

Abstract
Acquired resistance to aminoglycoside antibiotics primarily results from deactivation by three families of aminoglycoside-modifying enzymes. Here, we report the kinetic mechanism and structure of the aminoglycoside phosphotransferase 2″-IVa (APH(2″)-IVa), an enzyme responsible for resistance to aminoglycoside antibiotics in clinical enterococcal and staphylococcal isolates. The enzyme operates via a Bi-Bi sequential mechanism in which the two substrates (ATP or GTP and an aminoglycoside) bind in a random manner. The APH(2″)-IVa enzyme phosphorylates various 4,6-disubstituted aminoglycoside antibiotics with catalytic efficiencies (kcat/Km) of 1.5 × 103 to 1.2 × 106 (M−1 s−1). The enzyme uses both ATP and GTP as the phosphate source, an extremely rare occurrence in the phosphotransferase and protein kinase enzymes. Based on an analysis of the APH(2″)-IVa structure, two overlapping binding templates specifically tuned for hydrogen bonding to either ATP or GTP have been identified and described. A detailed understanding of the structure and mechanism of the GTP-utilizing phosphotransferases is crucial for the development of either novel aminoglycosides or, more importantly, GTP-based enzyme inhibitors which would not be expected to interfere with crucial ATP-dependent enzymes.
Keywords: aminoglycoside resistance, crystal structure, GTP-dependent, ATP-dependent, mechanism
Introduction
Aminoglycoside antibiotics are bactericidal agents used for the treatment of serious infections caused by various Gram-negative and Gram-positive microorganisms.1 Structurally, most clinically used aminoglycosides contain a characteristic aminocyclitol ring, usually 2-deoxystreptamine, tethered through glycosidic bonds to amino sugars at positions 4 and 5, or 4 and 6 (Fig. 1). In addition to these 4,5-disubstituted and 4,6-disubstituted antibiotics, several atypical aminoglycosides are produced by soil microorganisms. Streptomycin (Fig. 1), the most important representative of atypical aminoglycosides has a streptidine core, and has been used for the treatment of tuberculosis for more than 60 years. Another atypical aminoglycoside, spectinomycin, has a streptamine core and is used to treat acute gonococcal infections, particularly in areas with drug-resistant strains.2 Following decades of intensive use, the clinical importance of aminoglycosides has somewhat declined because of their toxicity and, more importantly, the spread of aminoglycoside-resistant microorganisms. In recent years, however, there has been a renewed interest in this class of antibiotics because of the emergence of “superbugs,” microorganisms resistant to the majority of available antimicrobial agents, and improved schemes for administration of aminoglycosides, resulting in a decrease of their toxicity.3
Figure 1.
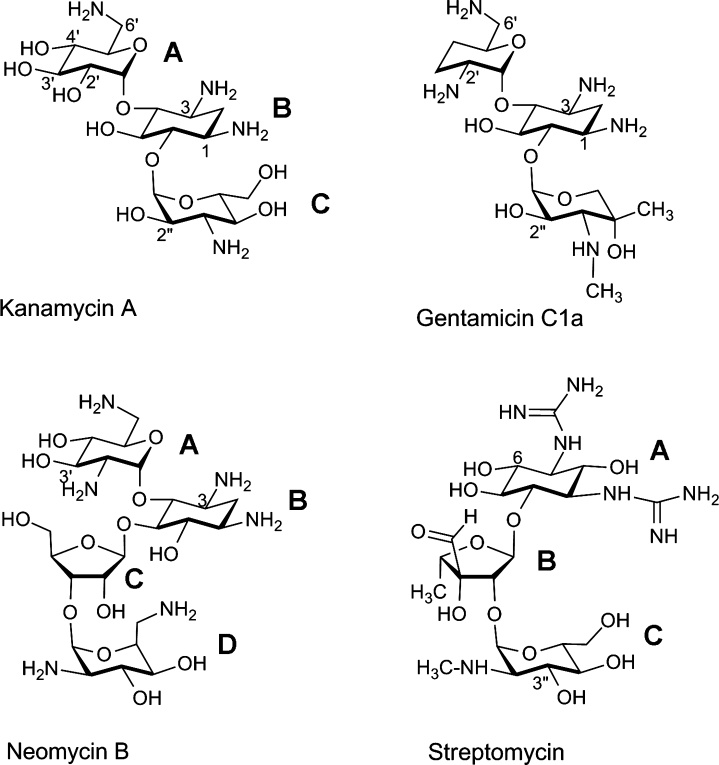
Structures of representative aminoglycoside antibiotics. Kanamycin A and gentamicin C1a are 4,6-disubstituted aminoglycosides, neomycin B is a 4,5-disubstituted aminoglycoside, and streptomycin is an atypical aminoglycoside. The rings are labeled B (the central deoxystreptamine ring in the first three molecules, and the ribose moiety in streptomycin), and A, C, and D if necessary, for the substituted glycan rings. The functional groups available for modification by various aminoglycoside-modifying enzymes are numbered.
The major mechanism of resistance to aminoglycoside antibiotics in clinical bacterial isolates is the production of aminoglycoside-modifying enzymes.4–7 Three families of aminoglycoside-modifying enzymes are recognized based on the nature of the chemical modification of the antibiotic they catalyze. They all are bisubstrate enzymes that transfer functional groups from one of their substrates to the aminoglycoside, thus compromising the binding of the modified drug to its target, the bacterial ribosome.8 Aminoglycoside acetyltransferases (AACs) use acetyl-coA as the second substrate to acetylate the 1, 3, 2′, or 6′ amino groups of these antibiotics (Fig. 1). Two other families of aminoglycoside-modifying enzymes use nucleotide triphosphates as the second substrates. The aminoglycoside nucleotidyltransferases (ANTs), also known as adenylyltransferases, modify hydroxyl groups at the 6, 4′, 2″, or 3″ positions of aminoglycosides (Fig. 1) by transferring the nucleoside moiety. Two enzymes that have been studied in detail (ANT-4′ and ANT-2″) are also able to transfer the nucleotide monophosphate group of various nucleotide triphosphates to aminoglycoside hydroxyl groups.9,10
Aminoglycoside phosphotransferases (APHs) modify the 2-deoxystreptamine antibiotics by phosphorylation of their hydroxyl groups at Positions 4, 6, 3′, 2″, 3″, 7″ (Fig. 1), or at Position 9 of spectinomycin11). The enzymes are named according to the reaction they catalyze, the position on the aminoglycoside that they modify, their resistance profile (which nucleotides and aminoglycosides they will bind) indicated by a Roman numeral, and their genetic variability indicated by a lower-case letter. It has been widely accepted that APHs use ATP as the source for phosphate; however, recently we demonstrated that GTP is also a substrate for some of these enzymes,12 either in preference to ATP or with a similar propensity.
