

Summary
Mdm2 is the major negative regulator of the p53 pathway. Here we report that Mdm2 is rapidly degraded after DNA damage and that phosphorylation of Mdm2 by Casein Kinase I (CKI) at multiple sites triggers its interaction with, and subsequent ubiquitination and destruction, by SCFβ-TRCP. Inactivation of either β-TRCP or CKI results in accumulation of Mdm2 and decreased p53 activity, and resistance to apoptosis induced by DNA damaging-agents. Moreover, SCFβ-TRCP-dependent Mdm2 turnover also contributes to the control of repeated p53 pulses in response to persistent DNA damage. Our results provide insight into the signaling pathways controlling Mdm2 destruction and further suggest that compromised regulation of Mdm2 results in attenuated p53 activity, thereby facilitating tumor progression.
Introduction
Tumors often arise through gain of function of certain oncogenes concomitantly with loss of key tumor suppressors. The p53 protein is considered an important tumor suppressor because its function is compromised in over 50% of human tumors (Hollstein et al., 1991). The ability of p53 to suppress tumor development is primarily attributed to its activity as a transcription factor that can activate a wide variety of downstream target genes that are responsible for p53-dependent cell cycle arrest or apoptosis upon a variety of cellular stresses (Harms et al., 2004; Levine, 1997), such as p21 (Bunz et al., 1998; el-Deiry et al., 1993), Gadd45, 14-3-3 (Chan et al., 1999; Wang et al., 1999) and Bax (Zhang et al., 2000). Because of its crucial role in response to DNA damage, p53 is critical for maintaining the integrity of the genome (Daujat et al., 2001; Harper and Elledge, 2007; Levine et al., 2004).
Due to the important role of p53 in regulating cell proliferation and survival, its activity has to be tightly regulated (Toledo and Wahl, 2006). In normal unstressed cells, p53 activity must be kept low because inappropriate activation of p53 promotes premature senescence or apoptosis (Blaydes and Wynford-Thomas, 1998; Mendrysa et al., 2003). On the other hand, p53 destruction must be quickly disabled to allow for the rapid establishment of the p53 stress response (Toledo and Wahl, 2006). Central to this regulatory mechanism is the Mdm2 protein, a major negative regulator of p53, which promotes p53 ubiquitination and subsequent destruction in unstressed cells (Haupt et al., 1997; Midgley and Lane, 1997). The Mdm2 protein’s N-terminus interacts with p53 and its C-terminal ring-finger domain, which possesses the ubiquitin E3 ligase activity, ubiquitinates p53 (Kussie et al., 1996). The importance of Mdm2 in p53 regulation is further illustrated by the fact that the embryonic lethal phenotype in Mdm2 knockout mice can be partially rescued by further inactivation of the p53 protein (Montes de Oca Luna et al., 1995).
Previous studies showed that binding of Mdm2 to p53 is subject to many layers of regulation. Among them, the DNA damage-induced ATM/ATR/CHK kinase cascade plays a pivotal role. In response to stress such as DNA damage, activation of the ATM/ATR/CHK kinase pathway results in p53 phosphorylation, which disrupts the interaction between Mdm2 and p53, allowing p53 to escape Mdm2 mediated proteolysis and become stabilized (Chehab et al., 2000; Harper and Elledge, 2007; Hirao et al., 2000). It was reported that in response to DNA damage signals, the Mdm2 protein was quickly degraded (Stommel and Wahl, 2004), allowing p53 to accumulate and become fully activated. However, the molecular mechanisms still remain unclear. Although it is proposed that Mdm2 undergoes auto-ubiquitination when cells are challenged with DNA damage agents (Stommel and Wahl, 2004; Stommel and Wahl, 2005), a recent study utilizing transgenic mice provided definitive evidence that the E3 ligase activity of Mdm2 is not required for the proper destruction of Mdm2 (Itahana et al., 2007). This suggests that the destruction of Mdm2 is controlled by an as yet unknown pathway. The goal of this study was to delineate the molecular mechanisms governing Mdm2 ubiquitination and destruction.
Results
Mdm2 stability is controlled by β-TRCP
In agreement with previous reports (Itahana et al., 2007; Stommel and Wahl, 2004), we found that Mdm2 became unstable after DNA damage treatment in various types of cells (Figure 1A and data not shown). Furthermore, we found that Mdm2 protein abundance fluctuated during the cell cycle (Figure 1B). Consistent with a more recent study showing that the E3-ligase activity of Mdm2 is not required for Mdm2 destruction (Itahana et al., 2007), we found that the degradation of the ring-finger mutant Mdm2C464A, which is defective in its E3 ubiquitin ligase activity, was still accelerated by a variety of DNA-damaging agents (data not shown). This data suggested that an unknown pathway controls Mdm2 destruction in response to genotoxic stress.
Figure 1. Mdm2 stability is controlled by β-TRCP.
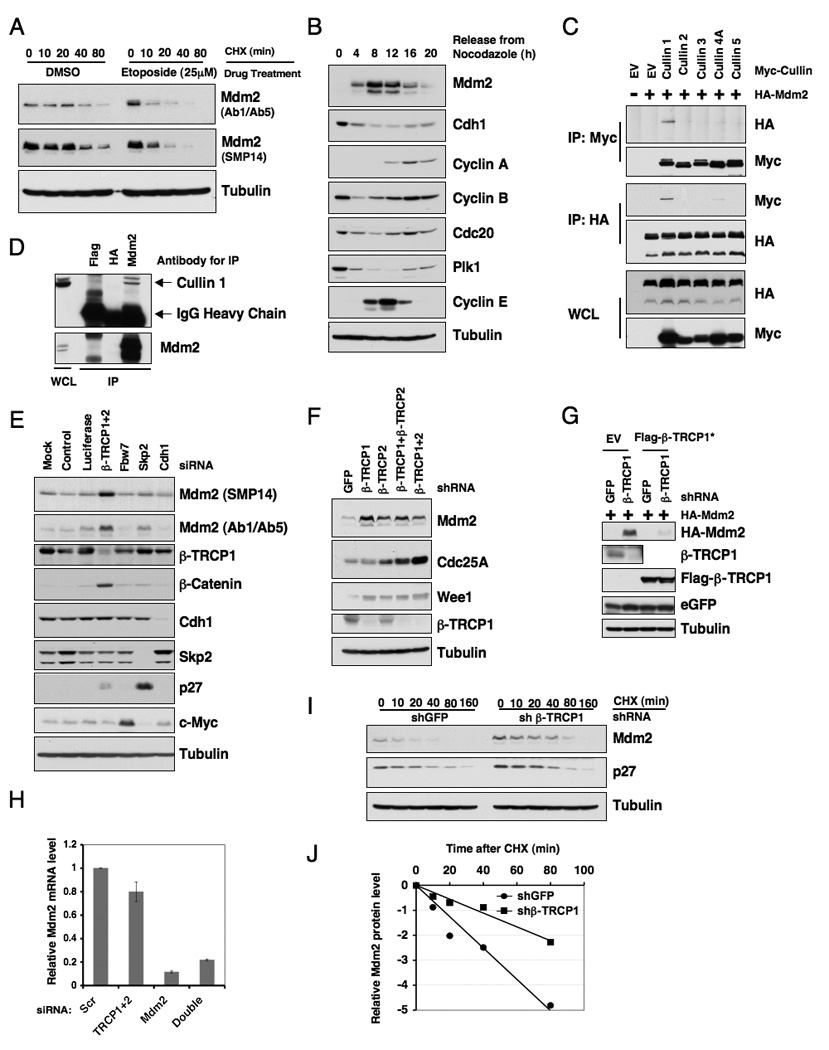
A. U2OS cells were treated with 25 µM etoposide for 1.5 hours before 20 µg/ml cycloheximide was added. At the indicated time points, whole cell lysates were prepared and immunoblots were probed with the indicated antibodies.
B. Immunoblot analysis of HeLa cells synchronized by growth in nocodazole, and then released for the indicated periods of time.
C. Immunoblot analysis of whole cell lysates (WCL) and immunoprecipitates (IP) derived from 293T cells transfected with HA-Mdm2 and various Myc-tagged Cullin constructs. Twenty hours post-transfection, cells were treated with 10 µM MG132 overnight before harvesting.
D. Immunoblot analysis of HeLa cell whole cell lysates (WCL) and anti-Mdm2 immunoprecipitates (IP). Flag- and HA-agarose beads were used as negative controls for the immunoprecipitation procedure. Cells were treated with 10 µM MG132 overnight before harvesting.
E. Immunoblot analysis of HeLa cells transfected with the indicated siRNA oligonucleotides. The control lane is scrambled E2F-1 siRNA; Luciferase, siRNA against firefly luciferase; β-TRCP1+2, siRNA oligo that can deplete both β-TRCP1 and β-TRCP2 isoforms. siRNA, short interfering RNA.
F. Immunoblot analysis of HeLa cells transfected with the indicated shRNA constructs.
G. Immunoblot analysis of 293T cells transfected with the indicated shRNA constructs together with HA-Mdm2 and jellyfish GFP (eGFP, as transfection controls). Where indicated, Flag-β-TRCP1*, which is engineered to resist the shβ-TRCP1 effect, was included in the transfection.
H. Real-time RT-PCR analysis to examine the relative Mdm2 mRNA expression levels in U2OS cells transfected with the indicated siRNA oligonucleotides. Three independent sets of experiments were performed to generate the error bar. The error bars represent +/− SD.
I. HeLa cells were transfected with the indicated shRNA constructs. Twenty hours post-transfection, cells were split into 60 mm dishes, and after another 20 hours, treated with 20 µg/ml cycloheximide. At the indicated time points, whole cell lysates were prepared and immunoblots were probed with the indicated antibodies.
J. Quantification of the band intensities in I. Mdm2 band intensity was normalized to tubulin, then normalized to the t=0 controls. See also Figure S1.
Multisubunit Cullin-Ring complexes comprise the largest known class of E3 ubiquitin ligases (Petroski and Deshaies, 2005). We first examined whether a specific Cullin-Ring complex is involved in controlling Mdm2 destruction. We found that Mdm2 specifically interacts with Cullin 1, but not other members of the Cullin family we examined (Figure 1C). We also detected the interaction between endogenous Mdm2 and endogenous Cullin 1 (Figure 1D). Depletion of endogenous Cullin 1 led to an increase in Mdm2 abundance (Figure S1A), indicating that the Cullin 1 pathway might play a role in the regulation of Mdm2 stability. Consistent with that Mdm2C464A can still be degraded, we found that Mdm2C464A binds to Cullin 1 as efficiently as wild-type Mdm2 (Figure S1B). These data together suggest that the Cullin 1 complex might be the E3 ligase responsible for Mdm2 stability independent of its autoubiquitination.
