving a 15 μM final concentration. Samples were sealed and equilibrated for 17 hours at 22 °C before measurement of intrinsic protein fluorescence in a Fluostar Optima plate reader as above. The same samples were then used to measure intrinsic protein fluorescence in the micro-capillary device for a direct comparison.
Equilibrium denaturation of mutant FKBP-12 (F99L)
Samples were prepared and analyzed as above for wild type, except that 25 μL of 100 μM FKBP-12 F99L in 50 mM Tris pH 7.5, 1 mM DTT was added to each well. Also, each concentration of GdnHCl was obtained using 0 and 4.1 M GdnHCl stocks in 50 mM Tris pH 7.5, 1 mM DTT, added to give a final volume of 225 μL and 11.1 μM protein. For rapamycin binding the 0 and 4.1 M GdnHCl stock solutions both contained 22.5 μM rapamycin (from a 54 mM stock in EtOH), giving a 20 μM final concentration.
Determination of thermodynamic parameters
Two-state protein denaturation was assumed, as previously observed for FKBP-12.40 Data for the observed fluorescence (Fobs) as a function of denaturant concentration ([D]) were fit using SigmaPlot 10.0 (Systat Software, Hounslow, UK) to Eq. (2) to obtain mG, C1/2, F0N, F0U, mN and mU with errors,47 where R is the gas constant (1.987 cal K−1 mol−1), T is 298 K, C1/2 (M) is the denaturant concentration at which 50% of the protein is denatured, F0N and F0U are the respective fluorescence signals of the native and unfolded states at 0 M denaturant, mG (kcal mol−1 M−1) is the slope (dG/d[D]) of the free energy of unfolding as a function of denaturant concentration, mN is the slope (dFN/d[D]) for the native state baseline, and mU the corresponding parameter for the unfolded state. The free energy of protein denaturation in water ΔGH2O was calculated as mGC1/2, using the fitted mG, where we set the denaturant dependent free energy (ΔGobs) to zero in Eq. (3) when [D] is equal to C1/2.47
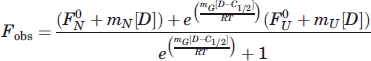 |
(2) |
| (3) |
Values of ΔGH2O were used to calculate the change in free energy of the mutant or ligand bound protein, ΔΔGX-WT, relative to un-liganded wild-type FKBP-12. The change in free energy of unfolding upon ligand binding, ΔΔGbinding, was calculated for each mutant from the values of ΔGH2O for liganded FKBP-12 relative to ΔGH2O for non-liganded FKBP-12. At saturating total ligand [LTOT]≫[PTOT], where [PTOT] is the total protein concentration, the dissociation constant Kd for binding to protein can be deduced from Eq. (4) and ΔΔGbinding,45 assuming the free ligand concentration [L] ≈ [LTOT].
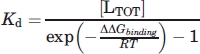 |
(4) |
Under non-saturating conditions the free ligand [L] and protein [P] concentrations must first be determined from [LTOT] and [PTOT] (see Supporting Information) giving Eq. (5) to determine the Kd.
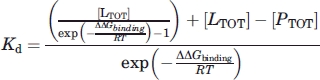 |
(5) |
Errors in Kd are mainly from mG values obtained by curve fitting to Eq. (2). As we do not expect this parameter to vary between mutants of FKBP-12 (our measurements are consistent with this assumption), we have used the values 〈mG〉, averaged over the two variants, to calculate for both liganded and un-liganded proteins, more accurate (ΔG〈mG〉) values for ΔGH2O.
Conclusions
Our experiments show for the first time that the intrinsic fluorescence of proteins can be used to accurately determine the thermodynamic parameters of protein stability for samples at low nanolitre volumes in a capillary with dimensions that are also typical of the channels used in microfluidic chips. Such a technique, easily implementable as an integrated biochip including on-board optics and microfluidics, could be used in the high-throughput formulation of protein therapeutics, or the screening of large libraries of protein engineered variants for improved stability. This is particularly facilitated by the use of flowing protein samples which both avoids the photobleaching effects associated with static samples and allows continuous processing with existing microfluidic mixer technologies. Indeed, we have demonstrated here [Fig. 1(b)] that sequences of bubbles containing various fluid mixtures can be prepared and then passed through a 1.5 nL fluorescence probe region. The dynamic range of the technique also permits measurements at protein concentrations from low physiologically relevant levels to the higher concentrations used in biopharmaceutical formulations. Finally, the associated microfluidics naturally enable serial or parallel testing of solution-based fluorescence alongside assays, such as SPR or micromechanical bending,48 for interactions with surface-immobilized targets. For example, a single solution can be prepared to flow past a cantilever assay array and subsequently a fluorescence assay region.
