

Abstract
The number of physical conditions and chemical agents induce accumulation of misfolded proteins creating proteotoxic stress. This leads to activation of adaptive pro-survival pathway, known as heat shock response (HSR), resulting in expression of additional chaperones. Several cancer treatment approaches, such as proteasome inhibitor Bortezomib and hsp90 inhibitor geldanamycin, involve activation of proteotoxic stress. Low efficacy of these therapies is likely due to the protective effects of HSR induced in treated cells, making this pathway an attractive target for pharmacological suppression. We found that the anti-malaria drugs quinacrine (QC) and emetine prevented HSR in cancer cells, as judged by induction of hsp70 expression. As opposed to emetine, which inhibited general translation, QC did not affect protein synthesis, but rather suppressed inducible HSF1-dependent transcription of the hsp70 gene in a relatively selective manner. The treatment of tumor cells in vitro with a combination of non-toxic concentrations of QC and proteotoxic stress inducers resulted in rapid induction of apoptosis. The effect was similar if QC was substituted by siRNA against hsp70, suggesting that the HSR inhibitory activity of QC was responsible for cell sensitization to proteotoxic stress inducers. QC was also found to enhance the antitumor efficacy of proteotoxic stress inducers in vivo: combinatorial treatment with 17-DMAG + QC resulted in suppression of tumor growth in two mouse syngeneic models. These results reveal that QC is an inhibitor of HSF1-mediated HSR. As such, this compound has significant clinical potential as an aimed at exploiting the cytotoxic potential of proteotoxic stress.
Keywords: proteasomal inhibitors, Hsp90, geldanamycin, bortezomib, hyperthermia, heat shock, apoptosis
Introduction
The accumulation of unfolded proteins that results from heat shock, hypoxia, exposure to heavy metal ions or agents decreasing proteasome and chaperone activities creates proteotoxic stress.1 Cells respond to this stress by induction of an ancient signal transduction pathway known as heat shock response (HSR). In mammalian cells, HSR is largely mediated by activation of the transcription factor HSF1. HSF1 normally resides in the cytoplasm in an inactive form bound to hsp90.2–6 Under conditions of proteotoxic stress, HSF1 becomes activated and translocates to the nucleus where it stimulates expression of a set of heat shock response genes including those encoding inducible chaperones such as hsp70 and hsp27.7–9 The newly synthesized chaperones serve to alleviate proteotoxic stress by promoting protein refolding, preventing protein aggregation and targeting unfolded proteins for proteasome-mediated degradation.10,11
In addition to synthesis of inducible chaperones, cells have two other interconnected mechanisms to overcome proteotoxic stress: ubiquitin-mediated proteasomal degradation and downregulation of translation.12 Proteasomal degradation of cellular proteins is an important mechanism of regulation for numerous cellular processes.13 Acting to degrade ubiquitinated protein substrates,14,15 proteasomes maintain cellular protein homeostasis by eliminating improperly folded proteins.12,16 Translational attenuation occurring in response to proteotoxic stress17,18 is mediated by phosphorylation of translation initiation factor eIF2α17 which is essential for cap-dependent mRNA translation.19 Inhibition of translation through this mechanism occurs in cells treated with proteasomal inhibitors or infected by virus.20,21
In normal cells, HSF1 is only engaged as a stress response mechanism under conditions of proteotoxic stress. However, in tumor cells, which are often characterized by an increased rate of protein misfolding, this factor is frequently found to be constitutively active.22 HSF-1 activity is not only a reflection of the transformed phenotype, but appears to be essential for the process of malignant transformation. This was demonstrated by the finding that HSF1-deficient mice show a dramatically reduced rate of tumor development.23 These observations place HSF1 among important anticancer treatment targets and provide strong rationale for the search for HSF1 inhibitors.
Agents or treatments inducing proteotoxic stress have been considered for anticancer therapy. Arsenic trioxide and the proteasomal inhibitor bortezomib are conventional anticancer drugs approved for treatment of leukemia.24,25 However, hyperthermia and the hsp90 inhibitor geldanamycin have not become conventional treatments due to insufficient anticancer efficacy.26–28 The limited efficacy of some proteotoxic treatments might be due to effective protection of tumor cells by the induction of HSR.29 In this regard, specific inhibitors of the HSF1 pathway may be useful not only as single agents, but also in combination with proteotoxic treatments.
To identify HSF1 inhibitors, we analyzed known anti-malaria drugs since, similar to cancer cells, the malaria parasite has to overcome proteotoxic stress to survive. This stress results from exposure of the parasite to heat shock as it moves between cold-blooded hosts (mosquitoes) and warm-blooded hosts (further complicated by fever). This growth condition requires constant synthesis of additional chaperones.30 We hypothesized that some empirically developed anti-malaria drugs might target this vitally important protective pathway. Considering the high degree of evolutionary conservation of HSR, such drugs might be capable of similar activity in mammalian cells. In fact, emetine and its derivatives were shown to suppress HSR caused by proteasome inhibitors.31 However, emetine is a general inhibitor of translation which limits its practical applications. Here, we report that another anti-malaria drug, quinacrine (QC), can suppress HSF1-mediated HSR with no effect on general protein synthesis. We describe the HSF1 inhibitory activity of QC and show that blockade of HSR in this manner greatly enhances the antitumor efficacy of proteotoxic stress inducers. These results provide strong support for clinical use of QC as an anticancer drug.
Results
Aminoacridines prevent activation of hsp70 in response to proteasome inhibition
Upregulation of the inducible form of hsp70 is a hallmark of HSR following proteotoxic stress, such as that generated by proteasome inhibition. We tested the effect of several anti-malaria drugs on synthesis of hsp70 activated by inhibition of proteasomes by the small molecule inhibitor MG132 in cultured HeLa cells. While quinine and chloroquine were not active in this assay at concentrations up to 20 μM, emetine and quinacrine (QC) suppressed hsp70 synthesis in response to MG132 (Fig. 1A). 9-aminoacridine (9AA), which is closely related in structure to QC, had a similar inhibitory effect on MG132-induced hsp70 expression (Fig. 1B). For both QC and 9AA, the inhibition was concentration-dependent, reaching a plateau between 10 μM and 20μM (data not shown).
Figure 1.

Quinacrine and 9AA prevent hsp70 synthesis in cells treated with proteasome inhibitors. (A) Immunoblotting with anti-hsp70 antibody of protein extracts from HeLa cells treated for 5 h with the proteasome inhibitor MG132 (MG) (5 μM) in combination with the anti-malaria drugs emetine (EM) (1 μM), chloroquine (CQ) (20 μM), quinine (Q) (20 μM), or quinacrine (QC) (20 μM). Lane 1 contains extract from untreated HeLa cells. Pirin expression was examined as a protein loading control (L.c.). (B) Protein extracts from HeLa cells treated for 5 h with MG132 (5 μM) alone or in combination with 20 μM QC or 9AA were examined by immunoblotting using an antibody specific for hsp70. Negative controls included untreated HeLa cells (lane 1) and cells treated with QC or 9AA in the absence of MG132 (lanes 6 and 7). Pirin was examined as a protein loading control (L.c.). (C) emetine, but not QC or 9AA, inhibits general protein synthesis. S35-labeled proteins from HeLa cells left untreated (lane 1) or treated for 4 h with QC (20 μM), 9AA (20 μM), or emetine (EM) (1 μM) were analyzed by electrophoresis and autoradiography. (D) Aminoacridines do not affect inhibition of proteasomes by MG132. Results of an in vitro proteasome activity assay using extracts of HeLa cells treated for 4 h with the indicated combinations of MG132 (5 μM), QC (20 μM) and 9AA (20 μM). Proteasome activity is shown relative to that in untreated cells (set at 100%). (E) QC inhibits activation of hsp70A1 transcription by proteasome inhibitors in HeLa cells. 10 μg of total RNA from untreated HeLa cells (lane 1) or treated for 5 h with 0.1 μM Bortezomib (BZ) or 5 μM MG132 (MG) alone or in combination with 20 μM QC were analyzed by northern blotting with hsp70A1 (top) and GAPDH (bottom) probes. GAPDH was used to control for the specificity of QC’s inhibitory activity and for RNA loading. (F) Aminoacridines do not prevent induction of metallothionein gene expression by ZnCl2. HeLa cells were left untreated (lane 1) or treated for 5 h with 200 μM ZnCl2 (Zn) alone or in combination with 20 μM QC or 9AA. 10 μg of total RNA were analyzed by northern blotting with metallothionein MT1 (MT), hsp70A1 and GAPDH probes.