Next, we sought to explore which F-box protein complexed with Cullin 1 is responsible for Mdm2 destruction. Since Mdm2 expression fluctuates during the cell cycle (Figure 1B), we started our investigation by using a panel of siRNAs targeting different F-box proteins (as well as Cdh1 as a negative control) that have been demonstrated to participate in cell cycle regulation. We found that depletion of β-TRCP, but not other F-box proteins, led to a significant accumulation of Mdm2 (Figure 1E). We further found that unlike other reported β-TRCP substrates, such as Cdc25A (Moshe et al., 2004), inactivation of either β-TRCP1 or β-TRCP2 is sufficient to upregulate Mdm2 (Figure 1F and Figure S1C). Additionally, ectopic expression of β-TRCP1 engineered to resist shTRCP1 or shTRCP1+2 treatment suppressed Mdm2 upregulation (Figure 1G and Figure S1D), indicating that depletion of endogenous β-TRCP might be causative for Mdm2 upregulation. Because depletion of β-TRCP did not increase Mdm2 mRNA level (Figure 1H), the observed increase in Mdm2 abundance is mainly through post-transcriptional mechanisms. In support of this idea, using cycloheximide, we demonstrated that the half-life of the endogenous Mdm2 protein was extended after depletion of the β-TRCP1 protein (Figure 1I–J). Furthermore, β-TRCP-mediated Mdm2 destruction occurred primarily in the late G1-S phase, as indicated by the marked increase in Mdm2 abundance after depletion of β-TRCP in this specific cell cycle window (Figure S1E).
Mdm2 interacts with β-TRCP1 in a phosphorylation-dependent manner
Consistent with a role for β-TRCP in regulating Mdm2 stability, under ectopic overexpression conditions, Mdm2 interacts with both β-TRCP1 and β-TRCP2 (Figure 2A). Furthermore, we detected the interaction between endogenous Mdm2 and endogenous β-TRCP1 (Figure 2B). The interaction between Mdm2 and β-TRCP1 is largely reduced if the C-terminal WD40 repeats motif of β-TRCP1, which has been shown to mediate the interaction with most of its substrates (Wu et al., 2003), is mutated (Figure 2C). We also found that Mdm2C464A interacts with β-TRCP1 as well (Figure S2) and that DNA damage treatment enhanced the interaction between endogenous Mdm2 and endogenous β-TRCP1 (Figure 2D). In keeping with previous reports (Frescas and Pagano, 2008; Petroski and Deshaies, 2005), we found that phosphatase treatment greatly reduced the interaction between Mdm2 and β-TRCP1 (Figure 2E), supporting the idea that Mdm2 interacts with β-TRCP1 in a phosphorylation-dependent manner.
Figure 2. Mdm2 interacts with β-TRCP in a phosphorylation dependent manner.
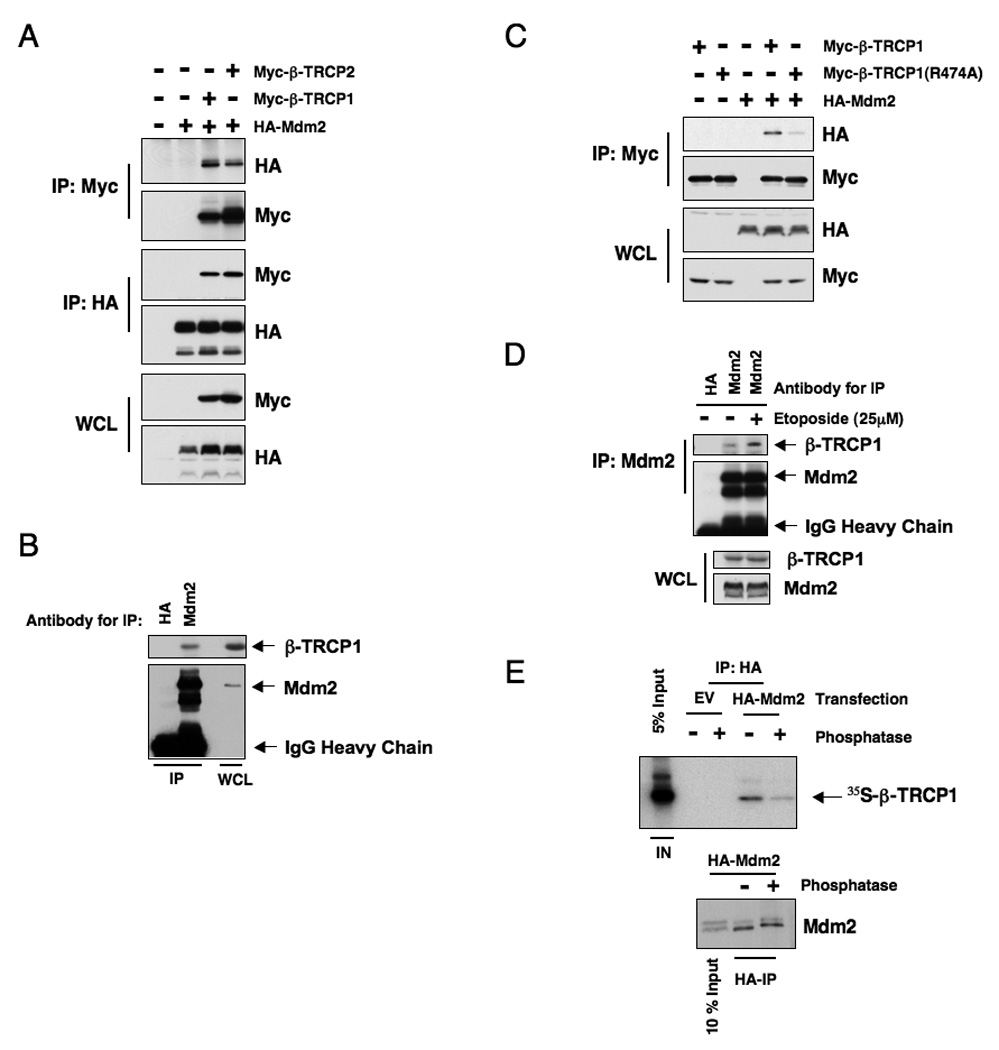
A. Immunoblot analysis of whole cell lysates (WCL) and immunoprecipitates (IP) derived from 293T cells transfected with HA-Mdm2 and Myc-tagged β-TRCP constructs. Twenty hours post-transfection, cells were treated with 10 µM MG132 overnight before harvesting.
B. Immunoblot analysis of HeLa cell whole cell lysates (WCL) and anti-Mdm2 immunoprecipitates (IP). HA-agarose beads were used as a negative control for the IP. Cells were treated with 10 µM MG132 overnight before harvesting.
C. Immunoblot analysis of whole cell lysates (WCL) and immunoprecipitates (IP) derived from 293T cells transfected with HA-Mdm2 and the indicated Myc-tagged β-TRCP constructs. Twenty hours post-transfection, cells were treated with 10 µM MG132 overnight before harvesting.
D. Immunoblot analysis of HeLa cell whole cell lysates (WCL) and anti-Mdm2 immunoprecipitates (IP). HA-agarose beads were used as a negative control for the IP. Cells were treated with 10 µM MG132 overnight before harvesting. Where indicated, cells were treated with 25 µM etoposide (or DMSO as a negative control) for 30 minutes before harvesting.
E. Autoradiograms showing recovery of 35S-labeled β-TRCP1 protein bound to Mdm2 protein immunoprecipitated from HeLa cells transfected with the indicated HA-Mdm2 constructs. Where indicated, the whole cell lysates were treated with λ-phosphatase prior to immunoprecipitation. IN, input (10% or 5% as indicated). See also Figure S2
Casein Kinase I controls Mdm2 stability
It has been suggested that most β-TRCP1 downstream targets need to be properly modified by a combination of kinases within their degron sequences prior to their recognition and destruction by the SCFβ-TRCP complex (Cardozo and Pagano, 2004; Jin et al., 2003). We therefore sought to identify the kinase that phosphorylates Mdm2 and triggers its interaction with β-TRCP1. Examination of the Mdm2 protein sequence did not reveal a canonical degron sequence (DSGxxS) that could be recognized by the SCFβ-TRCP E3 ligase complex (Cardozo and Pagano, 2004). Thus, we used the PESTfinder program to identify two strong PEST sequences, which are typically associated with protein instability where putative degron sequences might reside, within the Mdm2 protein sequence (Figure S3A). The Scansite program predicted that most of the putative phosphorylation sites within these two PEST sequences are either Casein Kinase I (CKI) or Casein Kinase II (CKII) sites. Hence, we began to investigate the potential role of CKI or CKII in Mdm2 stability control.
In support of the notion that CKI is the upstream kinase triggering β-TRCP1-mediated Mdm2 destruction, we found that GST-Mdm2 cannot be recognized by β-TRCP1 in vitro unless it is phosphorylated by purified CKI, but not CKII (Figure 3A). Using co-immunoprecipitation and GST pull-down assays we found that Mdm2 interacted with CKIα1, CKIα2, CKIδand CKIε (Figure S3B–C) (Winter et al., 2004). We also detected the interaction between endogenous Mdm2 and endogenous CKIδ (Figure 3B), further supporting the physiological role of CKIδin regulating Mdm2 stability. Consistent with this finding, overexpression of CKIδ and CKIε, and to less extent CKIα1 and CKIα2, reduced Mdm2 protein level (Figure 3C). On the other hand, expression of CKIγ1 and CKIγ2, or a kinase-dead mutant of CKIδ (K38R) (Peters et al., 1999) failed to promote Mdm2 destruction (Figure S3E–G).
Figure 3. Casein Kinase I is involved in regulating Mdm2 stability.
