

Abstract
Numerous epidemiological studies documented that obesity is a risk factor for breast cancer development in postmenopausal women. Leptin, the key player in the regulation of energy balance and body weight control also acts as a growth factor on certain organs in both normal and disease state. In this study, we analyzed the role of leptin and the molecular mechanism(s) underlying its action in breast cancer cells that express both short and long isoforms of leptin receptor. Leptin increased MCF7 cell population in the S-phase of the cell cycle along with a robust increase in CYCLIN D1 expression. Also, leptin induced Stat3-phosphorylation-dependent proliferation of MCF7 cells as blocking Stat3 phosphorylation with a specific inhibitor, AG490, abolished leptin-induced proliferation. Using deletion constructs of CYCLIN D1 promoter and chromatin immunoprecipitation assay, we show that leptin induced increase in CYCLIN D1 promoter activity is mediated through binding of activated Stat3 at the Stat binding sites and changes in histone acetylation and methylation. We also show specific involvement of coactivator molecules, histone acetyltransferase SRC1, and mediator complex in leptin-mediated regulation of CYCLIN D1 promoter. Importantly, silencing of SRC1 and Med1 abolished the leptin induced increase in CYCLIN D1 expression and MCF7 cell proliferation. Intriguingly, recruitment of both SRC1 and Med1 was dependent on phosphorylated Stat3 as AG490 treatment inhibited leptin-induced recruitment of these coactivators to CYCLIN D1 promoter. Our data suggest that CYCLIN D1 may be a target gene for leptin mediated growth stimulation of breast cancer cells and molecular mechanisms involve activated Stat3-mediated recruitment of distinct coactivator complexes.
The prevalence of obesity in the developed world has reached epidemic proportions in recent years. Obesity is a known risk factor for breast cancer (1). Obese breast cancer patients exhibit a higher risk for lymph node metastasis, larger tumor burden, and mortality when compared with non-obese breast cancer patients (2). Thus, it is very important to understand the manner in which obesity adversely affects the prognosis of breast cancer (both estrogen receptor (ER)2 positive and ER negative) to devise appropriate new approaches to their treatment. At present a biological explanation for risk associated between obesity and breast cancer is not known.
Obesity is now considered as an endocrine disorder (3) mediating its functions through adipocytokines (4) acting by endocrine, paracrine, and autocrine modes (5). Considerable efforts in adipocyte biology have emphasized the important role of adipocytokines in diverse processes such as energy metabolism, inflammation, and most importantly human cancer (6). Leptin, a product of the obese (OB) gene is a neuroendocrine hormone that has attracted attention since its identification in 1995 (7, 8). It is a multifunctional peptide hormone with wide ranging biological activities including appetite regulation, bone formation, reproductive function, and angiogenesis (9). Leptin circulates as a 16-kDa protein partially bound to plasma proteins (10) and exerts its actions through its specific cell surface receptors present in a variety of tissues (10). The leptin signaling is thought to be transmitted mainly by the JAK-STAT (Janus kinase/signal transducers and activators of transcription) pathway (11, 12).
Stat proteins were identified as mediators of cytokine signaling. Stat3 proteins get phosphorylated on a single tyrosine residue (Tyr705) by receptor-associated tyrosine kinases such as JAK kinases (13) and activated Stat3 transcription factors translocate into the nucleus to activate target genes. The molecular basis of gene activation by transcription factors involves the recruitment of different coactivator complexes to create an open chromatin conformation. Histone acetyltransferase activity containing coactivators include members of p160/steroid receptor coactivator (SRC) family. These proteins are thought to function in part by associating with potent histone acetyl-transferases such as the p300/CREB-binding protein and ultimately target the HAT activity to specific promoters (14). Another type of coactivator complex is the TRAP/Mediator complex, which binds through its anchor subunit Med1 and possibly facilitates the recruitment of RNA polymerase II (15). It is shown that histone acetylation is a prerequisite for Mediator complex recruitment and function (16). Transcription activation by Stat proteins relies on interactions with the coactivator CBP/p300 and requires its histone acetyltransferase activity (HAT) activity (17). Stat proteins can recognize a conserved element in the promoter regions of target genes and activate transcription. Many Stat3 target genes are key components of the regulation of cell cycle progression from G1 to S phase (13).
In recent years, a large body of evidence has shown that disruption of cell cycle control mechanisms is a common pathway in human cancer and overexpression of CYCLIN D1 is one of the most commonly observed alterations. CYCLIN D1 promoter contains multiple regulatory elements, including binding sites for AP-1, Stats, NF-κB, E2F, Oct1, Sp1, Myc/Max, Egr, Ets, CRE, and TCF/LEF (19). Overexpression of D-type cyclins and cyclin E shortens the G1 phase and accelerates S phase entry indicating that their overexpression would result in increased proliferation in cancer (20). Indeed now there is conclusive evidence that CYCLIN D1 overexpression is associated with the ER positive, slow-growing, more differentiated phenotype of breast cancer (21). In the present study, we examined the molecular mechanisms eliciting the biological effects of leptin in breast carcinoma cells. We show that leptin modulates cell cycle and augments proliferation via activation of the JAK/Stat3 axis in breast carcinoma cells. Leptin-mediated transactivation of CYCLIN D1 promoter involves recruitment of activated Stat3 and distinct coactivator protein containing histone acetyltransferase activity and anchor subunit of mediator complex.
EXPERIMENTAL PROCEDURES
Antibodies
Antibodies for short and long form of leptin receptors OB-R (C-20), OB-R (B-3), OB-R (H-300), Med1, Med12, Med24, SRC1, and SRC2 were purchased from Santa Cruz Biotechnology, Inc. Antibodies for pStat3 (phospho-Stat3-Tyr705), anti-Stat3, and CYCLIN D1 were purchased from Cell Signaling. Antibodies for SRC3, Ac H3, Ac H4, H3-K4, and H3-K9 were purchased from Upstate Biotechnology.
Cell Culture, Reagents, and Treatments
The human breast cancer cell lines, MCF-7, T47D, ZR75, MDA-MB-231, MDA-MB-468, and HCC-1806 were grown in Dulbecco’s modified Eagle’s medium supplemented with 5% fetal bovine serum (HyClone) and 2 μM L-glutamine (Invitrogen). For treatment, cells were seeded at a density of 1 × 106/100-mm tissue culture dish. After 24 h of serum starvation, the culture media were changed to serum-free media containing treatments as indicated. Cultures were treated with human recombinant leptin (Sigma) at 100 ng/ml. In other sets of experiments, cells were treated with STAT3 inhibitor AG 490 (Calbiochem) at 100 μM, PI 3-kinase inhibitor LY294002 (Cell Signaling) at 10 μM, mitogen-activated protein kinase inhibitor PD098059 (Sigma) at 10 μM for the indicated durations.
