

Abstract
Myogenic differentiation in adult muscle is normally suppressed and can be activated by myogenic cues in a subset of activated satellite cells. The switch mechanism that turns myogenesis on and off is not defined. In the present study, we demonstrate that tissue inhibitor of metalloproteinase 3 (TIMP3), the endogenous inhibitor of TNFα-converting enzyme (TACE), acts as an on–off switch for myogenic differentiation by regulating autocrine TNFα release. We observed that constitutively expressed TIMP3 is transiently downregulated in the satellite cells of regenerating mouse hindlimb muscles and differentiating C2C12 myoblasts. In C2C12 myoblasts, perturbing TIMP3 downregulation by overexpressing TIMP3 blocks TNFα release, p38 MAPK activation, myogenic gene expression and myotube formation. TNFα supplementation at a physiological concentration rescues myoblast differentiation. Similarly, in the regenerating soleus, overexpression of TIMP3 impairs release of TNFα and myogenic gene expression, and delays the formation of new fibers. In addition, downregulation of TIMP3 is mediated by the myogenesis-promoting microRNA miR-206. Thus, TIMP3 is a physiological regulator of myogenic differentiation.
Keywords: Muscle regeneration, Gene expression, TNFα converting enzyme, miR-206
Introduction
Adult myogenesis is essential to muscle regeneration. Key regulators of this process are still being identified (Wagers and Conboy, 2005). Skeletal muscle comprises terminally differentiated muscle fibers that have the capacity to regenerate in response to disease, injury or training. Muscle regeneration entails the activation, proliferation and differentiation of mononucleated satellite cells (muscle stem cells) that are associated with muscle fibers. Myogenic differentiation is a carefully controlled process that is normally suppressed until it is activated at an appropriate time in a subset of proliferating satellite cells. The remaining satellite cell pool stays undifferentiated and serves as the reserve for future regeneration events (Charge and Rudnicki, 2004). Although myogenic gene expression requires the reactivation of the myogenic program involving the expression of such transcription factors as Pax7, Myf5, MyoD, myogenin, MRF4 and MEF2, it is clear now that epigenetic regulations also have a pivotal role in mediating myogenesis in regenerating muscle (Guasconi and Puri, 2009).
Before myogenic gene expression, the SWI/SNF chromatin-remodeling complex first has to be activated to allow access of myogenic transcription factors to the muscle-specific gene promoters. Activation of the SWI/SNF chromatin-remodeling complex is mediated by coordinated activation of both p38 MAPK and AKT (Serra et al., 2007). Blockade of either kinase abolishes myogenesis (Cuenda and Cohen, 1999; de Angelis et al., 2005; Jiang et al., 1999; Perdiguero et al., 2007; Puri et al., 2000; Wu et al., 2000; Zetser et al., 1999). It has been known for sometime that myogenic activation of AKT is induced by IGF-I (Lawlor et al., 2000; Rommel et al., 2001; Tureckova et al., 2001). However, the signaling mechanism of myogenic activation of p38, particularly in adult muscle, emerged only recently. We demonstrated recently in adult muscle that myogenic activation of p38 requires TNFα-receptor-mediated signaling (Chen et al., 2005). In addition, we showed that in response to diverse myogenic cues, myoblasts release autocrine TNFα, which is crucial to myogenic activation of the MKK6–p38 pathway and ensuing myogenesis (Chen et al., 2007; Zhan et al., 2007). Moreover, TNFα-converting enzyme (TACE, also known as ADAM17), the disintegrin metalloproteinase (Black, 2002) that cleaves plasma membrane-anchored pro-TNFα (26 kDa) to release free TNFα (17 kDa), is rate limiting for myogenic activation of p38 (Zhan et al., 2007). These findings revealed a new signaling paradigm through which myogenic cues are transduced to activate myogenic gene expression via the activation of p38. In the current study, we address the question of how myogenic cues stimulate TACE release of TNFα.
TACE activity is normally repressed by its physiological inhibitor tissue inhibitor of metalloproteinase 3 (TIMP3). TIMP3 is a member of the tissue inhibitor of metalloproteinase family that uniquely inhibits TACE (Amour et al., 1998). As a transmembrane protein, TACE is structurally related to the matrix metalloproteinases (MMPs) (Black, 2002). TIMP3 appears to inhibit TACE in the same way the TIMPs inhibit MMPs: by chelating the extracellular active-site zinc with its N-terminus (Gomis-Ruth et al., 1997; Lee et al., 2005). TIMP3 is the only one of four TIMPs that binds to the extracellular matrix (Mohammed et al., 2003) and possesses an amino acid sequence (PFG) necessary for inhibiting TACE (Lee et al., 2005). TIMP3 suppresses inflammation (Black, 2004; Smookler et al., 2006) and impedes cell migration (van der Laan et al., 2003). These effects of TIMP3 could be attributed to its inhibition of TACE release of TNFα, which mediates inflammation (Tracey and Cerami, 1992) and stimulates the chemotactic response (Torrente et al., 2003). Because TIMP3 is constitutively expressed in muscle cells of mice (Leco et al., 1994) and humans (Apte et al., 1994), we hypothesized that it has a physiological role in suppressing myogenesis as an inhibitor of TACE, and that it has to be downregulated in response to myogenic cues to allow TACE release of autocrine TNFα and the ensuing activation of p38-dependent myogenesis. The present study demonstrates that TIMP3 is downregulated in regenerating mouse muscle, particularly, myogenic progenitor cells (MPCs). Furthermore, downregulation of TIMP3 is required for release of TNFα, activation of p38 and ensuing myogenesis. We also demonstrate that the downregulation of TIMP3 is mediated by microRNA-206 (miR-206). These data support a physiological role for TIMP3 in the suppression and activation of myogenesis.
Results
TIMP3 is downregulated in satellite cells of regenerating muscle and differentiating C2C12 myoblasts
To test our hypothesis, we analyzed the level of TIMP3 in regenerating mouse soleus muscle. We used the cardiotoxin injury model of regeneration in adult mice, in which cardiotoxin causes tissue necrosis and ensuing regeneration (Hawke and Garry, 2001). As shown in Fig. 1A, western blot analysis confirmed a constitutive expression of TIMP3 in the uninjured soleus. However, TIMP3 levels were rapidly downregulated after injury (within 1 day), reaching the lowest level on day 3. It is noteworthy that day 3 is the time myogenic differentiation takes place in this model (Chen et al., 2005; Chen et al., 2007). To investigate whether TIMP3 is downregulated during muscle regeneration induced by a different stimulus, we examined TIMP3 levels in functionally overloaded soleus by severing the synergistic gastrocnemius muscle according to an established procedure (Fluck et al., 1999). Although the functional overloading model has been widely used as a model of muscle hypertrophy, previous studies showed that satellite cell activation (Phelan and Gonyea, 1997; Rosenblatt et al., 1994) and myogenic differentiation (Serrano et al., 2008) are essential for the positive muscle remodeling induced by overloading. Therefore, the overloading model can also be used as a muscle regeneration model. As shown in Fig. 1B, TIMP3 was also downregulated in overloaded soleus, although the downregulation was not as pronounced and a trough was reached on day 5. Therefore, TIMP3 is transiently downregulated in muscle during muscle regeneration induced by diverse stimuli. To determine whether this takes place in satellite cells at the gene expression level, we isolated satellite cells from cardiotoxin-injured hindlimb muscles and analyzed Timp3 mRNA using real-time PCR. Similarly to TIMP3 protein levels in the injured soleus, Timp3 mRNA in satellite cells was downregulated on day 1, and reached the trough on day 3. Then, on day 10 when regeneration is largely completed, the Timp3 mRNA level recovered to 80% of the pre-injury level (Fig. 1C). Considering the reduction in satellite cell number on day 10 after myogenic differentiation, the Timp3 mRNA level in satellite cells was not inconsistent with the TIMP3 protein level observed in soleus on day 10 (Fig. 1A). These data reveal that expression of TIMP3 in satellite cells is transiently downregulated in regenerating muscle at the time myogenesis takes place.
