

Abstract
Synapsins are a family of synaptic vesicle proteins that are important for neurotransmitter release. Here we have used triple knock-out (TKO) mice lacking all three synapsin genes to determine the roles of synapsins in the release of two monoamine neurotransmitters, dopamine and serotonin. Serotonin release evoked by electrical stimulation was identical in substantia nigra pars reticulata slices prepared from TKO and wild-type mice. In contrast, release of dopamine in response to electrical stimulation was approximately doubled in striatum of TKO mice, both in vivo and in striatal slices, in comparison to wild-type controls. This was due to loss of synapsin III, because deletion of synapsin III alone was sufficient to increase dopamine release. Deletion of synapsins also increased the sensitivity of dopamine release to extracellular calcium ions. Although cocaine did not affect the release of serotonin from nigral tissue, this drug did enhance dopamine release. Cocaine-induced facilitation of dopamine release was a function of external calcium, an effect that was reduced in TKO mice. We conclude that synapsins play different roles in the control of release of dopamine and serotonin, with release of dopamine being negatively regulated by synapsins, specifically synapsin III, while serotonin release appears to be relatively independent of synapsins. These results provide further support for the concept that synapsin function in presynaptic terminals varies according to the neurotransmitter being released.
Introduction
Synapsins are phosphoproteins that bind to the cytosolic surface of synaptic vesicles and represent almost 10% of the total protein content of synaptic vesicles. There are three mammalian synapsin genes (synapsin I, II, and III), with alternative splicing creating 10 known synapsin isoforms. Synapsins are differentially expressed in nerve terminals, with synapsins I and II present in most areas of the adult brain while synapsin III expression is more restricted (Pieribone et al., 2002). Synapsins are thought to play a role in segregating vesicles into a reserve pool that is located away from the active zone, where exocytosis occurs (Greengard et al., 1993). This hypothesis is supported by observations showing that perturbing synapsins by microinjection of anti-synapsin antibodies (Pieribone et al., 1995) or synapsin peptides (Hilfiker et al., 2005) disrupts a distal, reserve pool of vesicles, sparing vesicles in the active zone. It is thought that mobilization of vesicles from the reserve pool is triggered by calcium-dependent phosphorylation of synapsins (Llinás et al., 1991; Greengard et al., 1993; Chi et al., 2003). More recent evidence suggests that synapsins are also important regulators of exocytosis, vesicle recycling, and vesicle integrity at the active zone (Fdez and Hilfiker, 2006; Evergren et al., 2007).
Synapsin knock-out (KO) mice have provided further insights into the role of synapsins in the regulation of neurotransmitter release. While deletion of individual synapsin genes (Ferreira et al., 2000; Feng et al., 2002) or genes for both synapsins I and II (Rosahl et al., 1995; Lonart and Simsek-Duran, 2006) affects synaptic vesicle pool dynamics, interpretation of results from such genotypes are hindered by possible compensation caused by remaining synapsin isoforms and differential expression of synapsins across brain regions. These problems have been solved by generating triple knock-out (TKO) mice that lack all three known synapsin genes. TKO mice exhibit reduced reserve pools in both glutamatergic and GABAergic nerve terminals, evident as changes in vesicle distribution and in neurotransmitter release during trains of action potentials (Gitler et al., 2004). However, when using single action potentials as stimuli to monitor release from the readily releasable pool of synaptic vesicles, release of GABA from this pool is reduced, while release of glutamate is not. This suggests that the function of synapsins at presynaptic terminals varies according to the type of neurotransmitter used.
Here we have extended the analysis to the release of dopamine (DA) and serotonin (5-HT), comparing release of these two monoamine neurotransmitters in synapsin TKO mice both in brain slices and in vivo. DA release was enhanced in the absence of synapsins, while 5-HT release from brain slices was identical in the two genotypes. The altered DA release in TKO mice appeared to be due to deletion of synapsin III since release was similarly altered in mice with a synapsin III deletion. The dependence of DA release on extracellular calcium was found to differ between TKO and wild-type (WT) mice, with TKO mice being more sensitive to calcium. The ability of cocaine to increase release of DA was reduced in synapsin TKO mice, supporting the assertion that synapsins contribute to the presynaptic actions of cocaine (Venton et al., 2006). In contrast, cocaine did not affect release of 5-HT in either genotype. These findings demonstrate that synapsins play different regulatory roles in nerve terminals releasing DA and 5-HT.
Materials and Methods
Animals.
Homozygous mutant synapsin TKO, synapsin III knock-out (S3KO), and WT male and female mice were bred at Duke University as previously described (Gitler et al., 2004). Mice were age and sex matched and experiments were conducted with protocols approved by the Institutional Animal Care and Use Committees of both Duke University and the University of North Carolina at Chapel Hill.
Electrochemistry.
Cylindrical carbon-fiber microelectrodes (6 μm diameter, Thornel T-650; Amoco) were fabricated as previously described (Bath et al., 2000). A carbon fiber was aspirated into glass capillary (1.2 mm × 0.68 mm, 4 inches; AM Systems). The electrodes were pulled on a vertical capillary puller (Narishige) and the excess carbon fiber was trimmed to 50 μm. Waveform application, current monitoring, and stimulus application were all controlled by locally written software (Tarheel CV, Labview) through a home-built potentiostat (UEI, UNC electronics shop).
For DA detection, the carbon-fiber electrode was held at −0.40 V versus an Ag/AgCl reference electrode between voltage scans. Detection in brain slices was achieved by application of a triangular waveform: −0.40 V to 1.00 V to −0.40 V with a scan rate of 600 V/s and repeated at 60 Hz. This waveform provided high temporal resolution (Bath et al., 2000). The waveform for in vivo use employed the same voltages, but was at 300 V/s and was repeated at 10 Hz; this method provided improved sensitivity (Baur et al., 1988). The current at the peak oxidation potential for DA (0.50–0.70 V vs Ag/AgCl) was used to evaluate DA concentration changes.
For 5-HT detection the electrode potential between scans was 0.20 V vs Ag/AgCl. The cyclic voltammogram was obtained with an initial scan to 1.00 V, back to −0.10 V, and then scanned to 0.20 V at 1000 V/s applied at 10 Hz. The positive rest potential minimizes electrode fouling from 5-HT reduction products (Jackson et al., 1995). In addition, the rapid scan rate enables side-reactions characteristic of electrochemical detection of 5-HT to be outrun. For both DA and 5-HT, the electrodes were calibrated in a flow injection system with a 1 μm bolus of the analyte of interest. Calcium ions inversely reduce the sensitivity of electrodes in a linear manner. These changes in electrode sensitivity were accounted for in calibration (Kume-Kick and Rice, 1998).
Slice experiments.
Mice were anesthetized with ether and decapitated. The brain was rapidly removed and placed on ice. Coronal brain slices (300 μm) containing the caudate–putamen or substantia nigra, pars reticulata (SNr), were prepared in ice-cold artificial CSF (aCSF) using a Lancer Vibratome (World Precision Instruments). The aCSF contained the following (in mm): 20 HEPES, 2.4 CaCl2, 1.2 MgCl2, 1.2 NaH2PO4, 2.45 KCl, 126 NaCl, 11 glucose, and 25 NaHCO3. All reagents were obtained from Sigma-Aldrich. Cocaine, picrotoxin, CNQX, and mecamylamine were dissolved in doubly deionized water at a concentration of 2 mm. Final dilutions were made in aCSF and applied to the slice for 35 min. before measurements. The pH was adjusted to 7.4 and the aCSF was saturated with 95% O2/5% CO2. Slices were superfused in aCSF at 37°C for 35–40 min. before recording. Recordings were performed on a microscope (Nikon Fn-1, Gibraltar Stage) fitted with a slice perfusion chamber (Warner Instruments).
Neurotransmitter release was evoked by local electrical stimulation delivered with a tungsten bipolar stimulating electrode placed on the surface of the slice (FHC). The stimulus consisted of a computer generated, biphasic (2 ms per phase), constant current (350 μA) pulse. A single stimulation pulse was used to evoke striatal DA release in the caudate–putamen while a 20 pulse, 100 Hz stimulation train was used to evoke 5-HT release in the SNr. The current pulse was optically isolated from the preparation (NL 800; Neurolog, Medical Systems). Stimuli were given at regular intervals. Neurotransmitter release was detected with a carbon-fiber microelectrode placed 75–100 μm into the slice at a distance of 100–200 μm from the stimulating electrode. Electrode placements were made with micromanipulators and the aid of the microscope.