RNA Isolation and Reverse Transcription-PCR
Total cellular RNA was extracted using the RNeasy Mini Kit (Qiagen) and quantified by UV absorption. RNA integrity was confirmed using formaldehydeagarose gel electrophoresis and ethidium bromide staining. Complementary DNA (cDNA) was synthesized using a first strand cDNA synthesis kit (Invitrogen). The synthesized cDNA was used as a template for PCR amplification. A semiquantitative PCR amplification was carried out using specific primers designed to amplify leptin receptors, OB-RB (long isoform) and OB-RT (short isoform). The primers were as follows: OB-RB sense, 5′-TCACCCAGTGATTACAA-GCT-3′; OB-RB antisense, 5′-CTGGAGAACTCTGATGT-CCG-3′; OB-RT sense, 5′-CATTTTATCCCCATTGAGAAG TA-3′; OB-RT antisense, 5′-CTGAAAATTAAGTCCTTGTG-CCCAG-3′. PCR generated 1071- and 273-bp fragments of the OB-RB and OB-RT genes, respectively. In addition, specific primers for the 18 S RNA were used as control. The primers were sense 5′-GAGGGAGCCTGAGAAACGG-3′ and antisense 5′-GTCGGGAGTGGGTAATTTGC-3′. PCR products were resolved by 1.5% agarose gel electrophoresis and visualized by ethidium bromide staining.
Immunoprecipitation of OB-RB and OB-RT
Immunoprecipitation of long and short forms of leptin receptor was performed essentially following our previously published protocol (22) using specific antibodies; OB-R (C-20) (specific for long form of leptin receptor) or OB-R (H-300) (for both long and short form of leptin receptor) followed by immunoblotting with mouse monoclonal OB-R (B-3) antibody (specific for both long and short forms of leptin receptor).
Western Blot
Whole cell lysates were prepared by scraping cells in 250 μl of ice-cold modified RIPA buffer (50 mM Tris-Cl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1% Nonidet P-40, 0.25% sodium deoxycholate, 1 mM phenylmethylsulfonyl fluoride, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 1 mM Na3VO4 and 1 mM NaF) (22). The lysate was rotated 360° for 1 h at 4 °C followed by centrifugation at 12,000 × g for 10 min at 4 °C to clear the cellular debris. Proteins were quantified using the Bradford protein assay kit (Bio-Rad). Equal amounts of proteins were resolved on SDS-polyacrylamide gels and transferred to nitro-cellulose membranes, and Western blot analyses were performed using the previously described antibodies. Immunodetection was performed using enhanced chemiluminescence (ECL system, Amersham Biosciences Inc.) according to the manufacturer’s instructions.
Cell Viability Assay
Cell viability assay was performed (22) by estimating reduction of XTT (2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxyanilide), using a commercially available kit (Roche) following the manufacturer’s instructions. MCF7 cells were plated in 96-well plates at an initial density of 4 × 103 cells/well for 24 h. After 24 h of serum starvation, the culture media were changed to serum-free media containing treatments as indicated. XTT labeling reagent was added to each culture well to attain a final concentration of 0.3 mg/ml. After 4 h exposure at 37 °C, absorbance was measured at 450 and 690 nm using a 96-well plate reader (SPECTRAmax PLUS, Molecular Devices, CA). Pilot experiments verified that the cell densities used in experiments performed were within the linear range of the XTT assay. A standard curve was prepared using cell densities from 1 × 103 to 1 × 106, and the results were calculated with respect to the number of cells.
Plasmids and Luciferase Assays
Human CYCLIN D1 promoter reporter constructs used for luciferase assays –1745-CD1-Luc, –674-CD1-Luc, –261-CD1-Luc, and –143-CD1-Luc were a kind gift from Dr. Richard Pestell (Georgetown University, Washington, D. C.) (23). Wild-type Stat3 (pCAGGS-HA-Stat3) and mutated Stat3 in pCAGGS vector, pCAGGS-HA-Stat3F, and pCAGGS-HA-Stat3D were kindly provided by Dr. Toshio Hirano (Osaka University, Japan) (24). The plasmid expressing a constitutively active form of Stat3 (Stat3C-FLAG) was a kind gift from Dr. James E. Darnell (The Rockefeller University, New York) (25). Overexpression constructs for SRC1, SRC2, and SRC3 were a kind gift from Dr. Myles Brown (Harvard Medical School, Boston, MA) (26). Overexpression construct for PBP/Med1 was a kind gift from Dr. Janardan K. Reddy (Northwestern University, Chicago, IL) (27). Constructs pSilencer 2.1-U6 hygro Med1 and pSHAG-SRC1a were kindly provided by Dr. P. Lefebvre (Faculte de Medecine Henri Warembourg, France) (28). Cells were transfected with various expression vectors and luciferase constructs either alone or in combination as indicated in the figures using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Forty-eight hours post-transfection, the cells were harvested and luciferase and Renilla activities were measured using a dual luciferase kit (Promega, Madison, WI). The relative firefly luciferase activities were calculated by normalizing transfection efficiency according to Renilla luciferase activities. The experiments were performed in triplicate, and similar results were obtained from at least three independent experiments.
Fluorescence-activated Cell Sorting (FACS) Analysis/Cellular DNA Flow Cytometry Using PI (S-phase Analysis)
FACS analysis of MCF7 cells was carried out using Cellular DNA Flow Cytometry analysis (Roche), as per the manufacturer’s instructions. Cells were grown and treated as described previously. Cells were harvested, adjusted to equal cell numbers, fixed in 70% ethanol for 30 min at −20 °C, and suspended in 500 μl of phosphate-buffered saline containing RNase A (5 Prime →3 Prime, Inc., Boulder, CO) (250 mg/liter) for 30 min at 4 °C. Fixed cells were stained with PI (100 mg/liter) before FACS analysis as described elsewhere (29). Briefly, after excitation of PI at 485 nm, red fluorescence from PI was collected through a 580-nm long pass filter and recorded to measure cellular DNA content. After counting 3.5 × 104 cells, the number of cells in each phase of cell cycles (G0/G1, S, and G2/M) was analyzed to assess the respective DNA content. The MCF7 cell cycle profile was determined using a BD Biosciences FACScan, and data were analyzed using ModFit LT 3.1.