Fig. 1.

TIMP3 expression is downregulated in satellite cells during muscle regeneration. (A) TIMP3 is downregulated in cardiotoxin-injured muscle. Mouse soleus was injured by direct injection of cardiotoxin. At indicated times, soleus was collected for western blot analysis of TIMP3 content, with α-tubulin monitored as a loading control. (B) TIMP3 is downregulated in functionally overloaded muscle. Mouse soleus that was overloaded for indicated durations was analyzed by western blot for TIMP3 content. (C) Timp3 mRNA is downregulated in satellite cells isolated from cardiotoxin-injured muscle. Mouse hindlimb muscles were excised and satellite cells were isolated at indicated times before and after cardiotoxin injection. Total RNA extracted from satellite cells was analyzed for Timp3 mRNA level using real time PCR. *P<0.05 significant difference from Day 0 determined by ANOVA. All results are means ± s.e.
To determine whether TIMP3 is downregulated during in vitro myogenesis, we monitored TIMP3 protein and mRNA levels in the mouse myoblast cell line C2C12. As shown in Fig. 2A, we observed constitutive expression of TIMP3 in C2C12 myoblasts. After induction of differentiation by switching the myoblasts from serum-rich growth medium (GM) into low-serum differentiation medium (DM), the TIMP3 level was downregulated from within 24 hours to at least 48 hours, when differentiation was initiated. In addition, a decline in the level of Timp3 mRNA, which took place slightly ahead of the decline in the TIMP3 protein level, was observed (Fig. 2B). These results indicate that downregulation of TIMP3 takes place during in vitro myogenesis. The above in vivo and in vitro data consistently indicate that TIMP3 expression undergoes a downregulation in MPCs that are exposed to myogenic cues.
Fig. 2.

TIMP3 expression is downregulated during in vitro myogenesis. (A) TIMP3 is downregulated in differentiating myoblasts. C2C12 myoblasts were induced to differentiate by switching GM to DM. At indicated times, myoblasts were collected for determination of TIMP3 content by western blot analysis. TIMP3 bands were quantified by densitometry analysis and normalized to β-actin. (B) Timp3 mRNA is downregulated in differentiating myoblasts. Total RNA was isolated from differentiating C2C12 myoblasts at indicated times, and Timp3 mRNA was determined by real-time PCR. *P<0.05, significant difference compared with 0 hour of differentiation determined by ANOVA. Results are means ± s.e.
Downregulation of TIMP3 is required for TNFα release, myogenesis and muscle regeneration
To understand the physiological function of the downregulation of TIMP3, we perturbed TIMP3 downregulation by overexpressing TIMP3 in C2C12 myoblasts using a previously described Timp3-encoding adenovirus (Baker et al., 1998). Western blot analysis confirmed the overexpression of TIMP3 24 hours after adenovirus infection and that overexpression of TIMP3 was sustained until at least 72 hours, after incubation in DM (Fig. 3A). The resistance of overexpressed TIMP3 to downregulation during myoblast differentiation suggests that the mechanism of the downregulation acts at the level of gene expression. To evaluate the effect of sustained TIMP3 on TNFα release by differentiating myoblasts, the concentration of TNFα in the cell culture medium was monitored by ELISA. Similarly to our previous report (Chen et al., 2007), TNFα concentration in DM increased during the first 48 hours of differentiation (Fig. 3B), indicating an increase in the release of TNFα by differentiating myoblasts. The increase in TNFα release was reciprocal to the TIMP3 downregulation shown in Fig. 2A. In differentiating myoblasts that overexpressed TIMP3, TNFα release was blocked, whereas overexpressed GFP did not alter TNFα release (Fig. 3B). These results indicate that the downregulation of TIMP3 relieves its inhibition of TACE and permits the release of TNFα.
Fig. 3.

TIMP3 overexpression suppresses TNFα release by differentiating myoblasts. (A) Overexpressed TIMP3 is resistant to downregulation in differentiating C2C12 myoblasts. C2C12 myoblasts were infected with recombinant adenovirus encoding Timp3 or GFP at 200 MOI. After 24 hours, myoblasts were switched from GM to DM (0 hours). At indicated times thereafter, myoblasts were collected for western blot analysis of TIMP3 content. (B) Overexpressed TIMP3 suppresses release of TNFα by differentiating myoblasts. The culture medium was collected from C2C12 myoblasts infected with recombinant adenovirus encoding TIMP3 or GFP at indicated times, concentrated and analyzed for TNFα content by using ELISA. *P<0.05, significant difference from control at the matching time points as indicated by ANOVA. Results are means ± s.e.
To assess the effect of TIMP3 downregulation on myogenesis, we determined the effect of TIMP3 overexpression on the differentiation of C2C12 myoblasts. We observed that overexpression of TIMP3 in C2C12 myoblasts blocked p38 activation, and suppressed expression of myogenin and MHC (Fig. 4A), whereas GFP overexpression did not alter these differentiation markers significantly. Considering that TACE has several substrates (Black, 2002) and TIMP3 also inhibits some other members of the ADAM family such as ADAM10 (Amour et al., 2000) and ADAM12S (Loechel et al., 2000), it is necessary to verify whether the observed inhibition of myogenesis by overexpressed TIMP3 was indeed due to the inhibition of TACE release of TNFα. We supplemented the DM with mouse recombinant TNFα at 0.05 ng/ml, which is a physiological concentration in injured muscle and stimulates myogenesis (Chen et al., 2007), and observed a restoration in p38 activation and expression of myogenin and MHC that were blocked by overexpressed TIMP3 (Fig. 4B). To further verify the effect of TIMP3 on C2C12 differentiation, we monitored the formation of myotubes by differentiating C2C12 myoblasts using a morphological approach. As shown in Fig. 5, after incubation in DM for 5 days, multinucleated myotubes formed in controls, but cells that overexpressed TIMP3 remained as mononucleated myoblasts. GFP overexpression did not alter myotube formation. Furthermore, consistent with data presented in Fig. 4, TNFα supplementation (0.05 ng/ml) rescued myotube formation. These results indicate that TIMP3 acts as a physiological suppressor of myogenesis via inhibiting TNFα release by TACE, and the downregulation of TIMP3 permits release of TNFα to activate myogenesis.