The APH(2″) enzymes are a family of four distantly related aminoglycoside phosphotransferases, which modify the 2″-hydroxyl of aminoglycoside antibiotics.13–16 These clinically important enzymes have been identified mainly in Gram-positive staphylococcal and enterococcal isolates, and produce resistance to most available aminoglycoside antibiotics.17 We recently reported the structure of the first APH(2″) enzyme, both as the binary gentamicin-APH(2″)-IIa complex and the ternary complex containing β,γ-methylene adenosine triphosphate (AMPPCP) and the inhibitor streptomycin.18 The enzyme folds into two structural domains, an N-terminal β-sheet domain comprising ˜100 residues, and a larger C-terminal domain composed predominantly of α-helices. The ATP-binding site is sandwiched between the N-terminal and C-terminal domains, with the aminoglycoside substrate-binding site located in a cleft in the C-terminal domain. There are three additional aminoglycoside phosphotransferase structures, APH(9) (spectinomycin phosphotransferase)11 and two from the APH(3′) family of enzymes,19,20 and despite a very low sequence identity between these APH(9), APH(3″), and APH(2″) families, there is a marked degree of structural similarity.11,18 Here, we report the X-ray structure, substrate profile, and kinetic mechanism of a second member of the APH(2″) subfamily, the APH(2″)-IVa aminoglycoside phosphotransferase, and for the first time we report the determinants of NTP recognition by these clinically important enzymes.
Results
Aminoglycoside substrate profile
The antibiotic substrate profile for APH(2″)-IVa with ATP as the second substrate (Table I) indicates that all 4,6-disubstituted aminoglycosides tested (kanamycin A, tobramycin, netilmicin, dibekacin, isepamicin, amikacin, and arbekacin) are substrates of the enzyme. The turnover numbers (kcat) for APH(2″)-IVa are in the range of 0.15–1.8 s−1. These numbers are significantly lower than those reported for the APH(2″)-IIa enzyme (kcat values 5.7–45 s−1),21 and similar to the kcat values of APH(2″)-Ia (0.2–1.1 s−1)22 and APH(2″)-IIIa (0.5–0.8 s−1).23 The Km values of APH(2″)-IVa for kanamycin A, tobramycin, netilmicin, and dibekacin are in the low micromolar range (<1.0–3.3 μM), similar to the Km values reported for the APH(2″)-IIa and APH(2″)-IIIa and lower than those reported for APH(2″)-Ia.22
Table I.
Steady State Kinetic Parameters for APH(2″)-IVa
| Substrate | kcat (s−1) | Km (μM) | kcat/Km (M−1 s−1) |
|---|---|---|---|
| 4,6-Disubstituted aminoglycosides | |||
| Kanamycin A | 0.92 ± 0.01 | 3.3 ± 0.3 | (2.8 ± 0.3) × 105 |
| Dibekacin | 1.23 ± 0.03 | 2.0 ± 0.3 | (6.2 ± 0.9) × 105 |
| Tobramycin | 1.80 ± 0.03 | 1.5 ± 0.1 | (1.2 ± 0.1) × 106 |
| Netilmicin | 0.64 ± 0.01 | <1 | >(6.4 ± 0.6) × 105 |
| Amikacin | 0.15 ± 0.01 | 98.0 ± 15 | (1.5 ± 0.3) × 103 |
| Isepamicin | 0.59 ± 0.02 | 17.0 ± 0.7 | (3.5 ± 0.2) × 104 |
| Arbekacin | 0.35 ± 0.01 | 18.0 ± 1.0 | (1.9 ± 0.1) × 104 |
| Nucleotide triphosphates | |||
| ATPa | 1.0 ± 0.1 | 310 ± 13 | (3.2 ± 0.3) × 103 |
| GTPa | 1.2 ± 0.1 | 330 ± 20 | (3.6 ± 0.4) × 103 |
All parameters were determined in 100 mM MES, pH 6.6, 80 mM NaCl, 10 mM MgCl2, and 20 mM KCl at 25°C. For the phosphorylation of aminoglycosides, ATP was used as the phosphate donor at a concentration of 2 mM.
Km and kcat of ATP and GTP were determined with a saturating concentration of kanamycin A (200 μM).
The dissociation constants for isepamicin (Kia) and ATP (Kib) were calculated from initial velocity studies. These data (Kia = 15 ± 1 μM and Kib = 352 ± 42 μM) were later used to determine the dissociation constants of substrate analogs from the binary enzyme-aminoglycoside or enzyme-NTP complex (Kis) or the ternary enzyme-aminoglycoside-NTP complex (Kii).
Neamine differs from the other 4,6-disubstituted aminoglycosides in that it lacks the hexose ring linked to the 6 position of the 2-deoxystreptamine, and is thus devoid of the 2″-hydroxyl. Not surprisingly, neamine is not a substrate for APH(2″)-IVa, but an inhibitor with dissociation constants of 25 ± 7 and 19 ± 3 μM for Kis and Kii, respectively (Table II). Of the four 4,5-disubstituted aminoglycosides studied, none is a substrate for the enzyme, but all are inhibitors. Neomycin is the best inhibitor among them (Kis = 6.2 ± 1.8 and Kii = 5.2 ± 0.9 μM), whereas butirosin is the worst (Kis = 105 ± 22 and Kii = 232 ± 63 μM). Lividomycin A and paromomycin have a very low affinity for free enzyme (Kis >100 μM) but have affinity in the low micromolar range for the binary enzyme-ATP complex (Kii = 7.6 ± 0.5 and 14 ± 2 μM, respectively). Since APH(2″)-IVa is expected to exist as a binary complex with ATP in a living cell (the concentration of ATP in E. coli is 10- to 30-fold above its Km value), both lividomycin A and paromomycin are expected to be potent inhibitors of the enzyme in vivo. Of the three other atypical aminoglycosides tested (spectinomycin, streptomycin, and hygromycin B), none is a substrate or potent inhibitor of APH(2″)-IVa (Table II).
Table II.
Dissociation Constants for APH(2″)-IVa
| Aminoglycoside | Kis (μM) | Kii (μM) |
|---|---|---|
| 4,5-Disubstituted aminoglycosides | ||
| Neomycin B | 6.2 ± 1.8 | 5.2 ± 0.9 |
| Butirosin | 105 ± 22 | 232 ± 63 |
| Lividomycin A | > 100 | 7.6 ± 0.5 |
| Paromomycin | > 100 | 14 ± 2 |
| Atypical aminoglycosides | ||
| Spectinomycin | 800 ± 200 | 1300 ± 400 |
| Streptomycin | 90 ± 19 | 172 ± 40 |
| Hygromycin B | 340 ± 80 | 210 ± 20 |
| Neamine | 25 ± 7 | 19 ± 3 |
Determined in 100 mM MES, pH 6.6, 80 mM NaCl, 10 mM MgCl2, and 20 mM KCl at 25°C.
Kis and Kii are the dissociation constants of the inhibitor from enzyme-inhibitor and enzyme-inhibitor-ATP complexes, respectively. For inhibition studies, a fixed Km level of isepamicin (15 μM) was used.