Chromatin Immunoprecipitation (ChIP)
ChIP analyses were performed using our published procedure (21, 30) with the following modifications. Chromatin samples were sonicated on ice three times for 10 s each (i.e. until the average length of sheared genomic DNA was 1 to 1.5 kb) followed by centrifugation for 10 min. The immunoprecipitated DNA was ethanol precipitated and resuspended in 25 μl of H2O. Total input samples were resuspended in 100 μl of H2O and diluted 1:100 before PCR analysis. Initially, PCR was performed with different numbers of cycles and/or dilutions of input DNA to determine the linear range of amplification; all results shown fall within this range. Following 28–30 cycles of amplification, PCR products were run on 1% agarose gel and analyzed by ethidium bromide staining. All ChIP assays were performed at least three times with similar results.
Statistical Analysis
All experiments were independently performed three times in triplicate. Data were analyzed using paired Student’s t test. Data were considered to be statistically significant if p < 0.05. Data are expressed as mean ± S.E.
RESULTS
Leptin Modulates Cell Cycle and Augments Proliferation via Activation of JAK/Stat3 Axis in Breast Cancer Cells
A disruption of cell cycle control mechanism due to overexpression of CYCLIN D1 is a common pathway in human breast cancer. Overexpression of CYCLIN D1 correlates with poor prognosis. The relationship between CYCLIN D1 and cancer through the regulation of fat metabolism remains to be determined. Cell cycle analysis revealed that the proportion of MCF7 cells was increased in S-phase by leptin treatment as compared with cells subjected to serum-free conditions (Fig. 1A). D-type cyclins are active in the G1 phase of the cell cycle. They complex with cyclin-dependent kinases to catalyze the transition from G1 to S phase of the cell cycle (31). Leptin modulates the cell cycle of breast carcinoma cells and one of the targets for leptin action may be CYCLIN D1 as leptin treatment induced up-regulation of CYCLIN D1 protein (Fig. 1B). The effect of leptin on the transcriptional activation of CYCLIN D1 was examined using a CYCLIN D1 promoter luciferase construct, –1745-CD1-Luc (the structure is shown in Fig. 1). As shown in Fig. 1C, MCF7 cells transfected with –1745-CD1-Luc demonstrate a 5–7-fold increase in luciferase expression in response to leptin, indicating that leptin positively regulates the activity of the CYCLIN D1 promoter.
FIGURE 1. Leptin dysregulates cell cycle, increases CYCLIN D1 expression, and increases proliferation.

A, leptin increased the fraction of MCF7 cells in S-phase of the cell cycle. Cells were synchronized by serum starvation in a medium containing 0.1% serum for 16 h. Serum-starved cells were exposed to serum-free (NS), 10% serum (S), or leptin (100 and 200 ng/ml). After 24 h, the distribution of MCF7 cells in the cell cycle was determined by flow cytometry using PI-stained nuclei. The results indicate the -fold increase of breast carcinoma cells in S phase after serum starvation, leptin, or 10% serum treatment. Mean ±S.E. are the results of three independent experiments performed in triplicate. *, p < 0.01, compared with serum-starved conditions. The table shows the distribution of MCF7 cells in the various phases of cell cycle. B, leptin increases CYCLIN D1 protein expression. MCF7 cells were grown to 80% confluence, serum starved for 16 h, and incubated in the presence of 100 ng/ml leptin for different durations of time as indicated. Cell lysates were prepared and normalized for protein content; 100 μg of protein was resolved on 10% SDS-PAGE, followed by immunoblot analysis with antibodies for CYCLIN D1. C, leptin induces transactivation of CYCLIN D1 promoter in a transient transfection assay. Diagrammatic representation of the CYCLIN D1 promoter showing two GAS consensus sites located at –481 and –247. MCF7 cells were transiently transfected with the human CYCLIN D1 construct (–1745-CD1-Luc) together with the internal control Renilla luciferase reporter as described under “Experimental Procedures,” and treated with or without leptin in serum-free media for different lengths of time as indicated. Cell lysates were assayed for luciferase activity and activities were normalized against the internal control. Data represents the mean of three experiments. *, p < 0.01, leptin-treated CYCLIN D1 construct (–1745-CD1-Luc) transfected compared with leptin-treated vector control. D, breast cancer cells express both long and short forms of leptin receptors. Total RNA was extracted from MCF7, T47D, ZR75, MDA-MB-231, MDA-MB-468, and HCC1806 cells and analyzed by reverse transcription PCR using specific primers for long isoform (Ob-Rb) and short isoform (OB-RT) leptin receptor. Primer set for 18 S RNA was used as a control. E, total protein was isolated from MCF7 cells and equal amounts of proteins were subjected to immunoprecipitation using specific antibodies for long and short forms of leptin receptor. Immunoprecipitation with IgG was included as a negative control. 25 μg of COLO 320DM cell lysate (Santa Cruz Biotechnology, Inc.) was included as positive control. Immunoprecipitates were resolved by SDS-PAGE and subjected to immunoblot analysis using a mouse monoclonal antibody against both long and short forms of leptin receptor. Long (OB-RB) and short (OB-RT) forms of leptin receptors were found to be present in MCF7 cells. F, leptin increases proliferation of MCF7 cells in an anchorage dependent proliferation assay. MCF7 cells were serum starved for 16 h followed by treatment with 50–200 ng/ml of leptin for 12, 24, and 48 h. XTT assays were then performed as described under “Experimental Procedures.” Leptin treatment increased proliferation of MCF7 cells in a time- and dose-dependent manner. *, p < 0.01, for different time and dose compared with respective untreated cells. The data represents mean ± S.E. and are the results of three independent experiments performed in triplicates.