Emetine is an inhibitor of general translation;32 therefore, its ability to suppress inducible hsp70 synthesis is likely a reflection of this property. However, QC and 9AA had no effect on overall protein synthesis (as shown by 35S-methionine and 35S-cysteine incorporation) at concentrations that were sufficient for complete suppression of hsp70 induction (Fig. 1C). This indicates that these aminoacridine-based compounds suppress inducible hsp70 synthesis through a different, more specific, mechanism. Neither QC nor 9AA affected the ability of MG132 to inhibit proteasome activity (Fig. 1D). Therefore, the drugs do not block hsp70 induction simply by interfering with the generation of proteotoxic stress via proteasome inhibition.
The inhibitory effects of QC on hsp70 induction were detectable at the mRNA level as shown by northern hybridization of RNA from HeLa cells treated with MG132 or another proteasomal inhibitor, bortezomib, with or without QC (Fig. 1E). The hybridization probe used in this experiment was specific to the inducible hsp70A1 mRNA and did not recognize the mRNA encoding the constitutive hsc70 protein. The results indicated that QC suppressed accumulation of hsp70 mRNA under conditions of proteasome inhibition. This effect was due to repression of transcription rather than stimulation of hsp70 mRNA degradation since addition of QC after the induction of HSR had no effect on hsp70 mRNA abundance (data not shown). The inhibitory effect of QC and 9AA on inducible gene expression was not universal. For example, ZnCl2-induced activation of metallothionein gene transcription was not affected by these compounds under conditions at which they abolished induction of hsp70 mRNA (Fig. 1F). Moreover, QC and 9AA both caused strong induction of transcription of p53 target genes.33
Quinacrine blocks induction of heat shock response by hsp90 inhibition or hyperthermia
Functional inactivation of hsp90 by geldanamycin or its derivative 17-DMAG induces proteotoxic stress and activates HSR including synthesis of hsp70.14,34–37 As shown in Figure 1 for proteotoxic stress induced by proteasome inhibitors, QC and 9AA also suppressed hsp70 induction in response to hsp90 inhibition and this occurred at the transcriptional level (Fig. 2A and B). Although QC and 9AA efficiently suppressed 17-DMAG-activated transcription of the hsp70A1 gene, they had no effect on the levels of several other (non-17-DMAG induced) mRNAs, including those for GAPDH, p65RelA and GRP94 (Fig. 2C).
Figure 2.
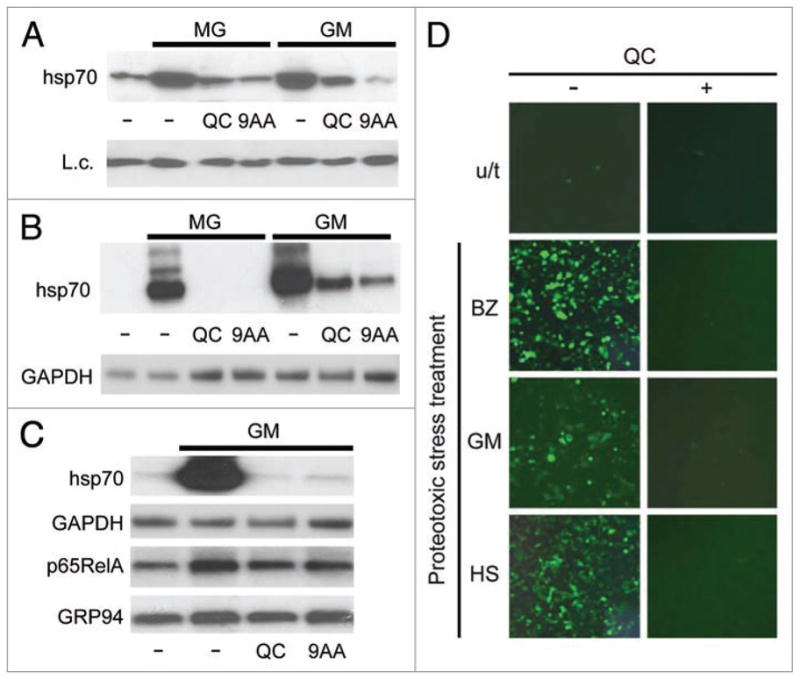
HSR induction by either MG132, 17-DMAG or heat shock is sensitive to aminoacridines. (A) Immunoblotting with anti-hsp70 antibody of protein extracts (10 μg) from HeLa cells treated for 5 h with 5 μM MG132 (MG) or 1 μM 17-DMAG (GM) alone or in combination with 20 μM QC or 9AA. Lane 1 contains extract from untreated HeLa cells. K18 expression was assessed as a protein loading control (L.c). (B) QC and 9AA suppress transcription of the hsp70A1 gene induced by 17-DMAG or MG132. Northern hybridization of RNA from untreated HeLa cells (lane 1), treated for 5 h with 1 μM 17-DMAG (GM) or 5 μM MG132 (MG) alone, and in combination with 20 μM QC or 9AA. GAPDH mRNA was analyzed to control for inhibitor specificity and RNA loading. (C) QC and 9AA do not change mRNA accumulation of genes that are not induced by 17-DMAG. Northern hybridization of RNA from HeLa cells treated for 5 h with 1 μM 17-DMAG (GM) alone or in combination with 20 μM QC or 9AA. GAPDH, p65RelA and GRP94 are genes whose expression is not affected by 17-DMAG. Hsp70A1 was included as a positive control for a 17-DMAG inducible gene. (D) QC suppresses HSF1-dependent expression of an EGFP reporter gene induced by proteotoxic stress. HeLa cells carrying an HSF1-EGFP reporter construct were left untreated, or treated with 0.1 μM of Bortezomib (BZ), 1 μM of 17-DMAG (GM), or 43°C for 1 h of heat shock (HS) alone or in combination with 20 μM QC. EGFP expression was assessed in 8 h by fluorescent microscopy.
QC was also effective in blocking HSR induced by hyperthermia as illustrated in Figure 2D. In this experiment, we directly assessed the activity of the HSF1 transcription factor which is known to be essential for induction of hsp70 expression during HSR. HSF1 reporter cells were generated by infecting HeLa cells with a lentiviral reporter construct containing the EGFP cDNA under the control of an HSF1-responsive promoter (see Methods). Upon treatment with bortezomib, 17-DMAG or hyperthermia, HSF1-dependent EGFP synthesis was induced, as detected by fluorescent microscopy. Co-treatment of the reporter cells with QC in addition to any of the three different proteotoxic stress inducers blocked EGFP accumulation.
Taken together, these observations indicate that the anti-malaria drug QC is a powerful inhibitor of HSR, regardless of how it is triggered, acting at the level of HSF1-mediated transcription.
Quinacrine affects HSF1 function downstream of nuclear translocation and DNA binding
Activation of HSF1-mediated transcription requires release of HSF1 from its complex with hsp90 in the cytoplasm. This is followed by formation of HSF1 homodimers and homotrimers which translocate into the nucleus and bind to specific HSF1 recognition sites within the promoters of heat shock-inducible genes. We studied the effect of QC and 9AA on this process in order to define the point at which these compounds act to inhibit HSF1 function. To assess the intracellular localization and DNA binding capacity of HSF1 complexes, we performed electrophoretic mobility shift assays (EMSA) using protein extracts purified from the cytoplasm or nuclei of cells subjected to proteotoxic stress alone or in combination with QC or 9AA. The results of these experiments are shown in Figure 3. Treatment of HeLa cells with MG132, heat shock, or 17-DMAG led to activation of specific HSF1 DNA binding activity in both cytoplasmic (Fig. 3A) and nuclear extracts (Fig. 3B). QC and 9AA did not interfere with or modify the strength of these effects at any concentration tested (up to 100 μM) (data not shown and Fig. 3B). These results indicate that inhibition of HSF1-dependent transcription by QC and 9AA occurs at a point downstream of its cytoplasmic activation, nuclear translocation, and DNA binding.
Figure 3.