Fig. 4.

TIMP3 overexpression suppresses myogenic gene expression via inhibition of TNFα release. (A) TIMP3 overexpression suppresses myogenic gene expression. C2C12 myoblasts were infected with recombinant adenovirus encoding TIMP3 or GFP at 200 MOI. After 24 hours, myoblasts were switched from GM to DM (0 hours). At indicated times thereafter, myoblasts were collected and analyzed for differentiation markers by western blot analysis. (B) TNFα supplementation rescues myogenesis impaired by TIMP3 overexpression. C2C12 myoblasts were infected and differentiated as described above. Mouse recombinant TNFα (0.05 ng/ml) was supplemented in DM as indicated. Differentiation markers in C2C12 myoblasts were analyzed at indicated times by western blot analysis.
Fig. 5.

TIMP3 overexpression suppresses myotube formation via inhibition of TNFα release. C2C12 myoblasts were infected and differentiated as described above. Mouse recombinant TNFα (0.05 ng/ml) was supplemented in DM as indicated. Cells were then stained with the Giemsa dye and examined under a microscope. Scale bar: 100 μm.
TIMP3 is known to promote apoptosis in some non-muscle cells (Drynda et al., 2005). To evaluate whether overexpressed TIMP3 promotes apoptosis in C2C12 myoblasts, which could affect differentiation due to lower myoblast density, we analyzed apoptosis in C2C12 myoblasts that overexpressed TIMP3 by using the TUNEL assay. No apoptosis was observed in C2C12 myoblasts that overexpressed TIMP3, whereas clear signs of apoptosis were observed in myoblasts that were treated with the apoptosis inducer TACS nuclease as the positive control (data not shown). We also assessed the effect of overexpressed TIMP3 on myoblast proliferation by counting the cell number 24 hours after adenovirus infection, but before the induction of differentiation. No difference in total cell number was seen between cells infected with Adv-GFP and Adv-TIMP3 (data not shown). Thus, we can rule out the possibility that TIMP3 overexpression affects myogenesis via promoting apoptosis or inhibiting myoblast proliferation.
To evaluate whether TIMP3 regulates myogenesis in vivo, we overexpressed TIMP3 in mouse soleus and gastrocnemius using adenovirus according to a previously described protocol by injecting adenovirus to hindlimb muscles of neonatal mice (Durbeej et al., 2003; Chen et al., 2007). At 6 weeks of age, a modest level of adenovirus-mediated TIMP3 overexpression was indicated by a mild increase in TIMP3 in the uninjured soleus (Day 0). On day 3 of injury, whereas endogenous TIMP3 in controls that overexpressed GFP dropped to 9% of the preinjury level, overexpressed TIMP3 reached 63% of the preinjury level (Fig. 6A). The resistance of overexpressed TIMP3 to downregulation again supports that the mechanism of the downregulation is at the level of gene expression. On day 3 of injury, when myogenesis takes place, we observed that overexpressed TIMP3 in the soleus suppressed TACE release of free TNFα (17 kDa) and myogenin expression as compared with soleus that overexpressed GFP (Fig. 6B). To assess the effect of adenovirus injection and TIMP3 overexpression on muscle regeneration, excised soleus and gastrocnemius from mice received adenovirus encoding GFP or TIMP3 were subjected to histology on day 0, 3 and 10 after cardiotoxin injection. H&E-stained muscle sections at day 0 showed largely normal histology in adenovirus-injected muscle at 6 weeks of age before injury (Fig. 6C). Immunohistochemical study of embryonic MHC (eMHC) expression on day 3 of injury revealed depressed expression of this muscle-regeneration marker in TIMP3-overexpressing muscle. Morphologically, newly regenerated muscle fibers with centralized nuclei prevailed in muscle overexpressing GFP, but not in muscle overexpressing TIMP3 (Fig. 6C). On day 10 of injury, muscle regeneration in GFP-overexpressing muscle was essentially complete. Regenerated muscle fibers were also formed in TIMP3-overexpressing muscle; however, newly formed muscle fibers were of much smaller size and some inflammatory infiltration remained, indicating incomplete regeneration (Fig. 6C). Thus, overexpressed TIMP3 delayed myogenesis and regeneration. These in vivo data support a crucial role for TIMP3 in mediating muscle regeneration via regulating the on–off switch of myogenesis.
Fig. 6.
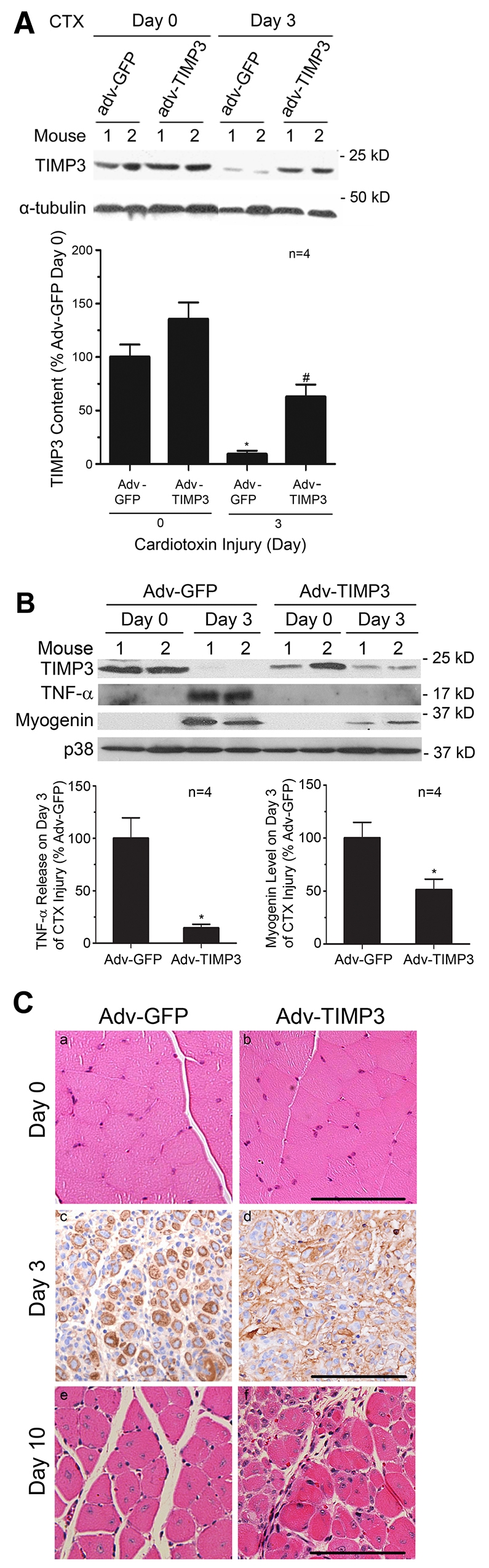
TIMP3 downregulation mediates myogenesis in regenerating muscle. (A) Overexpressed TIMP3 is resistant to downregulation in regenerating muscle. Adenovirus encoding TIMP3 or GFP was injected into the soleus of 3-day-old mice. At 6 weeks of age, soleus was injured by injection of cardiotoxin. TIMP3 levels in soleus on day 0 and day 3 of injury were evaluated by western blot analysis. Densitometry analysis was performed to quantify the results. *P<0.05 compared with Day 0 of Adv-GFP-injected muscle, and #P<0.05 compared with Day 0 and Day 3 Adv-GFP-injected muscle. (B) Overexpressed TIMP3 suppresses TNFα release and myogenesis. Levels of the released form of TNFα (17 kDa) and myogenin in mouse soleus overexpressing TIMP3 were evaluated on day 3 of injury by western blot analysis. **P<0.05 compared with soleus injected with Adv-GFP. Results are means ± s.e. (C) Adenovirus-mediated overexpression of TIMP3 impairs muscle regeneration. Adenovirus encoding TIMP3 or GFP was injected into the soleus of 3-day-old mice. At 6 weeks of age, paraffin-embedded cross-sections of excised muscle were prepared and subjected to H&E staining before (Day 0) and after cardiotoxin injection (Day 10), and immunohistochemical staining of eMHC was performed on sections collected 3 days after cardiotoxin injection (Day 3). Scale bars: 100 μm.