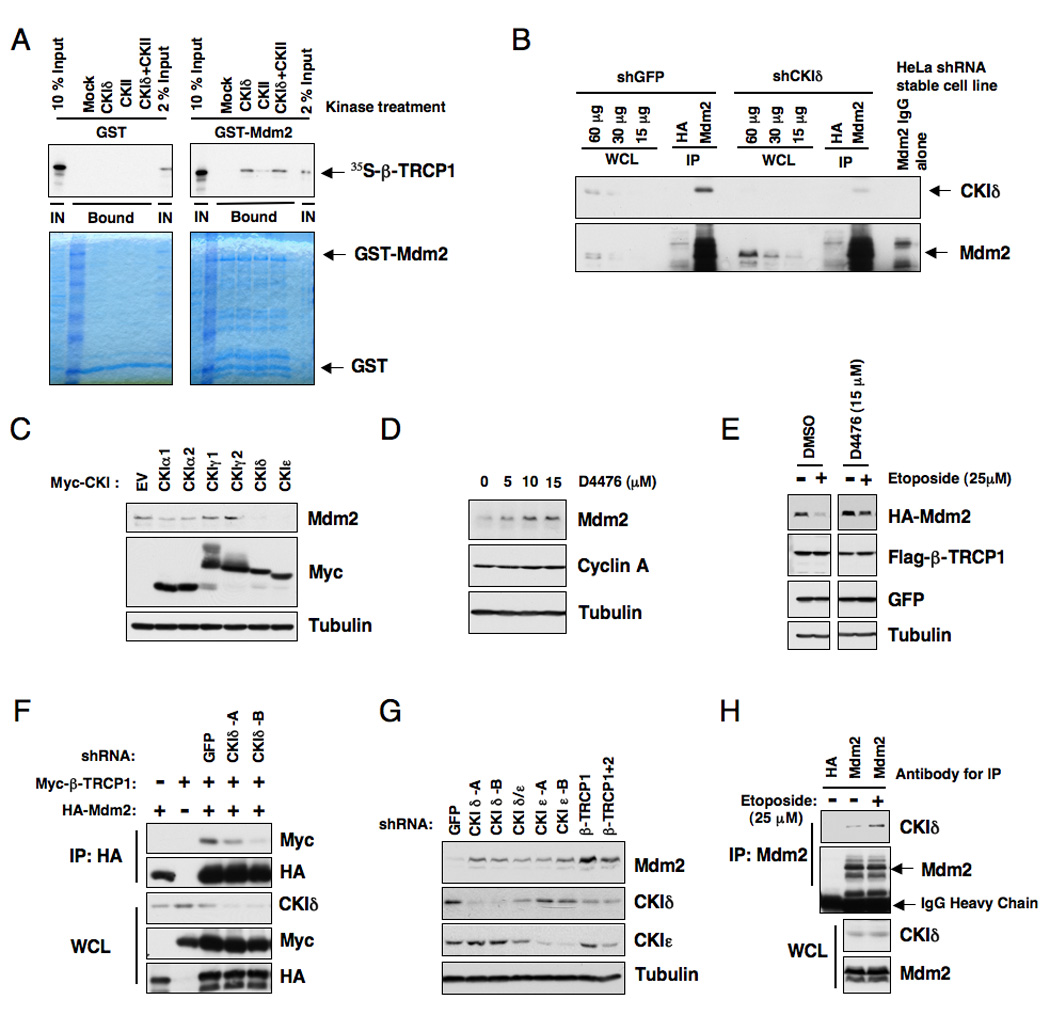
A. Autoradiograms showing recovery of 35S-labeled β-TRCP1 protein bound to GST-Mdm2 fusion proteins (GST protein as a negative control) incubated with the indicated kinase prior to the pull-down assays. IN, input (10% or 2% as indicated). 35S labeling was carried out by in vitro translation reaction with retic lysate.
B. Immunoblot analysis of whole cell lysates (WCL) and anti-Mdm2 immunoprecipitates (IP) derived from the indicated HeLa stable cell lines generated by infection with shGFP or shCKIδ lentiviral construct and subsequent selection with 1 µM puromycin to eliminate the non-infected cells. HA-agarose beads were used as a negative control for the immunoprecipitation procedure. Cells were treated with 10 µM MG132 overnight before harvesting.
C. Immunoblot analysis of whole cell lysates (WCL) derived from 293T cells transfected with various Myc-tagged CKI constructs to detect the changes in endogenous Mdm2 expression.
D. Immunoblot analysis of HeLa cells treated with the CKI inhibitor D4476 at the indicated concentrations for 12 hours.
E. HeLa cells were transiently transfected with the HA-Mdm2 plasmid together with Flag-β-TRCP1. Twenty-four hours post-transfection, cells were treated with the indicated CKI inhibitor for 4 hours and then incubated with 25 µM etoposide for an additional 1.5 hours. The whole cell lysates were recovered and immunoblots were performed with the indicated antibodies.
F. Immunoblot analysis of whole cell lysates (WCL) and immunoprecipitates (IP) derived from 293T cells transfected with HA-Mdm2 and Myc-tagged β-TRCP constructs. Where indicated, the shGFP or shCKIδ construct was included in the transfection. Twenty hours post-transfection, cells were treated with 10 µM MG132 overnight before harvesting.
G. Immunoblot analysis of HeLa cells transfected with the indicated shRNA constructs.
H. Immunoblot analysis of HeLa cell whole cell lysates (WCL) and anti-Mdm2 immunoprecipitates (IP). HA-agarose beads were used as a negative control for the IP. Cells were treated with 10 µM MG132 overnight before harvesting. Where indicated, cells were treated with 25 µM etoposide (or DMSO as control) for 30 minutes before harvesting. See also Figure S3.
Furthermore, inactivation of CKI by the small-molecule inhibitor D4476 (Rena et al., 2004), resulted in accumulation of endogenous Mdm2 (Figure 3D) as well as a moderate increase in resistance to etoposide-induced Mdm2 destruction (Figure 3E). Consistent with a role for CKI in Mdm2 stability control, inhibition of CKIδ activity by inhibitors (Figure S3H) or shRNA treatment (Figure 3F) significantly reduced the interaction between Mdm2 and β-TRCP1. Furthermore, depletion of CKI (mainly the CKIδ and CKIε isoforms) led to elevated Mdm2 abundance (Figure 3G and Figure S3I–J), and extended Mdm2 half-life (Figure S3K–L), indicating that CKIδ and CKIε might be the major isoforms regulating Mdm2 stability. DNA damage induced the interaction between endogenous Mdm2 and CKIδ (Figure 3H), providing a direct link between CKIδ and Mdm2 destruction after DNA damage response. In agreement with a previous report (Alsheich-Bartok et al., 2008), we found that CKIδ resides in the cytoplasm in non-stressed cells, but in response to DNA damage, a significant fraction of endogenous CKIδ translocated into the nucleus (Figure S3M–N).
Casein Kinase Iδ phosphorylates Mdm2 at multiple sites to trigger Mdm2/β-TRCP1 interaction
The absence of a canonical degron sequence in the Mdm2 protein sequence suggests that Mdm2 might contain multiple suboptimal degron sequences instead of one optimized degron. In support of this notion, using mass spectrometry analysis, we found that CKIδ phosphorylates GST-Mdm2 at multiple sites in vitro (Figure S4A–B). In addition to the CKI sites we identified, Mdm2 has also been reported to be phosphorylated by CKI at multiple sites in vivo (Figure S4D) (Winter et al., 2004). To further evaluate the physiological relevance of Mdm2 phosphorylation for its stability, we blocked the proteasome pathway with MG132 then analyzed the Mdm2 phosphorylation status with mass spectrometry. We found that proteasome inhibition resulted in increased phosphorylated Mdm2 species (Figure S4C), many of which overlapped with those identified in the in vitro CKIδ kinase assay (Figure S4B). Importantly, these phosphorylation sites resemble the DSGxxS degron site (Figure S4E).
Interestingly, the majority of the putative CKI sites reside in the two identified PEST sequences (Figure S3A and S4A–C). We named the PEST sequence containing the acidic domain the p2 region, and the PEST sequence containing the SQ cluster domain the p3 region (Figure S3A and S4A). We noticed that the Ser121 site (DSGTSVS) identified by mass spectrometry residing outside the two PEST sequences, however, most resembles the canonical degron sequence (Figure S4F). We named this region p1. To better understand the contribution of this putative degron to Mdm2 stability control, we developed phosphorylation-specific antibodies that recognize the phosphorylated state of Ser118 and Ser118/Ser121. Utilizing these two antibodies, we demonstrated that these two sites were indeed phosphorylated in vivo (Figure S4G–I). Furthermore, inhibition of CKIδ by either CKI inhibitors or shRNA treatment led to a reduction in phosphorylation on both Ser118 and Ser121 of Mdm2 (Figure S4J–K). On the other hand, depletion of β-TRCP1 resulted in a dramatic increase of phosphorylation on both Ser118 and Ser121 of Mdm2 (Figure S4K).
Although phosphorylation of both Ser118 and Ser121 within the p1 region is sufficient for a Mdm2 peptide to interact with β-TRCP1 (Figure S4L–N), mutation or deletion of these two putative CKI phosphorylation sites did not abolish the ability of Mdm2 to interact with β-TRCP in vivo (Figure S4O–P), indicating that Mdm2 might contain additional β-TRCP recognition motifs. To further understand their contributions to Mdm2 destruction, we created a panel of Mdm2 mutants where the DSG site (p1), acidic domain (p2), and the SQ cluster (p3) regions were deleted individually or in combination (Figure S4Q). We also generated mutants wherein the potential critical Ser/Thr sites for individual suboptimal degrons, including some experimentally identified putative CKI phosphorylation sites, were point-mutated (Figure 4A and Figure S4E, S4R).
Figure 4. Casein Kinase Iδ phosphorylates Mdm2 at multiple sites to trigger Mdm2 interaction with β-TRCP1.