To determine the amount of DA release from each genotype, the maximal release at four locations in a single slice was recorded over a 20 min period, and the values were averaged together. The responses from each genotype were pooled. To examine the effects of cocaine and Ca2+, measurements were made at a single site that exhibited maximal DA release. The amount of 5-HT released was determined in a similar fashion to DA with the exception that three locations were used and data were pooled.
In vivo experiments.
Mice were anesthetized with urethane (1.5 g/kg, i.p.) and placed in a stereotaxic frame. The coordinates relative to bregma for placement of the working electrode in the caudate–putamen were as follows (in mm): AP +1.1, ML +1.2, and DV −2.2 (Paxinos et al., 1985). The stimulating electrode was placed in the medial forebrain bundle at AP −2.4, ML +1.1, and DV −4.5. The dorsal–ventral placement of both the working and stimulating electrodes was adjusted in small increments to find maximal DA release. An Ag/AgCl reference electrode was inserted into the contralateral side of the brain.
HPLC.
Tissue samples were punched from slices (500 μm thickness) with a 2.5 mm diameter cork borer. The tissue was blotted dry, weighed, and homogenized with a wand sonicator (Fisher Scientific) in 200 μl of 0.1N perchloric acid spiked with 1 μm hydroquinone. Following centrifugation at 6000 rpm for 10 min, the supernatant was removed and filtered with a 0.2 μm syringe filter unit (Millex-LG). Injections (10 μl) were made onto a reverse-phase column (C-18, 5 μm, 4.6 × 250 mm, Waters symmetry 300). The mobile phase (prepared in HPLC grade water) contained 0.1 m citric acid, 0.1 mm EDTA, and 1 mm hexyl sodium sulfate, pH 3.5. Catecholamines were detected with a thin-layer radial electrochemical flow cell (BASi), with the working electrode at 700 mV versus the Ag/AgCl reference electrode. Catecholamine standards were prepared from 10 mm stock solutions in 0.1N perchloric acid.
Kinetic modeling.
Nonlinear regression was used to extract kinetic constants associated with release and uptake of DA from the extracellular space (Wu et al., 2001). Concentration traces were fit using a Michaelis-Menten-based model (Wightman et al., 1988). The nonlinear regression procedure used in conjunction with deconvolution of the measured responses from the electrode time constant (15–50 ms for a 60 Hz sampling rate) improves accuracy for release and uptake measurements (Garris and Wightman, 1995; Venton et al., 2002). The output of the regression is the amount of DA released for each stimulus pulse ([DA]p) during a train of stimuli and the Vmax for uptake. The Km value was fixed at 200 nm for DA (Near et al., 1988) and 170 nm for 5-HT (John et al., 2006). When competitive uptake inhibitors such as cocaine were used, Vmax was fixed at predrug values to determine the apparent Km.
Data analysis.
Data are shown as mean ± SEM. The n values are the number of animals used for a specific experiment. In addition to this replicate number, multiple locations and stimuli were used to reduce random error in the electrochemical measurements. Tests of significance included Student's t test, one-way ANOVA with Student–Newman–Keuls post hoc, two-way repeated-measures ANOVA with Bonferroni comparisons post hoc, and three-way repeated measures with Bonferroni comparisons post hoc where applicable.
Results
Serotonin release in substantia nigra brain slices
The influence of synapsins on electrically evoked release of 5-HT (Bunin et al., 1998) was evaluated by comparing 5-HT release in slices prepared from the brains of TKO and WT mice. In these experiments, we used local electrical stimulation with a stimulus train consisting of 20 biphasic pulses (2 ms each phase) of 350 μA at 100 Hz. 5-HT release was monitored by cyclic voltammetry and the voltammogram at the peak (Fig. 1, inset) was used to positively identify the released substance. 5-HT release evoked under such conditions was consistent over several hours if the stimulus trains were separated by intervals of 5 min or longer (Bunin et al., 1998). To minimize the contribution of spatial heterogeneity of 5-HT release sites, measurements were made at three different positions of the carbon-fiber electrode within each SNr slice and then averaged together.
Figure 1.

Representative traces of 5-HT release in coronal brain slices containing the SNr from WT and TKO mice evoked by a 100 Hz stimulation train (20 pulses, black line). Inset, The cyclic voltammogram is used for identification of 5-HT.
In slices from both genotypes, 5-HT release began promptly at the onset of the stimulus and ended as soon as the stimulus stopped, with the concentration of 5-HT returning to prestimulus values over the next 1–2 s (Fig. 1). Measurements of the peak concentration of released 5-HT ([5-HT]max) from 10 animals of each genotype showed that the mean [5-HT]max was 130 ± 8 nm in WT mice and 140 ± 7 nm in TKO mice; these values are not significantly different from each other (p > 0.05). The maximal rate of 5-HT uptake (Vmax) was evaluated in WT and TKO slices by using nonlinear regression to model the decay of the traces (see Materials and Methods). The Vmax of 5-HT uptake for WT and TKO animals was 450 ± 70 nm/s and 370 ± 80 nm/s, respectively. These values are not significantly different from each other (p > 0.05) and are similar to those reported previously for the SNr (John et al., 2006).
Tissue 5-HT content in the SNr was measured by HPLC. Tissue punches taken from TKO and WT mice (four animals per genotype) revealed that tissue levels of 5-HT were not different between genotypes (Table 1).
Table 1.
Neurotransmitter content from tissue samples containing either the dorsolateral striatum (DA) or the substantia nigra pars reticulata (5-HT)
| Animal | Striatal tissue DA content (μg/g) | SNr tissue 5-HT content (μg/g) |
|---|---|---|
| WT | 4.97 ± 0.96 | 1.03 ± 0.20 |
| TKO | 4.68 ± 0.60 | 0.74 ± 0.10 |
Data is from four animals of each genotype and is expressed as mean ± SEM. No statistical differences were found between genotypes for either neurotransmitter (p > 0.05, Student's t test).
Synapsins affect dopamine release in striatal brain slices
Local electrical stimulation was used to evoke DA release from terminals in the caudate–putamen of coronal brain slices from synapsin TKO and WT animals. In this case, a single biphasic current pulse (2 ms each phase) of 350 μA was used as a stimulus, with cyclic voltammetry used to positively identify the signal as DA (Fig. 2A, inset). The peak oxidation potential of DA occurs at ∼0.6 V versus Ag/AgCl, similar to that shown for the peak oxidation of 5-HT. However, the cyclic voltammograms for the two substances differ on the reverse scan, with the peak for reduction of the dopamine-o-quinone occurring at approximately −0.3 V with the waveform used, whereas the reduction peak following 5-HT oxidation occurs at more positive potentials (Jackson et al., 1995).
Figure 2.