Kinetic mechanism
The initial velocity pattern of the APH(2″)-IVa-catalyzed phosphorylation of isepamicin at several fixed concentrations of this antibiotic and variable concentration of ATP was determined to ascertain whether the enzyme proceeds via a sequential mechanism or a ping-pong mechanism. The double-reciprocal plots of 1/v versus 1/ [ATP] are shown in Supporting Information Figure S1. The observed intersection of lines to the left of the y-axis is characteristic of a sequential mechanism. To distinguish if substrates bind the enzyme randomly or in a specific order, inhibition studies with the dead-end inhibitors AMPPCP (a nonhydrolyzable ATP analog) and the aminoglycoside neomycin were performed. AMPPCP behaves as a noncompetitive inhibitor versus isepamicin (Kis = 2200 ± 700 μM and Kii = 2300 ± 300 μM) and a competitive inhibitor versus ATP (Supporting Information Table S1). Neomycin is a competitive inhibitor versus isepamicin but a noncompetitive inhibitor versus ATP (Kis = 6.2 ± 1.8 μM and Kii = 5.2 ± 0.9 μM). This dead-end inhibition pattern is indicative of the random binding of the substrates to the enzyme.24
Crystal structure
The APH(2″)-IVa structure has been solved in three crystal forms, two (forms I and II) in a related orthorhombic unit cell and a third (form III) in a monoclinic unit cell (Table III). The related APH(2″)-IIa structure18 was used as a molecular replacement search model. The APH(2″)-IVa molecule is composed of two major structural domains [Fig. 2(A)], an N-terminal domain (residues 1–99) and a C-terminal domain (residues 100–301), with the nucleotide-binding site located between these two domains. The C-terminal domain is further divided into the core subdomain (residues 100–144 and 190–254) and a helical subdomain (residues 145–189 and 255–301) [Fig. 2(A)]. A cleft between the core and the helical subdomains forms the putative aminoglycoside substrate-binding site.
Table III.
Structure Refinement Statistics
| Form I | Form II | Form III | |
|---|---|---|---|
| Space group | P212121 | P212121 | P21 |
| Unit cell dimensions | |||
| a (Å) | 50.06 | 46.38 | 75.94 |
| b (Å) | 63.61 | 62.59 | 65.14 |
| c (Å) | 101.34 | 96.49 | 78.49 |
| β (°) | — | — | 91.7 |
| Resolution range (Å) | 29.8–2.2 | 19.8–2.2 | 28.0–2.4 |
| Reflections, work/free | 15839/834 | 13815/727 | 26560/1417 |
| Rwork/Rfreea (%) | 18.7/24.3 | 17.8/27.3 | 19.3/26.3 |
| Number of refined atoms | |||
| Protein | 2461 | 2522 | 4940 |
| Water | 136 | 232 | 216 |
| Average B-factors | |||
| Protein | 42.6 | 22.7 | 42.6/44.0b |
| Water | 45.0 | 27.8 | 43.7 |
| RMS deviations | |||
| Bond lengths (A) | 0.007 | 0.008 | 0.008 |
| Bond angles (°) | 1.01 | 1.04 | 1.15 |
Rwork = Σ||Fo| − |Fc||/Σ|Fo|, where Fo and Fc are the observed and calculated structure factor amplitudes. Rfree is calculated using 5% of the reflections chosen randomly before refinement. The same Rfree set of reflections were used for the two P212121 data sets.
The two values refer to the two independent molecules in the asymmetric unit.
Figure 2.

(A) Ribbon representation of APH(2″)-IVa. The N-terminal domain is colored red and the C-terminal domain is in two shades of green representing the core subdomain (light green) and the helical subdomain (dark green). The numbering of the secondary structure elements is indicated. (B) Superposition of APH(2″)-IIa (pink) onto APH(2″)-IVa (green) based on all matching atoms. The location of the AMPPCP and gentamicin in APH(2″)-IIa are shown as ball-and-stick models (yellow). Figures 2–4, 5(A) and Supporting Information Figure S2 were prepared with the program PYMOL (http://pymol.sourceforge.net).
Superposition of the three apo APH(2″)-IVa crystal forms shows that forms I (P212121) and III (P21) are essentially identical, with an RMS difference (RMSD) of 0.5 Å for 297 matching Cα positions, whereas form II matches less well, with RMSD of 1.4 Å and 1.2 Å for superpositions with form I and form III, respectively. Inspection of the resultant superposition of form I onto form II (Supporting Information Figure S2A) shows that although the N-terminal and core subdomains appear to be in roughly the same relative orientations, three of the helices (α5, α6, and α10) in the helical subdomain are in a different position in the two structures. When the helical subdomain is removed from the superposition calculation, the N-terminal domain and the core subdomains match significantly better with an RMSD of 0.5 Å for 207 matching Cα atoms. Helix α9, also in the helical subdomain, is a long helix forming part of the substrate-binding site, and has a marked kink just over halfway along at residues Tyr270 and Trp271 [Fig. 2(A)]. This is similar to that described for the equivalent helix in APH(2″)-IIa.18 The first half of this helix matches well in the two crystal forms but beyond Trp271 the form II helix α9 becomes slightly less kinked and subsequently deviates from the path of the helix in form I. This, in turn, puts helix α10 in a different position (Supporting Information Figure S2B). Furthermore, helices α4 and α5 in form II are shifted from their positions in form I by a rotation of about 13° and a translation of over 2.5 Å. These two helices are linked to the core subdomain through long flexible loops and appear to move as a rigid unit. This structural re-orientation of the helical subdomain is most likely due to the compression of the unit cell, yet it clearly shows that the helical subdomain of APH(2″)-IVa is inherently flexible. Such flexibility of the helical subdomain has not been seen before in the APH enzymes.
The form I structure was superimposed onto the gentamicin-APH(2″)-IIa structure, giving an RMSD of 1.5 Å. Subsequent inspection of the superimposed models shows that the relative orientations of the three structural domains are similar in APH(2″)-IIa and APH(2″)-IVa [Fig. 2(B)]. The residues responsible for gentamicin binding in APH(2″)-IIa are essentially intact in the APH(2″)-IVa enzyme, including a cluster of acidic residue, two serine residues from the core subdomain, and a conserved tryptophan from the helical subdomain (Fig. 3). There is one striking difference, however, in that apart from the putative catalytic residue (Asp197), all the other acidic residues are glutamates in APH(2″)-IVa as opposed to aspartates in APH(2″)-IIa.
Figure 3.
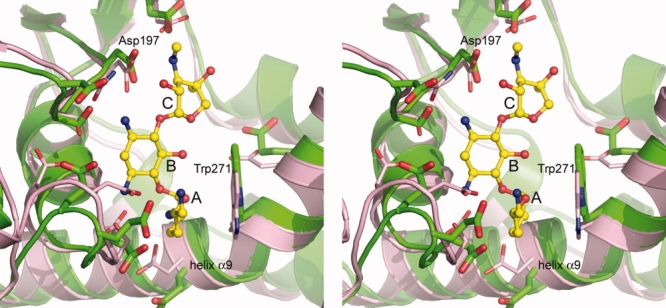
Stereoview of the superposition of APH(2″)-IIa (pink) onto APH(2″)-IVa (green) in the vicinity of the putative aminoglycoside substrate-binding site. The location of the bound gentamicin in APH(2″)-IIa is shown as a yellow ball-and-stick model, and the three rings have been labeled A, B, and C (see Fig. 1).