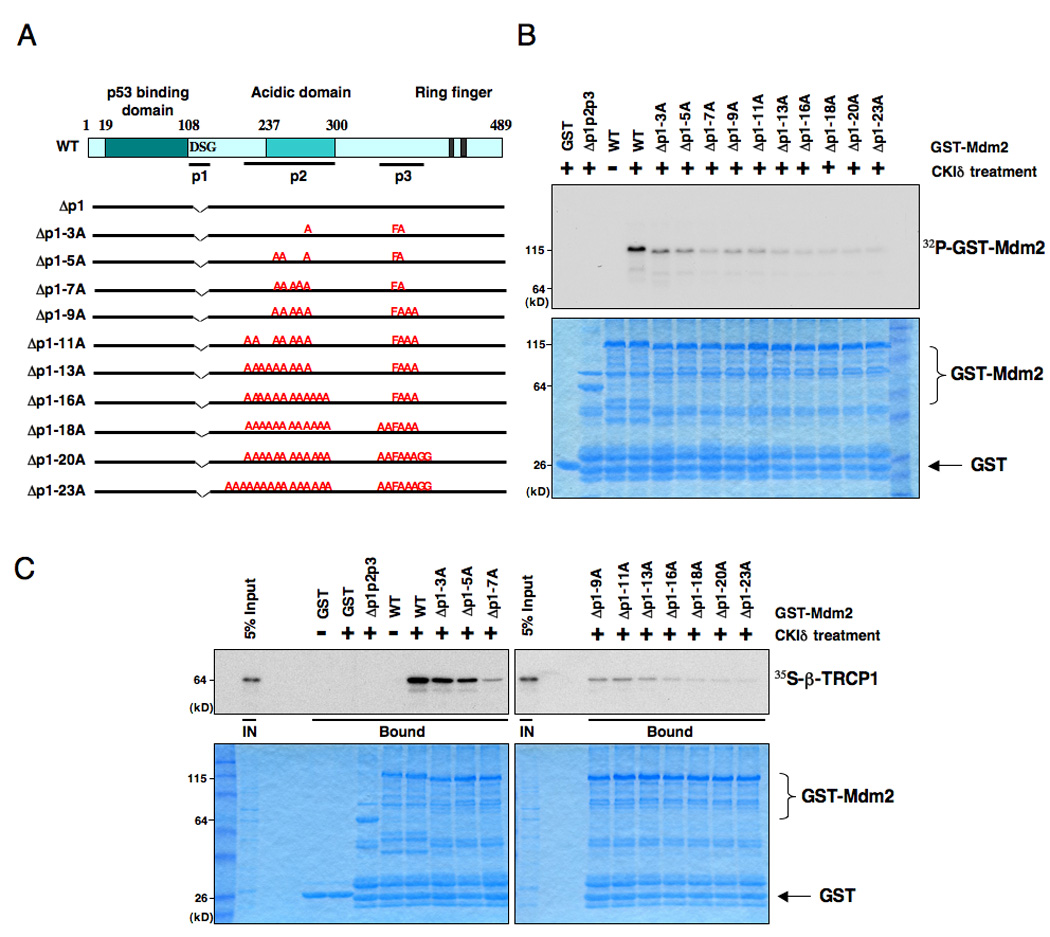
A. Illustration of the various Mdm2 mutants generated for this study.
B. Sequential inactivation of the potentially critical Ser/Thr sites for individual suboptimal degrons including the identified major CKIδ phosphorylation sites leads to progressive reduction of in vitro Mdm2 phosphorylation signals. Purified CKIδ protein (from New England Biolabs) was incubated with 5 µg of the indicated GST-Mdm2 proteins in the presence of γ-32P-ATP. The kinase reaction products were resolved by SDS-PAGE and phosphorylation was detected by autoradiography.
C. Consequently, the recovered in vitro β-TRCP1/Mdm2 interaction is reduced in a step-wise manner after sequential inactivation of the identified Ser/Thr phosphorylation sites in Mdm2, indicating that phosphorylation of Mdm2 at multiple sites by CKIδ triggers its interaction with β-TRCP1 in vitro. Autoradiograms showing recovery of 35S-labeled β-TRCP1 protein bound to the indicated GST-Mdm2 fusion proteins (GST protein as a negative control) incubated with CKIδ prior to the pull-down assays. IN, input (5% as indicated). See also Figure S4.
Using in vitro kinase assays, we found that deletion of the p2 region greatly reduced the ability of CKIδ to phosphorylate Mdm2 (Figure S4S). We also found that deletion of either the p1 or p3 regions further reduced the phosphorylation by CKIδ, indicating that CKIδ also phosphorylates Mdm2 in the DSG and SQ cluster regions (Figure S4S). More interestingly, we observed a progressive reduction of the CKIδ -mediated phosphorylation after mutating the major phosphorylation sites identified by mass spectrometry analysis in a step-wise fashion (Figure 4B). These Mdm2 mutants interact with CKIδ with similar efficacy as the wild-type Mdm2 (Figure S4T–U), which indicates that the loss of phosphorylation of these mutants is not due to loss of their interaction with the CKIδ kinase. Altogether, these results support a possible model that CKIδ phosphorylates Mdm2 at multiple sites, which closely resembles the way GSK3 and PKA phosphorylate the Gli3 oncoprotein (Jia et al., 2005; Tempe et al., 2006). Importantly, we found that the ability of various Mdm2 species to interact with β-TRCP1 correlated very well with their CKIδ phosphorylation status (Figure 4C and Figure S4V). Taken together, this data indicates that phosphorylation of Mdm2 by CKIδat multiple sites is critical for triggering Mdm2’s interaction with β-TRCP.
Casein Kinase Iδ phosphorylates Mdm2 at multiple sites to trigger Mdm2 destruction
Next, we explored whether β-TRCP promotes Mdm2 destruction in a CKIδ phosphorylation-dependent manner. We found that the degree of decrease in CKIδ-mediated phosphorylation of the various Mdm2 mutants correlated with their resistance to β-TRCP-induced proteolysis (Figure 5A and 5C). Inactivation of the E3-ligase activity of Mdm2 by mutating its ring-finger domain (C464A) did not interfere with the ability of β-TRCP and CKIδ to degrade the Mdm2 protein (Figure 5B).
Figure 5. Casein Kinase Iδ phosphorylates Mdm2 at multiple sites to trigger Mdm2 destruction by β-TRCP1.
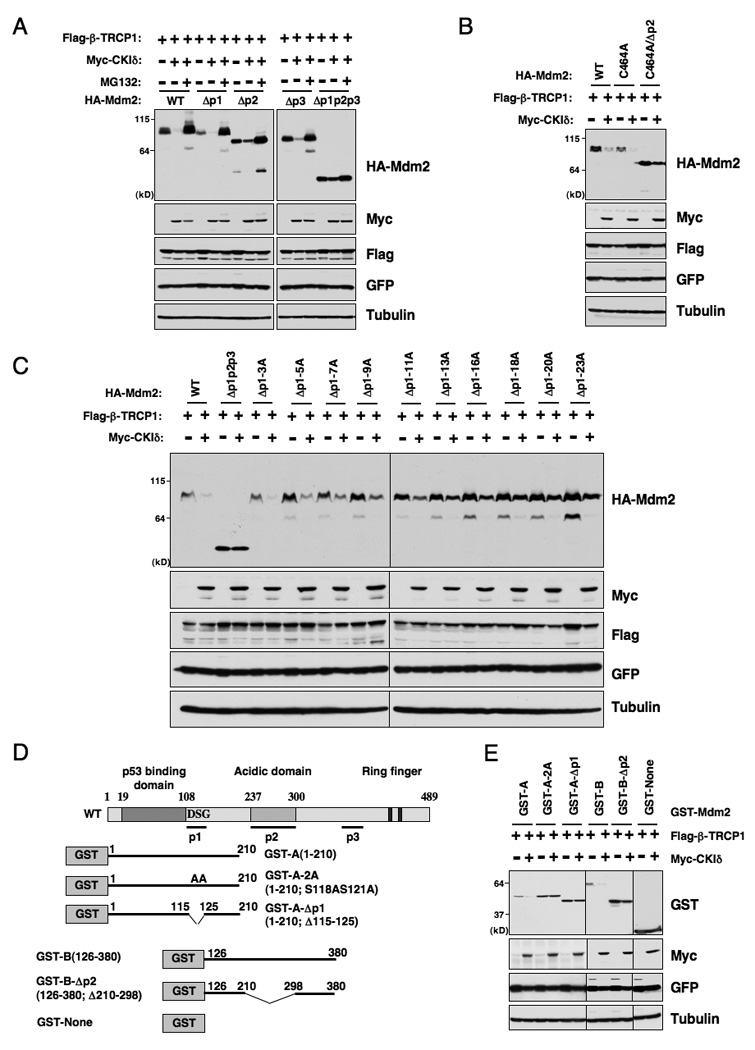
A. Immunoblot analysis of HeLa cells transfected with the indicated HA-Mdm2 and Flag-β-TRCP1 plasmids in the presence or absence of Myc-CKIδ. A plasmid encoding GFP was used as a negative control for transfection efficiency. Where indicated, the proteasome inhibitor MG132 (10 µM) was added.
B. Immunoblot analysis of HeLa cells transfected with various HA-Mdm2C464A constructs (with wild-type Mdm2 as a positive control) and Flag-β-TRCP1 plasmids in the presence or absence of Myc-CKIδ. A plasmid encoding GFP was used as a negative control for transfection efficiency.
C. Immunoblot analysis of HeLa cells transfected with the indicated HA-Mdm2 and Flag-β-TRCP1 plasmids in the presence or absence of Myc-CKIδ. A plasmid encoding GFP was used as a negative control for transfection efficiency.
D. Illustration of the various GST-Mdm2 constructs used in E and Figure S5.
E. Immunoblot analysis of HeLa cells transfected with the indicated GST-Mdm2 and Flag-β-TRCP1 plasmids in the presence or absence of Myc-CKIδ. A plasmid encoding GFP was used as a negative control for transfection efficiency. See also Figure S5.
Degrons can serve to recruit E3 ligases when fused to artificial substrates. To test whether the identified Mdm2 degron sequences can function outside the context of Mdm2, the p1 or p2 region was fused with the GST protein and their response to β-TRCP1-mediated protein degradation was examined. GST-p1 and GST-p2 were susceptible to β-TRCP-mediated destruction (Figure 5D–E and Figure S5) whereas those with key CKI phosphorylation sites mutated or deleted were not (Figure 5E and Figure S5). These results strongly support the hypothesis that phosphorylation of Mdm2 by CKIδ at the p1 and p2 regions are both sufficient and required to trigger Mdm2 destruction.
Mdm2 protein abundance is controlled by both auto- and β-TRCP-dependent ubiquitination pathways during cell cycle progression
In agreement with a previous report (Gu et al., 2003), we found that Mdm2C464A was more stable than WT-Mdm2 during the cell cycle (Figure 6A). However, Mdm2C464A was still unstable in the early G1 phase. Importantly, the Δp1p2p3 mutant, which cannot be phosphorylated by CKIδ, was more stable than WT-Mdm2 in most cell cycle phases and additional C464A mutation moderately increased its abundance (Figure 6A). These results suggest that both self-ubiquitination and β-TRCP-mediated ubiquitination play important roles in mediating the timely destruction of Mdm2 during cell cycle progression. However, Δp1p2p3/C464A was still unstable in certain cell cycle phases, indicating that there might be additional unknown pathway(s) involved in Mdm2 stability control. The half-life of Mdm2 was greatly extended after inactivation of either self-ubiquitination (C464A-Mdm2) or β-TRCP-mediated (Δp1p2p3-Mdm2) ubiquitination (Figure S6A). Furthermore, depletion of endogenous β-TRCP via shRNA treatment resulted in a sharp increase in the abundance of both the wild-type and C464A mutant Mdm2 proteins (Figure 6B), indicating that both could still be degraded by the β-TRCP pathway.
Figure 6. β-TRCP controls Mdm2 protein abundance during the cell cycle progression, and promotes the ubiquitination of the Mdm2 protein.