We have used the technique to perform a detailed biophysical analysis of the interaction of FKBP-12 with rapamycin and to specifically determine the energetic contribution of the interaction between a single residue and the rapamycin molecule. We show that the technique can be used to measure the interaction of small molecule drugs with a protein, without the need for protein or ligand labeling. This has significant potential for the high-throughput and low volume screening of combinatorial compound libraries against target proteins for drug discovery. The absence of a need for labeling makes the technique generically applicable to any protein target with intrinsic fluorescence, particularly from tryptophan residues.
References
- 1.Bleicher KH, Bohm HJ, Muller K, Alanine AI. Hit and lead generation: beyond high-throughput screening. Nat Rev Drug Discov. 2003;2:369–378. doi: 10.1038/nrd1086. [DOI] [PubMed] [Google Scholar]
- 2.Shah NS, Wright A, Bai GH, Barrera L, Boulahbal F, Martin-Casabona N, Drobniewski F, Gilpin C, Havelkova M, Lepe R, Lumb R, Metchock B, Portaels F, Rodrigues MF, Rusch-Gerdes S, Van DA, Vincent V, Laserson K, Wells C, Cegielski JP. Worldwide emergence of extensively drug-resistant tuberculosis. Emerg Infect Dis. 2007;13:380–387. doi: 10.3201/eid1303.061400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jain KK. Proteomics: delivering new routes to drug discovery—part 2. Drug Discov Today. 2001;6:829–832. doi: 10.1016/s1359-6446(01)01912-2. [DOI] [PubMed] [Google Scholar]
- 4.Jain KK. Proteomics: delivering new routes to drug discovery—part 1. Drug Discov Today. 2001;6:772–774. doi: 10.1016/s1359-6446(01)01913-4. [DOI] [PubMed] [Google Scholar]
- 5.Hong J, Edel JB, Demello AJ. Micro- and nanofluidic systems for high-throughput biological screening. Drug Discov Today. 2009;14:134–146. doi: 10.1016/j.drudis.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 6.Aucamp JP, Cosme AM, Lye GJ, Dalby PA. High-throughput measurement of protein stability in microtiter plates. Biotechnol Bioeng. 2005;89:599–607. doi: 10.1002/bit.20397. [DOI] [PubMed] [Google Scholar]
- 7.Aucamp JP, Martinez-Torres RJ, Hibbert EG, Dalby PA. A microplate-based evaluation of complex denaturation pathways: structural stability of Escherichia coli transketolase. Biotechnol Bioeng. 2008;99:1303–1310. doi: 10.1002/bit.21705. [DOI] [PubMed] [Google Scholar]
- 8.Matulis D, Kranz JK, Salemme FR, Todd MJ. Thermodynamic stability of carbonic anhydrase: measurements of binding affinity and stoichiometry using ThermoFluor. Biochemistry. 2005;44:5258–5266. doi: 10.1021/bi048135v. [DOI] [PubMed] [Google Scholar]
- 9.Edgell MH, Sims DA, Pielak GJ, Yi F. High-precision, high-throughput stability determinations facilitated by robotics and a semiautomated titrating fluorometer. Biochemistry. 2003;42:7587–7593. doi: 10.1021/bi034063g. [DOI] [PubMed] [Google Scholar]
- 10.Geysen HM, Schoenen F, Wagner D, Wagner R. Combinatorial compound libraries for drug discovery: an ongoing challenge. Nat Rev Drug Discov. 2003;2:222–230. doi: 10.1038/nrd1035. [DOI] [PubMed] [Google Scholar]
- 11.Dalby PA. Optimising enzyme function by directed evolution. Curr Opin Struct Biol. 2003;13:500–505. doi: 10.1016/s0959-440x(03)00101-5. [DOI] [PubMed] [Google Scholar]
- 12.Grant Y, Matejtschuk P, Dalby PA. Rapid optimisation of protein freeze-drying formulations using ultra scale-down and factorial design of experiment in microplates. Biotechnol Bioeng. 2009;104:957–964. doi: 10.1002/bit.22448. [DOI] [PubMed] [Google Scholar]
- 13.Kerby MB, Lee J, Ziperstein J, Tripathi A. Kinetic measurements of protein conformation in a microchip. Biotechnol Prog. 2006;22:1416–1425. doi: 10.1021/bp060111n. [DOI] [PubMed] [Google Scholar]