Discussion
APH(2″)-mediated aminoglycoside resistance
In the presence of ATP, APH(2″)-IVa is capable of phosphorylating a number of 4,6-disubstituted aminoglycosides, including kanamycin A, tobramycin, netilmicin, dibekacin, isepamicin, amikacin, and arbekacin, with Km values in the low micromolar range. Thus, APH(2″)-IVa has a substrate profile similar to that of APH(2″)-IIa.21 The other two APH(2″) enzymes, however, have resistance profiles at the two extremes; APH(2″)-Ia is capable of phosphorylating both 4,6- and 4,5-disubstituted compounds,22 whereas APH(2″)-IIIa has a much narrower substrate profile, as some of the 4,6-disubstituted aminoglycosides, like amikacin and isepamicin, are not phosphorylated by this enzyme.23 The very low Km values measured for most substrates indicate that APH(2″)-IVa (along with the other APH(2″) enzymes) would reach saturation at low micromolar concentrations of the antibiotics in the bacterial cell, a condition that is expected in enterococci (the frequent hosts for these enzymes), which are characterized by low penetration rates for aminoglycosides because of their facultative anaerobic metabolism.17 For the semisynthetic aminoglycosides (isepamicin, amikacin, and arbekacin), with bulky substituents at the N1 position of the deoxystreptamine ring, the Km values are significantly higher (17 μM for isepamicin, 18 μM for arbekacin, and 98 μM for amikacin). Because of these higher Km values, the catalytic efficiencies (kcat/Km) with these three substrates are 8- to 800-fold lower than for other tested 4,6-disubstituted aminoglycosides. As a result, the enzyme does not confer resistance to isepamicin, amikacin, or arbekacin when expressed in E. coli.12
In the APH(2″)-IIa enzyme, the neamine portion of the gentamicin substrate (rings A and B in Fig. 3) forms the majority of the hydrogen bonding and electrostatic interactions with the protein, sitting in a specificity pocket atop the first half of helix α9 and forming contacts with residues from the core subdomain and the helical domain. The Trp265 side chain packs against the A ring and the positioning of this residue is facilitating by the kinking of the helix. Based on the structural analysis of the APH(2″)-IVa binding pocket, it is highly likely that gentamicin would bind in a comparable fashion and form similar interactions with this enzyme. The neamine specificity pocket is lined with acidic residues and the side chain of a conserved tryptophan residue (Trp271 in APH(2″)-IVa residue numbering), and it is not surprising that neamine is a low micromolar inhibitor of APH(2″)-IVa. Given the changes in these acidic residues from predominantly aspartate in APH(2″)-IIa to glutamate in APH(2″)-IVa, this may result in the positioning of the aminoglycoside somewhat more toward the nucleotide-binding site.
The similarity of the APH(2″)-IVa and APH(2″)-IIa binding sites also provides a structural rationale for the similarity of the resistance profiles of these two aminoglycoside phosphotransferases. The 4,6-disubstituted aminoglycosides have a more elongated structure compared with the 4,5-disubstituted molecules (Fig. 1), such that when the neamine moiety is anchored in the specificity pocket, the C ring containing the 2″-hydroxyl group projects upward toward the catalytic base (Asp197, Figure 3) and the nucleotide-binding pocket. In the 4,5-disubstituted aminoglycosides, the C ring would sit at a significantly more acute angle relative to the A ring, and would be unable to present a C ring hydroxyl group in a similar position where it could potentially be phosphorylated.
As noted above, APH(2″)-Ia shows the broadest resistance profile of all the APH(2″) enzymes.22 This would imply either that the specificity pocket for the neamine moiety might have a slightly different structure, which allows the C ring of the 4,5-disubstituted molecules more freedom to move toward the catalytic aspartate residue, or that the enzyme binds the aminoglycoside substrates in a completely different manner. There is certainly precedent for a completely different binding conformation from the structures of the two APH(3′) enzymes,20,25 where the kanamycin molecule in these enzymes is bound essentially backward with the B ring projecting toward the helical subdomain rather than toward the core subdomain. However, analysis of the sequences of the APH(2″) enzymes shows that although the overall level of similarity is low to moderate (between 20 and 30%), the distribution of acidic residues in the four enzymes is very similar (Supporting Information Figure S3). Residues that are involved in gentamicin binding in APH(2″)-IIa, and implicated in substrate binding in APH(2″)-IVa, are all completely conserved in the APH(2″)-Ia and APH(2″)-IIIa enzymes.18 This implies that the substrate-binding sites in the four APH(2″) phosphotransferases might be similar, with small changes to account for the observed differences in their resistance profiles. Further analysis of these differences must await the elucidation of the structures of the remaining two enzymes.
Kinetic mechanism of APH(2″)-IVa
The APH enzymes catalyze a Bi-Bi reaction in which two substrates (aminoglycoside and NTP) are consumed and two products (phosphorylated aminoglycoside and NDP) are formed on catalysis. The transfer of the γ-phosphoryl group of ATP or GTP to various hydroxyl groups of aminoglycoside antibiotics can proceed via two alternative mechanisms. In a double-displacement mechanism (also called a ping-pong mechanism), the NTP binds to the enzyme first followed by transfer of its γ-phosphoryl group to the enzyme and, subsequently, release of the product, NDP. Next, the aminoglycoside binds to the enzyme and is phosphorylated. Alternatively, in a single-displacement or sequential mechanism, both substrates must bind to the enzyme active site to form a ternary complex prior to phosphate transfer. APH(2″)-IVa operates via a sequential mechanism in which the two substrates (NTP and aminoglycoside) bind in a random manner. All aminoglycoside-modifying enzymes studied to date (APH(3′)-Ia, APH(3′)-IIa, APH(2″)-Ia, APH(2″)-IIa, and APH(2″)-IIIa) operate via the sequential mechanism,21,23,26–33 and use a random mechanism for turnover chemistry,21,23,31,33,34 with the exception of APH(3′)-IIIa, which has been shown to use a Theorell-Chance mechanism with compulsory ordered substrate binding and product release.35
ATP and GTP recognition
As we have shown previously, APH(2″)-IVa can use both ATP and GTP as phosphate donors for the reaction with various aminoglycoside antibiotics.12 As the concentration of ATP in the bacterial cell is 2- to 3-fold higher than the concentration of GTP,36,37 APH(2″)-IVa is likely to prefer ATP as the second substrate in vivo. The Km value for ATP for APH(2″)-IVa (310 ± 13 μM at pH 6.6) is significantly higher than the Km values for the best NTP substrates in the other three APH(2″)s; for APH(2″)-Ia the Km value for GTP is 3.5 ± 0.2 μM,12 for APH(2″)-IIa the Km for ATP is 16.3 ± 1.6 μM,21 and for APH(2″)-IIIa the Km for GTP is 4.5 ± 0.5 μM.23 Despite the very high Km value for ATP, APH(2″)-IVa is expected to be saturated in vivo, given the concentration of ATP in the bacterial cell is 3.6–9.6 mM.36,37 The APH(2″)-IIa enzyme also can use both ATP and GTP with a slight preference for the former (Km values for ATP and GTP are 16 and 70 μM, respectively).12 This is in distinct contrast to the APH(2″)-Ia (Km values for ATP and GTP are 879 and 3.5 μM, respectively) and APH(2″)-IIIa (Km values for ATP and GTP are 1600 and 4 μM, respectively) enzymes, which show a substantial preference for GTP.12 In APH(2″)-IIa, the AMPPCP is bound between the N-terminal domain and the upper part of the core subdomain, sandwiched between four hydrophobic residues, Val40 and Met85 from the N-terminal domain, and two isoleucine residues (Ile199 and Ile209) from the core subdomain, and firmly held in place through hydrogen bonding interactions with the adenine and triphosphate moieties. Two hydrogen bonds anchor the adenine ring to the main chain of the interdomain linker; the N1 imino nitrogen accepts a hydrogen bond from the amide nitrogen of Ile88, and the N6 amino nitrogen donates a hydrogen bond to the carbonyl oxygen of Lys86 [Fig. 4(A)].18 From our analysis of the hydrogen bonding pattern and local structure in this region, this would seem to be typical for the APH(2″) phosphotransferases, and is highly reminiscent of the Watson–Crick hydrogen bonding pattern observed between A-T pairs in DNA.38
Figure 4.