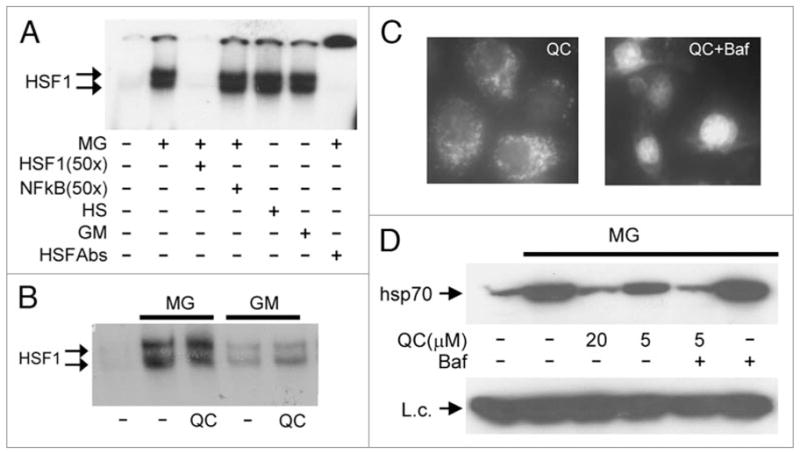
Aminoacridines act downstream of HSF1 activation and translocation to the nucleus. (A) Treatment of HeLa cells with MG132, 17-DMAG, or heat shock induces HSF1 DNA binding activity. Electrophoretic mobility shift assays (EMSA) were performed with cytoplasmic extracts from HeLa cells left untreated, treated with 5 μM MG132 (MG) or 1 μM 17-DMAG (GM) for 4 h, or treated with heat shock (43°C) for 1 h. Complex formation between HSF1 and a p32-labeled oligonucleotide probe containing an HSF1 binding site was inhibited by a 50x excess of the same unlabeled oligonucleotide (lane 3), but not by a similar excess of an unlabeled oligonucleotide containing an NFκB binding site (lane 4). The specificity of the detected complex was further confirmed by its super-shift in the presence of anti-HSF1 antibody (lane 7). (B) QC does not affect nuclear HSF1 DNA binding activity induced by proteotoxic stress. EMSA was performed as in (A) with a labeled HSF1-specific oligonucleotide probe and nuclear extracts from HeLa cells left untreated (lane 1), or treated for 4 h with 5 μM MG132 (MG) or 1 μM 17-DMAG (GM) alone or in combination with 20 μM QC. (C) Bafilomycin increases the nuclear concentration of QC. HeLa cells were treated with 5 μM QC alone or in combination with 0.5 μM of bafilomycin for 1 h. The intracellular localization of QC was analyzed by UV-microscopy with a blue filter. (D) Bafilomycin increases the HSR inhibitory activity of QC. HeLa cells were left untreated (lane 1) or treated for 5 h with the indicated combinations of 5 μM MG132 (MG), 0.5 μM bafilomycin (Baf), and variable amounts of QC. Whole cell protein extracts were analyzed by western blotting with anti-hsp70 antibody. pirin was examined as a protein loading control (L.c.).
Our finding that QC and 9AA affect HSF1 downstream of its nuclear translocation suggests that the compounds themselves are localized, at least in part, in the nucleus. This is supported by results obtained using experimental modulation of their intracellular distribution. It was previously shown by others that bafilomycin, a lysosome-targeted antibiotic, can significantly alter the intracellular distribution of lysosome-tropic compounds, including quinacrine and chloroquine, by inhibiting lysosomal H+-ATPase.38 Indeed, treatment of HeLa cells with bafilomycin resulted in a switch in the intracellular localization of QC from predominantly lysosomal to predominantly nuclear (Fig. 3C) as judged by fluorescent microscopy (both QC and 9AA are naturally fluorescent compounds). We then tested whether bafilomycin treatment would affect the sensitivity of hsp70 induction to QC. Bafilomycin increased the efficacy of QC as an HSR inhibitor in MG132-treated HeLa cells, as shown in Figure 3D. In the presence of the antibiotic, 5 μM QC inhibited hsp70 synthesis as effectively as 20 μM QC in the absence of the antibiotic. These results indicate that the inhibitory activity of QC against HSF1-mediated transcription depends on its nuclear concentration.
Combination of quinacrine with proteotoxic stress induces apoptosis
Heat shock response (HSR) is an adaptive pro-survival response that can protect cells from a variety of toxic conditions. Rapid accumulation of inducible forms of chaperones, such as hsp70 and hsp27, has been shown to prevent cell death under conditions of heat shock or treatment with inhibitors of proteasomes or hsp90.10,39,40 Therefore, we expected that the ability of QC and 9AA to prevent induction of proteins encoded by HSF1-responsive genes could greatly increase the cytotoxicity of proteotoxic stresses. To test this hypothesis, we treated HeLa cells with 17-DMAG or bortezomib alone or in combination with QC and analyzed cell viability and induction of apoptosis. As shown in Figure 4A and B, QC greatly enhanced the toxicity of 17-DMAG and bortezomib, respectively. Combined treatment with concentrations of QC that caused less than a 50% reduction in cell viability and practically non-toxic concentrations of 17-DMAG or bortezomib resulted in a dramatic reduction in the number of growing cells. We next investigated whether the impact of the drug treatments on the number of viable cells was due to induction of apoptosis. Apoptosis was monitored by the appearance of caspase-specific cleavage products of keratin 18 and PARP. Consistent with the results of the cell viability assays, combined treatment with QC and either bortezomib or 17-DMAG strongly activated caspase-mediated protein cleavage that was barely detectable when the drugs were used alone (Fig. 4C and D). The caspase inhibitor ZVAD-FMK blocked this effect completely, indicating that the proteolytic cleavage events were caspase-specific (Fig. 4E).
Figure 4.
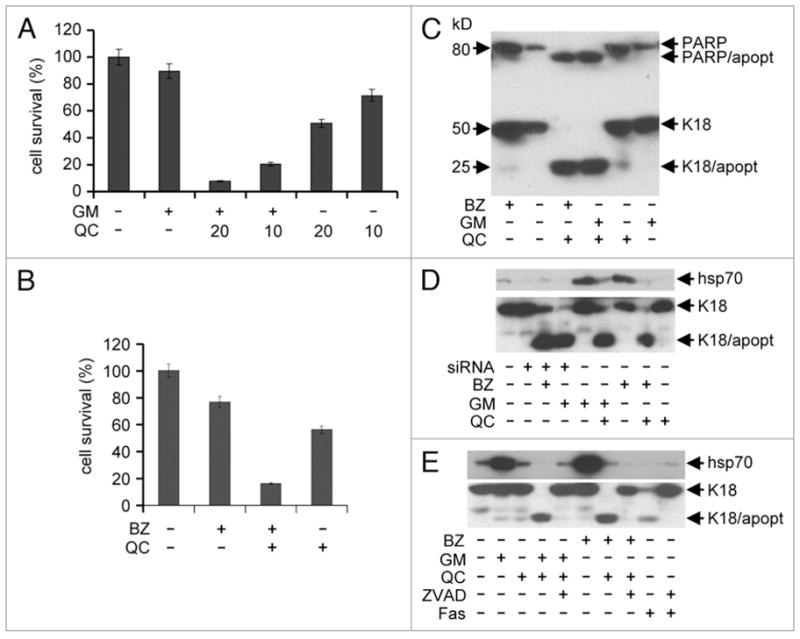
QC enhances the cytotoxicity of proteotoxic stress inducers. (A) The toxicity of drug combinations was measured using a cell viability assay. HeLa cells were treated for 4 h with the indicated combinations of 17-DMAG (1 μM) and QC (10 or 20 μM). Cell viability was assessed 72 h later by methylene blue staining. The data shown are the average of three experiments. (B) The toxicity of bortezomib/QC treatment was assessed using a cell viability assay as in (A). HeLa cells were treated for 4 h with 0.1 μM bortezomib (BZ) alone or in combination with 10 μM QC. The data shown are the average of 2 experiments. (C) Immunoblotting analysis of apoptosis in HeLa cells treated with combinations of bortezomib (BZ) (0.1 μM), 17-DMAG (GM) (1 μM) and QC (10 μm). Full-length PARP and K18 proteins as well as apoptosis-specific proteolytic fragments (PARP/apopt and K18/apopt) are indicated. (D) siRNA-mediated knockdown of Hsp70 sensitizes cells to bortezomib and 17-DMAG. Immunoblotting of proteins from hsp70 siRNA (siRNA)-expressing HeLa cells treated with the indicated concentrations of 0.1 μM bortezomib (BZ), 20 μM of QC and 1 μM 17-DMAG (GM). Protein extracts were prepared from cells transfected with hsp70 siRNA for 24 h and treated with bortezomib and DMAG overnight. Expression of hsp70 and apoptosis-specific cleavage of K18 was assessed with the corresponding antibodies. Control HeLa cells were untreated with drugs (lane 1), or treated overnight with amount of drugs indicated above (lanes 5–9). (E) The caspase inhibitor ZVAD-FMK protects cells from apoptosis induced by combinations of proteotoxic stress and aminoacridines. Immunoblotting analysis of hsp70 protein levels and apoptosis-specific cleavage of K18 in HeLa cells treated overnight with the indicated combinations of 1 μM 17-DMAG (GM), 0.1 μM bortezomib (BZ), 20 μM QC and 20 μM ZVAD-FMK (ZVAD). Treatment of cells with anti-Fas antibody (Fas, lane 9) and the combination of anti-Fas antibody and ZVAD-FMK (lane 10) provided positive controls for induction of apoptosis and the protective effect of ZVAD-FMK, respectively.