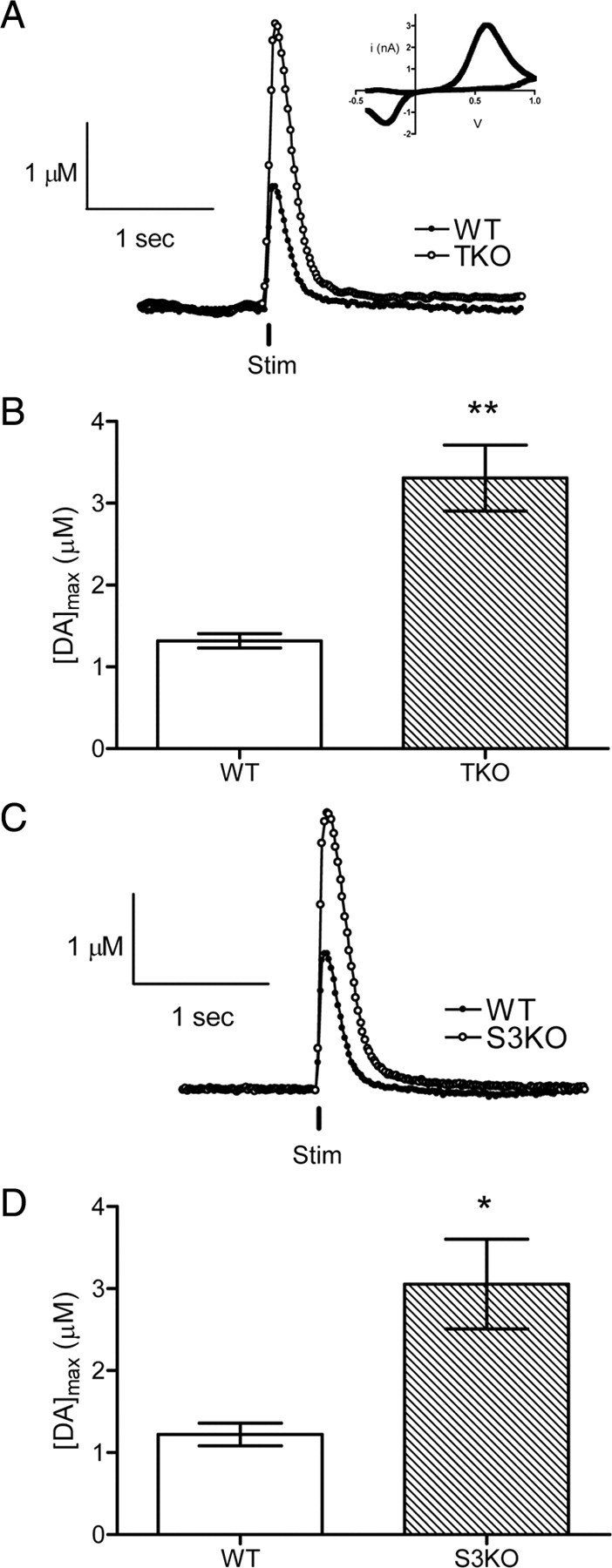
DA release evoked by a single stimulation pulse (vertical black line) in striatal brain slices from WT or TKO animals. A, Representative responses of stimulated DA release; the time of stimulation is shown by the vertical bar. Inset, The cyclic voltammogram identifies the released neurotransmitter as DA. B, Mean [DA]max from multiple locations of each genotype show an approximate twofold difference in striatal DA release (**p < 0.01, n = 8 TKO, n = 6 WT). C, Representative traces of DA release evoked by a single stimulus pulse in WT or S3KO animals. Time of stimulus is shown as a vertical black line. D, Measurements of DA release at multiple locations of each genotype indicate that S3KO slices have approximately double the amount of DA release measured in WT slices (*p < 0.05, n = 5 S3KO, n = 5 WT).
DA release was evoked every 5 min at four different sites per slice, using the single biphasic pulse described above, and the values of maximal DA release ([DA]max) determined at each site were pooled for each genotype. In every experiment, DA release was observed at the onset of stimulation and then declined back to prestimulus values within 1 s after cessation of the stimulation (Fig. 2A). The [DA]max was significantly greater (p < 0.01) in brain slices from TKO mice than in slices prepared from WT mice, being 1.31 ± 0.09 μm (n = 6) for WT and 3.31 ± 0.40 μm (n = 8) for TKO slices (Fig. 2B). The Vmax for DA uptake was evaluated with the same procedure described above for 5-HT uptake. The analysis revealed that the Vmax values for DA uptake were not significantly different between genotypes, at 6.00 ± 0.40 μm/s for WT and 6.20 ± 0.50 μm/s for TKO samples. These values are slightly higher than those reported previously in WT mice (Johnson et al., 2006).
Because synapsins I and II are lacking in DA terminals (Bogen et al., 2006), it is likely that synapsin III is the isoform that controls DA release. To test this prediction, DA release was compared in caudate–putamen slices of S3KO and WT animals. Release of DA in S3KO brain slices was significantly higher (p < 0.05) than in WT slices (Fig. 2C), with [DA]max being 3.06 ± 0.55 μm (n = 5) in S3KO and 1.22 ± 0.13 μm in WT slices (n = 5) (Fig. 2D). DA uptake values were not different between genotypes, 5.97 ± 0.58 μm/s for S3KO (n = 5) and 6.16 ± 0.38 μm/s for WT slices (n = 5). A comparison of the mean [DA]max in TKO and S3KO groups showed that these values are not statistically different. The similar amounts of DA release in TKO and S3KO slices indicate that a synapsin III gene product is responsible for regulating DA release.
To determine whether the previously reported changes in release of glutamate or GABA (Gitler et al., 2004) underlie the changes in DA release observed in TKO animals, we repeated our measurements in the presence of CNQX, to block AMPA receptors, or picrotoxin to block GABAA receptors. CNQX increased DA release while picrotoxin depressed it; however, the difference in [DA]max between genotypes remained unchanged (supplemental Table S1, available at www.jneurosci.org as supplemental material). Thus, the difference between TKO and WT mice does not arise from differential regulation of DA release by glutamate or GABA. Because spontaneous release of acetylcholine also regulates DA release (Zhou et al., 2001), we further measured DA release in the presence of mecamylamine, an antagonist of nicotinic acetylcholine receptors. Consistent with previous reports, we found that mecamylamine reduced DA release. However, the degree of reduction in DA release was not different between genotypes (supplemental Table S1, available at www.jneurosci.org as supplemental material). Thus, the increase in DA release in synapsin TKO and S3KO mice also is not a result of altered cholinergic signaling at DA terminals.
Dopaminergic neurons are known to release only a small fraction of their total stores to individual stimulus pulses (Gubernator et al., 2009). To determine whether the increased release of DA observed in TKO mice was due to the striatum containing greater amounts of DA, we used HPLC to analyze the DA content of striatal tissue. No significant differences in the amounts of tissue DA in the striatum were found between the two genotypes (Table 1), indicating that the increased amount of DA release found in TKO mice was not due to larger DA stores, but instead is due to electrical stimulation releasing a larger fraction of these stores.
Given that synaptic depression is altered in synapsin TKO mice (Gitler et al., 2004), we asked whether loss of synapsins affects DA release during repetitive stimulation. With either 0.3 s or 3 min intervals between stimuli, the amount of DA released per stimulus was depressed during repetitive stimulation and no differences in this activity-dependent decrement in DA release were observed between slices from WT and TKO mice (Fig. 3). At stimulus intervals of 5 min, the amount of DA released per stimulus was constant in WT slices but there was a small reduction in DA release that was not significant (main effect of genotype, F = 5.12, p = 0.054) in TKO slices after six or more stimuli. This might reflect the smaller size of the reserve pool of DA in TKO neurons (Venton et al., 2006).
Figure 3.

The effects of repetitive stimuli on DA release. Single stimuli were applied at intervals of 0.3 s, 3 min, and 5 min, and the amount of DA released in response to each stimulus was normalized to the amount of DA released in response to the first stimulus.
Synapsins also affect striatal dopamine release in vivo
We next assessed whether synapsins influence the amount of DA released in vivo by measuring DA release in the brains of anesthetized TKO and WT mice. In these experiments, DA release in the caudate–putamen was evoked by electrical stimulation of the medial forebrain bundle, rather than by the local electrical stimulation that was used in our brain slice experiments. As reported previously, the smaller amounts of DA released per stimulus in vivo made it necessary to deliver repeated stimuli to elicit detectable amounts of DA release (Venton et al., 2006). Consistent with the results of our slice experiments, 5 min interstimulus intervals were required to observe consistent amounts of DA release in response to each stimulus. Figure 4A shows averaged measurements of DA release measured in TKO and WT mice in response to a brief train of 20 Hz stimuli (0.5 s duration, 300 μA; n = 8 for each genotype). At this frequency, trains of activity in WT animals led to a concentration change that approached steady state during the stimulus, a response that has previously been documented in rats in vivo (Wightman et al., 1988).
Figure 4.