Leptin elicits its biological functions through binding to its receptors that mediate a downstream signal by activating multiple signaling pathways (10). We next examined the expression of leptin receptors in ER positive (MCF7, T47D, and ZR75), ER negative (MDA-MB-231 and MDA-MB-468), and triple negative ER, PR, Her2 negative (HCC1806) breast cancer cells. The expression of leptin receptor mRNA and protein was examined using reverse transcriptase-PCR and Western blot analysis. A predicted PCR product of OB-RB (long isoform) was obtained as 1071 bp and OB-RT (short isoform) as 273 bp by specific primers (Fig. 1D) in all breast cancer cells examined. Immunoprecipitation was performed using specific antibodies: OB-R (C-20) (recognizes only long form of leptin receptor) and OB-R (H-300) (recognizes both long and short form of leptin receptor) followed by Western blot analysis using mouse monoclonal OB-R (B-3) (recognizes both long and short form of leptin receptor). Immunoprecipitates with specific antibodies show the presence of both long and short forms of leptin receptor in MCF7 cells, whereas IgG controls do not (Fig. 1E). Our studies show that leptin dysregulates cell cycle via overexpression of CYCLIN D1 (Fig. 1, A–C). Overexpression of CYCLIN D1 might lead to unchecked cell cycle progression and possibly tumorigenic growth. Therefore, we next examined the effect of leptin on breast carcinoma cell proliferation using the XTT assay. For these experiments, MCF7 cells were serum starved for 16 h followed by treatment with various concentrations of leptin for different periods of time. Leptin stimulated the growth of MCF7 cells in a time- and dose-dependent manner (Fig. 1F). Substantial stimulation was observed at all time intervals after treatment of cells at 100 ng/ml leptin, whereas higher concentrations were not particularly more stimulatory.
To gain insight into the signaling mechanism underlying the biological effect of leptin on breast carcinoma cells, we next examined the changes in signal transduction pathways plausibly involved in mediating leptin action. Previous studies have demonstrated that leptin activates JAK, which in turn phosphorylates and activates Stats in other systems (22, 29). Total cellular proteins were extracted from cells treated with 100 ng/ml leptin for various time periods and lysates were immunoblotted with a specific phosphotyrosine Stat3 antibody. Stat3 phosphorylation was stimulated by 100 ng of leptin in a time-dependent manner resulting in an increase in Stat3 phosphorylation within 15 min of treatment (Fig. 2A). Immunoblots were reprobed with antibodies against Stat3 showing that the increase in Stat3 phosphorylation was not due to the increased Stat3 protein expression (Fig. 2A).
FIGURE 2. Leptin stimulates the phosphorylation of Stat3.

A, MCF7 cells show increased phosphorylation of Stat3 in response to leptin treatment. Breast cancer cells (MCF7) were cultured, serum starved for 16 h, and treated with leptin (100 ng/ml) for various intervals of time. Time 0 represents the absence of leptin or untreated cells (U). Cell lysates were prepared and quantified for protein content. A total of 100 μg of protein was resolved on 10% SDS-PAGE, followed by immunoblot analysis with specific antibodies against total or phosphorylated forms of Stat3. B, AG490 inhibits phosphorylation of Stat3 and leptin treatment cannot rescue it. MCF7 cells were cultured, serum starved for 16 h, and treated with leptin (100 ng/ml) (L). For combined treatment, cells were pretreated with 100 μM AG490 (AG or AG + L), 10 μM PD98059 (PD or PD + L), or 10 μM LY294002 (LY or LY + L) for 45 min followed by leptin treatment. Untreated controls are designated as U. Cell lysates were prepared and quantified for protein content. A total of 100 μg of protein was resolved on 10% SDS-PAGE, followed by immunoblot analysis with specific antibodies against total or phosphorylated forms of pStat3, Stat3. C, leptin fails to induce proliferation of MCF7 cells in the absence of JAK/Stat3 phosphorylation. Cells were treated with leptin and AG490 as described earlier and XTT assays were performed as described under “Experimental Procedures.” The data represent experiments performed three times in triplicate. *, p < 0.001, compared with untreated (U) control cells; #, p < 0.005, compared with leptin (L) treatment. D, cotransfection with both wild-type Stat3 and constitutively active Stat3 augments the leptin induced increase in CYCLIN D1 transactivation. Diagrammatic representation of the CYCLIN D1 promoter showing two GAS consensus sites located at –481 and –247. MCF7 cells were transiently cotransfected with the human CYCLIN D1 construct (–1745-CD1-Luc) and wild-type Stat3 (Stat3WT), constitutively active Stat3 (Stat3C), and dominant negative Stat3 (Stat3F and Stat3D) as indicated together with the internal control Renilla luciferase reporter as described under “Experimental Procedures,” and treated with or without leptin in serum-free media. Cell lysates were assayed for luciferase activity and activities were normalized against the internal control. Data represents the mean of three experiments.
Next, to investigate if activation of JAK/Stat3 axis is directly involved in leptin-induced proliferation of breast carcinoma cells, we studied the effect of pharmacological inhibitors of JAK/Stat (AG490), extracellular signal-regulated kinase (ERK) (PD098059), and PI 3-kinase (LY294002) on leptin-induced growth stimulation. Treatment of cells with AG490 decreased the phosphorylation of Stat3 protein significantly without affecting the expression of total Stat3 protein (Fig. 2B), whereas PD098059 and LY294002 did not affect the phosphorylation of Stat3. Importantly, simultaneous treatment with leptin and AG490 could not restore the level of phosphorylation as achieved by treatment with leptin alone. Examination of cell proliferation in these treatment conditions clearly showed that blocking Stat3 phosphorylation significantly reduced the growth stimulation of MCF7 cells by leptin (Fig. 2C), indicating that the activation of Stat3 is essential for the cell proliferative effect of leptin in breast carcinoma.
The JAK/Stat3 Pathway Is Important for Leptin-mediated Transactivation of CYCLIN D1 Promoter
Studies of cytokine signaling dependent gene expression showed that Stat proteins are signaling molecules with dual functions (32). Stat proteins not only transmit a signal from the cell surface to the nucleus but also directly participate in gene regulation (32). Our data showed that leptin uses the JAK/Stat3 pathway to augment breast cancer cell proliferation and increases CYCLIN D1 expression. Because the expression of CYCLIN D1 is elevated in Stat3-C (constitutively activated Stat3) expressing cells (25, 33), it is plausible that activated Stat3 in response to leptin modulates transcriptional regulation of CYCLIN D1 in breast cancer cells. CYCLIN D1 promoter contains binding sites for a number of transcription factors (19, 34). The consensus Stat binding motifs (TTC-NNNGAA) at –247 and –481 were of particular interest for these studies because these consensus sites have previously been shown to be sites for cytokine action mediated by Stats in other systems. Therefore, the effect of overexpression of wild-type, dominant-negative and constitutively active Stat3 on leptin signaling to the CYCLIN D1 promoter was examined. Cotransfection with wild-type Stat3 (HA-Stat3 WT) significantly increased CYCLIN D1 promoter luciferase activity, whereas cotransfection with dominant negative mutants carrying either phenylalanine substitution at Tyr705 (Stat3F) or mutations at the positions important for DNA binding (Stat3D) efficiently inhibited leptin-mediated transactivation of CYCLIN D1 promoter (Fig. 2D). We also found that leptin induced luciferase activity driven by the CYCLIN D1 promoter was augmented by cotransfection of the constitutively active form of Stat3 (Stat3C) (Fig. 2D). Collectively, the results showed the involvement of Stat3 in leptin-mediated transactivation of the CYCLIN D1 gene in breast cancer cells.