To address whether inhibition of hsp70 induction plays a critical role in QC-mediated cell sensitization to bortezomib and 17-DMAG, we compared the effect of QC with that of an alternative method of hsp70 suppression. HeLa cells were transfected with siRNA specifically designed against the inducible form of hsp70. The transfected cells were unable to synthesize inducible hsp70 upon treatment with proteasome or hsp90 inhibitors (Fig. 4D). The inability to synthesize hsp70 under conditions of proteotoxic stress activated an apoptotic response similar to that caused by combined treatment with QC and proteotoxic stress inducing agents (Fig. 4D). The similarity of the biological effects of siRNA-mediated knockdown of hsp70 and QC treatment with respect to cell sensitivity to bortezomib and 17-DMAG suggests that the combined toxicity of QC and proteotoxic stress inducers is mediated by suppression of HSR by QC.
Anti-tumor effect of combination of quinacrine with hsp90 inhibitor
Our finding that combination of QC with proteotoxic stress-inducing drugs has a strong toxic effect on HeLa cells in vitro suggests that such combinations could have significant potential as anticancer therapies. To investigate this possibility, we tested the effect of QC/17-DMAG combination treatment on tumor growth in vivo using two syngeneic mouse tumor models: MCA205 fibrosarcoma and B-16 melanoma. We focused on 17-DMAG since it is a proteotoxic stress inducer that has shown limited antitumor efficacy as a monotherapy.41 First, we analyzed the effect of 17-DMAG and QC on induction of HSR (Fig. 5A) and cell viability (Fig. 5B) in MCA205 and B-16 cells in vitro. Both cell lines responded to the drugs in a manner similar to HeLa cells, thereby validating use of these models to test the anti-tumor activity of the QC/17-DMAG combination. To confirm that the drugs have a similar effect on HSR induction in tumors growing in vivo, MCA205 cells were implanted in C57BL/6 mice and the resulting tumors were treated with a single intra-tumor injection of PBS (control), 25 μg 17-DMAG alone, or 1.25 mg QC with 25 μg 17-DMAG. Five hours later, mice were sacrificed and RNA prepared from the isolated tumors was analyzed for hsp70 induction. As shown in Figure 5C, 17-DMAG treatment induced hsp70 synthesis in tumors grown in vivo and QC prevented this induction. To assess the effect of the drug combination on tumor growth in vivo, mice carrying MCA205 (Fig. 5D) or B-16 (Fig. 5E) tumors were treated by intra-tumor injection with QC and 17-DMAG alone or in combination (scheduling details see in figure legend and in Methods). The size of the tumors was measured regularly (every 2 days) after drug injection. In both experimental models, QC and 17-DMAG applied as single agents had minor antitumor effects (the delay in tumor growth), whereas in combination they completely prevented tumor growth. Tumor sizes were measured up to 24 days after implantation. Tumors with single drug injection started to overgrow the tumors with combinatorial drug injections from day 16 for B-16 cells and from day 18 for MCA205 cells (Fig. 5A and B). The combined QC/17-DMAG treatment not only prevented tumor growth, but actually led to tumor regression, with tumors shrinking from ~50 mm3 in size to undetectable. These findings are consistent with our in vitro results demonstrating that combination of QC with proteotoxic stress inducers leads to apoptosis.
Figure 5.
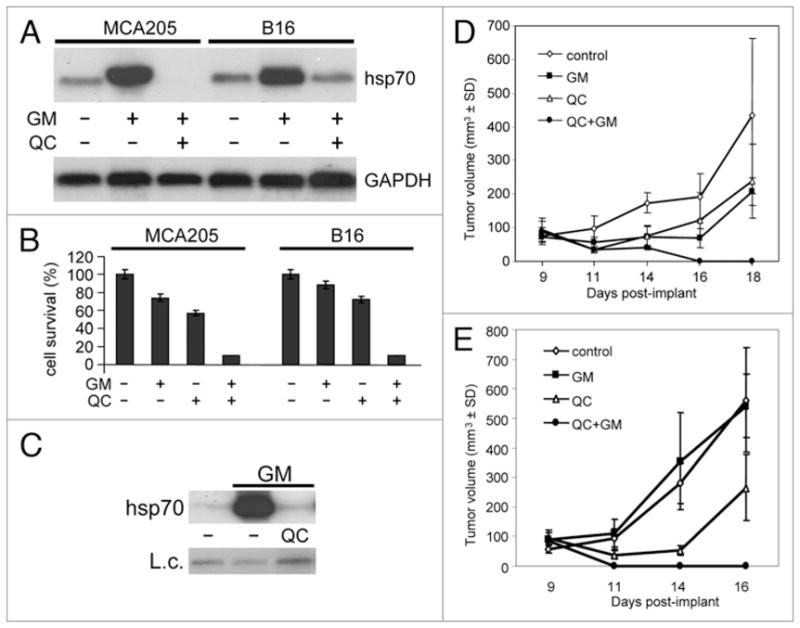
Effect of 17-DMAG and quinacrine on tumor cell growth in vitro and in vivo. (A) QC suppresses hsp70 synthesis in response to proteotoxic stress in MCA205 and B-16 cells. RNA from control cells and cells treated for 5 h with 1 μM 17-DMAG (GM) in the presence or absence of 20 μM QC was analyzed by northern hybridization with hsp70A1 and GAPDH probes. (B) Results of cell viability assay of MCA205 and B-16 cells treated for 4 h with 1 μM 17-DMAG (GM) in combination with 20 μM QC. Cells were harvested, diluted 1:50 and assayed for cell viability by methylene blue staining 72 h later. The data shown are the average of two experiments. (C) Intra-tumor injection of QC blocks 17-DMAG-induced hsp70 expression. C57BL/6 mice carrying MCA205 tumors were given a single intra-tumor injection of PBS (control), 25 μg 17-DMAG (GM), or 1.25 mg QC + 25 μg 17-DMAG. Five hours later, mice were sacrificed. RNA prepared from the tumors was analyzed by northern hybridization with a hsp70A1 specific probe. Hybridization with a GAPDH probe was used as an RNA loading control (L.c.). The data shown is from a single tumor for each treatment. These are representative data of two experiments. (D and E) C57BL/6 mice were injected with MCA205 (D) or B-16 (E) cells as described in Methods. Animals (n = 5 per group) began receiving intra-tumor drug injections on day 9 after tumor implant. The control group received PBS injections on days 9, 10, 11 and 12. Mice in group 2 (QC) were injected with 1.25 mg QC on days 9 and 10. Mice in group 3 (GM) received 25 μg 17-DMAG on days 9, 10, 11 and 12. Mice in group 4 (QC + GM) were injected with 1.25 mg QC + 25 μg 17-DMAG on days 9 and 10, and 25 μg 17-DMAG on days 11 and 12. Average tumor volume within each group is shown.