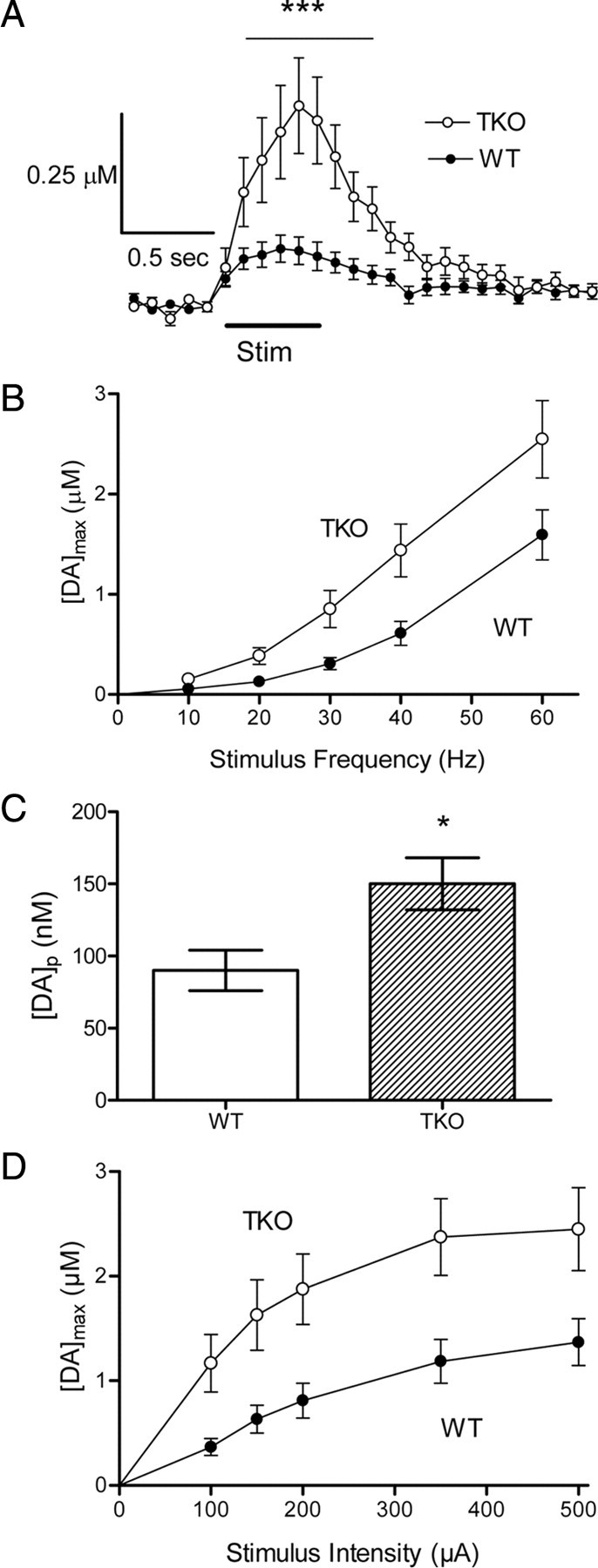
Electrically stimulated DA release in the striatum measured in vivo. A, Average release profiles measured in WT and TKO mice (n = 8 for each) in response to 0.5 s stimulus train (300 μA, 20 Hz; ***p < 0.001). B, [DA]max values exhibit similar frequency dependencies but release is uniformly greater in TKO animals. C, [DA]p measured from the frequency-dependent release shown in B (*p < 0.05). D, Although similar responses to stimulus intensity (60 Hz stimulations) are found in both genotypes, DA release is greater in TKO animals.
The larger amounts of DA released from TKO terminals led to the different shapes of the DA concentration profiles seen in Figure 4A, where the DA concentration measured during stimulation more closely approached a steady state during the stimulation in WT than in TKO animals. Transmission at dopaminergic synapses is a balance between DA release and uptake: because of this balance, DA concentration in WT animals reaches a steady plateau during the stimulus train. In contrast, the DA concentration continually increases when larger amounts of DA are released, because DA transport becomes saturated and thereby permits continued accumulation of extracellular DA (Wightman et al., 1988). Such behavior for DA release in the TKO mice presumably is due to the larger amounts of DA release in these mice.
Given that repetitive stimulation was required to elicit measurable DA release, we next evaluated how loss of synapsins affected the frequency dependence of DA release. At all stimulus frequencies tested, DA release was greater in TKO animals than in WT mice (Fig. 4B) (main effect of genotype, F = 6.86, p < 0.05). At a stimulus intensity of 350 μA, the data could be described by a model that assumed that the amount of DA released per stimulus pulse ([DA]p) in vivo was constant. [DA]p was found to be 90 ± 14 nm for WT and 150 ± 18 nm for TKO mice (Fig. 4C). This difference between the two genotypes is significant (p < 0.05). Although DA release from TKO animals approached a plateau at the same stimulus intensity (350 μA) as in WT animals, the amount of DA released during 60 Hz stimulation was higher at all stimulus intensities tested (Fig. 4D) (main effect of genotype, F = 8.69, p < 0.05). Saturation of the relationship between DA release and stimulus intensity is thought to arise from saturating stimuli maximally depolarizing the membrane potential and maximally releasing DA from a restricted pool available for immediate release in response to such depolarization (Wiedemann et al., 1990). Given that the ability of the stimuli to depolarize should be similar between the two genotypes, the greater amount of DA released at saturation suggests a larger pool of readily releasable DA in TKO mice.
Calcium dependence of dopamine release
Given the essential role of calcium ions (Ca2+) in triggering neurotransmitter release (Katz, 1969; Augustine et al., 1987; Neher, 1998), we next determined whether loss of synapsins affected the calcium dependence of DA release. For this purpose, caudate–putamen slices first were equilibrated in conventional aCSF containing 2.4 mm Ca2+ and DA release produced by electrical stimuli (single biphasic pulse, 2 ms each phase, 350 μA) was measured at a single location. This location was maintained throughout the experiment while the Ca2+ concentration ([Ca2+]) was varied. Treatment with aCSF lacking Ca2+ was then used to remove Ca2+ from the slice and the extracellular [Ca2+] was subsequently increased incrementally from 0.5 to 5 mm. At each [Ca2+], slices were allowed to equilibrate for 35 min and then were stimulated to evoke DA release. To compare the absolute amount of DA released at each [Ca2+] between genotypes, DA release at each [Ca2+] was divided by the DA release at 2.4 mm Ca2+ in WT slices and the resulting ratio was termed “relative dopamine release.”
We found that the Ca2+ dependence of DA release differed between slices from TKO and WT animals. At all Ca2+ concentrations examined, DA release was significantly higher in slices from TKO mice than in WT slices (p < 0.001, n = 5 for each genotype) (Fig. 5A). To quantify the relationship between external [Ca2+] and DA release, the data shown in Figure 5A were replotted in logarithmic coordinates (Dodge and Rahamimoff, 1967; Augustine and Charlton, 1986). In this case, the sigmoidal relationship between [Ca2+] and DA release is linearized, with the slope of the line representing the amount of “cooperativity” in Ca2+ triggering DA release and the [Ca2+] required for half-maximal DA release reflecting the apparent Ca2+ affinity, KCa2+ (Dodge and Rahamimoff, 1967). To determine these two parameters, we fit the data with the Hill equation. Although the shapes of these logarithmic plots were very similar for DA release from both genotypes, we found that the TKO data were shifted toward lower [Ca2+] (Fig. 5B). This leftward shift was due to a significant difference in the apparent KCa2+ between the two genotypes: 2.80 ± 0.27 mm for WT and 1.34 ± 0.04 mm for TKO animals (p < 0.001). The Hill coefficient was similar for both genotypes at 2.11 ± 0.20 for WT animals and 2.73 ± 0.22 for TKO animals (p > 0.05). These values for the Hill coefficient are similar to those previously reported for depolarization-evoked DA release from synaptosomes (Nachshen and Sanchez-Armass, 1987). Thus, we conclude that the absence of synapsins raises the apparent Ca2+ affinity of the DA release process without changing the degree of cooperativity.
Figure 5.