The presence of consensus Stat binding sites in the CYCLIN D1 promoter, and the role of Stat3 in the leptin response, suggests that these sites may be involved in leptin control of CYCLIN D1 expression. To assess the involvement of these sites, we used deletion constructs of CYCLIN D1 promoter. The reporter gene activities induced by Stat3WT and Stat3C were observed in –1745-CD1-Luc and –674-CD1-Luc, whereas they were severely reduced for the –261-CD1-Luc and –143-CD1-Luc constructs (Fig. 3A). These results indicate the involvement of these two consensus sites in leptin-mediated transactivation of the CYCLIN D1 gene promoter. Although our findings clearly demonstrate the role of Stat3 in leptin-mediated regulation of the CYCLIN D1 promoter, we further sought to determine that Stat3 directly participates in leptin-mediated CYCLIN D1 gene regulation using the chromatin immunoprecipitation assay. Using specific antibodies against Stat3, formaldehyde cross-linked protein-chromatin complexes were immunoprecipitated from MCF7 cells cultured with or without leptin. The resulting precipitated genomic DNA was then analyzed by PCR using primers spanning the Stat3 binding elements in the promoter region of the CYCLIN D1 gene. As shown in Fig. 3B, ChIP analysis with anti-Stat3 antibodies revealed that Stat3 is associated with the CYCLIN D1 promoter in the presence of leptin. Interestingly, we also observed a significant increase in histone acetylation at the CYCLIN D1 promoter as evidenced by ChIP analysis using anti-acetylated histone H3 and H4 antibodies (Fig. 3B) in response to leptin treatment. Genes in the repressed state have been shown to be associated with methylation of K9-dimethylated H3, whereas the active state is associated with increased methylation of K4-dimethylated H3 (35). Intriguingly, we observed increased H3-K4 methylation and decreased H3-K9 methylation in response to leptin treatment indicating active chromatin conformation in response to leptin (Fig. 3C).
FIGURE 3. The JAK/Stat3 pathway is important for leptin-mediated transactivation of CYCLIN D1 promoter.

A, deletion of GAS sites in CYCLIN D1 promoter results in decreased transactivation even in the presence of leptin and constitutively active Stat3. Diagrammatic representation of the truncated CYCLIN D1 promoter constructs showing two GAS consensus sites located at –481 and –247. MCF7 cells were transiently cotransfected with the truncated human CYCLIN D1 constructs (–1745-CD1-Luc, –674-CD1-Luc, –261-CD1-Luc, and –143-CD1-Luc) along with either wild-type Stat3 (Stat3WT) or constitutively active Stat3 (Stat3C) in the presence of leptin as indicated together with the internal control Renilla luciferase reporter as described under “Experimental Procedures.” Cell lysates were assayed for luciferase activity and activities were normalized against the internal control. Data represents the mean of three experiments. B and C, chromatin immunoprecipitation assay showed the recruitment of Stat3 and acetylation of histones on the CYCLIN D1 promoter. Soluble chromatin was prepared from MCF7 cells treated and untreated with leptin as described under “Experimental Procedures” and immunoprecipitated with 5 μg of specific antibodies against pStat3, Ac.H3, Ac.H4, H3-K9, and H3-K4 overnight at 4 °C. The immune complexes were pulled down with protein A-agarose/salmon sperm DNA beads and washed extensively as described under “Experimental Procedures” and cross-linking was reversed. The purified DNA was analyzed by PCR using primers spanning the GAS sites (–481 and –247) at the CYCLIN D1 promoter. EBAG9 was used as a control gene.
Leptin-activated Stat3 Transcription Factor Recruits Distinct Coactivator Complexes
Gene activation by DNA binding transcription factors involves the recruitment of different coactivator complexes to modify or remodel chromatin at target promoters. HAT activity containing coactivators such as p160/SRC proteins (SRC1, SRC2, and SRC3) and p300 get recruited first and rapidly induce histone acetylation, followed by the recruitment of TRAP/Mediator complex via its anchor subunit Med1 (16). Transcriptional activation by Stat proteins relies on interactions with HAT activity containing coactivators (17). To determine whether coactivators might be involved in the regulation of the CYCLIN D1 gene by leptin-activated Stat3, MCF7 cells were cotransfected with the CYCLIN D1 reporter construct containing two Stat3 consensus binding sites, Stat3 together with different coactivator proteins in the presence and absence of leptin (Fig. 4A). Stat3 up-regulated CYCLIN D1 reporter gene activity up to 10-fold in response to leptin. This activation was further increased in the presence of SRC1 and Med1 expression vectors (Fig. 4A), whereas SRC2 and SRC3 did not induce any changes in the transactivation levels. Similar results were found in the absence of transfected Stat3 plasmid indicating that leptin-activated endogenous Stat3 can also activate CYCLIN D1 promoter activity in the presence of SRC1 and Med1 (Fig. 4B). The expression levels of SRC1, SRC2, SRC3, and Med1 were comparable by Western blot analysis (Fig. 4C). Having shown that both SRC1 and Med1 coactivator proteins potentiate leptin-activated Stat3 transactivation of the CYCLIN D1 gene promoter, we next examined the recruitment of SRC proteins and Med1 to the Stat3-responsive region of the CYCLIN D1 promoter. Chromatin immunoprecipitation analysis with specific antibodies to Stat3, SRC1, SRC2, SRC3, and Med1 showed that Stat3, SRC1, and Med1 are specifically recruited to the promoter region of the CYCLIN D1 gene in response to leptin (Fig. 4D), whereas SRC2 and SRC3 show no recruitment. Mediator complex is a multisubunit coactivator complex with Med1 acting as the anchor subunit (16). To determine whether Med1 binding to CYCLIN D1 promoter in response to leptin represents the recruitment of mediator complex we performed chromatin immunoprecipitation using antibodies against Med12 and Med24. Leptin does induce the recruitment of mediator complex as we found that both Med12 and Med24 bind to the CYCLIN D1 promoter in a leptin-dependent manner (Fig. 4E). Next, to unequivocally determine the importance of recruitment of SRC1 and Med1 coactivator proteins to the CYCLIN D1 promoter during leptin-mediated growth stimulation of MCF7 cells, we used a gene silencing approach to silence both SRC1 and Med1 (Fig. 5A). We observed that silencing of SRC1 and Med1 inhibits the leptin induced increase in CYCLIN D1 transactivation (Fig. 5B). As shown in Fig. 5C, silencing of SRC1 and Med1 negatively affect leptin-induced proliferation of MCF7 cells further underscoring the importance of these specific coactivators in biological effects of leptin.