Discussion
Proteotoxic stress is induced by the accumulation of misfolded proteins that occurs under a variety of stressful conditions. These conditions include physical assaults, such as heat shock and hypoxia, and chemical stimuli, such as heavy metal ions, analogues of amino acids, and inhibitors of proteasome and chaperone activities.1,3,10,42 Cells can effectively mitigate proteotoxic stress by activating heat shock response (HSR), one of the most evolutionarily ancient cytoprotective signal transduction pathways. HSR involves the synthesis of additional chaperones, such as hsp70 and hsp27, in mammalian cells and numerous chaperones in cells of other origins.43–45 HSR was initially detected in insects46–49 and later described for other types of cellular organisms.50–52 The additional chaperones synthesized during HSR counterbalance the excess of denatured or unfolded proteins and stabilize protein homeostasis in cells.12 Other cellular mechanisms that serve to decrease the toxicity of stress are GCN2- or PERK-dependent attenuation of translation and synthesis of several stress-related proteins. Attenuation of general translation helps minimize proteotoxic pressure by reducing the amount of newly synthesized proteins.53,54
Despite the presence of HSR and other mitigating mechanisms, the cytotoxic potential of proteotoxic stress has led to exploration of its usefulness as an approach to anticancer treatment. In some cases, the clinical potential of proteotoxic stress inducers has been borne out. The proteasome inhibitor bortezomib received FDA approval in June 2008 as an anti-cancer drug and is showing strong effects against multiple myeloma. It is also noteworthy that probably the oldest chemotherapeutic anti-cancer agent, arsenic trioxide (used in China 2,000 years ago), is a powerful inducer of proteotoxic stress55 and is approved for use in the US against acute promyelocytic leukemia.24 However, other proteotoxic stress-inducing treatments have been disappointing as anticancer therapies. Hyperthermia remains in the arsenal of oncology, although with unpredictable efficacy. The hsp90 inhibitor geldanamycin and its derivatives showed promise in experimental models, but extensive testing in humans was largely unsuccessful.27 In these cases, it is possible that protective cellular mechanisms such as HSR limit the effectiveness of the treatment. In addition to purely empirical observations, the rationale for use of proteotoxic stress to eradicate cancer cells is supported by numerous indications that tumor cells frequently acquire constitutively active HSR.56 “Addiction” of tumor cells to the HSF1-mediated HSR pathway is not completely understood; however, it likely stems from the higher incidence of protein mis-folding in transformed cells as compared to normal cells.22,23 This phenomenon might be associated with the generally higher rate of translation in tumor cells and other changes in their metabolism, including the necessity to proliferate under hypoxic conditions that stimulate protein denaturation.22,23 Constitutive use of the protective power of HSF1-mediated HSR by tumor cells potentially puts them at a disadvantage under conditions of exposure to additional proteotoxic stress, since their remaining inducible protective capacity might be less than that of normal cells. However, it is clear that this logic is applicable only to a minority of cancer cells. It seems likely that the insufficient anticancer efficacy of some proteotoxic stress-inducing treatments, such as hyperthermia and geldanamycin, can be explained by the cyto-protective effects of HSF1-mediated HSR, further inducibility of which may be retained even in tumors with acquired constitutive HSR activity. This suggests that powerful and specific HSF1 inhibitors could radically improve the anticancer efficacy of proteotoxic stress inducers.
The need for HSF1 inhibitors is supported by a recent remarkable work that demonstrated the vital importance of HSF1 function for the process of malignant transformation.23 HSF1 deficiency in mice was shown to be associated with a dramatic reduction in cancer incidence, even in animals carrying pro-cancerous genetic alterations. The conclusion drawn from this study that HSF1 activity is an essential condition for transformation greatly increases the value of this pathway as a target for cancer treatment and prevention.
To identify inhibitors of HSR for potential use as anticancer drugs, we chose to investigate anti-malaria drugs before resorting to broad screening of chemical compound libraries. Our reasoning for taking this approach was based on the characteristics of malaria and the biology of Plasmodium falciparum replication. To suppress Plasmodium growth, the infected organism induces fever and heat shock in the form of febrile episodes.57,58 Fever is a protective mechanism against infections, especially for malaria.59–62 However, it is frequently insufficient to completely cure the disease. This is due, at least in part, to the ability of both uninfected and infected host cells to survive at elevated temperatures by inducing HSR. Moreover, P. falciparum induces its own heat shock proteins,30 which allows the organism to survive the elevated temperatures (fever) experienced during its change of hosts. Therefore, in principle, malaria could be treated by decreasing the efficacy of heat shock protein synthesis to make infected cells and P. falciparum more sensitive to fever. This idea led us to hypothesize that some existing anti-malaria drugs might act as inhibitors of HSR. Our prediction appeared to be correct for QC, a 9AA-based compound that was widely and successfully used as an anti-malaria treatment in the past, although with a previously unknown mechanism of action. We found that another anti-malaria drug, emetine, also suppressed hsp70 synthesis, but that it did so in a non-specific manner by acting as a general inhibitor of translation. In contrast to emetine, QC did not inhibit translation. Rather, the dramatic effect of QC on hsp70 synthesis was due to reduced HSF1-dependent transcription. This effect appears to be relatively selective since QC did not have the same effect on a number of other signal transduction pathways analyzed.
The mechanism underlying QC-mediated modulation of transcription is an interesting and important question. This mechanism will be addressed in a separate manuscript, in which we will present evidence of non-genotoxic DNA intercalation as the basis for the biological effects of QC. Here, based on our published observations, we can state that the effect of this drug on HSF1-mediated transcription is not unique. In our earlier work, we described the ability of QC to inhibit NFκB-mediated transcription in a way very similar to its effect on HSF1:33 in both cases, the drug did not interfere with activation, nuclear translocation, or DNA binding of the transcription factor, but blocked transcription initiation. The similarity of the two scenarios suggests that in both cases QC employs a similar mechanism that requires its nuclear localization. This is supported by the results of our experiments with the lysosomal poison bafilomycin (Fig. 3). It was recently reported that QC inhibits the induction of transcription of the genes encoding metalloproteinases MMP1 and MMP8 in response to phorbol ester treatment.63 This indicates that the inhibitory effects of QC are broader than “just” NFκB and HSF1. At the same time, the effects of QC on inducible transcription are clearly selective, since the drug does not block, but actually stimulates, p53-mediated transcription.33 Moreover, QC has no effect on metallothionein gene induction under the same conditions as when it blocks HSR (Fig. 1). The fact that the effects of QC are limited to certain specific signaling pathways might provide an explanation for its remarkable safety as a drug that can be used for months at high doses with no serious side effects.
The expectation that pharmacological inhibition of HSF1-mediated HSR might dramatically increase the efficacy of proteotoxic stresses as anticancer treatments has been fully met in our experiments using mouse tumor models. QC used in combination with the hsp90 inhibitor geldanamycin demonstrated radical antitumor effects under conditions where neither of the drugs administered alone showed decisive therapeutic efficacy. Sensitization of cells to hyperthermia, MG132 and geldanamycin by hsp70 knockdown was previously shown using genetic manipulations.10,39,40 It has now been demonstrated using a pharmacological approach with QC as an HSF1 inhibitor. A similar effect was described for combinatorial treatment of cancer cells with cisplatin and geldanamycin, where cisplatin affected hsp70 synthesis in geldanamycin treated cells.29 This encouraging result with QC projects new anticancer applications for this old anti-malaria drug which may provide an opportunity to reincarnate those proteotoxic stress inducers that have not demonstrated sufficient anticancer efficacy in the past (e.g., hyperthermia and geldanamycin).
It is noteworthy that everything that we have learned in our extensive studies of the biological activity and underlying mechanism of action of QC strongly supports its potential for clinical anticancer use. In fact, the unique multi-targeted mechanism of action of QC suggests that it may be a great improvement over other drugs/treatment strategies. Not only does QC inhibit HSF, but it is also a powerful inducer of p53 and inhibitor of NFκB.33 Thus, QC modulates all three of the most universal anticancer treatment targets in the desired directions. Moreover, QC demonstrates a complete lack of genotoxicity and remarkable safety profile that is supported by decades of human use. Taken together, these properties provide strong justification for testing the anticancer properties of QC and for development of new drugs with QC-like characteristics but improved efficacy and pharmacological characteristics for anticancer applications.
Materials and Methods
Ethics statement
Experiments with mice were conducted strictly in accordance with the protocol approved by the Institutional IACUC of the Cleveland Clinic.
Cell culture, lentiviral vectors, siRNA transfection and drugs
HeLa cells were cultured in Dulbecco modified Eagle’s medium (Invitrogen/Gibco BRL, Carlsbad, CA) supplemented with 10% fetal calf serum. Murine melanoma B16 and fibro-sarcoma MCA205 cells were grown in complete RPMI-1640 medium supplemented with 2 mM L-glutamine, 100 μg/ml streptomycin, 100 U/ml penicillin, 50 μg/ml Gentamicin (Life Technologies, Grand Island, NY). HSF1-specific EGFP expression was generated by insertion of 6 copies of HSF1 regulatory elements (HSE) 5′ CAG AAC GTT CTA G 3′ upstream of minimal CMV promoter into PTR-mCMV-EGFP lentiviral vector. Cells were infected by lentivirus with MOI 10 in the presence of 4 μg/ml of polybrene over night. Efficiency of infection was detected by EGFP expression in response to 43°C for 60 min of heat shock. Between 70 and 90% of the cells were infected by virus and expressed GFP in response to heat shock.
HeLa cells were transfected with hsp70 siRNA (Santa Cruz Biotechnology, Santa Cruz, CA) for 24 h to inhibit hsp70 expression during additional treatments with drugs. Control cells were transfected with not relevant siRNA-A (Santa Cruz Biotechnology, Santa Cruz, CA). Cells were treated as described in the text with the proteasome inhibitors MG132 (Calbiochem, San Diego, CA) and bortezomib (LC Laboratories, Woburn, MA), the hsp90 inhibitor 17-DMAG (LC Laboratories, Woburn, MA), quinacrine, emetine, quinine, chloroquine, and 9AA (all from Sigma, St. Louis, MO). Conditions for hyperthermia/heat shock treatment were incubation at 43°C for 1 h.