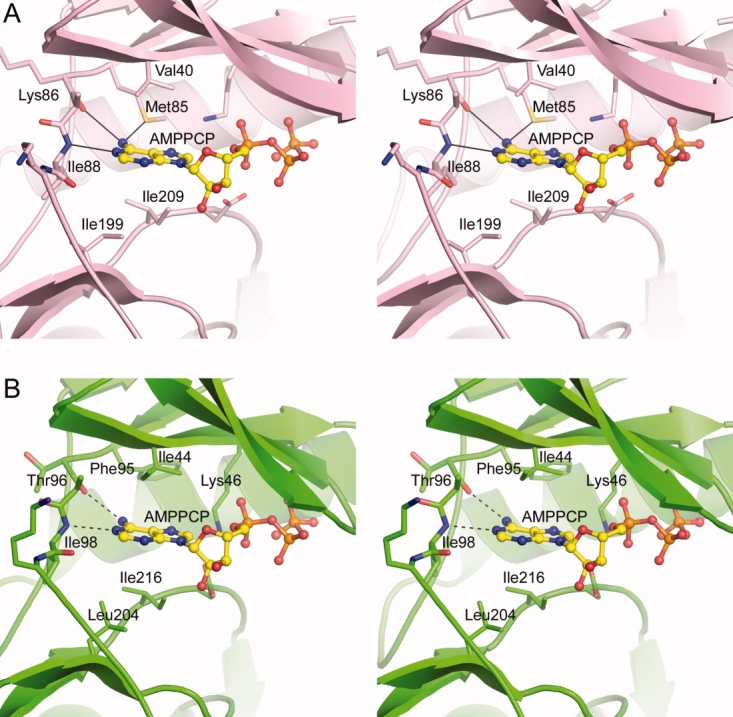
A: Stereoview of the nucleotide-binding site in APH(2″)-IIa18 showing the AMPPCP (yellow ball-and-stick model) and some of the residues which form the adenine-binding pocket. B: Stereoview of the putative nucleotide-binding site in APH(2″)-IVa showing the residues in equivalent positions to those in APH(2″)-IIa. The two potential hydrogen bonds that would anchor the adenine moiety are shown as dashed lines.
An AMPPCP was modeled into the APH(2″)-IVa nucleotide-binding site [Fig. 4(B)], based on a superposition of APH(2″)-IIa onto APH(2″)-IVa. The adenine-binding pocket in APH(2″)-IVa is delineated by four hydrophobic residues Ile44, Phe95, Leu204, and Ile216. A short piece of polypeptide linking the N-terminal domain and the core subdomain forms the back wall of the adenine-binding pocket, and based on the structural similarities between APH(2″)-IVa and APH)2″)-IIa, it is likely that the carbonyl oxygen of Thr96 and the amide nitrogen of Ile98 of this interdomain linker would play a role in binding the adenine moiety through two hydrogen bonding interactions to the N6 amine nitrogen and the N1 imine nitrogen on the AMPPCP, respectively. The side chain of Lys46 projects toward the position of the triphosphate group; this residue is completely conserved in all phosphotransferases and kinases, and plays a key role in anchoring the triphosphate and stabilizing the negative charge.
A GTP molecule was also modeled into the active site of APH(2″)-IVa, such that the triphosphate moiety occupies approximately the same location as the triphosphate in AMPPCP. This would be vitally important with respect to enzyme activity, because the reaction catalyzed by APH(2″)-IVa results in the highly efficient transfer of the γ-phosphate onto the acceptor hydroxyl group on the aminoglycoside. Hence, the orientation of the γ-phosphate relative to the accepting hydroxyl group is extremely critical. When GTP is modeled in this way, the guanine ring roughly overlaps with the position of the adenine ring in the AMPPCP model, and positions the guanine O6 ketone oxygen in the same location as the adenine N6 amine nitrogen. However, the guanine could not form a viable hydrogen bonding network with the interdomain linker in this orientation, primarily because interactions could not be made between the guanine O6 atom and the carbonyl oxygen of Thr96 (since both atoms are hydrogen bond acceptors), and between the guanine N1 amine nitrogen and the main chain nitrogen of Ile98 (since both atoms are hydrogen bond donors and in this orientation the hydrogen atoms on the two nitrogen atoms point directly toward each other). Therefore, a shift of the guanine group by ˜2 Å away from Thr96 (primarily by small rotations of the bonds linking the ribose and the triphosphate) would allow the guanine O6 atom to accept a hydrogen bond from the amide nitrogen of Ile98, and the N1 amino nitrogen would be in a position to donate a hydrogen bond to the carbonyl oxygen of Ile98 [Fig. 5(A)]. In this regard, the amide nitrogen and carbonyl oxygen of Ile98 would form a hydrogen-bonding template specific for GTP.
Figure 5.

A: The nucleotide-binding pocket in APH(2″)-IVa (green sticks) showing the interdomain linker (left) and the conserved hydrophobic residues, which flank the nucleotide (Ile216 is partially occluded by the nucleotide and not labeled). The location of the ATP (magenta sticks) is based on the superposition of APH(2″)-IVa and APH(2″)-IIa. The GTP is shown as yellow sticks. B: Schematic representation of the purine nucleotide-binding scaffold in the APH(2″) phosphotransferases. The four colored residues (N, N+1, N+2, and N+3) belong to the interdomain linker and the structure of this linker is highly conserved in the phosphotransferases. The adenine- and guanine-specific hydrogen bonding templates are indicated, along with potential hydrogen bonds, with arrows indicating whether the nucleotide is accepting or donating the hydrogen bond.