FIGURE 4. Leptin-activated Stat3 transcription factor recruits distinct coactivator complexes.

A and B, cotransfection with SRC1 and Med1 further increases the transactivation of the CYCLIN D1 reporter in the presence of both constitutively active Stat3 and leptin. MCF7 cells were transiently cotransfected with the human CYCLIN D1 construct (–1745-CD1-Luc) and expression constructs of SRC1, SRC2, SRC3, and Med1 along with (A) or without (B) Stat3 as indicated together with the internal control Renilla luciferase reporter as described under “Experimental Procedures” and treated with or without leptin in serum-free media. Cell lysates were assayed for luciferase activity and activities were normalized against the internal control. Data represents the mean of three experiments. C, MCF7 cells transiently transfected with the expression constructs of SRC1, SRC2, SRC3, and Med1 as described under “Experimental Procedures” and cell lysates were prepared and equal protein samples were subjected to Western blot analysis using specific antibodies as indicated. D and E, chromatin immunoprecipitation assay showed the recruitment of Stat3, SRC1 Med1, Med12, and Med24 on the CYCLIN D1 promoter. Soluble chromatin was prepared from MCF7 cells treated and untreated with leptin as described under “Experimental Procedures” and immunoprecipitated with 5 μg of specific antibodies against pStat3, SRC1, SRC2, SRC3, Med1, Med12, and Med24 overnight at 4 °C. The immune complexes were pulled down with protein A-agarose/salmon sperm DNA beads and washed extensively as described under “Experimental Procedures” and cross-linking was reversed. The purified DNA was analyzed by PCR using primers spanning the GAS sites (–481 and –247) at the CYCLIN D1 promoter. Primers against EBAG9 were used as control.
FIGURE 5. Silencing of SRC1 and Med1 causes a decrease in leptin-induced transactivation of CYCLIN D1 promoter and proliferation of MCF7 cells.

A, transfection of pSHAG-SRC1 and pSilencer-Med1 efficiently silences SRC1 and Med1. MCF7 cells were transfected with pSHAG, pSHAG-SRC1, pSilencer, and pSilencer-Med1, as described under “Experimental Procedures,” cell lysates were prepared and equal protein samples were subjected to Western blot analysis using specific antibodies as indicated. B, MCF7 cells were cotransfected with the human CYCLIN D1 construct (–1745-CD1-Luc) and pSHAG, pSHAG-SRC1, pSilencer, and pSilencer-Med1 as indicated in the presence of leptin together with the internal control Renilla luciferase reporter as described under “Experimental Procedures.” Cell lysates were assayed for luciferase activity and activities were normalized against the internal control. Data represents the mean of three experiments. C, MCF7 cells were transfected with pSHAG, pSHAG-SRC1, pSilencer, and pSilencer-Med1 and serum starved for 24 h followed by treatment with leptin for 24 h. XTT assays were then performed as described under “Experimental Procedures.” Leptin treatment increased proliferation of MCF7 cells in vector-transfected controls, whereas pSHAG-SRC1 and pSilencer-Med1 showed a significant decrease. *, p < 0.01, for vector controls to silencing plasmids. The data represents mean ±S.E. and are the results of three independent experiments performed in triplicates.
Next we sought to determine whether Stat3 mediates the recruitment of specific coactivator complexes to CYCLIN D1 promoter in response to leptin. To this end, MCF7 cells were treated with either leptin alone or along with the JAK/Stat phosphorylation inhibitor AG490 and subjected to chromatin immunoprecipitation analysis using specific antibodies for Stat3, SRC1, and Med1. Our results clearly show that pretreatment of MCF7 cells with AG490 abrogates leptin induced binding of Stat3 to CYCLIN D1 promoter. Interestingly, inhibition of Stat3 binding with AG490 resulted in inhibition of SRC1 and Med1 recruitment to the CYCLIN D1 promoter indicating that recruitment of both SRC1 and Med1 was dependent on phosphorylated Stat3 as AG490 treatment inhibited leptin-induced recruitment of these coactivators to the CYCLIN D1 promoter (Fig. 6A). These treatments did not affect the endogenous levels of Stat3, SRC1, and Med1 (Fig. 6B). Our data suggest that CYCLIN D1 may be a target gene for leptin-mediated growth stimulation of breast cancer cells and molecular mechanisms involve activated Stat3-mediated recruitment of distinct coactivator complexes (Fig. 7).
FIGURE 6. Stat3 mediates the recruitment of specific coactivator complexes to CYCLIN D1 promoter in response to leptin.
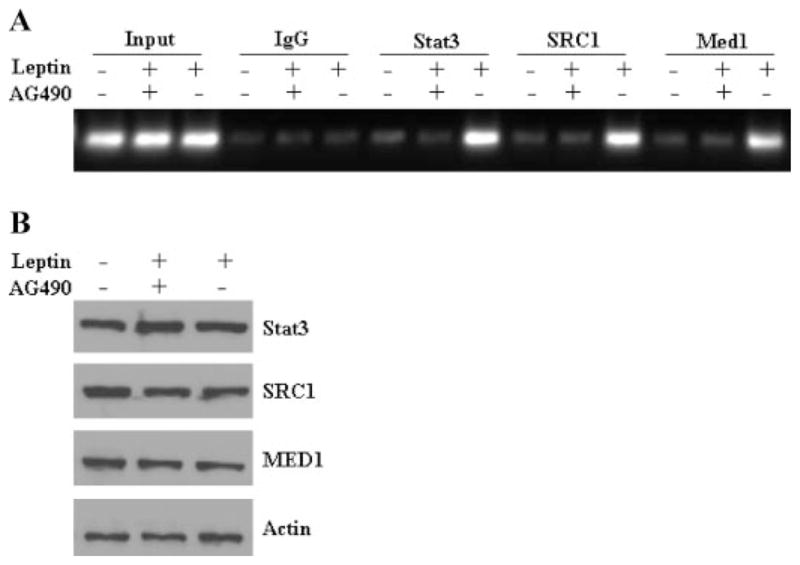
A, soluble chromatin was prepared from MCF7 cells pretreated with AG490 and treated and untreated with leptin as described under “Experimental Procedures” and immunoprecipitated with 5 μg of specific antibodies against pStat3, SRC1, and Med1 overnight at 4 °C. The immune complexes were pulled down with protein A-agarose/salmon sperm DNA beads and washed extensively as described under “Experimental Procedures,” and cross-linking was reversed. The purified DNA was analyzed by PCR using primers spanning the GAS sites (–481 and –247) at the CYCLIN D1 promoter. B, MCF7 cells were pretreated with AG490 and treated and untreated with leptin as described under “Experimental Procedures,” cell lysates were prepared and equal protein samples were subjected to immunoblot analysis using specific antibodies as indicated.