Western immunoblotting
Total protein extracts from HeLa cells were prepared in RIPA buffer (150 mM NaCl, 1% SDS, 10 mM Tris (pH 8.0), 1% sodium deoxycholate, 1% NP-40) containing a protease inhibitor cocktail (Sigma, St. Louis, MO). Protein extracts were separated by electrophoresis in 4–20% gradient polyacrylamide gels with SDS (Invitrogen/Novex, Carlsbad, CA) and then transferred to nylon PVDF membranes (Amersham, Piscataway, NJ). Membranes were pre-incubated over-night in 5% milk solution, and then incubated for 1 h in 1% milk solution with primary antibodies at 1:1,000 dilutions. The following primary antibodies were used: rabbit anti-hsp70A1 inducible form (Assay Designs/StressGen, Victiria, BC, Canada) and rat anti-HSF1 (Assay Designs/StressGen, Victiria, BC, Canada). Controls for protein loading were rat anti-pirin antibody (gift of Dr. E.L. Winnacker, Institut für Biochemie der Ludwig-Maximilians-Universität München, Germany.) and rabbit anti-keratin 18 antibody (a gift from Dr. R. Oshima, Burnham research Institute, CA). Apoptosis was analyzed by detection of caspase-specific cleavage of K18 (antibody from Dr. R. Oshima) and PARP (antibody from Santa Cruz Biotechnology, Santa Cruz, CA). HRP-conjugated secondary anti-rabbit, anti-rat and anti-mouse antibodies were purchased from Santa Cruz Biotechnology. Detection reagent (ECL) was from Perkin Elmer (Shelton, CT).
Northern blotting and hybridization
Total RNA (10 μg) from HeLa, B-16 or MCA205 cells was analyzed by northern blot hybridization with probes specific to the hsp70A1, MT1, p65RelA, GRP94 and GAPDH genes. A PCR fragment was generated from the hsp70A1 cDNA (Invitrogen, Carlsbad, CA) with primers specific for the hsp70A1 coding sequence (A1s: 5′ CCA CCA TCC CCA CCA AGC AGA C3′; A1a: 5′ CAT GAA CCA TCC TCT CCA CCT 3′). All other hybridization probes were generated by restriction endonuclease digestion from cDNA clones (ORIGENE).
In vivo 35S-protein labeling
HeLa cells were treated with drugs and their combinations according to the protocols of experiments. The regular culture medium was changed to a methionine/cysteine-free medium supplemented with 35S-methionine and 35S-cysteine (50 μCi/ml) (New England Nuclear/Perkin Elmer, Shelton, CT) and the cells were incubated for additional 60 min in the presence of drugs. Cytoplasmic protein extracts were prepared according to the Dignam protocol.64 Labeled proteins were analyzed by electrophoresis in 10% polyacrilimide gel and autoradiography.
In vitro assay for proteasome activity
To determine the efficiency of the proteasome inhibitors MG132 and Bortezomib alone and in combination with QC and 9AA, we treated HeLa cells with the indicated drugs for 4 h, purified cytoplasmic protein extracts,64 and analyzed proteasome activity using fluorochromic proteasome substrate 1 (Calbiochem, San Diego, CA).
Electrophoretic mobility shift assay (EMSA)
HSF1 DNA binding activity was detected with EMSA. Briefly, cytoplasmic and nuclear protein extracts were purified using the Digman protocol64 from control HeLa cells and HeLa cells treated with MG132 or 17-DMAG alone, or in combination with QC or 9AA. 10 μg of protein extracts were analyzed with a 32P-labeled double-stranded oligonucleotide (5′ TCG AGC TAG AAG CTT CTA GAA GCT TCT AGC 3′) specific for HSF1 binding.65 For competition assay, a 50x excess of unlabeled oligonucleotides were added to protein extracts together with 32P-labeled double-stranded oligonucleotides. Protein extracts were pre-incubated with HSF1 specific antibodies (Assay Designs/StressGen, Victoria, BC, Canada) for 15 min at room temperature before to add P32-labeled double-stranded oligonucleotide for super shift assay.
Cell viability assay
To determine the cytotoxicity of drugs and drug combinations, we used a cell viability assay. Cells were grown to 75–80% confluency and then treated for 4 h with various combinations of 1 μM 17-DMAG, 0.1 μM bortezomib and 10 μM or 20 μM QC. Cells were collected by trypsinization and a 1:50 dilution was seeded in a 6-well plate. Cell viability was determined 72 h later by methylene blue staining after fixation of the cells with 10% formaldehyde. Methylene blue was extracted by 0.1 M HCl and its absorbance was measured at 560 nm.
In vivo assay for tumor growth in mice
MCA205 and B-16 cells were detached from plastic Petri dishes using 0.5% Trypsin-EDTA, washed and resuspended in sterile PBS. Cells were injected intra-dermally into the shaved abdominal area of C57BL/6 mice (3 × 105 cells in a single injection/mouse). Tumors were measured in two dimensions and tumor volume was calculated according to the formula: mm3 = tumor length × (tumor width).2 On day 8 after implantation, tumors were typically ~50 mm3 in size. Tumor-bearing mice were divided into four groups of five animals each and began receiving intra-tumor drug injections on day 9. The control group received PBS injections on days 9, 10, 11 and 12. Mice in group 2 were injected with 1.25 mg QC in PBS33 on days 9 and 10. Mice in group 3 received 25 μg 17-DMAG in PBS66 on days 9, 10, 11 and 12. Mice in group 4 were injected with 1.25 mg QC + 25 μg 17-DMAG on days 9 and 10, and 25 μg 17-DMAG on days 11 and 12. We used only two injections of QC, because according to our in vitro studies, this drug is stable inside of cells. It may be detected in cells as long as 72 h after treatment and the change of medium. Tumors were measured every 2 days after drugs injections for 24 days. Animals with tumors volume 500 mm3 and larger were euthanized.
Acknowledgments
We are grateful to Robert Oshima for his gift of anti-keratin 18 antibodies and Patricia Baker for help in manuscript preparation.
N.N. designed, supervised and conducted most of the experiments. N.N. and A.V.G. developed the concept and wrote the manuscript. L.N. assisted in performing the experiments. A.n.G. and R.L.F. designed and performed the in vivo experiments. K.V.G. and A.l.G. provided chemicals and designed the EMCA experiments. A.K.B. designed the S35-methionine labeling experiments and provided reagents. A.K. created the HSF1-responsive reporter cell lines. A.A. designed the experiments analyzing proteasome activity and apoptosis and provided the reagents. N.N., K.V.G., A.K.B., A.A., R.L.F., and A.V.G. reviewed, analyzed and discussed all of the data. N.N., A.A., R.L.F., A.V.G., and A.K.B. wrote the manuscript. A.V.G. submitted the manuscript.
This work was supported by grants from NIH to A.K.B. (AI26585), A.V.G. (CA60730 and CA88071), and A.A. (CA81504).