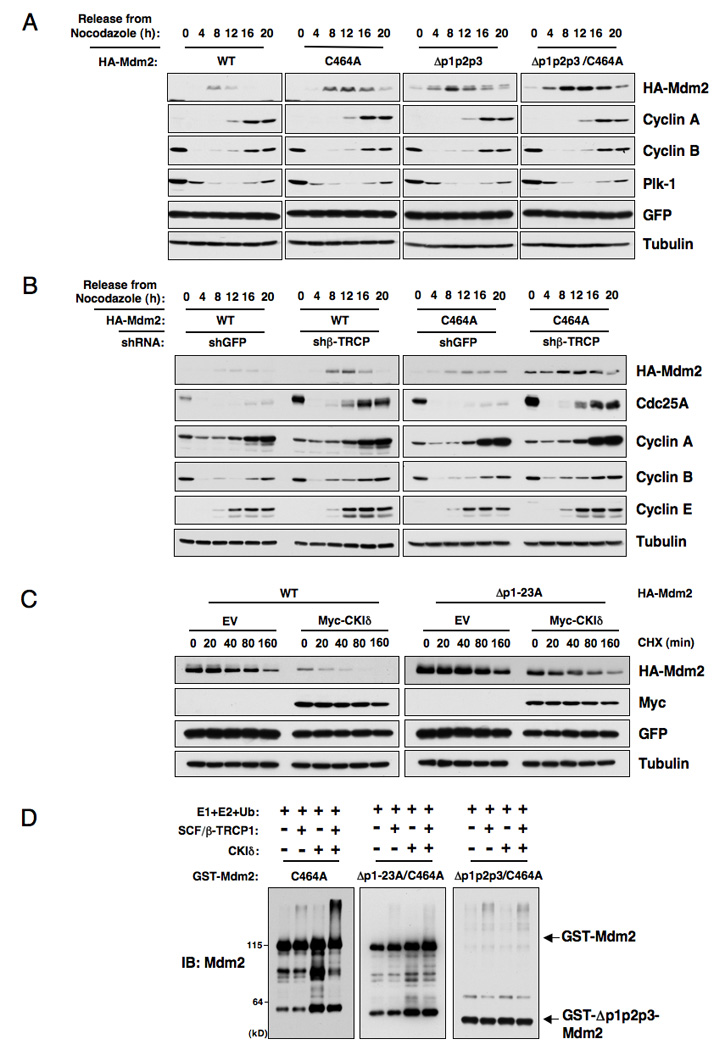
A. Immunoblot analysis of HeLa cells transfected with the indicated HA-Mdm2 plasmids, synchronized by growth in nocodazole, and then released for the indicated periods of time.
B. Immunoblot analysis of HeLa cells transfected with the indicated HA-Mdm2 plasmids together with the shβ-TRCP1+2 construct (or shGFP as a negative control), synchronized by growth in nocodazole, and then released for the indicated periods of time.
C. HeLa cells were transfected with the indicated HA-Mdm2 constructs together with the Flag-β-TRCP1 and Myc-CKIδ plasmids. Twenty hours post-transfection, cells were split into 60 mm dishes, and after another 20 hours, treated with 20 µg/ml cycloheximide. At the indicated time points, whole-cell lysates were prepared and immunoblots were probed with the indicated antibodies.
D. SCF/β-TRCP1 promotes Mdm2 ubiquitination in vitro in a CKI-phosphorylation-dependent manner. Purified CKIδ protein (from New England Biolabs, or kinase buffer as a negative control) was incubated with 5 µg of the indicated GST-Mdm2 proteins in the presence of ATP. The kinase reaction products were incubated with purified E1, E2, ubiquitin and/or SCF/β-TRCP1 as indicated in 30°C for 45 minutes. The ubiquitination reaction products were resolved by SDS-PAGE and probed with the indicated antibodies. See also Figure S6.
β-TRCP promotes Mdm2 turnover and ubiquitination
The observed progressive increase in Mdm2 stability after sequential mutation of multiple potential CKI sites (Figure 5C) might be due to inefficient β-TRCP-dependent proteolysis. In support of this idea, co-expression of CKIδ and β-TRCP1 resulted in a marked reduction in the half-life of wild-type (Figure 6C) and Mdm2C464A (Figure S6B), while it did not affect the half-life of the Δp1-23A (Figure 6C) and Δp1p2p3 Mdm2 mutants (Figure S6B). Importantly, the Δp1-23A Mdm2 mutant interacts with p53 similarly to wild-type Mdm2 (Figure S6C) and degrades p53 as efficiently as wild-type Mdm2 (Figure S6D). Collectively, these results indicate that these mutations did not alter the function of the Δp1-23A Mdm2 mutant, and that the observed stability increase in the Δp1p2p3 Mdm2 mutant might not be due to gross conformational changes.
Consistent with this result, co-expression of CKIδ and β-TRCP promoted the ubiquitination of the Mdm2C464A protein, but ubiquitination of the CKI-phosphorylation defective (Δp1p2p3) Mdm2C464A mutant was greatly reduced (Figure S6E). More importantly, purified SCF/β-TRCP E3 ligase complex could promote the ubiquitination of GST-Mdm2C464A protein in vitro in a CKI-phosphorylation-dependent manner, while its ability to ubiquitinate CKI-phosphorylation-defective Mdm2C464A mutants (Δp1p2p3 and Δp1-23A) is significantly reduced (Figure 6D).
β-TRCP promotes DNA damage dependent turnover of Mdm2
Next, we asked how manipulation of the β-TRCP-mediated Mdm2 destruction pathway affects p53 activity and its cellular functions in response to DNA damage. As shown in Figure 7A and Figure S7A–B, inactivation of β-TRCP partially blocked the destruction of Mdm2 in response to DNA damaging agents. Inactivation of CKIδ by shRNA treatment also partially blocked DNA damage-induced destruction of Mdm2 (Figure S7B). To further understand the role of β-TRCP1 in regulating the Mdm2/p53 pathway, we performed β-TRCP1 siRNA experiments in the U2OS cell line. As shown in Figure 7B, inactivation of β-TRCP led to increased Mdm2 abundance in the late G1-S phase, and this resulted in decreased p53 protein (Peschiaroli et al., 2006) and p53’s downstream targets p21 and Bax (Figure 7B). Correspondingly, this resulted in elevated resistance to DNA damage-induced apoptosis (Figure 7C). Using p53−/− HCT116 cells or p53 siRNA treated U2OS cells, we demonstrated that β-TRCP-depletion-induced resistance to apoptosis is partially through the p53 pathway (Figure S7C–G). Mdm2 might be required for this phenotype because further depletion of Mdm2 induced apoptosis in cells whose endogenous β-TRCP has been depleted (Figure 7C and Figure S7H–I). However, the lack of a full recovery in the β-TRCP/Mdm2 double siRNA treated sample to levels of apoptosis observed in the Mdm2 single siRNA treated sample indicate that other potential β-TRCP1 substrate(s) (Ding et al., 2007) might also influence the ability of β-TRCP to regulate the cellular apoptotic pathway independent of Mdm2. Importantly, depletion of endogenous β-TRCP resulted in elevated Mdm2 expression in p53−/− HCT116 cells as well, indicating that this process is p53-independent (Figure S7G). Furthermore, consistent with increased Mdm2 expression, depletion of endogenous β-TRCP in WT-HCT116 cells resulted in reduced p53 and subsequent p21 expression (Figure S7G). As a result, it led to increased BrdU incorporation (Figure S7J) and a moderate increase in xenografted tumor growth (Figure S7K).
Figure 7. Depletion of β-TRCP results in resistance to apoptosis by affecting the Mdm2/p53 pathway.
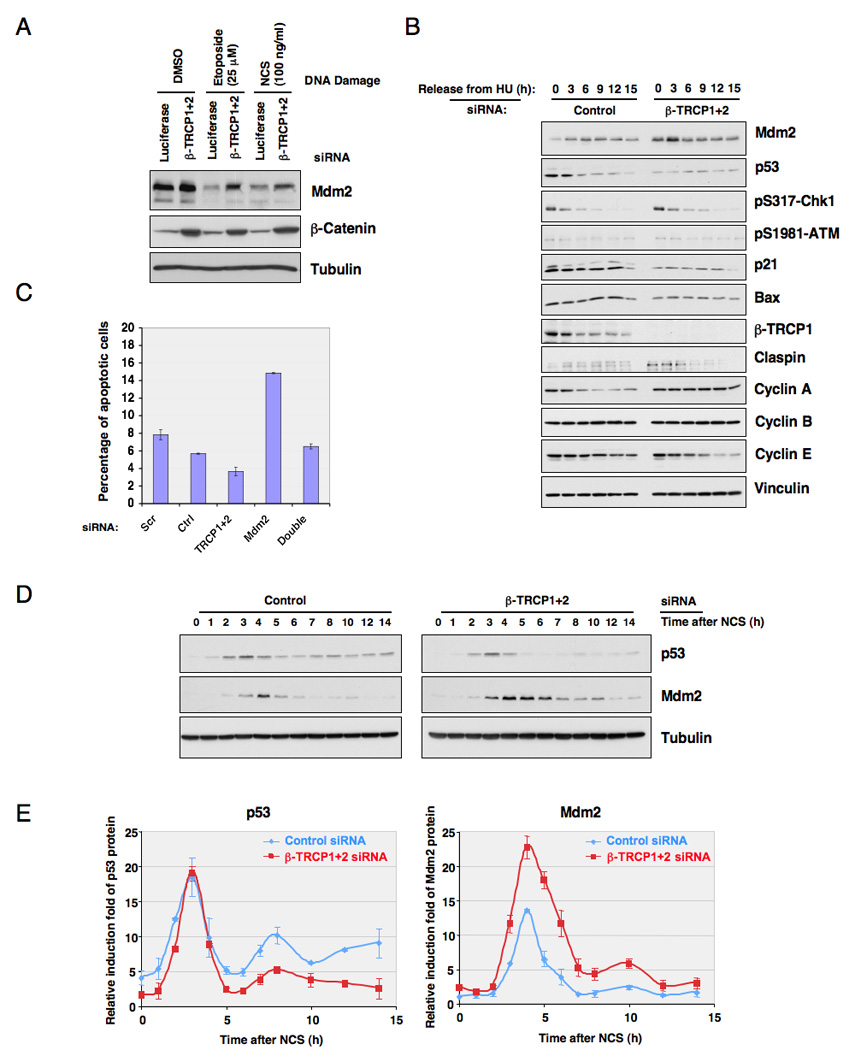
A. Immunoblot analysis of HeLa cells transfected with the indicated siRNA oligos. Twenty hours post-transfection, cells were treated with the indicated DNA damage drugs for 3 hours before harvesting.
B. Immunoblot analysis of U2OS cells transfected with the indicated siRNA oligos, after synchronization with hydroxyurea and release.