- 14.Hertzog DE, Michalet X, Jager M, Kong XX, Santiago JG, Weiss S, Bakajin O. Femtomole mixer for microsecond kinetic studies of protein folding. Anal Chem. 2004;76:7169–7178. doi: 10.1021/ac048661s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Srisa-Art M, Kang DK, Hong J, Park H, Leatherbarrow RJ, Edel JB, Chang SI, Demello AJ. Analysis of protein-protein interactions by using droplet-based microfluidics. Chembiochem. 2009;10:1605–1611. doi: 10.1002/cbic.200800841. [DOI] [PubMed] [Google Scholar]
- 16.Hanninen P, Soini A, Meltola N, Soini J, Soukka J, Soini E. A new microvolume technique for bioaffinity assays using two-photon excitation. Nat Biotechnol. 2000;18:548–550. doi: 10.1038/75421. [DOI] [PubMed] [Google Scholar]
- 17.Li Q, Seeger S. Label-free detection of protein interactions using deep UV fluorescence lifetime microscopy. Anal Biochem. 2007;367:104–110. doi: 10.1016/j.ab.2007.04.050. [DOI] [PubMed] [Google Scholar]
- 18.Quigley WW, Dovichi NJ. Capillary electrophoresis for the analysis of biopolymers. Anal Chem. 2004;76:4645–4658. doi: 10.1021/ac040100d. [DOI] [PubMed] [Google Scholar]
- 19.Jaspe J, Hagen SJ. Do protein molecules unfold in a simple shear flow? Biophys J. 2006;91:3415–3424. doi: 10.1529/biophysj.106.089367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schulze P, Ludwig M, Kohler F, Belder D. Deep UV laser-induced fluorescence detection of unlabeled drugs and proteins in microchip electrophoresis. Anal Chem. 2005;77:1325–1329. doi: 10.1021/ac048596m. [DOI] [PubMed] [Google Scholar]
- 21.Lapidus LJ, Yao SH, Mcgarrity KS, Hertzog DE, Tubman E, Bakajin O. Protein hydrophobic collapse and early folding steps observed in a microfluidic mixer. Biophys J. 2007;93:218–224. doi: 10.1529/biophysj.106.103077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang C, Zhao S, Yuan H, Xiao D. Determination of excitatory amino acids in biological fluids by capillary electrophoresis with optical fiber light-emitting diode induced fluorescence detection. J Chromatogr B Analyt Technol Biomed Life Sci. 2006;833:129–134. doi: 10.1016/j.jchromb.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 23.Lee J, Tripathi A. Measurements of label free protein concentration and conformational changes using a microfluidic UV-LED method. Biotechnol Prog. 2007;23:1506–1512. doi: 10.1021/bp0701970. [DOI] [PubMed] [Google Scholar]
- 24.Hoffmann A, Kane A, Nettels D, Hertzog DE, Baumgartel P, Lengefeld J, Reichardt G, Horsley DA, Seckler R, Bakajin O, Schuler B. Mapping protein collapse with single-molecule fluorescence and kinetic synchrotron radiation circular dichroism spectroscopy. Proc Natl Acad Sci USA. 2007;104:105–110. doi: 10.1073/pnas.0604353104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Neuweiler H, Johnson CM, Fersht AR. Direct observation of ultrafast folding and denatured state dynamics in single protein molecules. Proc Natl Acad Sci USA. 2009;106:18569–18574. doi: 10.1073/pnas.0910860106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Orte A, Craggs TD, White SS, Jackson SE, Klenerman D. Evidence of an intermediate and parallel pathways in protein unfolding from single-molecule fluorescence. J Am Chem Soc. 2008;130:7898–7907. doi: 10.1021/ja709973m. [DOI] [PubMed] [Google Scholar]
- 27.Rai JS, Henley MJ, Ratan HL. Mammalian target of rapamyca new target in prostate cancer. Urol Oncol. 2009;28:134–138. doi: 10.1016/j.urolonc.2009.03.023. [DOI] [PubMed] [Google Scholar]
- 28.Heitman J, Movva NR, Hall MN. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science. 1991;253:905–909. doi: 10.1126/science.1715094. [DOI] [PubMed] [Google Scholar]
- 29.Gaumann A, Schlitt HJ, Geissler EK. Immunosuppression and tumor development in organ transplant recipients: the emerging dualistic role of rapamycin. Transpl Int. 2008;21:207–217. doi: 10.1111/j.1432-2277.2007.00610.x. [DOI] [PubMed] [Google Scholar]
- 30.Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–395. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kay JE. Structure-function relationships in the FK506-binding protein (FKBP) family of peptidylprolyl cis-trans isomerases. Biochem J. 1996;314:361–385. [PMC free article] [PubMed] [Google Scholar]