References
- 1.Kampinga HH. Chaperones in preventing protein denaturation in living cells and protecting against cellular stress. Handb Exp Pharmacol. 2006:1–42. doi: 10.1007/3-540-29717-0_1. [DOI] [PubMed] [Google Scholar]
- 2.Chang YS, Lee LC, Sun FC, Chao CC, Fu HW, Lai YK. Involvement of calcium in the differential induction of heat shock protein 70 by heat shock protein 90 inhibitors, geldanamycin and radicicol, in human non-small cell lung cancer H460 cells. J Cell Biochem. 2006;97:156–65. doi: 10.1002/jcb.20623. [DOI] [PubMed] [Google Scholar]
- 3.Kawazoe Y, Nakai A, Tanabe M, Nagata K. Proteasome inhibition leads to the activation of all members of the heat-shock-factor family. Eur J Biochem. 1998;255:356–62. doi: 10.1046/j.1432-1327.1998.2550356.x. [DOI] [PubMed] [Google Scholar]
- 4.Kim D, Kim SH, Li GC. Proteasome inhibitors MG132 and lactacystin hyperphosphorylate HSF1 and induce hsp70 and hsp27 expression. Biochem Biophys Res Commun. 1999;254:264–8. doi: 10.1006/bbrc.1998.9840. [DOI] [PubMed] [Google Scholar]
- 5.Morimoto RI, Sarge KD, Abravaya K. Transcriptional regulation of heat shock genes. A paradigm for inducible genomic responses. J Biol Chem. 1992;267:21987–90. [PubMed] [Google Scholar]
- 6.Shen HY, He JC, Wang Y, Huang QY, Chen JF. Geldanamycin induces heat shock protein 70 and protects against MPTP-induced dopaminergic neurotoxicity in mice. J Biol Chem. 2005;280:39962–9. doi: 10.1074/jbc.M505524200. [DOI] [PubMed] [Google Scholar]
- 7.Davenport EL, Moore HE, Dunlop AS, Sharp SY, Workman P, Morgan GJ, et al. Heat shock protein inhibition is associated with activation of the unfolded protein response pathway in myeloma plasma cells. Blood. 2007;110:2641–9. doi: 10.1182/blood-2006-11-053728. [DOI] [PubMed] [Google Scholar]
- 8.Davenport EL, Morgan GJ, Davies FE. Untangling the unfolded protein response. Cell Cycle. 2008;7:865–9. doi: 10.4161/cc.7.7.5615. [DOI] [PubMed] [Google Scholar]
- 9.Mathew A, Mathur SK, Jolly C, Fox SG, Kim S, Morimoto RI. Stress-specific activation and repression of heat shock factors 1 and 2. Mol Cell Biol. 2001;21:7163–71. doi: 10.1128/MCB.21.21.7163-7171.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Noonan EJ, Place RF, Giardina C, Hightower LE. Hsp70B’ regulation and function. Cell Stress Chaperones. 2007;12:393–402. doi: 10.1379/CSC-278e.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shringarpure R, Catley L, Bhole D, Burger R, Podar K, Tai YT, et al. Gene expression analysis of B-lymphoma cells resistant and sensitive to bortezomib. Br J Haematol. 2006;134:145–56. doi: 10.1111/j.1365-2141.2006.06132.x. [DOI] [PubMed] [Google Scholar]
- 12.Imai J, Yashiroda H, Maruya M, Yahara I, Tanaka K. Proteasomes and molecular chaperones: cellular machinery responsible for folding and destruction of unfolded proteins. Cell Cycle. 2003;2:585–90. [PubMed] [Google Scholar]
- 13.Wang J, Maldonado MA. The ubiquitin-proteasome system and its role in inflammatory and autoimmune diseases. Cell Mol Immunol. 2006;3:255–61. [PubMed] [Google Scholar]
- 14.Stebbins CE, Russo AA, Schneider C, Rosen N, Hartl FU, Pavletich NP. Crystal structure of an Hsp90-geldanamycin complex: targeting of a protein chaperone by an antitumor agent. Cell. 1997;89:239–50. doi: 10.1016/s0092-8674(00)80203-2. [DOI] [PubMed] [Google Scholar]
- 15.Weissman AM. Regulating protein degradation by ubiquitination. Immunol Today. 1997;18:189–98. doi: 10.1016/s0167-5699(97)84666-x. [DOI] [PubMed] [Google Scholar]
- 16.Stravopodis DJ, Margaritis LH, Voutsinas GE. Drug-mediated targeted disruption of multiple protein activities through functional inhibition of the Hsp90 chaperone complex. Curr Med Chem. 2007;14:3122–38. doi: 10.2174/092986707782793925. [DOI] [PubMed] [Google Scholar]
- 17.Cowan JL, Morley SJ. The proteasome inhibitor, MG132, promotes the reprogramming of translation in C2C12 myoblasts and facilitates the association of hsp25 with the eIF4F complex. Eur J Biochem. 2004;271:3596–611. doi: 10.1111/j.0014-2956.2004.04306.x. [DOI] [PubMed] [Google Scholar]
- 18.Rahmani M, Davis EM, Bauer C, Dent P, Grant S. Apoptosis induced by the kinase inhibitor BAY 43–9006 in human leukemia cells involves downregulation of Mcl-1 through inhibition of translation. J Biol Chem. 2005;280:35217–27. doi: 10.1074/jbc.M506551200. [DOI] [PubMed] [Google Scholar]
- 19.Proud CG. eIF2 and the control of cell physiology. Semin Cell Dev Biol. 2005;16:3–12. doi: 10.1016/j.semcdb.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 20.Clemens MJ. Translational control in virus-infected cells: models for cellular stress responses. Semin Cell DevBiol. 2005;16:13–20. doi: 10.1016/j.semcdb.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 21.He B. Viruses, endoplasmic reticulum stress, and interferon responses. Cell Death Differ. 2006;13:393–403. doi: 10.1038/sj.cdd.4401833. [DOI] [PubMed] [Google Scholar]
- 22.Solimini NL, Luo J, Elledge SJ. Non-oncogene addiction and the stress phenotype of cancer cells. Cell. 2007;130:986–8. doi: 10.1016/j.cell.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 23.Dai C, Whitesell L, Rogers AB, Lindquist S. Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell. 2007;130:1005–18. doi: 10.1016/j.cell.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Munshi NC, Tricot G, Desikan R, Badros A, Zangari M, Toor A, et al. Clinical activity of arsenic trioxide for the treatment of multiple myeloma. Leukemia. 2002;16:1835–7. doi: 10.1038/sj.leu.2402599. [DOI] [PubMed] [Google Scholar]
- 25.Hayden PJ, Mitsiades CS, Anderson KC, Richardson PG. Novel therapies in myeloma. Curr Opin Hematol. 2007;14:609–15. doi: 10.1097/MOH.0b013e3282f0e948. [DOI] [PubMed] [Google Scholar]
- 26.Perez CA, Pajak T, Emami B, Hornback NB, Tupchong L, Rubin P. Randomized phase III study comparing irradiation and hyperthermia with irradiation alone in superficial measurable tumors. Final report by the Radiation Therapy Oncology Group. Am J Clin Oncol. 1991;14:133–41. doi: 10.1097/00000421-199104000-00008. [DOI] [PubMed] [Google Scholar]
- 27.McCollum AK, TenEyck CJ, Stensgard B, Morlan BW, Ballman KV, Jenkins RB, et al. P-Glycoprotein-mediated resistance to Hsp90-directed therapy is eclipsed by the heat shock response. Cancer Res. 2008;68:7419–27. doi: 10.1158/0008-5472.CAN-07-5175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nowakowski GS, McCollum AK, Ames MM, Mandrekar SJ, Reid JM, et al. A phase I trial of twice-weekly 17-allylamino-demethoxy-geldanamycin in patients with advanced cancer. Clin Cancer Res. 2006;12:6087–93. doi: 10.1158/1078-0432.CCR-06-1015. [DOI] [PubMed] [Google Scholar]
- 29.McCollum AK, Lukasiewicz KB, Teneyck CJ, Lingle WL, Toft DO, Erlichman C. Cisplatin abrogates the geldanamycin-induced heat shock response. Mol Cancer Ther. 2008;7:3256–64. doi: 10.1158/1535-7163.MCT-08-0157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Polla BS. Heat shock proteins in host-parasite interactions. Immunol Today. 1991;12:38–41. doi: 10.1016/S0167-5699(05)80011-8. [DOI] [PubMed] [Google Scholar]
- 31.Zaarur N, Gabai VL, Porco JA, Jr, Calderwood S, Sherman MY. Targeting heat shock response to sensitize cancer cells to proteasome and Hsp90 inhibitors. Cancer Res. 2006;66:1783–91. doi: 10.1158/0008-5472.CAN-05-3692. [DOI] [PubMed] [Google Scholar]
- 32.Grollman AP. Inhibitors of protein biosynthesis. V. Effects of emetine on protein and nucleic acid biosynthesis in HeLa cells. J Biol Chem. 1968;243:4089–94. [PubMed] [Google Scholar]