C. U2OS cells were transfected with the indicated siRNA oligos. Twenty hours post-transfection, cells were treated with hydroxyurea for 16 hours, and then treated with 1.7 µM doxorubicin for 2 hours before release to normal medium. Twelve hours later, cells were recovered for FACS analysis for detection of the apoptotic (sub-G1) cell population. The error bars represent +/− SD.
D. Immunoblot analysis of MCF7 cells transfected with the indicated siRNA oligos. Twenty hours post-transfection, cells were treated with NCS, and whole cell lysates were collected at the indicated time points.
E. Quantification of the band intensity in D. p53 and Mdm2 band intensities were normalized to tubulin, then normalized to the t=0 controls. Three independent sets of experiments were performed to generate the error bars. The error bars represent +/− SD. See also Figure S7
Previous studies have revealed that p53 levels oscillate in MCF7 cells subjected to DNA damage (Batchelor et al., 2008; Hu et al., 2007). These oscillations reflect pulsatile signaling of the ATM/Chk2 pathway and negative feedback on p53 including the Mdm2-p53 feedback loop, wherein p53 induction by DNA damage is accompanied by increased Mdm2 expression, which in turn promotes subsequent p53 turnover. In this process, negative feedback by Mdm2 contributes to the reduction in p53 levels after the initial pulse, but is not sufficient to trigger continuous p53 pulses (Batchelor et al., 2008). As expected, DNA damage resulted in oscillations of both p53 and Mdm2 expression in MCF7 cells (Figure 7D). While the first p53 pulse following DNA damage was normal, depletion of β-TRCP resulted in increased Mdm2 levels and the second pulse of p53 was substantially reduced (Figure 7D–E). Furthermore, we found that inhibition of CKIδ by inhibitors (Figure S7L) or shRNA treatment (Figure S7M) also attenuated the p53 pulse in response to continuous DNA damage. Thus, CKIδ /SCFβ-TRCP-dependent Mdm2 turnover contributes to the control of p53 dynamics in response to DNA damage which has been suggested to be important for the tumor suppressor function of p53 (Hu et al., 2007).
Discussion
The data presented here provides evidence for a molecular mechanism by which CKI-dependent phosphorylation of Mdm2 at multiple sites triggers SCFβ-TRCP-mediated Mdm2 destruction (Figure 8). It is well recognized that most F-box proteins, including β-TRCP, interact with their ubiquitin substrates only after they are properly phosphorylated (Petroski and Deshaies, 2005). Therefore, the destruction of these substrates is regulated primarily at their phosphorylation by appropriate kinases. In the case of Mdm2, specific CKI isoforms control the timing of Mdm2 turnover. In support of this idea, blocking CKI activity resulted in a marked increase in the steady-state abundance of Mdm2. It is known that CKI activity is subject to many layers of regulation (Giamas et al., 2007; Knippschild et al., 2005; Swiatek et al., 2004). Here we report the nuclear localization of CKIδ and its interaction with Mdm2 was enhanced in response to DNA damage. However, additional studies are required to fully understand the molecular mechanisms by which CKIδ activity is regulated in response to DNA damage and during cell cycle progression.
Figure 8. Proposed model for the CKI/SCFβ-TRCP pathway that controls cell cycle regulated and DNA damage-dependent Mdm2 turnover.
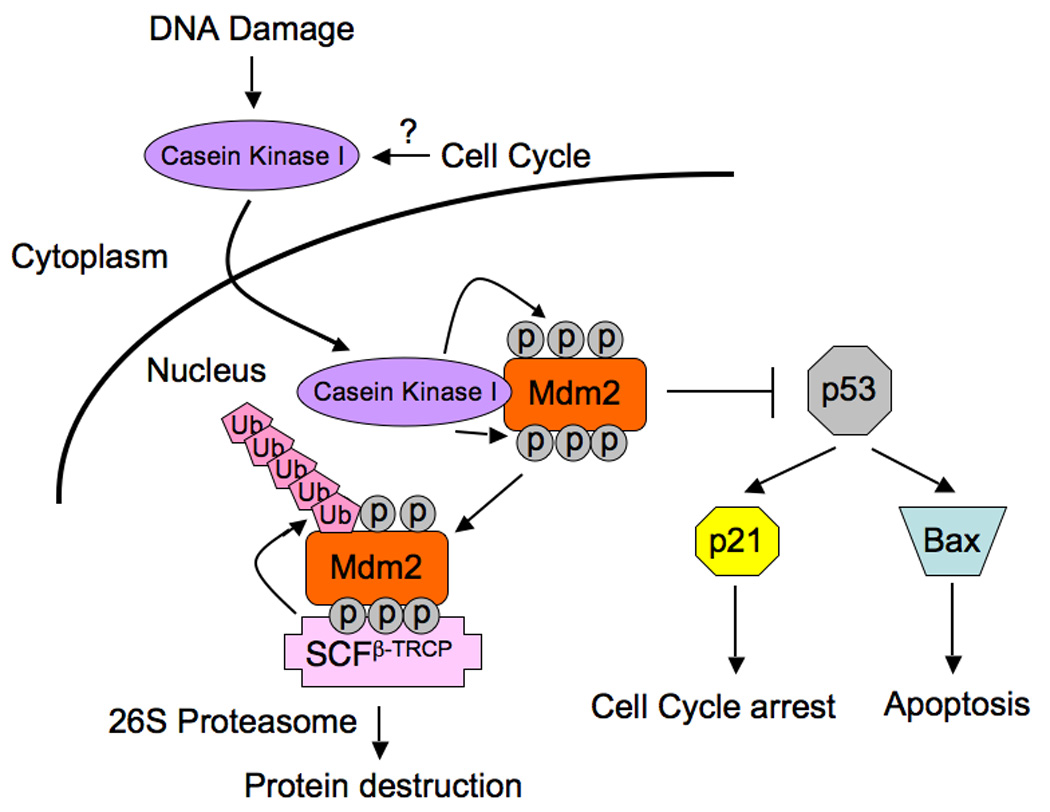
DNA damage and possibly cell cycle stimulatory signals trigger the translocation of Casein Kinase I from the cytoplasm, where it resides in non-stressed conditions, into the nucleus. This results in enhanced interaction between CKIδ and Mdm2, and subsequently multisite phosphorylation of Mdm2 by CKIδ. Phosphorylation of these multiple suboptimal degron sequences function combinatorially to facilitate Mdm2 recognition and destruction by the SCFβ-TRCP pathway, thereby setting up a switch for Mdm2 destruction in response to DNA damage and during the cell cycle. Inactivation of either β-TRCP or CKI results in accumulation of Mdm2 and decreased p53 activity, thus perturbing the proper establishment of the p53 stress response and subsequently facilitates tumor progression.
We demonstrated that the major CKI phosphorylation sites are located in the Mdm2 acidic domain (p2), while other minor CKI sites are located in the N-terminal DSG site (p1) and the C-terminal SQ cluster (p3) region. More interestingly, sequential inactivation of the potentially critical Ser/Thr sites for suboptimal degrons in Mdm2 resulted in a progressive reduction of CKI-mediated phosphorylation (Figure 4B), which correlated well with reduced interaction of Mdm2 with β-TRCP1 (Figure 4C), and reduced destruction of Mdm2. Furthermore, individual mutation of each phosphorylation site did not show considerable protection against β-TRCP1/CKIδ-mediated destruction (data not shown). This suggests that, in contrast to many known β-TRCP1 substrates that contain one optimized canonical degron sequence, such as Emi-1 or Claspin, the Mdm2 protein contains many suboptimal degron sequences that are phosphorylated by CKI. Other β-TRCP1 substrates, including Cdc25A (Kanemori et al., 2005) and Gli3 (Pan et al., 2006; Tempe et al., 2006; Wang and Li, 2006), are also known to contain multiple degron sequences. A similar mechanism has been described for SCFCdc4-dependent turnover of the Sic1 Cdk inhibitor (Nash et al., 2001). Multi-site phosphorylation likely represents a more general mechanism for creating switch-like transitions via SCF-dependent turnover pathways (Ferrell, 2001; Harper, 2002). Our studies suggested that the cumulative, multisite phosphorylation of Mdm2 by CKIδ might set up a switch for Mdm2 destruction in response to DNA damage and during the cell cycle.
It was reported previously that Mdm2 auto-ubiquitination plays an important role in DNA-damage triggered Mdm2 destruction (Stommel and Wahl, 2004). However, in agreement with a recent report (Itahana et al., 2007), we found that Mdm2C464A could be efficiently degraded in response to various types of DNA damage agents (data not shown). More importantly, co-expression of CKIδand β-TRCP1 promotes destruction of Mdm2C464A, but not Δp2- Mdm2C464A, as efficiently as the wild-type Mdm2 (Figure 5B). These findings suggest that CKI-mediated phosphorylation, rather than the ring-finger domain, might be critical for Mdm2 destruction after DNA damage. Nevertheless, Mdm2 auto-ubiquitination also plays a critical role in regulating the steady state level of Mdm2 during the cell cycle, as Mdm2C464A was more stable than wild-type Mdm2 in synchronous cell cycle release experiments (Figure 6A). Consistent with gain-of-function experiments, depletion of β-TRCP1 also stabilized Mdm2C464A (Figure 6B). Taken together, these data indicate that constitutive Mdm2 destruction during the cell cycle is tightly regulated by both auto-ubiquitination and β-TRCP1-mediated ubiquitination pathways.
Mdm2 is an oncoprotein that is frequently overexpressed in tumors, which enhances cellular transformation (Momand et al., 2000). Although amplification of the MDM2 locus has been found in some cases, the molecular mechanism leading to elevated Mdm2 expression in most tumors remains unclear. Our studies suggested that disruption of the Mdm2 destruction pathway could potentially contribute to increased Mdm2 abundance. This could be achieved by inactivation of its E3 ligase β-TRCP1 (Frescas and Pagano, 2008; Nakayama and Nakayama, 2005), inactivation of its modifying enzyme CKI, or by acquiring mutations that disrupt its phosphorylation by CKI. Similarly, both c-Myc gene amplification and mutations within the degron sequence are thought to contribute to c-Myc overexpression in cancers (Welcker et al., 2004). In this regard, it is interesting that alterations in β-TRCP1 have been reported in many types of cancers (Nakayama and Nakayama, 2005). Our data suggest that a compromised Mdm2 destruction pathway might lead to enhanced Mdm2 oncogenic activity by promoting p53 destruction and thus facilitating tumor progression. Further studies may reveal the extent to which the CKI/β-TRCP pathway and its linkage to Mdm2 is disrupted in cancer.