The ability of phosphotransferases and protein kinases to use both ATP and GTP is extremely rare, with the α subunit of casein kinase (CK2α) from human and Zea mays being one of the only kinases showing such a propensity.39 In this enzyme, both ATP and GTP are bound with almost equal affinity (Km for ATP = 13 μM; Km for GTP = 22 μM) and analysis of the binding pocket shows that on GTP binding, the guanine ring moves ˜2 Å in the pocket to use different hydrogen bonding partners on the interdomain linker, in what is termed a hydrogen-bonding frame-shift.39
Our structural analysis of the nucleotide-binding pocket in the APH(2″) phosphotransferases reveals the presence of distinct ATP- and GTP-binding templates, superimposed onto one overall purine nucleotide hydrogen bonding scaffold. A generalized form of this scaffold is shown in Figure 5(B). The scaffold has two overlapping binding templates, an adenine-specific template comprising the carbonyl oxygen of residue N and the amide nitrogen of residue N + 2, and a guanine-specific template comprising the amide nitrogen and carbonyl oxygen of residue N + 2. Our discovery of the two overlapping NTP-binding templates (one for ATP and one for GTP) in the nucleotide-binding site of APHs opens new avenues for the design of inhibitors of these enzymes targeting their GTP-binding domain, which could then be administered together with aminoglycoside antibiotics. An inhibitor based on the GTP-binding template would not be expected to effect the activity of human protein kinase, disrupt vital processes and give rise to serious side effect and toxicity, since these protein kinases are ATP-specific.
Materials and Methods
Cloning of the aph(2″)-IVa gene and purification of the APH(2″)-IVa phosphotransferase
For protein expression, the gene for the APH(2″)-IVa enzyme was recloned into the expression vector pET22b(+) (Novagen) between the NdeI and HindIII restriction sites. E. coli BL21 (DE3) harboring the pET22b(+) vector, with the cloned aph(2″)-IVa gene, were used to overexpress the enzyme for protein purification as described earlier.12
Kinetic studies
Enzyme activity was monitored by coupling the release of ADP or GDP from the APH(2″)-IVa-catalyzed phosphorylation of the aminoglycoside to pyruvate kinase and lactate dehydrogenase, as previously described.34 Reaction mixtures containing 100 mM MES (pH 6.6), 10 mM MgCl2, 20 mM KCl, 4 mM phospho(enol)pyruvate, 200 μM β-nicotinamide adenine dinucleotide (reduced form), 20 U/mL pyruvate kinase, 25 U/mL lactate dehydrogenase, 0.1–2 mM ATP or GTP, the substrate analog (during inhibition experiments), and APH(2″)-IV (100–300 nM) were initiated by the addition of the aminoglycoside substrate, and the change in absorbance monitored at 340 nm.
All spectrophotometric data were collected on a Cary 50 spectrophotometer (Varian) at 25°C. Analyses were performed using the nonlinear regression program Prism 5 (GraphPad Software) using data obtained from at least three independent experiments.
The parameters kcat and Km for NTP and aminoglycoside substrates were measured in reactions where the second substrate was fixed at saturating levels. The steady-state velocities (v) were determined from the linear phase of the reaction time courses and were plotted as a function of the variable substrate. The data were fit nonlinearly with the Michaelis–Menten equation,
| (1) |
where kcat = Vmax/[E], Vmax is the maximum velocity, [E] is the enzyme concentration, [S] and Km are the concentration and the Michaelis constant of the variable substrate.
The dissociation constants (Ks and Kia for isepamicin, and Kib for ATP) of the substrates from enzyme substrate complex (EA or EB) were determined by plotting the initial velocities as a function of the concentration of ATP (100 μM to 2 mM) at several fixed concentrations of isepamicin (5–200 μM) and fit nonlinearly with Eq. ( 2), corresponding to a random mechanism, as described by Morrison.40 We assumed a rapid equilibrium mechanism (where KiaKb = KibKa) based on the fact that the initial velocities as a function of one substrate at subsaturating concentrations of the other substrate were hyperbolic and not sigmoidal, which would have indicated a steady-state mechanism.41
| (2) |
where Vmax is the maximum velocity and Ka and Kb are the Michaelis constants (Km) for substrates A (isepamicin) and B (ATP).
The inhibitor dissociation constants, Kis (dissociation of the enzyme-inhibitor complex, EI) and Kii (dissociation of the enzyme-inhibitor-substrate complexes, EIA or EIB) of substrate analogues (aminoglycosides or AMPPCP) were determined by plotting the initial velocities at a fixed Km level of substrate A (300 μM for ATP or 15 μM for isepamicin) as a function of substrate B at several fixed concentrations of inhibitor (analog of substrate A) and fitting nonlinearly with Eq. ( 3) (noncompetitive inhibition) or Eq. (4) (competitive inhibition) as described by Morrison.40
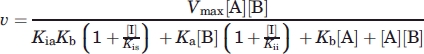 |
(3) |
 |
(4) |
where A and B are the substrates, I is the inhibitor, and Kic is as described in Eq. ( 5).
 |
(5) |
Crystal structure determination
Crystals of apo APH(2″)-IVa were prepared as previously described.42 The apo-enzyme crystallized in three different forms, two related orthorhombic forms in space group P212121 with cell parameters a = 50.06 Å, b = 63.61 Å, c = 101.34 Å (form I) and a = 46.38 Å, b = 62.59 Å, c = 96.49 Å (form II), and a monoclinic form (form III) in space group P21 with unit cell parameters a = 75.94 Å, b = 65.14 Å, c = 78.49 Å, β = 91.7°. Form II crystals appear to be a more compact form of the form I crystals, with a unit cell volume ˜13% smaller than that of form I and only 38% solvent content, and this could be possibly due to some degree of dehydration.42 Additional complexes were also prepared and submitted to crystallization trials, including ternary complexes with the nonhydrolyzable ATP and GTP analogs adenosine-5′-(β,γ-imido)triphosphate (AMPPNP) and guanosine-5′-(β,γ-imido)triphosphate (GMPPNP), and substrates gentamicin and kanamycin but these have yet to yield diffraction quality crystals.