- 33.Gurova KV, Hill JE, Guo C, Prokvolit A, Burdelya LG, Samoylova E, et al. Small molecules that reactivate p53 in renal cell carcinoma reveal a NFkappaB-dependent mechanism of p53 suppression in tumors. Proc Natl Acad Sci USA. 2005;102:17448–53. doi: 10.1073/pnas.0508888102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chiosis G, Caldas LE, Solit D. Heat shock protein-90 inhibitors: a chronicle from geldanamycin to today’s agents. Curr Opin Investig Drugs. 2006;7:534–41. [PubMed] [Google Scholar]
- 35.Drysdale MJ, Brough PA, Massey A, Jensen MR, Schoepfer J. Targeting Hsp90 for the treatment of cancer. Curr Opin Drug Discov Devel. 2006;9:483–95. [PubMed] [Google Scholar]
- 36.Schulte TW, Neckers LM. The benzoquinone ansamycin 17-allylamino-17-demethoxygeldanamycin binds to HSP90 and shares important biologic activities with geldanamycin. Cancer Chemother Pharmacol. 1998;42:273–9. doi: 10.1007/s002800050817. [DOI] [PubMed] [Google Scholar]
- 37.Sharp S, Workman P. Inhibitors of the HSP90 molecular chaperone: current status. Adv Cancer Res. 2006;95:323–48. doi: 10.1016/S0065-230X(06)95009-X. [DOI] [PubMed] [Google Scholar]
- 38.Weber SM, Levitz SM. Chloroquine interferes with lipopolysaccharide-induced TNFalpha gene expression by a nonlysosomotropic mechanism. J Immunol. 2000;165:1534–40. doi: 10.4049/jimmunol.165.3.1534. [DOI] [PubMed] [Google Scholar]
- 39.Demidenko ZN, Vivo C, Halicka HD, Li CJ, Bhalla K, Broude EV, et al. Pharmacological induction of Hsp70 protects apoptosis-prone cells from doxorubicin: comparison with caspase-inhibitor- and cycle-arrest-mediated cytoprotection. Cell Death Differ. 2006;13:1434–41. doi: 10.1038/sj.cdd.4401812. [DOI] [PubMed] [Google Scholar]
- 40.Guo F, Rocha K, Bali P, Pranpat M, Fiskus W, Boyapalle S, et al. Abrogation of heat shock protein 70 induction as a strategy to increase antileukemia activity of heat shock protein 90 inhibitor 17-allylamino-demethoxy geldanamycin. Cancer Res. 2005;65:10536–44. doi: 10.1158/0008-5472.CAN-05-1799. [DOI] [PubMed] [Google Scholar]
- 41.Xiao L, Lu X, Ruden DM. Effectiveness of hsp90 inhibitors as anti-cancer drugs. Mini Rev Med Chem. 2006;6:1137–43. doi: 10.2174/138955706778560166. [DOI] [PubMed] [Google Scholar]
- 42.Voellmy R. On mechanisms that control heat shock transcription factor activity in metazoan cells. Cell Stress Chaperones. 2004;9:122–33. doi: 10.1379/CSC-14R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Arya R, Mallik M, Lakhotia SC. Heat shock genes—integrating cell survival and death. J Biosci. 2007;32:595–610. doi: 10.1007/s12038-007-0059-3. [DOI] [PubMed] [Google Scholar]
- 44.Latchman DS. HSP27 and cell survival in neurones. Int J Hyperthermia. 2005;21:393–402. doi: 10.1080/02656730400023664. [DOI] [PubMed] [Google Scholar]
- 45.Jaattela M. Escaping cell death: survival proteins in cancer. Exp Cell Res. 1999;248:30–43. doi: 10.1006/excr.1999.4455. [DOI] [PubMed] [Google Scholar]
- 46.Costlow N, Lis JT. High-resolution mapping of DNase I-hypersensitive sites of Drosophila heat shock genes in Drosophila melanogaster and Saccharomyces cerevisiae. Mol Cell Biol. 1984;4:1853–63. doi: 10.1128/mcb.4.9.1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bonner JJ, Pardue ML. The effect of heat shock on RNA synthesis in Drosophila tissues. Cell. 1976;8:43–50. doi: 10.1016/0092-8674(76)90183-5. [DOI] [PubMed] [Google Scholar]
- 48.Spradling A, Penman S, Pardue ML. Analysis of drosophila mRNA by in situ hybridization: sequences transcribed in normal and heat shocked cultured cells. Cell. 1975;4:395–404. doi: 10.1016/0092-8674(75)90160-9. [DOI] [PubMed] [Google Scholar]
- 49.McKenzie SL, Henikoff S, Meselson M. Localization of RNA from heat-induced polysomes at puff sites in Drosophila melanogaster. Proc Natl Acad Sci USA. 1975;72:1117–21. doi: 10.1073/pnas.72.3.1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Subjeck JR, Shyy TT. Stress protein systems of mammalian cells. Am J Physiol. 1986;250:1–17. doi: 10.1152/ajpcell.1986.250.1.C1. [DOI] [PubMed] [Google Scholar]
- 51.Lindquist S. The heat-shock response. Annu Rev Biochem. 1986;55:1151–91. doi: 10.1146/annurev.bi.55.070186.005443. [DOI] [PubMed] [Google Scholar]
- 52.Neidhardt FC, VanBogelen RA, Vaughn V. The genetics and regulation of heat-shock proteins. Annu Rev Genet. 1984;18:295–329. doi: 10.1146/annurev.ge.18.120184.001455. [DOI] [PubMed] [Google Scholar]
- 53.Jiang HY, Wek RC. Phosphorylation of the alpha-subunit of the eukaryotic initiation factor-2 (eIF2alpha) reduces protein synthesis and enhances apoptosis in response to proteasome inhibition. J Biol Chem. 2005;280:14189–202. doi: 10.1074/jbc.M413660200. [DOI] [PubMed] [Google Scholar]
- 54.Neznanov N, Dragunsky EM, Chumakov KM, Neznanova L, Wek RC, Gudkov AV, et al. Different effect of proteasome inhibition on vesicular stomatitis virus and poliovirus replication. PLoS ONE. 2008;3:1887. doi: 10.1371/journal.pone.0001887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim YH, Park EJ, Han ST, Park JW, Kwon TK. Arsenic trioxide induces Hsp70 expression via reactive oxygen species and JNK pathway in MDA231 cells. Life Sci. 2005;77:2783–93. doi: 10.1016/j.lfs.2005.04.024. [DOI] [PubMed] [Google Scholar]
- 56.Powers MV, Clarke PA, Workman P. Dual targeting of HSC70 and HSP72 inhibits HSP90 function and induces tumor-specific apoptosis. Cancer Cell. 2008;14:250–62. doi: 10.1016/j.ccr.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 57.Kwiatkowski D, Bate CA, Scragg IG, Beattie P, Udalova I, Knight JC. The malarial fever response—pathogenesis, polymorphism and prospects for intervention. Ann Trop Med Parasitol. 1997;91:533–42. doi: 10.1080/00034989760905. [DOI] [PubMed] [Google Scholar]
- 58.Senanayake N, Roman GC. Neurological complications of malaria. Southeast Asian J Trop Med Public Health. 1992;23:672–80. [PubMed] [Google Scholar]
- 59.Smith T, Felger I, Tanner M, Beck HP. Premunition in Plasmodium falciparum infection: insights from the epidemiology of multiple infections. Trans R Soc Trop Med Hyg. 1999;93:59–64. doi: 10.1016/s0035-9203(99)90329-2. [DOI] [PubMed] [Google Scholar]
- 60.Pavithra SR, Kumar R, Tatu U. Systems analysis of chaperone networks in the malarial parasite Plasmodium falciparum. PLoS Comput Biol. 2007;3:1701–15. doi: 10.1371/journal.pcbi.0030168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Acharya P, Kumar R, Tatu U. Chaperoning a cellular upheaval in malaria: heat shock proteins in Plasmodium falciparum. Mol Biochem Parasitol. 2007;153:85–94. doi: 10.1016/j.molbiopara.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 62.Silva MD, Cooke BM, Guillotte M, Buckingham DW, Sauzet JP, Le Scanf C, et al. A role for the Plasmodium falciparum RESA protein in resistance against heat shock demonstrated using gene disruption. Mol Microbiol. 2005;56:990–1003. doi: 10.1111/j.1365-2958.2005.04603.x. [DOI] [PubMed] [Google Scholar]
- 63.Stuhlmeier KM, Pollaschek C. Quinacrine but not chloroquine inhibits PMA induced upregulation of matrix metalloproteinases in leukocytes: quinacrine acts at the transcriptional level through a PLA2-independent mechanism. J Rheumatol. 2006;33:472–80. [PubMed] [Google Scholar]
- 64.Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475–89. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Trinklein ND, Chen WC, Kingston RE, Myers RM. Transcriptional regulation and binding of heat shock factor 1 and heat shock factor 2 to 32 human heat shock genes during thermal stress and differentiation. Cell Stress Chaperones. 2004;9:21–8. doi: 10.1379/481.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Harrison EM, Sharpe E, Bellamy CO, McNally SJ, Devey L, Garden OJ, et al. Heat shock protein 90-binding agents protect renal cells from oxidative stress and reduce kidney ischemia-reperfusion injury. Am J Physiol Renal Physiol. 2008;295:397–405. doi: 10.1152/ajprenal.00361.2007. [DOI] [PubMed] [Google Scholar]