FIGURE 7. Schematic model of leptin-induced CYCLIN D1 up-regulation.
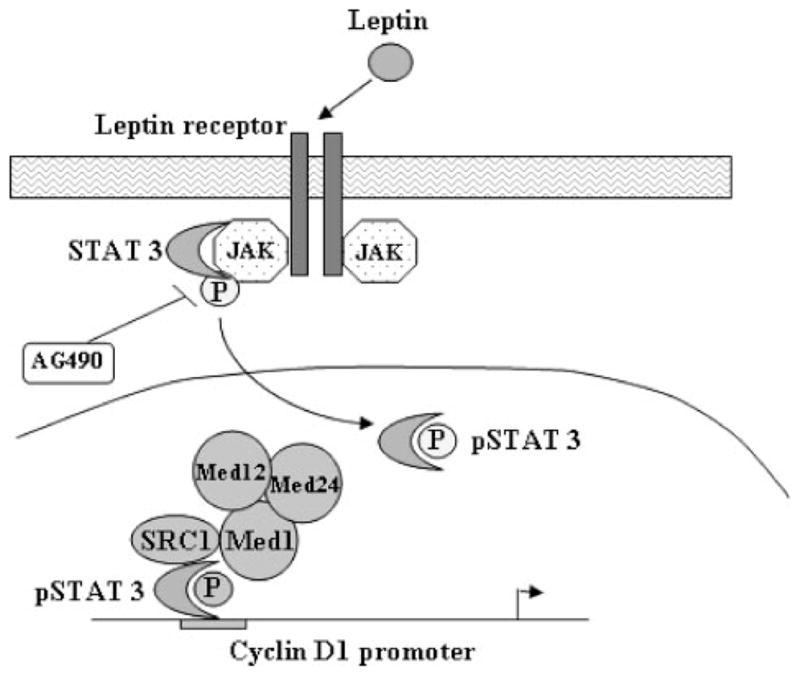
Leptin treatment leads to phosphorylation of Stat3. Phosphorylated Stat3 up-regulates the CYCLIN D1 gene via recruiting distinct coactivators, histone acetyltransferase, SRC1, and Med1 to CYCLIN D1 promoter.
DISCUSSION
A compelling body of evidence has identified obesity as a risk factor for various disease states including cancer (36). Obesity is considered as an endocrine disorder mediating its biological effects via adipocytokines secreted primarily from white adipose tissue (3–5). Studies have shown that adipose tissue can directly influence tumor growth. Mature rat adipocytes promoted the proliferation of breast carcinoma cells in a three-dimensional collagen matrix (37). Microarray analysis of MCF7 breast cancer cells treated with conditioned media from either murine fibroblasts or murine adipocytes revealed up-regulation of genes involved in invasion, proliferation, and metastasis and down-regulation of p18 and BARD1, a cell cycle checkpoint inhibitor and tumor suppressor, respectively (38). One important adipocytokine is leptin. Genetically obese leptin-deficient MMTV-TGF-α/Lepob Lepob and leptin-receptor-deficient MMTV-TGF-α/Leprdb Leprdb female mice do not develop mammary tumors, providing additional evidence that leptin and its cognate receptor may be involved in mammary tumorigenesis (39, 40). In a direct comparison, leptin receptors were not detectable in normal mammary epithelial cells by immunohistochemistry, whereas carcinoma cells showed positive staining for Ob-R in 83% of cases (41). Leptin has been characterized as a growth factor for breast cancer but the molecular mechanisms are largely unknown.
Research examining the molecular pathogenesis of cancer has presented disruption of the cell cycle in human tumors as a critical interface between hormonal signaling and tumorigenesis (21, 31). Thus, in the present study, effect of leptin on growth stimulation and alteration of cell cycle of breast carcinoma cells was studied. The dysregulation of cell cycle control in cancer is condensed into a model where disruption of two parallel pathways is required for the occurrence of cancer. These two parallel pathways include either the p16 (INK4a) pathway or the p19 (ARF) pathway. Our studies show that leptin led to overexpression of CYCLIN D1 thus inactivating one arm of the signaling pathway leading to unchecked cell cycle progression and possibly tumorigenic growth. Investigating the signaling pathways involved in leptin induced dysregulation of the cell cycle; we found that activation of the JAK/Stat pathway regulates transactivation of the CYCLIN D1 gene. In a parallel study, Leslie et al. (42) reported that CYCLIN D1 is transcriptionally regulated by Stat3 and is required for transformation. Our studies showed that leptin-activated Stat3 binds to its cognate binding sites in the CYCLIN D1 promoter leading to hyperacetylation and overexpression of the CYCLIN D1 gene. Importantly, leptin-activated Stat3 also recruited distinct coactivator proteins containing histone acetyltransferase activity, SRC1 to the promoter region of CYCLIN D1 promoting histone acetylation. Also, specific recruitment of mediator complex was observed in a leptin-dependent manner. Silencing of either SRC1 or Med1 led to inhibition of growth stimulation of breast carcinoma cells as a biological effect of inhibition of the CYCLIN D1 gene. In summary, our studies show for the first time that leptin-induced growth stimulation of breast cancer cells involves recruitment of histone acetyltransferases and mediator complex to CYCLIN D1 promoter via activation of Stat3.