The APH(2″)-IVa structure was solved by molecular replacement using the homologous APH(2″)-IIa structure (PDB code 3HAM)18 as a search model against the form III data. The program CHAINSAW from the CCP4 suite,43 guided by the sequence alignment of APH(2″)-IVa and APH(2″)-IIa, was used to truncate the APH(2″)-IIa model such that the conserved residues were retained and nonconserved residues truncated to alanine. Molecular replacement calculations were performed using the program MOLREP44 against the data from all three forms, and analysis of the solutions indicated that form III gave the best initial model for refinement (rotation peaks of 2.2 σ and a correlation coefficient of 0.26). This model was partially refined using the program REFMAC45 from the CCP4 suite, with all side chains added to the electron density according to the APH(2″)-IVa sequence. The Rfree at this stage was 0.34 and this partial model was used to once again calculate molecular replacement solutions from the form I and form II data. The form I data gave a very strong solution (rotation peak greater than 4 σ and a correlation coefficient of 0.65), which was subsequently refined with REFMAC, while the form II data failed to give a solution that would refine (the Rfree following 10 cycles of REFMAC refinement hung at 0.56). The partial form III model was therefore broken into the three structural domains, the N-terminal domain, the core subdomain, and the helical subdomain, and the molecular replacement carried out first searching for the core subdomain, then the N-terminal domain (with the core subdomain fixed) and finally the helical subdomain (with the core subdomain and the N-terminal domain fixed). The third step failed to find a solution, so a composite model comprising the N-terminal domain and the core subdomain was used for initial refinement. The correlation coefficient from molecular replacement for this composite model was 0.46 and after 10 cycles of REFMAC refinement the Rfree dropped to 0.42. The helical subdomain was built in fragments over the course of three rebuilding cycles with COOT.46 Final refinement on all three apo APH(2″)-IVa crystal forms was carried out with the program PHENIX.47 Table III summarizes the refinement results for the three crystal forms.
Accession numbers
The atomic coordinates and structure factors have been deposited in the Protein Data Bank with accession codes 3N4T (form I), 3N4U (form II), and 3N4V (form III).
Acknowledgments
The Stanford Synchrotron Radiation Lightsource (SSRL), a national user facility, is operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences (BES).
Glossary
Abbreviations:
- AAC
aminoglycoside acetyltransferase
- ANT
aminoglycoside nucleotidyltransferase
- APH
aminoglycoside phosphotransferase
- AMPPCP
β,γ-methylene adenosine triphosphate.
References
- 1.Edson RS, Terrell CL. The aminoglycosides. Mayo Clin Proc. 1991;66:1158–1164. doi: 10.1016/s0025-6196(12)65798-x. [DOI] [PubMed] [Google Scholar]
- 2.Chen PL, Hsieh YH, Lee HC, Ko NY, Lee NY, Wu CJ, Chang CM, Lee CC, Ko WC. Suboptimal therapy and clinical management of gonorrhoea in an area with high-level antimicrobial resistance. Int J STD AIDS. 2009;20:225–228. doi: 10.1258/ijsa.2008.008286. [DOI] [PubMed] [Google Scholar]
- 3.Durante-Mangoni E, Grammatikos A, Utili R, Falagas ME. Do we still need the aminoglycosides? Int J Antimicrob Agents. 2009;33:201–205. doi: 10.1016/j.ijantimicag.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 4.Wright GD. Aminoglycoside-modifying enzymes. Curr Opin Microbiol. 1999;2:499–503. doi: 10.1016/s1369-5274(99)00007-7. [DOI] [PubMed] [Google Scholar]
- 5.Shaw KJ, Rather PN, Hare RS, Miller GH. Molecular genetics of aminoglycoside resistance genes and familial relationships of the aminoglycoside-modifying enzymes. Microbiol Rev. 1993;57:138–163. doi: 10.1128/mr.57.1.138-163.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vakulenko SB, Mobashery S. Versatility of aminoglycosides and prospects for their future. Clin Microbiol Rev. 2003;16:430–450. doi: 10.1128/CMR.16.3.430-450.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith CA, Baker EN. Aminoglycoside antibiotic resistance by enzymatic deactivation. Curr Drug Targets Infect Dis. 2002;2:143–160. doi: 10.2174/1568005023342533. [DOI] [PubMed] [Google Scholar]
- 8.Moazed D, Noller HF. Interaction of antibiotics with functional sites in 16S ribosomal RNA. Nature. 1987;327:389–394. doi: 10.1038/327389a0. [DOI] [PubMed] [Google Scholar]
- 9.Gates CA, Northrop DB. Substrate specificities and structure-activity relationships for the nucleotidylation of antibiotics catalyzed by aminoglycoside nucleotidyltransferase 2″-I. Biochemistry. 1988;27:3820–3825. doi: 10.1021/bi00410a045. [DOI] [PubMed] [Google Scholar]
- 10.Le Goffic F, Martel A, Capmau ML, Baca B, Goebel P, Chardon H, Soussy CJ, Duval J, Bouanchaud DH. New plasmid-mediated nucleotidylation of aminoglycoside antibiotics in Staphlococcus aureus. Antimicrob Agents Chemother. 1976;10:258–264. doi: 10.1128/aac.10.2.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fong DH, Lemke CT, Hwang J, Xiong B, Berghuis AM. Structure of the antibiotic resistance factor spectinomycin phosphotransferase from Legionella pneumophila. J Biol Chem. 2010;285:9545–9555. doi: 10.1074/jbc.M109.038364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Toth M, Chow JW, Mobashery S, Vakulenko SB. Source of phosphate in the enzymic reaction as a point of distinction among aminoglycoside 2″-phosphotransferases. J Biol Chem. 2009;284:6690–6696. doi: 10.1074/jbc.M808148200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chow JW, Zervos MJ, Lerner SA, Thal LA, Donabedian SM, Jaworski DD, Tsai S, Shaw KJ, Clewell DB. A novel gentamicin resistance gene in Enterococcus. Antimicrob Agents Chemother. 1997;41:511–514. doi: 10.1128/aac.41.3.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferretti JJ, Gilmore KS, Courvalin P. Nucleotide sequence analysis of the gene specifying the bifunctional 6′-aminoglycoside acetyltransferase 2″-aminoglycoside phosphotransferase enzyme in Streptococcus faecalis and identification and cloning of gene regions specifying the two activities. J Bacteriol. 1986;167:631–638. doi: 10.1128/jb.167.2.631-638.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kao SJ, You I, Clewell DB, Donabedian SM, Zervos MJ, Petrin J, Shaw KJ, Chow JW. Detection of the high-level aminoglycoside resistance gene, aph(2″)-Ib, in Enterococcus faecium and Escherichia coli. Antimicrob Agents Chemother. 2000;44:2876–2879. doi: 10.1128/aac.44.10.2876-2879.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsai S, Zervos MJ, Clewell DB, Donabedian SM, Sahm DF, Chow JW. A new high-level gentamicin resiatance gene, aph(2″)-Id, in Enterococcus spp. Antimicrob Agents Chemother. 1998;42:1229–1232. doi: 10.1128/aac.42.5.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chow JW. Aminoglycoside resistance in enterococci. Clin Infect Dis. 2000;31:586–589. doi: 10.1086/313949. [DOI] [PubMed] [Google Scholar]