- 32.Michnick SW, Rosen MK, Wandless TJ, Karplus M, Schreiber SL. Solution structure of FKBP, a rotamase enzyme and receptor for FK506 and rapamycin. Science. 1991;252:836–839. doi: 10.1126/science.1709301. [DOI] [PubMed] [Google Scholar]
- 33.Van Duyne GD, Standaert RF, Karplus PA, Schreiber SL, Clardy J. Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J Mol Biol. 1993;229:105–124. doi: 10.1006/jmbi.1993.1012. [DOI] [PubMed] [Google Scholar]
- 34.Van Duyne GD, Standaert RF, Karplus PA, Schreiber SL, Clardy J. Atomic structure of FKBP-FK506, an immunophilin-immunosuppressant complex. Science. 1991;252:839–842. doi: 10.1126/science.1709302. [DOI] [PubMed] [Google Scholar]
- 35.Egan DA, Logan TM, Liang H, Matayoshi E, Fesik SW, Holzman TF. Equilibrium denaturation of recombinant human FK binding protein in urea. Biochemistry. 1993;32:1920–1927. doi: 10.1021/bi00059a006. [DOI] [PubMed] [Google Scholar]
- 36.Main ER, Fulton KF, Jackson SE. Folding pathway of FKBP12 and characterisation of the transition state. J Mol Biol. 1999;291:429–444. doi: 10.1006/jmbi.1999.2941. [DOI] [PubMed] [Google Scholar]
- 37.Main ER, Fulton KF, Jackson SE. Context-dependent nature of destabilizing mutations on the stability of FKBP12. Biochemistry. 1998;37:6145–6153. doi: 10.1021/bi973111s. [DOI] [PubMed] [Google Scholar]
- 38.The PyMOL Molecular Graphics System. San Carlos, CA: DeLano Scientific; 2002. [Google Scholar]
- 39.Jackson SE, Fersht AR. Folding of chymotrypsin inhibitor 2. 1. Evidence for a two-state transition. Biochemistry. 1991;30:10428–10435. doi: 10.1021/bi00107a010. [DOI] [PubMed] [Google Scholar]
- 40.Main ER, Jackson SE. Does trifluoroethanol affect folding pathways and can it be used as a probe of structure in transition states? Nat Struct Biol. 1999;6:831–835. doi: 10.1038/12287. [DOI] [PubMed] [Google Scholar]
- 41.Pace CN, Mcgrath T. Substrate stabilization of lysozyme to thermal and guanidine hydrochloride denaturation. J Biol Chem. 1980;255:3862–3865. [PubMed] [Google Scholar]
- 42.Main ERG. 2000. Studies on the immunosuppressant binding protein FKBP12 and the nuclear/steroid receptors vitamin D3 and oestrogen. PhD Dissertation. University of Cambridge. [Google Scholar]
- 43.Bierer BE, Mattila PS, Standaert RF, Herzenberg LA, Burakoff SJ, Crabtree G, Schreiber SL. Two distinct signal transmission pathways in T lymphocytes are inhibited by complexes formed between an immunophilin and either FK506 or rapamycin. Proc Natl Acad Sci USA. 1990;87:9231–9235. doi: 10.1073/pnas.87.23.9231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Banaszynski LA, Liu CW, Wandless TJ. Characterization of the FKBP.rapamycin.FRB ternary complex. J Am Chem Soc. 2005;127:4715–4721. doi: 10.1021/ja043277y. [DOI] [PubMed] [Google Scholar]
- 45.Tang L, Hopper ED, Tong Y, Sadowsky JD, Peterson KJ, Gellman SH, Fitzgerald MC. H/D exchange- and mass spectrometry-based strategy for the thermodynamic analysis of protein-ligand binding. Anal Chem. 2007;79:5869–5877. doi: 10.1021/ac0700777. [DOI] [PubMed] [Google Scholar]
- 46.Carter PJ, Winter G, Wilkinson AJ, Fersht AR. The use of double mutants to detect structural changes in the active site of the tyrosyl-tRNA synthetase (Bacillus stearothermophilus) Cell. 1984;38:835–840. doi: 10.1016/0092-8674(84)90278-2. [DOI] [PubMed] [Google Scholar]
- 47.Pace CN. Determination and analysis of urea and guanidine hydrochloride denaturation curves. Methods Enzymol. 1986;131:266–280. doi: 10.1016/0076-6879(86)31045-0. [DOI] [PubMed] [Google Scholar]
- 48.Ndieyira JW, Watari M, Barrera AD, Zhou D, Vogtli M, Batchelor M, Cooper MA, Strunz T, Horton MA, Abell C, Rayment T, Aeppli G, Mckendry RA. Nanomechanical detection of antibiotic-mucopeptide binding in a model for superbug drug resistance. Nat Nanotechnol. 2008;3:691–696. doi: 10.1038/nnano.2008.275. [DOI] [PubMed] [Google Scholar]