miR-206 mediates TIMP3 downregulation
MicroRNAs are known to negatively regulate the expression of their target genes (Jackson and Standart, 2007) and have an important role in myogenesis (Rao et al., 2006). Using the search software at http://www.targetscan.org, two miR-1/206 targeting sites in the 3′ UTR of the Timp3 gene (ACAUUCCA and CAUUCCA) were identified. The two miR-1/206 targeting sites are conserved in human, mouse, rat, dog and chicken. Previously, miR-206 was found to downregulate connexin43 during myogenesis via two miR-1/206 targeting sites present in the 3′ UTR of the connexin43 gene (Cja1) (Anderson et al., 2006), which are nearly identical to those in Timp3 (ACAUUCCA and ACAUUCCA). Connexin43 is a major component of gap junctions and has been shown to be important for muscle regeneration and in vitro differentiation (Araya et al., 2005). Both miR-1 and miR-206 are induced during myogenesis (Rao et al., 2006; Yuasa et al., 2008) in a MyoD- and Myf5-dependent manner (Rosenberg et al., 2006; Sweetman et al., 2008), and promote myogenesis (Chen et al., 2006; Kim et al., 2006; Rosenberg et al., 2006). To evaluate whether the two microRNAs mediate TIMP3 downregulation, synthesized miR-1 or miR-206 was transfected into proliferating C2C12 myoblasts. After incubating the transfected myoblasts in GM for 24 hours, Timp3 gene expression was analyzed by real-time PCR. This protocol allows the evaluation of the effect of transfected microRNAs on Timp3 expression without the induction of differentiation, and thus avoids the influence of endogenous miR-1 and miR-206. Transfection of miR-1 resulted in a downregulation of Timp3 mRNA by 46%, whereas transfection of miR-206 downregulated Timp3 mRNA by 78%. Co-transfection of the two microRNAs did not alter Timp3 mRNA downregulation to levels seen with miR-206 alone (Fig. 7). These data indicate that miR-206 is highly efficient in mediating Timp3 downregulation, and that the less-efficient miR-1 does not compete with miR-206 in the regulation of Timp3 gene expression. Therefore, downregulation of TIMP3 is primarily mediated by miR-206 similarly to connexin43 (Anderson et al., 2006).
Fig. 7.
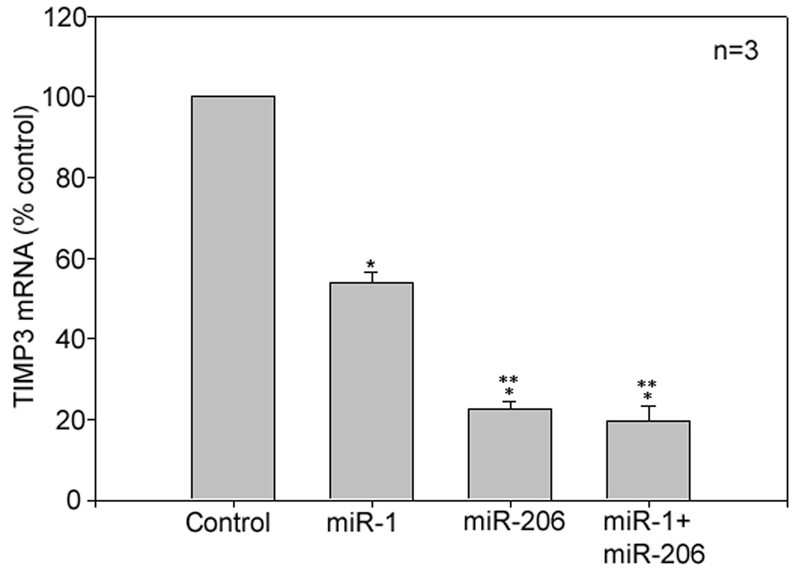
TIMP3 downregulation is mediated by miR-206. Synthesized miR-1 or miR-206 was transfected into C2C12 myoblasts. Random-sequenced RNA was transfected as control. The myoblasts were then incubated in GM for 24 hours. Total RNA was collected for analysis of Timp3 mRNA using real-time PCR. *P<0.05 compared with control and **P<0.05 compared with miR-1-transfected cells. Results are means ± s.e.
Discussion
We demonstrated in the present study that TIMP3 acts as a suppressor of myogenesis via its inhibition of TACE activity, and that TIMP3 downregulation mediated by miR-206 allows the initiation of myogenesis. These findings depict TIMP3 as a physiological regulator for myogenesis, and provide new opportunities for further exploration of the regulatory mechanisms that govern myogenesis.
Myogenesis is a process of terminal differentiation that needs to be suppressed most of the time. Even during muscle regeneration, muscle-released growth factors including FGF, HGF, EGF and PDGF inhibit myoblast differentiation while stimulating myoblast proliferation to prevent premature myogenesis (Hawke and Garry, 2001). After satellite cell activation and proliferation, only a subset of proliferating satellite cells goes through the differentiation route, the rest remain as undifferentiated reserve cells (Charge and Rudnicki, 2004). However, the mechanism through which myogenesis is suppressed is not well defined. The identification of constitutively expressed TIMP3 as a gatekeeper for myogenesis sheds new light on the mechanism that suppresses myogenesis. Moreover, we show that TIMP3 is also used as a switch for turning on myogenesis through the downregulation of its expression in satellite cells. The discovery of this regulatory mechanism for TACE release of autocrine TNFα in myogenic cells strengthens the concept that autocrine TNFα mediates myogenesis as a key activator of p38 MAPK (Chen et al., 2005; Chen et al., 2007; Zhan et al., 2007), and depicts a new paradigm in which adult myogenesis is mediated through the miR-206–TIMP3–TACE–TNFα–p38 signaling pathway.