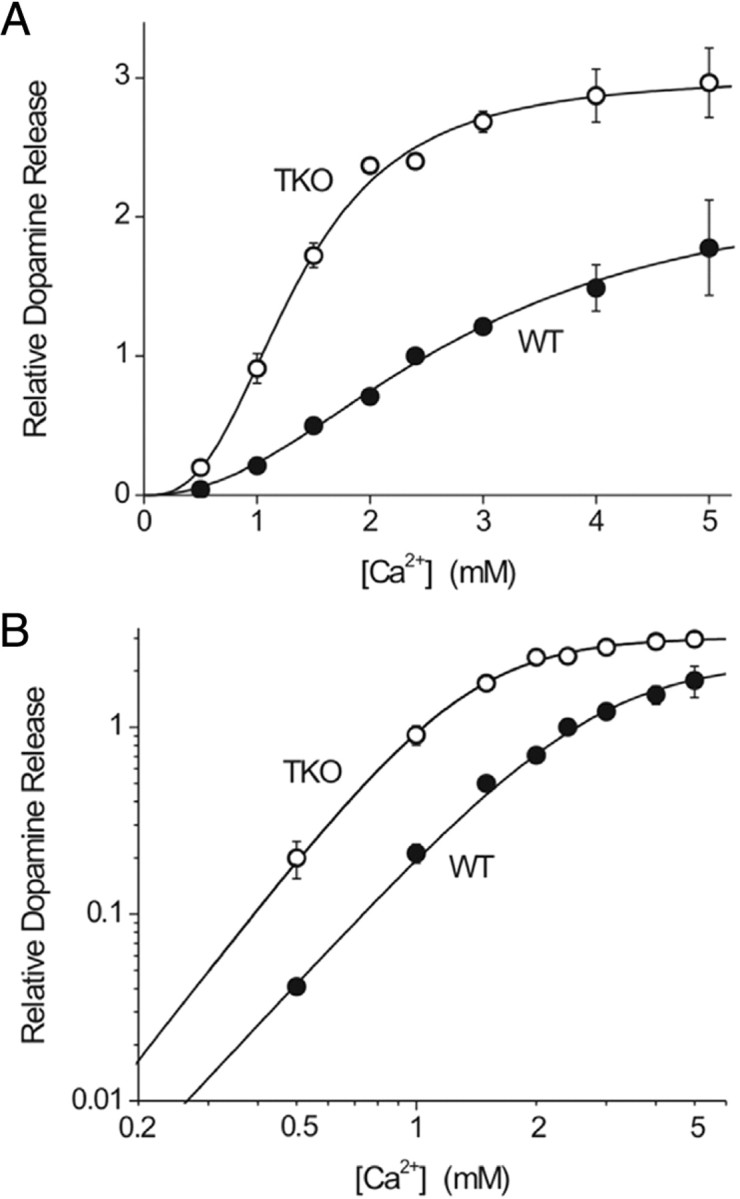
Calcium dependence of DA release in striatal slices. A, Relative [DA]max for TKO and WT animals at 0.5–5 mm extracellular [Ca2+]. TKO animals release significantly more DA at all [Ca2+]. B, Log-plot used to determine stoichiometric relationship between DA release and [Ca2+] using a fit to the Hill equation.
Effects of cocaine on dopamine and serotonin release
The ability of cocaine to enhance DA release in vivo is reduced in synapsin TKO mice (Venton et al., 2006). We therefore compared the effects of cocaine on release of 5-HT and DA in these mice. The effect of cocaine on 5-HT release in SNr brain slices was evaluated by a 35 min application of cocaine (10 μm), a concentration that causes competitive inhibition of 5-HT transport (John and Jones, 2007) and thereby increases the apparent value of Km. Cocaine treatment caused dramatic changes in the shape of the 5-HT concentration profile produced by electrical stimulation (20 biphasic pulses, 2 ms each phase, 100 Hz, 350 μA), prolonging the time course of these signals in slices from both WT and TKO mice (Fig. 6, left panels). This prolongation is due to cocaine inhibiting 5-HT uptake. To estimate the apparent Km for 5-HT uptake in the presence of cocaine (Jones et al., 1995), we again used nonlinear regression to evaluate the decaying phase of the 5-HT signal. The apparent Km values were 0.75 ± 0.06 μm for WT and 0.83 ± 0.09 μm for TKO animals in the presence of cocaine (n = 7 for WT, n = 6 for TKO), values that were larger than the predrug value of 0.17 μm (John et al., 2006). However, the Km values measured in the presence of cocaine for each genotype were not significantly different (p > 0.05) and were similar to previous measurements made for 5-HT uptake in the presence of 10 μm cocaine (John and Jones, 2007). In addition, cocaine treatment did not significantly (p > 0.05) alter the amount of 5-HT released by electrical stimulation in slices from either genotype: in the presence of 10 μm cocaine, the amplitude of 5-HT release was 110 ± 6% of predrug values in tissue from WT and 112 ± 8% of predrug values in tissue from TKO animals.
Figure 6.

Effects of cocaine on stimulated 5-HT and DA release and uptake in brain slices. Left traces, Representative responses of 5-HT release in SNr from WT (upper) and TKO (lower) mice in the presence and absence of 10 μm cocaine for 35 min. Right traces, Representative responses of DA release in striatal tissue from WT (upper) and TKO (lower) mice measured before and 35 min after the introduction of 2 μm cocaine.
We next considered the influence of synapsins on the ability of cocaine to increase DA release. The effect of cocaine on DA release evoked by electrical stimuli (single biphasic pulse, 2 ms each phase, 350 μA) was compared in caudate–putamen slices prepared from the two mouse genotypes. These slices were treated for 35 min with 2 μm cocaine, a dose known to inhibit DA uptake in mouse brain slices (Jones et al., 1995). Although uptake of DA was much faster than that of 5-HT (compare traces in left and right columns of Fig. 6), cocaine still caused a significant slowing in the time course of the DA signal produced by electrical stimulation (Fig. 6, right panels). This slowing indicates an inhibition of DA uptake. The apparent Km values for DA uptake after application of cocaine were 2.60 ± 0.09 μm for WT and 2.60 ± 0.15 TKO μm mice (n = 7 for each genotype). While these values are not significantly different from each other, they are substantially larger than the value of 0.2 μm measured in control conditions.
Cocaine treatment also caused an increase in the peak amplitude of the DA signals. This finding is similar to observations in rat (Jones et al., 1995) and mouse (Venton et al., 2006) brain tissue. The stimulatory effect of cocaine on DA was much greater in slices from WT mice than in slices from TKO mice (Fig. 6, right panels). On average, cocaine increased electrically evoked DA release by 89 ± 4% in slices from WT mice, but only 49 ± 3% in TKO slices (Fig. 6). This difference between the cocaine responses of the two genotypes is significant (p < 0.001; n = 4 for each genotype).
We also examined the influence of external Ca2+ on the ability of cocaine to enhance DA release. Over the entire range of [Ca2+] considered, cocaine caused a greater enhancement of DA release from WT than from TKO slices (p < 0.001). Figure 7A shows the relative DA release from both genotypes as a function of the external [Ca2+] after all release values were normalized to the value measured for WT samples at 2.4 mm Ca2+. The difference in Ca2+ sensitivity between WT and TKO animals shown in Figure 5 was diminished by 2 μm cocaine. Hill equation fits to these data indicate that the apparent Ca2+ affinity, KCa2+, was identical in the two genotypes, being 1.07 ± 0.15 mm for WT and 1.09 ± 0.13 mm for TKO. Likewise, the degree of cooperativity was identical, with Hill coefficients of 2.19 ± 0.43 for WT and 2.11 ± 0.34 for TKO. Even in the presence of cocaine, the maximum values for relative DA release were still higher in TKO mice. To further quantify the effects of cocaine on the Ca2+ dependence of DA release, we calculated the ratio of DA release measured in the presence and absence of cocaine relative to the value at 2.4 mm Ca2+ for each genotype (Fig. 7B). This ratio was significantly greater than 1 when [Ca2+] was below 2.4 mm for WT slices (p < 0.001, n = 6), while TKO slices only showed significant effects of cocaine at [Ca2+] of 1 mm or less (p < 0.001, n = 6). Therefore, we conclude that cocaine preferentially enhances DA release from WT animals and that this enhancement depends upon Ca2+ and synapsins.
Figure 7.

The effect of 2 μm cocaine on the Ca2+ dependence of DA release in striatal slices from WT and TKO mice. A, Relative [DA]max in TKO and WT animals as a function of extracellular [Ca2+] in the presence of 2 μm cocaine. Data are normalized relative to the amount of DA release measured in WT slices at 2.4 mm Ca2+. TKO slices release more DA at all [Ca2+], although little difference in the Ca2+ sensitivity of release is seen between the genotypes when cocaine is present. B, Ratio of release in the presence of cocaine to that in its absence as a function of external [Ca2+] for WT and TKO animals (n = 6, each genotype); measured release was normalized to release at 2.4 mm Ca2+ for each genotype. In slices from WT mice, cocaine significantly increased electrically stimulated DA release at all [Ca2+] below 2.4 mm; however, TKO animals are only facilitated at low levels of [Ca2+]. ***p < 0.001 indicates significant difference from 2.4 mm Ca2+.