Experimental Procedures
siRNAs
Human siRNA oligo which can deplete both β-TRCP1 and β-TRCP2 (sense, 5’-AAGUGGAAUUUGUGGAACAUC-3’) has been validated previously (Jin et al., 2003) and was purchased from Dharmacon. Human siRNA oligos against Cullin-1, Fbw7, Skp2 and Cdh1 have been described previously (Benmaamar and Pagano, 2005; Wei et al., 2004; Wei et al., 2005). Human siRNA oligos which can deplete Mdm2 (Mdm2: sense, 5’-AAGCCATTGCTTTTGAAGTTA-3’), or p53 (sense, 5’-AAGGATGCCCAGGCTGGGAAG-3’) were purchased from Dharmacon. Luciferase GL2 siRNA oligo was purchased from Dharmacon. As described previously, siRNA oligos were transfected into subconfluent cells using Oligofectamine or Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions (Wei et al., 2004; Wei et al., 2005). Transient transfection of the control and β-TRCP siRNA oligos into MCF7 cells was performed according to the manufacturer’s instructions (Amaxa Biosystems).
Cell Culture and Cell Synchronization
Cell culture including synchronization and transfection have been described (Wei et al., 2004). To perform U2OS cell synchronization, 2 mM hydroxyurea was added into U2OS cells cultured at subconfluence (around 20–25%) to arrest cells at the late G1 phase. After 18 hours, cells were washed three times with warm PBS and then freshly made DMEM medium containing 10% FBS was added. Lentiviral shRNA virus packaging and subsequent infection was performed as described previously (Boehm et al., 2005).
Xenografted Tumor Growth Assays
Six week old female nude mice (Taconic) were used for xenograft studies. Approximately 1.5 × 106 viable tumor cells were re-suspended in 50 µl PBS solution and injected subcutaneouly into the mice (n = 6 mice for each group) and the bioluminescent imaging and normalization were performed as described previously (Zhang et al., 2009). Briefly, for bioluminescent detection and quantification of the xenografted tumor cells, mice were given a single i.p. injection of a mixture of luciferin (50 mg/kg), ketamine (150 mg/kg), and xylazine (12 mg/kg) in sterile water. Five minutes later, mice were placed in a light-tight chamber equipped with a charge-coupled device IVIS imaging camera (Xenogen). Photons were collected for a period of 1–60 s, and images were obtained by using LIVING IMAGE 2.60.1 software (Xenogen) and quantified using IGOR Pro 4.09A image analysis software (WaveMatrics). The total photons from the β-TRCP1 shRNA tumor region of interest (ROI) were divided by the total photons from GFP shRNA tumor ROI and, for each mouse, normalized based on the natural log of the ratio of the initial bioluminescent imaging (day 0) for that mouse. Results were presented as mean ± standard error of the mean (SEM). All mice experiments were approved by the Harvard University and Dana-Farber Cancer Institute Institutional Animal Care and Use Committee, and the experiments were performed according to the relevant regulatory standards.
Mdm2 Binding Assays
Binding to immobilized GST proteins was performed as described before (Wei et al., 2004). Where indicated, the GST-Mdm2 proteins were incubated with CKI in the presence of ATP for 1 hour prior to the binding assays.
In vitro Kinase Assay
Casein Kinase I was purchased from New England Biolab. The Casein Kinase I in vitro kinase assays were performed according to the manufacturer’s instructions (New England Biolab). Briefly, 5 µg of indicated GST fusion proteins were incubated with purified active Casein Kinase I in the presence of 5 µCi [γ-32P] ATP and 200 µM cold ATP in the Casein Kinase I reaction buffer for 15–30 minutes. The reaction was stopped by the addition of SDS-containing lysis buffer, resolved on SDS-PAGE, and detected by autoradiography.
In vitro Ubiquitination Assay
The in vitro ubiquitination assays were performed as described previously (Jin et al., 2005). To purify SCF/β-TRCP1 complex, 293T cells were transfected with vectors encoding GST-β-TRCP1, Myc-Cul-1, Myc-Skp1 and HA-Rbx1. The SCF/β-TRCP1 (E3) complexes were purified from the whole cell lysates using GST-agarose beads. Prior to the in vitro ubiquitination assay, indicated GST-Mdm2 proteins were incubated with purified, recombinant active CKIδin the presence of ATP (with kinase reaction buffer as a negative control) in 30°C for 30 minutes. Afterwards, the kinase reaction products were incubated with purified SCF/β-TRCP1 (E3) complexes in the presence of purified, recombinant active E1, E2 (UbcH5a and UbcH3), ATP and ubiquitin. The reactions were stopped by the addition of 2X SDS-PAGE sample buffer and the reaction products were resolved by SDS-PAGE gel and probed with the indicated antibodies.
Additional experimental procedures are included in Supplemental Experimental Procedures
Significance
The Mdm2/p53 pathway is compromised in more than 50% of human cancers. The dynamics of the p53/Mdm2 pathway is influenced by DNA damage-induced turnover of the p53 ubiquitin ligase Mdm2 but the underlying molecular mechanisms remain unclear. We demonstrated that multisite-phosphorylation by CKI triggers both cell cycle and DNA damage-dependent Mdm2 destruction via SCFβ-TRCP. Furthermore, the Mdm2C464A mutant lacking intrinsic ubiquitin ligase activity could be efficiently degraded by SCFβ-TRCP. These findings reveal a pathway that controls the stability of Mdm2 independently of its own ubiquitin ligase activity, and thereby controls p53 activity in response to genotoxic stress. It further implies that defects in β-TRCP or CKI function may contribute to the elevated Mdm2 expression frequently found in tumors.
Highlights
Multisite-phosphorylation by CKI triggers Mdm2 destruction via SCFβ-TRCP. SCFβ-TRCP controls Mdm2 stability independently of its auto-ubiquitination process. Inactivation of β-TRCP results in elevated resistance to DNA damage-induced apoptosis. Mdm2 turnover by SCFβ-TRCP controls p53 activity in response to genotoxic stress.
Supplementary Material
Acknowledgments
We thank Alex Toker, Christoph Schorl, Ralph Scully, Alan Lau and Susan Glueck for critical reading of the manuscript, James DeCaprio, Jiandong Chen, Aart G. Jochemsen and William Hahn for providing reagents, and members of the Wei, Harper, Kaelin and Xiao labs for useful discussions. We also thank Ross Tomaino and Xuemei Yang for their kind help in the mass spectrometry analysis. W.W. is a Leukemia and Lymphoma Society Special Fellow, Kimmel Scholar and V Scholar. This work was supported in part by the Massachusetts Life Science Center New Investigator award (W.W.), and by NIH grants GM089763 to W.W., GM054137 and AG011085 to J.W.H. and 5R01CA076120-03 to W.G.K., X.L.A. was supported by a Pre-doctoral fellowship from NIH, Q.Z is supported by a postdoctoral fellowship from Terri Brodeur Breast Cancer Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alsheich-Bartok O, Haupt S, Alkalay-Snir I, Saito S, Appella E, Haupt Y. PML enhances the regulation of p53 by CK1 in response to DNA damage. Oncogene. 2008;27:3653–3661. doi: 10.1038/sj.onc.1211036. [DOI] [PubMed] [Google Scholar]
- Batchelor E, Mock CS, Bhan I, Loewer A, Lahav G. Recurrent initiation: a mechanism for triggering p53 pulses in response to DNA damage. Mol Cell. 2008;30:277–289. doi: 10.1016/j.molcel.2008.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benmaamar R, Pagano M. Involvement of the SCF complex in the control of Cdh1 degradation in S-phase. Cell Cycle. 2005;4:1230–1232. doi: 10.4161/cc.4.9.2048. [DOI] [PubMed] [Google Scholar]
- Blaydes JP, Wynford-Thomas D. The proliferation of normal human fibroblasts is dependent upon negative regulation of p53 function by mdm2. Oncogene. 1998;16:3317–3322. doi: 10.1038/sj.onc.1201880. [DOI] [PubMed] [Google Scholar]
- Boehm JS, Hession MT, Bulmer SE, Hahn WC. Transformation of human and murine fibroblasts without viral oncoproteins. Mol Cell Biol. 2005;25:6464–6474. doi: 10.1128/MCB.25.15.6464-6474.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497–1501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- Cardozo T, Pagano M. The SCF ubiquitin ligase: insights into a molecular machine. Nat Rev Mol Cell Biol. 2004;5:739–751. doi: 10.1038/nrm1471. [DOI] [PubMed] [Google Scholar]
- Chan TA, Hermeking H, Lengauer C, Kinzler KW, Vogelstein B. 14-3-3Sigma is required to prevent mitotic catastrophe after DNA damage. Nature. 1999;401:616–620. doi: 10.1038/44188. [DOI] [PubMed] [Google Scholar]
- Chehab NH, Malikzay A, Appel M, Halazonetis TD. Chk2/hCds1 functions as a DNA damage checkpoint in G(1) by stabilizing p53. Genes Dev. 2000;14:278–288. [PMC free article] [PubMed] [Google Scholar]
- Daujat S, Neel H, Piette J. MDM2: life without p53. Trends Genet. 2001;17:459–464. doi: 10.1016/s0168-9525(01)02369-1. [DOI] [PubMed] [Google Scholar]
- Ding Q, He X, Hsu JM, Xia W, Chen CT, Li LY, Lee DF, Liu JC, Zhong Q, Wang X, Hung MC. Degradation of Mcl-1 by beta-TrCP mediates glycogen synthase kinase 3-induced tumor suppression and chemosensitization. Mol Cell Biol. 2007;27:4006–4017. doi: 10.1128/MCB.00620-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- Ferrell JE., Jr Six steps to destruction. Nature. 2001;414:498–499. doi: 10.1038/35107180. [DOI] [PubMed] [Google Scholar]
- Frescas D, Pagano M. Deregulated proteolysis by the F-box proteins SKP2 and beta-TrCP: tipping the scales of cancer. Nat Rev Cancer. 2008;8:438–449. doi: 10.1038/nrc2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giamas G, Hirner H, Shoshiashvili L, Grothey A, Gessert S, Kuhl M, Henne-Bruns D, Vorgias CE, Knippschild U. Phosphorylation of CK1delta: identification of Ser370 as the major phosphorylation site targeted by PKA in vitro and in vivo. Biochem J. 2007;406:389–398. doi: 10.1042/BJ20070091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu L, Ying H, Zheng H, Murray SA, Xiao ZX. The MDM2 RING finger is required for cell cycle-dependent regulation of its protein expression. FEBS Lett. 2003;544:218–222. doi: 10.1016/s0014-5793(03)00502-7. [DOI] [PubMed] [Google Scholar]