TACE is activated by post-transcriptional modifications such as phosphorylation. For example, phorbol esters and ERK1/2 MAPK activate TACE (Fan et al., 2003; Reddy et al., 2000; Rousseau et al., 2008). We demonstrate here a mechanism that regulates TACE activity through the downregulation of TIMP3. We show that constitutively expressed TIMP3 in mouse muscle, satellite cells and C2C12 myoblasts is transiently downregulated in response to myogenic cues, and that this event is essential for myogenesis.
By overexpressing TIMP3 ectopically to counter the physiological downregulation of TIMP3 in C2C12 myoblasts, we demonstrate that TIMP3 suppresses TNFα release and myogenesis, and that its downregulation is required for TACE release of TNFα to activate p38-dependent myogenesis. Precaution was taken to rule out the possibility that the observed effect of overexpressed TIMP3 on myogenesis was due to its stimulation of apoptosis or inhibition of proliferation. The observed inability of overexpressed TIMP3 to stimulate apoptosis in C2C12 myoblasts is not surprising, because myocytes are known to be more resistant to apoptosis than many other cell types. We have shown previously that the effect of TNFα on myogenesis is dose-dependent. It stimulates myogenesis at physiological concentrations and inhibits myogenesis at pathological concentrations (Chen et al., 2007). The observed capacity of a physiological concentration of TNFα to rescue myoblast differentiation suppressed by overexpressed TIMP3 confirms that TIMP3 inhibits myogenesis via its inhibition of TNFα release by TACE. Thus, the downregulation of TIMP3 serves the purpose of permitting the initiation of myogenesis by the TACE–TNFα–p38 pathway.
Using a similar approach, we verified the regulator role of TIMP3 in myogenesis during muscle regeneration with biochemical and morphological evidence. The level of ectopically expressed TIMP3 we achieved was much lower in muscle than in myoblasts. For expression of recombinant adenovirus in adult muscle, it is necessary to inject adenovirus into neonatal muscle within several days of birth (Durbeej et al., 2003), which is not favorable for sustained high level of overexpression in adult muscle for several reasons. For example, rapid postnatal growth dilutes overexpressed protein; and over the prolonged period between viral infection and experimentation (~6 weeks), the level of overexpressed protein declines (Durbeej et al., 2003). The modest level of TIMP3 overexpression in muscle, which was the best we could achieve, maintained TIMP3 on day 3 of injury to 63% of the preinjury level. Despite the less than perfect retention of TIMP3, myogenesis and muscle regeneration were significantly delayed. The in vivo data from regenerating muscle complements the in vitro data from differentiating C2C12 myoblasts and support a physiological role for TIMP3 in regulating adult myogenesis.
It is noteworthy that the role of TIMP3 in adult myogenesis described here might not be as crucial in embryonic myogenesis. Myogenic activation of p38 during embryonic development might involve an alternative mechanism. The cell surface protein Cdo, which is only expressed during embryonic development (Kang et al., 1997), has been shown to activate p38 during primary myogenesis (Takaesu et al., 2006).
As with miR-1, miR-206 is induced in differentiating myoblasts (Rao et al., 2006) and regenerating muscle (Yuasa et al., 2008) by the muscle regulatory factors (MRFs) MyoD and Myf5 (Rosenberg et al., 2006; Sweetman et al., 2008); and has a crucial role in the promotion of myogenesis (Chen et al., 2006; Kim et al., 2006). However, the mechanism through which miR-206 promotes myogenesis is just emerging, despite the fact that miR-206 is predicted to target a long list of genes which includes Timp3 (McCarthy, 2008). Our data indicate that miR-206 is the primary microRNA that downregulates TIMP3 expression during myogenesis. Thus, TIMP3 downregulation is part of the intrinsic myogenic program that activates myogenesis. In addition, our data provides a mechanism for miR-206 promotion of myogenesis: miR-206 promotes myogenesis by serving as an upstream signal for myogenic activation of p38, a key mediator of cell cycle exit (Perdiguero et al., 2007), chromatin remodeling (Simone et al., 2004) and the activation of myogenic gene expression (de Angelis et al., 2005; Lluis et al., 2005; Wu et al., 2000). Interestingly, of the four TIMP genes, only Timp3 contains the target sites of miR-1/206, which is consistent to the unique role of TIMP3 in myogenesis. MicroRNAs are known to negatively regulate the expression of their target mRNAs via the degradation of the bound mRNA target or by directly inhibiting translation of the target mRNA (Jackson and Standart, 2007). Our observation that transfected miR-206 downregulates the Timp3 mRNA level suggests that miR-206 exerts its effect on TIMP3 by degrading its mRNA.
Taken together, our data indicate that TIMP3 is a physiological suppressor of myogenesis and that it is downregulated in MPCs during myogenesis to allow TNFα release and subsequent p38 activation. In addition, we provide evidence that downregulation of TIMP3 in MPCs is mediated by miR-206, a known regulator of myogenesis. Thus, TIMP3 has an important role in the suppression and activation of myogenesis.
Materials and Methods
Animal use
Experimental protocols were approved in advance by the Institutional Animal Care and Use Committee (IACUC) at Baylor College of Medicine and the Animal Welfare Committee (AWC) at University of Texas Houston Health Science Center. Male mice 6–8 weeks old (B6;129SF2/J) were used for muscle regeneration models. The cardiotoxin injury model was created by injecting 10 μM cardiotoxin (Sigma-Aldrich Corporation) dissolved in PBS into soleus (100 μl), gastrocnemius (100 μl) or entire hindlimb muscles (400 μl) to induce necrotic injury, as previously described (Chen et al., 2005). Functional overloading of mouse soleus muscle was produced by the bilateral surgical ablation of the synergistic gastrocnemius muscle, as previously described (Fluck et al., 1999). Briefly, gastrocnemius muscle was exposed by a posterior longitudinal incision through the skin and biceps femoris muscle of each lower hindlimb under anesthesia. After the soleus and plantaris muscles were separated from the gastrocnemius muscle at the Achilles tendon, the distal two-thirds of both heads of each gastrocnemius muscle were excised. Sham operations (control) consisted of the same procedure, except for gastrocnemius excision. Incisions were closed using wound clips, with no postoperative complications being observed over the course of the study. At indicated time points, soleus was collected from euthanized mice for analyses. For muscle overexpression of TIMP3, infectious particles (1.5×109) of Ad5 cytomegalovirus encoding Timp3 (Baker et al., 1998) or Gfp cDNA in 25 μl PBS were injected into the hindlimb of 3- to 6-day-old mice longitudinally in the region of the soleus. The mice were allowed to grow to 6 weeks of age before being subjected to muscle regeneration study.