Discussion
Our comparison of DA and 5-HT release from WT and synapsin-deficient TKO mice revealed differences in the effect of synapsin deletion on the release of these two monoamine neurotransmitters. The amount of DA released by electrical stimuli was almost twice as large in TKO mice as in WT controls. In contrast, 5-HT release was unaltered in TKO mice. While cocaine blocked uptake of both 5-HT and DA, raising the apparent value of Km in both genotypes, it was able to enhance the release only of DA. The enhancement of DA release by cocaine was greater in WT than in TKO animals. The absence of synapsins increased the apparent Ca2+ affinity of the DA release process, an effect that was mimicked by cocaine in WT tissue. Thus, synapsins play very different roles in nerve terminals using these two biogenic amine neurotransmitters.
Synapsins regulate dopamine release
Greater DA release was observed in intact TKO mice, in striatal brain slices prepared from these mice, and in S3KO slices. This establishes a role for synapsins in general, and synapsin III in particular, in dopaminergic presynaptic terminals. Although DA release was enhanced, uptake rates and tissue DA content were similar in TKO and WT mice. Thus, loss of synapsins caused a change in the ability of stimuli to release DA, rather than producing a change in the overall amount of DA or altering the function of DAT. Despite the increased amount of DA release in TKO mice, the activity dependence of DA release during repetitive activity was minimally affected. This suggests that synapsins play only a minor role in replenishing the readily releasable pool of DA.
Synapsins have a wide range of actions at different nerve terminals. In the adult brain, loss of synapsins leads to a decrease in the amount of GABA released by single stimuli, an effect likely due to a reduction in the readily releasable pool of GABAergic vesicles (Gitler et al., 2004; Baldelli et al., 2007). This action of synapsins is thought to be mediated by synapsin I (Baldelli et al., 2007). While release of glutamate in response to single stimuli is unaffected by loss of synapsins, the kinetics of synaptic depression are accelerated (Gitler et al., 2004). This is thought to be due to the ability of synapsin IIa to maintain glutamatergic synaptic vesicles in a reserve pool (Gitler et al., 2008). Loss of synapsins increases the release of catecholamines from adrenal chromaffin cells by increasing the number of exocytotic events, an effect rescued by expression of synapsin IIa (Villanueva et al., 2006). These findings suggest that synapsins play different roles in nerve terminals using different types of neurotransmitters.
The results of our analysis of monoamine neurotransmitter release in synapsin TKO mice provide two additional lines of support for the hypothesis that synapsins play different roles in nerve terminals using different types of neurotransmitters. First, release of 5-HT in response to brief trains of stimuli is unaffected by the absence of synapsins. While it is possible that different synapsins can have opposing effects on 5-HT release that are neutralized by deleting all three synapsin genes, the most parsimonious explanation for our results is that synapsins do not regulate 5-HT release. This is similar to the case for acetylcholine release at neuromuscular junctions, where transmission reportedly is normal in TKO mice (Gaffield and Betz, 2007). The absence of an effect of synapsins on 5-HT release may be due to the fact that 5-HT is mainly stored in large dense-core vesicles (LDCVs) (Nirenberg et al., 1995), while glutamate, GABA, and DA are primarily stored in small synaptic vesicles (SSVs) (Nirenberg et al., 1997). In synapses that contain both SSVs and LDCVs, synapsin I reportedly associates only with SSVs (Navone et al., 1984). Thus, the lack of effect on 5-HT release in TKO brain slices indicates that synapsins do not regulate 5-HT release. Although synapsin I mRNA is expressed in the SNr (Allen Institute for Brain Discovery), it is possible that this protein is not expressed in serotonergic nerve terminals. Second, we found that release of DA in response to single stimuli or brief trains of stimuli is enhanced in TKO animals. This is most likely due to loss of synapsin III, because DA release was enhanced in slices from S3KO mice. These results are consistent with the observation that the striatum is one of the few locations where synapsin III is found in the adult mouse brain (Pieribone et al., 2002). Even though the mRNAs for both synapsins I and II are present in the striatum (Allen Institute for Brain Discovery), neither isoform colocalizes with the vesicular monoamine transporter 2 (VMAT2) so it is likely that synapsin III is the synapsin isoform present in dopaminergic terminals (Bogen et al., 2006). The fact that DA released by single, brief stimuli was enhanced suggests that synapsin III serves as a negative regulator of the readily releasable pool of dopaminergic vesicles. This is reminiscent of the observation that S3KO mice have an increased recycling pool of glutamatergic synaptic vesicles (Feng et al., 2002), suggesting a general role for synapsin III as a negative regulator of neurotransmitter release. The regulation of DA release by synapsin III is even more intriguing in light of work implicating synapsin III in schizophrenia, a disorder in which altered DA signaling is thought to be important (Vawter et al., 2002; Porton and Wetsel, 2007; Porton et al., 2010).
In summary, it seems clear that different synapsin isoforms play distinct roles in nerve terminals using different neurotransmitters. In some cases, synapsin I predominates, while in other cases synapsins II or III are the primary regulators of neurotransmitter release. Further, some synapsin isoform effects are limited to vesicles in the reserve pool, while in other cases they more directly influence exocytosis from the readily releasable pool of vesicles [see Hilfiker et al. (1998) and Coleman and Bykhovskaia (2009)]. Together, these findings extend the classical model of synapsins maintaining the reserve pool of vesicles through phosphorylation-sensitive tethers to the cytoskeleton (Greengard et al., 1993).
Synapsin removal alters calcium and cocaine responses
Increasing extracellular [Ca2+] enhances neurotransmitter release by increasing Ca2+ entry into presynaptic terminals and subsequent accumulation of Ca2+ within the terminal cytoplasm (Augustine and Charlton, 1986; Nachshen and Sanchez-Armass, 1987). We found that terminals from TKO mice were more sensitive to extracellular [Ca2+] than terminals from WT mice,. The higher saturation value of DA release reached in TKO animals (Fig. 5) provides another indication that synapsins serve as a negative regulator of the readily releasable pool of dopaminergic vesicles.
Cocaine inhibits the DA transporter, thereby increasing extracellular DA concentrations (Jones et al., 1996). Cocaine also facilitates DA release from presynaptic terminals (Lee et al., 2001) by a synapsin-dependent effect (Venton et al., 2006). The present work confirms this cocaine-induced enhancement of DA release and demonstrates that it is significantly less in tissue from synapsin TKO animals (Fig. 6). In WT tissue, cocaine enhances both DA release and Ca2+ sensitivity (compare Fig. 5A and Fig. 7A) to values similar to those obtained in TKO tissue without cocaine. It is unclear how cocaine affects the Ca2+ sensitivity of release; cocaine can increase intracellular Ca2+ levels in brain cells (Du et al., 2006), which would be expected to increase the sensitivity of DA release to extracellular Ca2+ levels. This effect is not produced by methylphenidate, indicating that cocaine could increase DA release via a DAT-independent pathway (Du et al., 2006). Cocaine also has been proposed to alter calcium influx through calcium channels (Han et al., 2002), which would also produce an increase in the sensitivity of DA release to extracellular Ca2+. The fact that cocaine can increase DA release in TKO slices at low [Ca2+] (Fig. 7B) indicates the presence of a second, minor pathway for enhancing DA release that is independent of synapsins. This effect may be minor because DA release is already close to saturation in TKO tissue.
Cocaine is also a potent inhibitor of the 5-HT transporter (Uhl et al., 2002; Uhl and Lin, 2003). Our measurements in the SNr confirm this action of cocaine by showing that the rate of uptake of 5-HT was lowered in the presence of the drug. However, unlike its effects on DA release, cocaine does not enhance 5-HT release (Fig. 6). Furthermore, there is no difference in the cocaine sensitivity of 5-HT uptake between WT mice and synapsin TKO mice. These distinctions between the actions of cocaine on dopaminergic and serotonergic neurons may reflect the different roles that synapsins play in terminals using DA and 5-HT.
In conclusion, the loss of synapsins alters DA release, but not 5-HT release, from presynaptic terminals. The similar enhancement of DA release that occurs in synapsin III KO mice emphasizes the predominant role of this synapsin isoform in regulating DA release. The ability of calcium to trigger exocytosis of DA was also enhanced by the absence of synapsins. Cocaine enhanced the Ca2+ sensitivity of DA release in WT slices to a level found in TKO tissue, further demonstrating the importance of synapsins for Ca2+ regulation of DA release. Together, our results indicate that synapsins have dramatically different functions at presynaptic terminals that employ different neurotransmitters. These findings should stimulate future efforts to understand the molecular mechanisms underlying the diverse actions of synapsins.