- 18.Young PG, Walanj R, Lakshmi V, Byrnes LJ, Metcalf P, Baker EN, Vakulenko S, Smith CA. The crystal structures of substrate and nucleotide complexes of Enterococcus faecium aminoglycoside-2″-phosphotransferase-IIa [APH(2″)-IIa] provide insights into substrate selectivity in the APH(2″) subfamily. J Bacteriol. 2009;191:4133–4143. doi: 10.1128/JB.00149-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hon WC, McKay GA, Thompson PR, Sweet RM, Yang DSC, Wright GD, Berghuis AM. Structure of an enzyme required for aminoglycoside antibiotic resistance reveals homology to eukaryotic protein kinases. Cell. 1997;89:887–895. doi: 10.1016/s0092-8674(00)80274-3. [DOI] [PubMed] [Google Scholar]
- 20.Nurizzo D, Shewry SC, Perlin MH, Brown SA, Dholakia JN, Fuchs RL, Deva T, Baker EN, Smith CA. The crystal structure of aminoglycoside-3′-phosphotransferase-IIa, an enzyme responsible for antibiotic resistance. J Mol Biol. 2003;327:491–506. doi: 10.1016/s0022-2836(03)00121-9. [DOI] [PubMed] [Google Scholar]
- 21.Toth M, Zajicek J, Kim C, Chow JW, Smith C, Mobashery S, Vakulenko S. Kinetic mechanism of enterococcal aminoglycoside phosphotransferase 2″-Ib. Biochemistry. 2007;46:5570–5578. doi: 10.1021/bi6024512. [DOI] [PubMed] [Google Scholar]
- 22.Daigle DM, Hughes DW, Wright GD. Prodigious substrate specificity of AAC(6′)-APH(2″), an aminoglycoside antibiotic resistance determinant in enterococci and staphylococci. Chem Biol. 1999;6:99–110. doi: 10.1016/S1074-5521(99)80006-4. [DOI] [PubMed] [Google Scholar]
- 23.Badarau A, Shi Q, Chow JW, Zajicek J, Mobashery S, Vakulenko S. Aminoglycoside 2″-phosphotransferase type IIIa from Enterococcus. J Biol Chem. 2008;283:7638–7647. doi: 10.1074/jbc.M709645200. [DOI] [PubMed] [Google Scholar]
- 24.Copeland RA. Enzymes: a practical introduction to structure, mechanism, and data analysis. 2nd ed. New York: Wiley; 2000. [Google Scholar]
- 25.Fong D, Berghuis AM. Substrate promiscuity of an aminoglycoside antibiotic resistance enzyme via target mimicry. EMBO J. 2002;21:2323–2331. doi: 10.1093/emboj/21.10.2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen-Goodspeed M, Vanhooke JL, Holden HM, Raushel FM. Kinetic mechanism of kanamycin nucleotidyltransferase from Staphylococcus aureus. Bioorg Chem. 1999;27:395–408. [Google Scholar]
- 27.Draker KA, Northrop DB, Wright GD. Kinetic mechanism of the GCN5-related chromosomal aminoglycoside acetyltransferase AAC(6′)-Ii from Enterococcus faecium: evidence of dimer subunit cooperativity. Biochemistry. 2003;42:6565–6574. doi: 10.1021/bi034148h. [DOI] [PubMed] [Google Scholar]
- 28.Golemi D, Maveyraud L, Vakulenko S, Samama JP, Mobashery S. Critical involvement of a carbamylated lysine in catalytic function of class D beta-lactamases. Proc Natl Acad Sci USA. 2001;98:14280–14285. doi: 10.1073/pnas.241442898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jana S, Deb JK. Kinetic mechanism of streptomycin adenylyltransferase from a recombinant Escherichia coli. Biotechnol Lett. 2005;27:519–524. doi: 10.1007/s10529-005-2544-9. [DOI] [PubMed] [Google Scholar]
- 30.Kim C, Hesek D, Zajicek J, Vakulenko SB, Mobashery S. Characterization of the bifunctional aminoglycoside-modifying enzyme ANT(3″)-Ii/AAC(6′)-IId from Serratia marcescens. Biochemistry. 2006;45:8368–8377. doi: 10.1021/bi060723g. [DOI] [PubMed] [Google Scholar]
- 31.Martel A, Masson M, Moreau N, Le Goffic F. Kinetic studies of aminoglycoside acetyltransferase and phosphotransferase from Staphylococcus aureus RPAL. Relationship between the two activities. Eur J Biochem. 1983;133:515–521. doi: 10.1111/j.1432-1033.1983.tb07494.x. [DOI] [PubMed] [Google Scholar]
- 32.McKay GA, Wright GD. Kinetic mechanism of aminoglycoside phosphotransferase type IIIa. Evidence for a Theorell-Chance mechanism. J Biol Chem. 1995;270:24686–24692. doi: 10.1074/jbc.270.42.24686. [DOI] [PubMed] [Google Scholar]
- 33.Siregar JJ, Miroshnikov K, Mobashery S. Purification, characterization, and investigation of the mechanism of aminoglycoside 3′-phosphotransferase type Ia. Biochemistry. 1995;34:12681–12688. doi: 10.1021/bi00039a026. [DOI] [PubMed] [Google Scholar]
- 34.Siregar JJ, Lerner SA, Mobashery S. Purification and characterization of aminoglycoside 3′-phosphotransferase type IIa and kinetic comparison with a new mutant enzyme. Antimicrob Agents Chemother. 1994;38:641–647. doi: 10.1128/aac.38.4.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McKay GA, Wright GD. Catalytic mechanism of enterococcal kanamycin kinase (APH(3′)-IIIa): viscosity, thio, and solvent isotope effects support a Theorell-Chance mechanism. Biochemistry. 1996;35:8680–8685. doi: 10.1021/bi9603884. [DOI] [PubMed] [Google Scholar]
- 36.Bennett BD, Kimball EH, Gao M, Osterhout R, van Dien SJ, Rabinowitz JD. Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nat Chem Biol. 2009;5:593–599. doi: 10.1038/nchembio.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Buckstein MH, He J, Rubin H. Characterization of nucleotide pools as a function of physiological state in Escherichia coli. J Bacteriol. 2008;190:718–726. doi: 10.1128/JB.01020-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Watson JD, Baker TA, Bell SP, Gann A, Levine M, Losick R. Molecular biology of the gene. 5th ed. San Francisco: Pearson Benjamin Cummings; 2004. [Google Scholar]
- 39.Niefind K, Putter M, Guerra B, Issinger OG, Schomburg D. CTP plus water mimic ATP in the active site of protein kinase CK2. Nat Struct Biol. 1999;6:1100–1103. doi: 10.1038/70033. [DOI] [PubMed] [Google Scholar]
- 40.Morrison JF. Encyclopedia of life sciences. Chichester: John Wiley & Sons; 2001. Enzyme activity: reversible inhibition. [Google Scholar]
- 41.Segel IH. Enzyme kinetics: behavior and analysis of rapid equilibrium and steady state enzyme systems. New York: Wiley; 1975. [Google Scholar]
- 42.Toth M, Vakulenko S, Smith CA. Purification, crystallization and preliminary X-ray analysis of Enterococcus casseliflavus aminoglycoside-2″-phosphotransferase-IVa [APH(2″)-IVa] Acta Crystallogr. 2010;F66:81–84. doi: 10.1107/S1744309109050039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Collaborative Computing Project No. 4. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 44.Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. J Appl Cryst. 1997;30:1022–1025. [Google Scholar]
- 45.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 46.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 47.Adams PD, Grosse-Kunstleve RW, Hung L-W, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. 2002;D58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]