Myogenic cell cultures
Murine C2C12 cells (ATCC) were cultured in growth medium (GM, DMEM supplemented with 10% fetal bovine serum and penicillin/streptomycin) at 37°C under 5% CO2. At 85% confluence, differentiation was induced by replacing GM with differentiation medium (DM, DMEM supplemented with 2% heat-inactivated horse serum and penicillin-streptomycin). Recombinant murine TNFα (BD Biosciences) was added to DM when indicated and replaced every 24 hours. To isolate satellite cells, hindlimb muscles were excised from 6- to 8-week-old mice and minced with razor blades in a minimal volume of Hanks Balanced Salt Solution supplemented with 2.5 mM of glutamine, 100 U/ml of penicillin and 100 μg/ml of streptomycin (HBSS+, Invitrogen). After removing the buffer by brief centrifugation, minced muscle was enzymatically dissociated in dissociation buffer (0.2% type II collagenase and 0.05% DNase in HBSS+) at 37°C for 30 minutes. The slurry was washed with HBSS+ and centrifuged at 1100 g for 5 minutes, and the supernatant was discarded. The pallet was triturated in 37°C DMEM supplemented with 10% horse serum, 1% glutamine and antibiotics by pipetting repeatedly. The triturate was passed through a 100 μM filter (Falcon) and cells were centrifuged at 1100 g for 5 minutes. The pallet was resuspended in HBSS+ and centrifuged through a density gradient made of 40% and 70% Percoll (Pharmacia, Piscataway, NJ) in PBS at 770 g for 20 minutes at room temperature. The band containing satellite cells at the interface between 40% and 70% Percoll layers was collected and washed in HBSS+. The purity of the satellite cell preparation was determined to be larger than 90% by immunofluorescence staining of quiescent satellite cells before injury with an antibody against CD34 (BD Bioscience) or activated satellite cells from day 3 of injury with an antibody against MyoD (MyoD C-20 from Santa Cruz Biotechnology).
Determination of TNFα concentration
TNFα concentration in DM and GM was determined by using an ELISA kit (R&D Systems) according to the manufacturer's protocol, after concentrating the medium with a spin concentrator from Millipore (10 kDa pore size).
Western blot analysis
Whole cell extracts or muscle extracts were subjected to western blot analysis as previously described (Chen et al., 2005). Antibody specific for TIMP3 was purchased from Chemicon. Antibodies against pan and phosphorylated p38 (T181/Y182) were obtained from Cell Signaling Technology (Danvers, MA). Antibodies against myogenin (F5D) and MHC (MF20) were purchased from the Developmental Studies Hybridoma Bank at the University of Iowa. All western blot experiments were repeated at least three times, and representative blots are shown.
Real-time PCR
Total RNA was extracted from isolated satellite cells or C2C12 myoblasts using RNA STAT-60 (Tel-Test) and dissolved in diethylpyro-carbonate (DEPC)-treated water. Total RNA was reverse transcribed into cDNA using 50 ng total RNA in 20 μl reaction mixture containing 200 U RevertAid M-MuLV reverse transcriptase (Fermentas). Real-time PCR was performed in a total reaction volume of 25 μl per reaction, containing 12.5 μl of SYBR green master mix (Bio-Rad), 10 pmol of each forward and reverse primer, 1 μl cDNA and nuclease-free water. Specific primer sets for mouse Timp3 (Forward: 5′-AAGGTACTAGAAACAGACTCCTCCAG-3′, and Reverse: 5′-TTGATACAGGACAAGAACTTGAGTG-3′) and glyceraldehyde-3-phosphate dehydrogenase (Gapdh, Forward: 5′-CATGGCCTTCCGTGTTCCTA-3′; Reverse: 5′-GCGGCACGTCAGATCCA-3′) were synthesized by Sigma-Genosys (Woodlands, TX). PCR runs were performed in triplicate with MyiQ real time PCR detection system (Bio-Rad, Salt Lake City, UT). Levels of Timp3 mRNA detected were calculated using the 2-ΔΔCt method and normalized to that of Gapdh.
Assay for apoptosis
The TUNEL assay (BD Biosciences) was carried out according to manufacturer's protocol.
Cell morphology
Cells were stained with the Giemsa dye. Cell images were acquired using MetaVue computer software and a Zeiss Axioplan 2 microscope coupled to a Photometrics CoolSNAP CCD camera with 20× objective lens; images were edited using Adobe Photoshop software.
MicroRNA transfection
Duplexes of miR-1 and miR-206 microRNA were purchased from Dharmacon and introduced into C2C12 myoblasts by electroporation using the Nucleofector system (Amaxa) at 5 μg per 100 μl of buffer. Myoblasts were then incubated in growth medium for 24 hours before being collected for analysis.
Histology
Excised soleus or gastrocnemius was fixed with 3.7% formaldehyde and embedded in paraffin. Cross sections were prepared and subjected to H&E staining or immunochemical staining. Immunohistochemical staining was performed using the MOM Immunodetection Kit (Vector Laboratories) according to manufacturer's protocol. Antibody against embryonic MHC (eMHC, F1.652) was purchased from the Developmental Studies Hybridoma Bank at the University of Iowa. An Olympus BX61 microscope was used to examine the histology. A SPOT Flex microscope digital camera coupled to the Spot Advanced software was used to take photos that were then edited with the Photoshop software.
Statistics
Values are expressed as means ± s.e. The one-way ANOVA and appropriate multiple comparison tests were used to analyze the multiple-group data using the SigmaStat software. Differences were regarded as significant at a level of P<0.05.
Acknowledgments
We thank Andrew H. Baker of University of Glasgow, UK for sharing adenovirus encoding TIMP3. This work was supported by Grant R01 AR049022 from the National Institute of Arthritis and Musculoskeletal and Skin Diseases to Y.-P.L. Deposited in PMC for release after 12 months.