Footnotes
This work was supported by National Institutes of Health Grants MH-067044 and NS-21624 (G.J.A.), MH70898 and MH082441 (W.C.W.), and NS-38879 (R.M.W.). We thank Drs. Barbara Porton and H. T. Kao at Brown University for providing the synapsin III mice.
References
- Augustine GJ, Charlton MP. Calcium dependence of presynaptic calcium current and post-synaptic response at the squid giant synapse. J Physiol. 1986;381:619–640. doi: 10.1113/jphysiol.1986.sp016347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustine GJ, Charlton MP, Smith SJ. Calcium action in synaptic transmitter release. Annu Rev Neurosci. 1987;10:633–693. doi: 10.1146/annurev.ne.10.030187.003221. [DOI] [PubMed] [Google Scholar]
- Baldelli P, Fassio A, Valtorta F, Benfenati Lack of synapsin I reduces the readily releasable pool of synaptic vesicles at central inhibitory synapses. J Neurosi. 2007;27:13520–13531. doi: 10.1523/JNEUROSCI.3151-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bath BD, Michael DJ, Trafton BJ, Joseph JD, Runnels PL, Wightman RM. Subsecond adsorption and desorption of dopamine at carbon-fiber microelectrodes. Anal Chem. 2000;72:5994–6002. doi: 10.1021/ac000849y. [DOI] [PubMed] [Google Scholar]
- Baur JE, Kristensen EW, May LJ, Wiedemann DJ, Wightman RM. Fast-scan voltammetry of biogenic amines. Anal Chem. 1988;60:1268–1272. doi: 10.1021/ac00164a006. [DOI] [PubMed] [Google Scholar]
- Bogen IL, Boulland JL, Mariussen E, Wright MS, Fonnum F, Kao HT, Walaas SI. Absence of synapsin I and II is accompanied by decreases in vesicular transport of specific neurotransmitters. J Neurochem. 2006;96:1458–1466. doi: 10.1111/j.1471-4159.2005.03636.x. [DOI] [PubMed] [Google Scholar]
- Bunin MA, Prioleau C, Mailman RB, Wightman RM. Release and uptake rates of 5-hydroxytryptamine in the dorsal raphe and substantia nigra reticulata of the rat brain. J Neurochem. 1998;70:1077–1087. doi: 10.1046/j.1471-4159.1998.70031077.x. [DOI] [PubMed] [Google Scholar]
- Chi P, Greengard P, Ryan TA. Synaptic vesicle mobilization is regulated by distinct synapsin I phosphorylation pathways at different frequencies. Neuron. 2003;38:69–78. doi: 10.1016/s0896-6273(03)00151-x. [DOI] [PubMed] [Google Scholar]
- Coleman WL, Bykhovskaia M. Synapsin I accelerates the kinetics of neurotransmitter release in mouse motor terminals. Synapse. 2009;63:531–533. doi: 10.1002/syn.20635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodge FA, Jr, Rahamimoff R. Co-operative action a calcium ions in transmitter release at the neuromuscular junction. J Physiol. 1967;193:419–432. doi: 10.1113/jphysiol.1967.sp008367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du C, Yu M, Volkow ND, Koretsky AP, Fowler JS, Benveniste H. Cocaine increases the intracellular calcium concentration in brain independently of its cerebrovascular effects. J Neurosci. 2006;26:11522–11531. doi: 10.1523/JNEUROSCI.3612-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evergren E, Benfenati F, Shupliakov O. The synapsin cycle: a view from the synaptic endocytic zone. J Neurosci Res. 2007;85:2648–2656. doi: 10.1002/jnr.21176. [DOI] [PubMed] [Google Scholar]
- Fdez E, Hilfiker S. Vesicle pools and synapsins: new insights into old enigmas. Brain Cell Biol. 2006;35:107–115. doi: 10.1007/s11068-007-9013-4. [DOI] [PubMed] [Google Scholar]
- Feng J, Chi P, Blanpied TA, Xu Y, Magarinos AM, Ferreira A, Takahashi RH, Kao HT, McEwen BS, Ryan TA, Augustine GJ, Greengard P. Regulation of neurotransmitter release by synapsin III. J Neurosci. 2002;22:4372–4380. doi: 10.1523/JNEUROSCI.22-11-04372.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira A, Kao HT, Feng J, Rapoport M, Greengard P. Synapsin III: developmental expression, subcellular localization, and role in axon formation. J Neurosci. 2000;20:3736–3744. doi: 10.1523/JNEUROSCI.20-10-03736.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaffield MA, Betz WJ. Synaptic vesicle mobility in mouse motor nerve terminals with and without synapsin. J Neurosci. 2007;27:13691–13700. doi: 10.1523/JNEUROSCI.3910-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garris PA, Wightman RM. Distinct pharmacological regulation of evoked dopamine efflux in the amygdala and striatum of the rat in vivo. Synapse. 1995;20:269–279. doi: 10.1002/syn.890200311. [DOI] [PubMed] [Google Scholar]
- Gitler D, Takagishi Y, Feng J, Ren Y, Rodriguiz RM, Wetsel WC, Greengard P, Augustine GJ. Different presynaptic roles of synapsins at excitatory and inhibitory synapses. J Neurosci. 2004;24:11368–11380. doi: 10.1523/JNEUROSCI.3795-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitler D, Cheng Q, Greengard P, Augustine GJ. Synapsin IIa controls the reserve pool of glutamatergic synaptic vesicles. J Neurosci. 2008;28:10835–10843. doi: 10.1523/JNEUROSCI.0924-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greengard P, Valtorta F, Czernik AJ, Benfenati F. Synaptic vesicle phosphoproteins and regulation of synaptic function. Science. 1993;259:780–785. doi: 10.1126/science.8430330. [DOI] [PubMed] [Google Scholar]
- Gubernator NG, Zhang H, Staal RG, Mosharov EV, Pereira DB, Yue M, Balsanek V, Vadola PA, Mukherjee B, Edwards RH, Sulzer D, Sames D. Fluorescent false neurotransmitters visualize dopamine release from individual presynaptic terminals. Science. 2009;324:1441–1444. doi: 10.1126/science.1172278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han W, Saegusa H, Zong S, Tanabe T. Altered cocaine effects in mice lacking Ca(v)2.3 (alpha(1E)) calcium channel. Biochem Biophys Res Commun. 2002;299:299–304. doi: 10.1016/s0006-291x(02)02632-3. [DOI] [PubMed] [Google Scholar]
- Hilfiker S, Schweizer FE, Kao HT, Czernik AJ, Greengard P, Augustine GJ. Two sites of action for synapsin domain E in regulating neurotransmitter release. Nat Neurosci. 1998;1:29–35. doi: 10.1038/229. [DOI] [PubMed] [Google Scholar]
- Hilfiker S, Benfenati F, Doussau F, Nairn AC, Czernik AJ, Greengard P. Structural domains involved in the regulation of transmitter release by synapsins. J Neurosci. 2005;25:2658–2669. doi: 10.1523/JNEUROSCI.4278-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson BP, Dietz SM, Wightman RM. Fast-scan cyclic voltammetry of 5-hydroxytryptamine. Anal Chem. 1995;67:1115–1120. doi: 10.1021/ac00102a015. [DOI] [PubMed] [Google Scholar]
- John CE, Jones SR. Voltammetric characterization of the effect of monoamine uptake inhibitors and releasers on dopamine and serotonin uptake in mouse caudate-putamen and substantia nigra slices. Neuropharmacology. 2007;52:1596–1605. doi: 10.1016/j.neuropharm.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John CE, Budygin EA, Mateo Y, Jones SR. Neurochemical characterization of the release and uptake of dopamine in ventral tegmental area and serotonin in substantia nigra of the mouse. J Neurochem. 2006;96:267–282. doi: 10.1111/j.1471-4159.2005.03557.x. [DOI] [PubMed] [Google Scholar]
- Johnson MA, Rajan V, Miller CE, Wightman RM. Dopamine release is severely compromised in the R6/2 mouse model of Huntington's disease. J Neurochem. 2006;97:737–746. doi: 10.1111/j.1471-4159.2006.03762.x. [DOI] [PubMed] [Google Scholar]