- Harms K, Nozell S, Chen X. The common and distinct target genes of the p53 family transcription factors. Cell Mol Life Sci. 2004;61:822–842. doi: 10.1007/s00018-003-3304-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper JW. A phosphorylation-driven ubiquitination switch for cell-cycle control. Trends Cell Biol. 2002;12:104–107. doi: 10.1016/s0962-8924(01)02238-3. [DOI] [PubMed] [Google Scholar]
- Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell. 2007;28:739–745. doi: 10.1016/j.molcel.2007.11.015. [DOI] [PubMed] [Google Scholar]
- Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–299. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- Hirao A, Kong YY, Matsuoka S, Wakeham A, Ruland J, Yoshida H, Liu D, Elledge SJ, Mak TW. DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science. 2000;287:1824–1827. doi: 10.1126/science.287.5459.1824. [DOI] [PubMed] [Google Scholar]
- Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- Hu W, Feng Z, Ma L, Wagner J, Rice JJ, Stolovitzky G, Levine AJ. A single nucleotide polymorphism in the MDM2 gene disrupts the oscillation of p53 and MDM2 levels in cells. Cancer Res. 2007;67:2757–2765. doi: 10.1158/0008-5472.CAN-06-2656. [DOI] [PubMed] [Google Scholar]
- Itahana K, Mao H, Jin A, Itahana Y, Clegg HV, Lindstrom MS, Bhat KP, Godfrey VL, Evan GI, Zhang Y. Targeted inactivation of Mdm2 RING finger E3 ubiquitin ligase activity in the mouse reveals mechanistic insights into p53 regulation. Cancer Cell. 2007;12:355–366. doi: 10.1016/j.ccr.2007.09.007. [DOI] [PubMed] [Google Scholar]
- Jia J, Zhang L, Zhang Q, Tong C, Wang B, Hou F, Amanai K, Jiang J. Phosphorylation by double-time/CKIepsilon and CKIalpha targets cubitus interruptus for Slimb/beta-TRCP-mediated proteolytic processing. Dev Cell. 2005;9:819–830. doi: 10.1016/j.devcel.2005.10.006. [DOI] [PubMed] [Google Scholar]
- Jin J, Ang XL, Shirogane T, Wade Harper J. Identification of substrates for F-box proteins. Methods Enzymol. 2005;399:287–309. doi: 10.1016/S0076-6879(05)99020-4. [DOI] [PubMed] [Google Scholar]
- Jin J, Shirogane T, Xu L, Nalepa G, Qin J, Elledge SJ, Harper JW. SCFbeta-TRCP links Chk1 signaling to degradation of the Cdc25A protein phosphatase. Genes Dev. 2003;17:3062–3074. doi: 10.1101/gad.1157503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanemori Y, Uto K, Sagata N. Beta-TrCP recognizes a previously undescribed nonphosphorylated destruction motif in Cdc25A and Cdc25B phosphatases. Proc Natl Acad Sci U S A. 2005;102:6279–6284. doi: 10.1073/pnas.0501873102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knippschild U, Gocht A, Wolff S, Huber N, Lohler J, Stoter M. The casein kinase 1 family: participation in multiple cellular processes in eukaryotes. Cell Signal. 2005;17:675–689. doi: 10.1016/j.cellsig.2004.12.011. [DOI] [PubMed] [Google Scholar]
- Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, Pavletich NP. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science. 1996;274:948–953. doi: 10.1126/science.274.5289.948. [DOI] [PubMed] [Google Scholar]
- Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- Levine AJ, Finlay CA, Hinds PW. P53 is a tumor suppressor gene. Cell. 2004;116:S67–S69. doi: 10.1016/s0092-8674(04)00036-4. 61 p following S69. [DOI] [PubMed] [Google Scholar]
- Mendrysa SM, McElwee MK, Michalowski J, O'Leary KA, Young KM, Perry ME. mdm2 Is critical for inhibition of p53 during lymphopoiesis and the response to ionizing irradiation. Mol Cell Biol. 2003;23:462–472. doi: 10.1128/MCB.23.2.462-473.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midgley CA, Lane DP. p53 protein stability in tumour cells is not determined by mutation but is dependent on Mdm2 binding. Oncogene. 1997;15:1179–1189. doi: 10.1038/sj.onc.1201459. [DOI] [PubMed] [Google Scholar]
- Momand J, Wu HH, Dasgupta G. MDM2--master regulator of the p53 tumor suppressor protein. Gene. 2000;242:15–29. doi: 10.1016/s0378-1119(99)00487-4. [DOI] [PubMed] [Google Scholar]
- Montes de Oca Luna R, Wagner DS, Lozano G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature. 1995;378:203–206. doi: 10.1038/378203a0. [DOI] [PubMed] [Google Scholar]
- Moshe Y, Boulaire J, Pagano M, Hershko A. Role of Polo-like kinase in the degradation of early mitotic inhibitor 1, a regulator of the anaphase promoting complex/cyclosome. Proc Natl Acad Sci U S A. 2004;101:7937–7942. doi: 10.1073/pnas.0402442101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama KI, Nakayama K. Regulation of the cell cycle by SCF-type ubiquitin ligases. Semin Cell Dev Biol. 2005;16:323–333. doi: 10.1016/j.semcdb.2005.02.010. [DOI] [PubMed] [Google Scholar]
- Nash P, Tang X, Orlicky S, Chen Q, Gertler FB, Mendenhall MD, Sicheri F, Pawson T, Tyers M. Multisite phosphorylation of a CDK inhibitor sets a threshold for the onset of DNA replication. Nature. 2001;414:514–521. doi: 10.1038/35107009. [DOI] [PubMed] [Google Scholar]
- Pan Y, Bai CB, Joyner AL, Wang B. Sonic hedgehog signaling regulates Gli2 transcriptional activity by suppressing its processing and degradation. Mol Cell Biol. 2006;26:3365–3377. doi: 10.1128/MCB.26.9.3365-3377.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peschiaroli A, Dorrello NV, Guardavaccaro D, Venere M, Halazonetis T, Sherman NE, Pagano M. SCFbetaTrCP-mediated degradation of Claspin regulates recovery from the DNA replication checkpoint response. Mol Cell. 2006;23:319–329. doi: 10.1016/j.molcel.2006.06.013. [DOI] [PubMed] [Google Scholar]
- Peters JM, McKay RM, McKay JP, Graff JM. Casein kinase I transduces Wnt signals. Nature. 1999;401:345–350. doi: 10.1038/43830. [DOI] [PubMed] [Google Scholar]
- Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol. 2005;6:9–20. doi: 10.1038/nrm1547. [DOI] [PubMed] [Google Scholar]
- Rena G, Bain J, Elliott M, Cohen P. D4476, a cell-permeant inhibitor of CK1, suppresses the site-specific phosphorylation and nuclear exclusion of FOXO1a. EMBO Rep. 2004;5:60–65. doi: 10.1038/sj.embor.7400048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stommel JM, Wahl GM. Accelerated MDM2 auto-degradation induced by DNA-damage kinases is required for p53 activation. Embo J. 2004;23:1547–1556. doi: 10.1038/sj.emboj.7600145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stommel JM, Wahl GM. A new twist in the feedback loop: stress-activated MDM2 destabilization is required for p53 activation. Cell Cycle. 2005;4:411–417. doi: 10.4161/cc.4.3.1522. [DOI] [PubMed] [Google Scholar]
- Swiatek W, Tsai IC, Klimowski L, Pepler A, Barnette J, Yost HJ, Virshup DM. Regulation of casein kinase I epsilon activity by Wnt signaling. J Biol Chem. 2004;279:13011–13017. doi: 10.1074/jbc.M304682200. [DOI] [PubMed] [Google Scholar]
- Tempe D, Casas M, Karaz S, Blanchet-Tournier MF, Concordet JP. Multisite protein kinase A and glycogen synthase kinase 3beta phosphorylation leads to Gli3 ubiquitination by SCFbetaTrCP. Mol Cell Biol. 2006;26:4316–4326. doi: 10.1128/MCB.02183-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo F, Wahl GM. Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat Rev Cancer. 2006;6:909–923. doi: 10.1038/nrc2012. [DOI] [PubMed] [Google Scholar]
- Wang B, Li Y. Evidence for the direct involvement of {beta}TrCP in Gli3 protein processing. Proc Natl Acad Sci U S A. 2006;103:33–38. doi: 10.1073/pnas.0509927103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XW, Zhan Q, Coursen JD, Khan MA, Kontny HU, Yu L, Hollander MC, O'Connor PM, Fornace AJ, Jr, Harris CC. GADD45 induction of a G2/M cell cycle checkpoint. Proc Natl Acad Sci U S A. 1999;96:3706–3711. doi: 10.1073/pnas.96.7.3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Ayad NG, Wan Y, Zhang GJ, Kirschner MW, Kaelin WG., Jr Degradation of the SCF component Skp2 in cell-cycle phase G1 by the anaphase-promoting complex. Nature. 2004;428:194–198. doi: 10.1038/nature02381. [DOI] [PubMed] [Google Scholar]
- Wei W, Jin J, Schlisio S, Harper JW, Kaelin WG., Jr The v-Jun point mutation allows c-Jun to escape GSK3-dependent recognition and destruction by the Fbw7 ubiquitin ligase. Cancer Cell. 2005;8:25–33. doi: 10.1016/j.ccr.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Welcker M, Orian A, Jin J, Grim JE, Harper JW, Eisenman RN, Clurman BE. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc Natl Acad Sci U S A. 2004;101:9085–9090. doi: 10.1073/pnas.0402770101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter M, Milne D, Dias S, Kulikov R, Knippschild U, Blattner C, Meek D. Protein kinase CK1delta phosphorylates key sites in the acidic domain of murine double-minute clone 2 protein (MDM2) that regulate p53 turnover. Biochemistry. 2004;43:16356–16364. doi: 10.1021/bi0489255. [DOI] [PubMed] [Google Scholar]
- Wu G, Xu G, Schulman BA, Jeffrey PD, Harper JW, Pavletich NP. Structure of a beta-TrCP1-Skp1-beta-catenin complex: destruction motif binding and lysine specificity of the SCF(beta-TrCP1) ubiquitin ligase. Mol Cell. 2003;11:1445–1456. doi: 10.1016/s1097-2765(03)00234-x. [DOI] [PubMed] [Google Scholar]
- Zhang L, Yu J, Park BH, Kinzler KW, Vogelstein B. Role of BAX in the apoptotic response to anticancer agents. Science. 2000;290:989–992. doi: 10.1126/science.290.5493.989. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Gu J, Li L, Liu J, Luo B, Cheung HW, Boehm JS, Ni M, Geisen C, Root DE, et al. Control of cyclin D1 and breast tumorigenesis by the EglN2 prolyl hydroxylase. Cancer Cell. 2009;16:413–424. doi: 10.1016/j.ccr.2009.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.