References
- Amour A., Slocombe P. M., Webster A., Butler M., Knight C. G., Smith B. J., Stephens P. E., Shelley C., Hutton M., Knauper V., et al. (1998). TNF-alpha converting enzyme (TACE) is inhibited by TIMP-3. FEBS Lett. 435, 39-44 [DOI] [PubMed] [Google Scholar]
- Amour A., Knight C. G., Webster A., Slocombe P. M., Stephens P. E., Knauper V., Docherty A. J., Murphy G. (2000). The in vitro activity of ADAM-10 is inhibited by TIMP-1 and TIMP-3. FEBS Lett. 473, 275-279 [DOI] [PubMed] [Google Scholar]
- Anderson C., Catoe H., Werner R. (2006). MIR-206 regulates connexin43 expression during skeletal muscle development. Nucleic Acids Res. 34, 5863-5871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apte S. S., Mattei M. G., Olsen B. R. (1994). Cloning of the cDNA encoding human tissue inhibitor of metalloproteinases-3 (TIMP-3) and mapping of the TIMP3 gene to chromosome 22. Genomics 19, 86-90 [DOI] [PubMed] [Google Scholar]
- Araya R., Eckardt D., Maxeiner S., Kruger O., Theis M., Willecke K., Saez J. C. (2005). Expression of connexins during differentiation and regeneration of skeletal muscle: functional relevance of connexin43. J. Cell Sci. 118, 27-37 [DOI] [PubMed] [Google Scholar]
- Baker A. H., Zaltsman A. B., George S. J., Newby A. C. (1998). Divergent effects of tissue inhibitor of metalloproteinase-1, -2, or -3 overexpression on rat vascular smooth muscle cell invasion, proliferation, and death in vitro. TIMP-3 promotes apoptosis. J. Clin. Invest. 101, 1478-1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black R. A. (2002). Tumor necrosis factor-alpha converting enzyme. Int. J. Biochem. Cell Biol. 34, 1-5 [DOI] [PubMed] [Google Scholar]
- Black R. A. (2004). TIMP3 checks inflammation. Nat. Genet. 36, 934-935 [DOI] [PubMed] [Google Scholar]
- Charge S. B., Rudnicki M. A. (2004). Cellular and molecular regulation of muscle regeneration. Physiol. Rev. 84, 209-238 [DOI] [PubMed] [Google Scholar]
- Chen J. F., Mandel E. M., Thomson J. M., Wu Q., Callis T. E., Hammond S. M., Conlon F. L., Wang D. Z. (2006). The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat. Genet. 38, 228-233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S. E., Gerken E., Zhang Y., Zhan M., Mohan R. K., Li A. S., Reid M. B., Li Y. P. (2005). Role of TNF-α signaling in regeneration of cardiotoxin-injured muscle. Am. J. Physiol. Cell Physiol. 289, C1179-C1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S. E., Jin B., Li Y. P. (2007). TNF-alpha regulates myogenesis and muscle regeneration by activating p38 MAPK. Am. J. Physiol. Cell Physiol. 292, C1660-C1671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuenda A., Cohen P. (1999). Stress-activated protein kinase-2/p38 and a rapamycin-sensitive pathway are required for C2C12 myogenesis. J. Biol. Chem. 274, 4341-4346 [DOI] [PubMed] [Google Scholar]
- de Angelis L., Zhao J., Andreucci J. J., Olson E. N., Cossu G., McDermott J. C. (2005). Regulation of vertebrate myotome development by the p38 MAP kinase-MEF2 signaling pathway. Dev. Biol. 283, 171-179 [DOI] [PubMed] [Google Scholar]
- Drynda A., Quax P. H., Neumann M., van der Laan W. H., Pap G., Drynda S., Meinecke I., Kekow J., Neumann W., Huizinga T. W., et al. (2005). Gene transfer of tissue inhibitor of metalloproteinases-3 reverses the inhibitory effects of TNF-alpha on Fas-induced apoptosis in rheumatoid arthritis synovial fibroblasts. J. Immunol. 174, 6524-6531 [DOI] [PubMed] [Google Scholar]
- Durbeej M., Sawatzki S. M., Barresi R., Schmainda K. M., Allamand V., Michele D. E., Campbell K. P. (2003). Gene transfer establishes primacy of striated vs. smooth muscle sarcoglycan complex in limb-girdle muscular dystrophy. Proc. Natl. Acad. Sci. USA 100, 8910-8915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan H., Turck C. W., Derynck R. (2003). Characterization of growth factor-induced serine phosphorylation of tumor necrosis factor-alpha converting enzyme and of an alternatively translated polypeptide. J. Biol. Chem. 278, 18617-18627 [DOI] [PubMed] [Google Scholar]
- Fluck M., Carson J. A., Gordon S. E., Ziemiecki A., Booth F. W. (1999). Focal adhesion proteins FAK and paxillin increase in hypertrophied skeletal muscle. Am. J. Physiol. 277, C152-C162 [DOI] [PubMed] [Google Scholar]
- Gomis-Ruth F. X., Maskos K., Betz M., Bergner A., Huber R., Suzuki K., Yoshida N., Nagase H., Brew K., Bourenkov G. P., et al. (1997). Mechanism of inhibition of the human matrix metalloproteinase stromelysin-1 by TIMP-1. Nature 389, 77-81 [DOI] [PubMed] [Google Scholar]
- Guasconi V., Puri P. L. (2009). Chromatin: the interface between extrinsic cues and the epigenetic regulation of muscle regeneration. Trends Cell. Biol. 19, 286-294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawke T. J., Garry D. J. (2001). Myogenic satellite cells: physiology to molecular biology. J. Appl. Physiol. 91, 534-551 [DOI] [PubMed] [Google Scholar]
- Jackson R. J., Standart N. (2007). How do microRNAs regulate gene expression? Sci. STKE 2007, re1 [DOI] [PubMed] [Google Scholar]
- Jiang B. H., Aoki M., Zheng J. Z., Li J., Vogt P. K. (1999). Myogenic signaling of phosphatidylinositol 3-kinase requires the serine-threonine kinase Akt/protein kinase B. Proc. Natl. Acad. Sci. USA 96, 2077-2081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang J. S., Gao M., Feinleib J. L., Cotter P. D., Guadagno S. N., Krauss R. S. (1997). CDO: an oncogene-, serum-, and anchorage-regulated member of the Ig/fibronectin type III repeat family. J. Cell Biol. 138, 203-213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H. K., Lee Y. S., Sivaprasad U., Malhotra A., Dutta A. (2006). Muscle-specific microRNA miR-206 promotes muscle differentiation. J. Cell Biol. 174, 677-687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawlor M. A., Feng X., Everding D. R., Sieger K., Stewart C. E., Rotwein P. (2000). Dual control of muscle cell survival by distinct growth factor-regulated signaling pathways. Mol. Cell. Biol. 20, 3256-3265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leco K. J., Khokha R., Pavloff N., Hawkes S. P., Edwards D. R. (1994). Tissue inhibitor of metalloproteinases-3 (TIMP-3) is an extracellular matrix-associated protein with a distinctive pattern of expression in mouse cells and tissues. J. Biol. Chem. 269, 9352-9360 [PubMed] [Google Scholar]
- Lee M. H., Rapti M., Murphy G. (2005). Total conversion of tissue inhibitor of metalloproteinase (TIMP) for specific metalloproteinase targeting: fine-tuning TIMP-4 for optimal inhibition of tumor necrosis factor-{alpha}-converting enzyme. J. Biol. Chem. 280, 15967-15975 [DOI] [PubMed] [Google Scholar]
- Lluis F., Ballestar E., Suelves M., Esteller M., Munoz-Canoves P. (2005). E47 phosphorylation by p38 MAPK promotes MyoD/E47 association and muscle-specific gene transcription. EMBO J. 24, 974-984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loechel F., Fox J. W., Murphy G., Albrechtsen R., Wewer U. M. (2000). ADAM 12-S cleaves IGFBP-3 and IGFBP-5 and is inhibited by TIMP-3. Biochem. Biophys. Res. Commun. 278, 511-515 [DOI] [PubMed] [Google Scholar]