- Jones SR, Garris PA, Wightman RM. Different effects of cocaine and nomifensine on dopamine uptake in the caudate-putamen and nucleus accumbens. J Pharmacol Exp Ther. 1995;274:396–403. [PubMed] [Google Scholar]
- Jones SR, O'Dell SJ, Marshall JF, Wightman RM. Functional and anatomical evidence for different dopamine dynamics in the core and shell of the nucleus accumbens in slices of rat brain. Synapse. 1996;23:224–231. doi: 10.1002/(SICI)1098-2396(199607)23:3<224::AID-SYN12>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Katz B. The release of neural transmitter substances. Liverpool, UK: Liverpool UP; 1969. [Google Scholar]
- Kume-Kick J, Rice ME. Dependence of dopamine calibration factors on media Ca2+ and Mg2+ at carbon-fiber microelectrodes used with fast-scan cyclic voltammetry. J Neurosci Methods. 1998;84:55–62. doi: 10.1016/s0165-0270(98)00087-9. [DOI] [PubMed] [Google Scholar]
- Lee TH, Balu R, Davidson C, Ellinwood EH. Differential time-course profiles of dopamine release and uptake changes induced by three dopamine uptake inhibitors. Synapse. 2001;41:301–310. doi: 10.1002/syn.1087. [DOI] [PubMed] [Google Scholar]
- Llinás R, Gruner JA, Sugimori M, McGuinness TL, Greengard P. Regulation by synapsin I and Ca(2+)-calmodulin-dependent protein kinase II of the transmitter release in squid giant synapse. J Physiol. 1991;436:257–282. doi: 10.1113/jphysiol.1991.sp018549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonart G, Simsek-Duran F. Deletion of synapsins I and II genes alters the size of vesicular pools and rabphilin phosphorylation. Brain Res. 2006;1107:42–51. doi: 10.1016/j.brainres.2006.05.092. [DOI] [PubMed] [Google Scholar]
- Nachshen DA, Sanchez-Armass S. Co-operative action of calcium ions in dopamine release from rat brain synaptosomes. J Physiol. 1987;387:415–423. doi: 10.1113/jphysiol.1987.sp016581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navone F, Greengard P, De Camilli P. Synapsin I in nerve terminals: selective association with small synaptic vesicles. Science. 1984;226:1209–1211. doi: 10.1126/science.6438799. [DOI] [PubMed] [Google Scholar]
- Near JA, Bigelow JC, Wightman RM. Comparison of uptake of dopamine in rat striatal chopped tissue and synaptosomes. J Pharmacol Exp Ther. 1988;245:921–927. [PubMed] [Google Scholar]
- Neher E. Vesicle pools and Ca2+ microdomains: new tools for understanding their roles in neurotransmitter release. Neuron. 1998;20:389–399. doi: 10.1016/s0896-6273(00)80983-6. [DOI] [PubMed] [Google Scholar]
- Nirenberg MJ, Liu Y, Peter D, Edwards RH, Pickel VM. The vesicular monoamine transporter 2 is present in small synaptic vesicles and preferentially localizes to large dense core vesicles in rat solitary tract nuclei. Proc Natl Acad Sci U S A. 1995;92:8773–8777. doi: 10.1073/pnas.92.19.8773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nirenberg MJ, Chan J, Liu Y, Edwards RH, Pickel VM. Vesicular monoamine transporter-2: immunogold localization in striatal axons and terminals. Synapse. 1997;26:194–198. doi: 10.1002/(SICI)1098-2396(199706)26:2<194::AID-SYN10>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C, Pennisi M, Topple A. Bregma, lambda and the interaural midpoint in stereotaxic surgery with rats of different sex, strain and weight. J Neurosci Methods. 1985;13:139–143. doi: 10.1016/0165-0270(85)90026-3. [DOI] [PubMed] [Google Scholar]
- Pieribone VA, Shupliakov O, Brodin L, Hilfiker-Rothenfluh S, Czernik AJ, Greengard P. Distinct pools of synaptic vesicles in neurotransmitter release. Nature. 1995;375:493–497. doi: 10.1038/375493a0. [DOI] [PubMed] [Google Scholar]
- Pieribone VA, Porton B, Rendon B, Feng J, Greengard P, Kao HT. Expression of synapsin III in nerve terminals and neurogenic regions of the adult brain. J Comp Neurol. 2002;454:105–114. doi: 10.1002/cne.10417. [DOI] [PubMed] [Google Scholar]
- Porton B, Wetsel WC. Reduction of synapsin III in the prefrontal cortex of individuals with schizophrenia. Schizophr Res. 2007;94:366–370. doi: 10.1016/j.schres.2007.04.016. [DOI] [PubMed] [Google Scholar]
- Porton B, Rodriguiz RM, Phillips LE, Gilbert JW, 4th, Feng J, Greengard P, Kao HT, Wetsel WC. Mice lacking synapsin III show abnormalities in explicit memory and conditioned fear. Genes Brain Behav. 2010;9:257–268. doi: 10.1111/j.1601-183X.2009.00555.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosahl TW, Spillane D, Missler M, Herz J, Selig DK, Wolff JR, Hammer RE, Malenka RC, Südhof TC. Essential functions of synapsins I and II in synaptic vesicle regulation. Nature. 1995;375:488–493. doi: 10.1038/375488a0. [DOI] [PubMed] [Google Scholar]
- Uhl GR, Lin Z. The top 20 dopamine transporter mutants: structure-function relationships and cocaine actions. Eur J Pharmacol. 2003;479:71–82. doi: 10.1016/j.ejphar.2003.08.058. [DOI] [PubMed] [Google Scholar]
- Uhl GR, Hall FS, Sora I. Cocaine, reward, movement and monoamine transporters. Mol Psychiatry. 2002;7:21–26. doi: 10.1038/sj.mp.4000964. [DOI] [PubMed] [Google Scholar]
- Vawter MP, Thatcher L, Usen N, Hyde TM, Kleinman JE, Freed WJ. Reduction of synapsin in the hippocampus of patients with bipolar disorder and schizophrenia. Mol Psychiatry. 2002;7:571–578. doi: 10.1038/sj.mp.4001158. [DOI] [PubMed] [Google Scholar]
- Venton BJ, Troyer KP, Wightman RM. Response times of carbon fiber microelectrodes to dynamic changes in catecholamine concentration. Anal Chem. 2002;74:539–546. doi: 10.1021/ac010819a. [DOI] [PubMed] [Google Scholar]
- Venton BJ, Seipel AT, Phillips PE, Wetsel WC, Gitler D, Greengard P, Augustine GJ, Wightman RM. Cocaine increases dopamine release by mobilization of a synapsin-dependent reserve pool. J Neurosci. 2006;26:3206–3209. doi: 10.1523/JNEUROSCI.4901-04.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villanueva M, Thornley K, Augustine GJ, Wightman RM. Synapsin II negatively regulates catecholamine release. Brain Cell Biol. 2006;35:125–136. doi: 10.1007/s11068-007-9015-2. [DOI] [PubMed] [Google Scholar]
- Wiedemann DJ, Basse-Tomusk A, Wilson RL, Rebec GV, Wightman RM. Interference by DOPAC and ascorbate during attempts to measure drug-induced changes in neostriatal dopamine with Nafion-coated, carbon-fiber electrodes. J Neurosci Methods. 1990;35:9–18. doi: 10.1016/0165-0270(90)90089-x. [DOI] [PubMed] [Google Scholar]
- Wightman RM, Amatore C, Engstrom RC, Hale PD, Kristensen EW, Kuhr WG, May LJ. Real-time characterization of dopamine overflow and uptake in the rat striatum. Neuroscience. 1988;25:513–523. doi: 10.1016/0306-4522(88)90255-2. [DOI] [PubMed] [Google Scholar]
- Wu Q, Reith ME, Wightman RM, Kawagoe KT, Garris PA. Determination of release and uptake parameters from electrically evoked dopamine dynamics measured by real-time voltammetry. J Neurosci Methods. 2001;112:119–133. doi: 10.1016/s0165-0270(01)00459-9. [DOI] [PubMed] [Google Scholar]
- Zhou FM, Liang Y, Dani JA. Endogenous nicotinic cholinergic activity regulates dopamine release in the striatum. Nat Neurosci. 2001;4:1224–1229. doi: 10.1038/nn769. [DOI] [PubMed] [Google Scholar]