- McCarthy J. J. (2008). MicroRNA-206: the skeletal muscle-specific myomiR. Biochim. Biophys. Acta 1779, 6826-6891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammed F. F., Smookler D. S., Khokha R. (2003). Metalloproteinases, inflammation, and rheumatoid arthritis. Ann. Rheum. Dis. 62, ii43-ii47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perdiguero E., Ruiz-Bonilla V., Gresh L., Hui L., Ballestar E., Sousa-Victor P., Baeza-Raja B., Jardi M., Bosch-Comas A., Esteller M., et al. (2007). Genetic analysis of p38 MAP kinases in myogenesis: fundamental role of p38alpha in abrogating myoblast proliferation. EMBO J. 26, 1245-1256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelan J. N., Gonyea W. J. (1997). Effect of radiation on satellite cell activity and protein expression in overloaded mammalian skeletal muscle. Anat. Rec. 247, 179-188 [DOI] [PubMed] [Google Scholar]
- Puri P. L., Wu Z., Zhang P., Wood L. D., Bhakta K. S., Han J., Feramisco J. R., Karin M., Wang J. Y. (2000). Induction of terminal differentiation by constitutive activation of p38 MAP kinase in human rhabdomyosarcoma cells. Genes Dev. 14, 574-584 [PMC free article] [PubMed] [Google Scholar]
- Rao P. K., Kumar R. M., Farkhondeh M., Baskerville S., Lodish H. F. (2006). Myogenic factors that regulate expression of muscle-specific microRNAs. Proc. Natl. Acad. Sci. USA 103, 8721-8726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy P., Slack J. L., Davis R., Cerretti D. P., Kozlosky C. J., Blanton R. A., Shows D., Peschon J. J., Black R. A. (2000). Functional analysis of the domain structure of tumor necrosis factor-alpha converting enzyme. J. Biol. Chem. 275, 14608-14614 [DOI] [PubMed] [Google Scholar]
- Rommel C., Bodine S. C., Clarke B. A., Rossman R., Nunez L., Stitt T. N., Yancopoulos G. D., Glass D. J. (2001). Mediation of IGF-1-induced skeletal myotube hypertrophy by PI(3)K/Akt/mTOR and PI(3)K/Akt/GSK3 pathways. Nat. Cell Biol. 3, 1009-1013 [DOI] [PubMed] [Google Scholar]
- Rosenberg M. I., Georges S. A., Asawachaicharn A., Analau E., Tapscott S. J. (2006). MyoD inhibits Fstl1 and Utrn expression by inducing transcription of miR-206. J. Cell Biol. 175, 77-85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblatt J. D., Yong D., Parry D. J. (1994). Satellite cell activity is required for hypertrophy of overloaded adult rat muscle. Muscle Nerve 17, 608-613 [DOI] [PubMed] [Google Scholar]
- Rousseau S., Papoutsopoulou M., Symons A., Cook D., Lucocq J. M., Prescott A. R., O'Garra A., Ley S. C., Cohen P. (2008). TPL2-mediated activation of ERK1 and ERK2 regulates the processing of pre-TNF alpha in LPS-stimulated macrophages. J. Cell Sci. 121, 149-154 [DOI] [PubMed] [Google Scholar]
- Serra C., Palacios D., Mozzetta C., Forcales S. V., Morantte I., Ripani M., Jones D. R., Du K., Jhala U. S., Simone C., et al. (2007). Functional interdependence at the chromatin level between the MKK6/p38 and IGF1/PI3K/AKT pathways during muscle differentiation. Mol. Cell 28, 200-213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano A. L., Baeza-Raja B., Perdiguero E., Jardi M., Munoz-Canoves P. (2008). Interleukin-6 is an essential regulator of satellite cell-mediated skeletal muscle hypertrophy. Cell Metab. 7, 33-44 [DOI] [PubMed] [Google Scholar]
- Simone C., Forcales S. V., Hill D. A., Imbalzano A. N., Latella L., Puri P. L. (2004). p38 pathway targets SWI-SNF chromatin-remodeling complex to muscle-specific loci. Nat. Genet. 36, 738-743 [DOI] [PubMed] [Google Scholar]
- Smookler D. S., Mohammed F. F., Kassiri Z., Duncan G. S., Mak T. W., Khokha R. (2006). Tissue inhibitor of metalloproteinase 3 regulates TNF-dependent systemic inflammation. J. Immunol. 176, 721-725 [DOI] [PubMed] [Google Scholar]
- Sweetman D., Goljanek K., Rathjen T., Oustanina S., Braun T., Dalmay T., Munsterberg A. (2008). Specific requirements of MRFs for the expression of muscle specific microRNAs, miR-1, miR-206 and miR-133. Dev. Biol. 321, 491-499 [DOI] [PubMed] [Google Scholar]
- Takaesu G., Kang J. S., Bae G. U., Yi M. J., Lee C. M., Reddy E. P., Krauss R. S. (2006). Activation of p38alpha/beta MAPK in myogenesis via binding of the scaffold protein JLP to the cell surface protein Cdo. J. Cell Biol. 175, 383-388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrente Y., El Fahime E., Caron N. J., Del, Bo R., Belicchi M., Pisati F., Tremblay J. P., Bresolin N. (2003). Tumor necrosis factor-alpha (TNF-alpha) stimulates chemotactic response in mouse myogenic cells. Cell Transplant. 12, 91-100 [DOI] [PubMed] [Google Scholar]
- Tracey K. J., Cerami A. (1992). Pleiotropic effects of TNF in infection and neoplasia: beneficial, inflammatory, catabolic, or injurious. Immunol. Ser. 56, 431-452 [PubMed] [Google Scholar]
- Tureckova J., Wilson E. M., Cappalonga J. L., Rotwein P. (2001). Insulin-like growth factor-mediated muscle differentiation: collaboration between phosphatidylinositol 3-kinase-Akt-signaling pathways and myogenin. J. Biol. Chem. 276, 39264-39270 [DOI] [PubMed] [Google Scholar]
- van der Laan W. H., Quax P. H., Seemayer C. A., Huisman L. G., Pieterman E. J., Grimbergen J. M., Verheijen J. H., Breedveld F. C., Gay R. E., Gay S., et al. (2003). Cartilage degradation and invasion by rheumatoid synovial fibroblasts is inhibited by gene transfer of TIMP-1 and TIMP-3. Gene Ther. 10, 234-242 [DOI] [PubMed] [Google Scholar]
- Wagers A. J., Conboy I. M. (2005). Cellular and molecular signatures of muscle regeneration: current concepts and controversies in adult myogenesis. Cell 122, 659-667 [DOI] [PubMed] [Google Scholar]
- Wu Z., Woodring P. J., Bhakta K. S., Tamura K., Wen F., Feramisco J. R., Karin M., Wang J. Y., Puri P. L. (2000). p38 and extracellular signal-regulated kinases regulate the myogenic program at multiple steps. Mol. Cell. Biol. 20, 3951-3964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuasa K., Hagiwara Y., Ando M., Nakamura A., Takeda S., Hijikata T. (2008). MicroRNA-206 is highly expressed in newly formed muscle fibers: implications regarding potential for muscle regeneration and maturation in muscular dystrophy. Cell Struct. Funct. 33, 163-169 [DOI] [PubMed] [Google Scholar]
- Zetser A., Gredinger E., Bengal E. (1999). p38 mitogen-activated protein kinase pathway promotes skeletal muscle differentiation. Participation of the Mef2c transcription factor. J. Biol. Chem. 274, 5193-5200 [DOI] [PubMed] [Google Scholar]
- Zhan M., Jin B., Chen S. E., Reecy J. M., Li Y. P. (2007). TACE release of TNF-alpha mediates mechanotransduction-induced activation of p38 MAPK and myogenesis. J. Cell Sci. 120, 692-701 [DOI] [PMC free article] [PubMed] [Google Scholar]