CYCLIN D1 transgenic animals show perturbed mammary gland development with increased proliferation and precocious lobuloalevolar development (43). Tumors appear in 75% of mice, but after a long latency period compared with oncogenes c-myc, Ha-ras, and c-neu, which induce tumors relatively early (21, 43). The ability of CYCLIN D1 overexpression to induce mammary carcinoma and the necessity for CYCLIN D1 function for cell cycle progression raises the question of whether CYCLIN D1 is necessary for tumor development. So far, the relationship between overexpression of CYCLIN D1 and breast cancer outcome has been controversial with studies reporting both positive and negative findings (44, 45). Interpretation is further complicated by reports indicating that CYCLIN D1 overexpression may actually lead to a worse clinical outcome by conferring resistance to endocrine treatments (46, 47). It was also suggested in a clinical study that the duration of response to tamoxifen was significantly longer in ER+ patients with low CYCLIN D1 than those with high CYCLIN D1 (46). Obese breast cancer patients having high serum levels of leptin exhibit a higher risk for mammary tumor metastasis and resistance toward hormonal therapy when compared with non-obese breast cancer patients (48). Our studies show that leptin treatment augments CYCLIN D1 expression along with increased breast carcinoma cell proliferation, partly explaining the molecular mechanisms linking obesity and breast tumorigenesis.
Our studies report that inhibition of the JAK/Stat pathway significantly reduced leptin-induced growth of breast carcinoma cells. Also, expression of the CYCLIN D1 gene was positively regulated by leptin-activated Stat3, acting as a transcription factor. The main domains of Stat3 protein include the tetramerization and leucine zipper at the N terminus, the DNA binding domain, and the SH2 transactivation domain at the C terminus. The SH2 region is responsible for the binding of Stat3 to the tyrosine-phosphorylated receptors and for the dimerization necessary for DNA binding and gene expression (49). STAT3 is activated by phosphorylation at tyrosine residue 705 leading to dimerization, nuclear translocation, and recognition of Stat3-specific DNA binding elements and activation of target gene transcription (13, 32, 49). This transcriptional activation of Stat3 proteins has been shown to require the recruitment of coactivators such as CREB-binding protein (CBP/p300) (17). A class of histone acetyltransferase activity containing coactivators includes members of p160/SRC family that function in part by associating with potent histone acetyltransferases, such as the p300/CREB-binding protein, ultimately targeting the HAT activity to specific promoters. Another class of coactivators is the TRAP/Mediator complex that binds through its anchor subunit Med1, and directly interacts with basal transcription machinery possibly facilitating the recruitment of RNA polymerase II. Histone acetylation seems to be a prerequisite for Mediator complex recruitment and function (16). Our studies show that overexpression of SRC1 and Med1 enhanced transcriptional activation of CYCLIN D1 by Stat3 proteins in the presence of leptin in transient transfection experiments. Using chromatin immunoprecipitation assays, we found that Stat3, SRC1, and Med1 are recruited to the CYCLIN D1 promoter following leptin stimulation.
Stat3 activation has been associated with transformation and tumor progression suggesting that Stat3 may be an attractive target for cancer therapy. Multiple strategies have been used to block Stat activation including indirect and direct approaches such as antisense methods, ectopic expression of dominant negative mutants, inhibition of upstream kinases, and phosphotyrosyl peptides (13). Tyrphostin AG490 has been implicated in inhibition of proliferation of human acute lymphocytic leukemia (50), in human and mouse myeloma cells (51, 52). Also, introduction of antisense Stat3 oligodeoxynucleotide specifically blocked expression of Stat3 mRNA in human head and neck squamous carcinoma cell lines inhibiting proliferation (53). In a recent study, blocking Stat3 activation using a transcription factor decoy approach decreased tumor growth (54). Virtual data base screening of nearly 429,000 compounds identified several candidate small molecule inhibitors for Stat3 function. Serial functional evaluation demonstrated that a small molecule inhibitor, STA21, was found to inhibit human breast cancer cells expressing constitutively active STAT3 (55). Our study implicating leptin-mediated activation of Stat3 in CYCLIN D1 up-regulation leading to cell cycle dys-regulation and breast carcinoma cell growth stimulation suggests that therapeutic approaches directed at Stat3 activation might prove useful in breast cancer patients with elevated leptin levels.
In summary, our data present the molecular mechanisms responsible for leptin-mediated breast cancer cell proliferation and altered cell cycle regulation establishing direct association between obesity and mammary carcinogenesis. Reagents blocking leptin activity might prove useful for breast cancer patients with elevated leptin levels. Such neutralization can be achieved with soluble leptin receptors that bind free leptin in the circulation, leptin antagonists that bind to, but do not activate leptin receptors and either specific anti-leptin receptor monoclonal antibodies that bind to the receptor preventing leptin signaling or anti-leptin antibodies. Importantly, recent development of leptin muteins (18) with antagonistic properties and other proteins blocking leptin activity present new possibilities for research and ultimately therapy.
Footnotes
This work was supported by United States Army Medical Research and Material Command Grant BC-030963 and Susan G. Komen Research Foundation Grant BCTR 0503526 (to D. S.) and National Institutes of Health Grants AA 12933 and DK 062092 (to F. A. A.).
The abbreviations used are: ER, estrogen receptor; OB, obese; STAT, signal transducers and activators of transcription; JAK, Janus kinase; SRC, steroid receptor coactivator; CREB, cAMP-response element-binding protein; HAT, histone acetyltransferase; PI 3-kinase, phosphatidylinositol 3-kinase; FACS, fluorescence-activated cell sorter; ChIP, chromatin immunoprecipitation; MMTV, murine mammary tumor virus; TGF, transforming growth factor; SH2, Src homology domain 2.
References
- 1.Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. N Engl J Med. 2003;348:1625–1638. doi: 10.1056/NEJMoa021423. [DOI] [PubMed] [Google Scholar]
- 2.Berclaz G, Li S, Price KN, Coates AS, Castiglione-Gertsch M, Rudenstam CM, Holmberg SB, Lindtner J, Erien D, Collins J, Snyder R, Thurlimann B, Fey MF, Mendiola C, Werner ID, Simoncini E, Crivellari D, Gelber RD, Goldhirsch A. Ann Oncol. 2004;15:875–884. doi: 10.1093/annonc/mdh222. [DOI] [PubMed] [Google Scholar]
- 3.Dizdar O, Alyamac E. Med Hypotheses. 2004;63:790–792. doi: 10.1016/j.mehy.2004.01.046. [DOI] [PubMed] [Google Scholar]
- 4.Hausman DB, DiGirolamo M, Bartness TJ, Hausman GJ, Martin RJ. Obes Rev. 2001;2:239–254. doi: 10.1046/j.1467-789x.2001.00042.x. [DOI] [PubMed] [Google Scholar]
- 5.Rajala MW, Scherer PE. Endocrinology. 2003;144:3765–3773. doi: 10.1210/en.2003-0580. [DOI] [PubMed] [Google Scholar]
- 6.Housa D, Housova J, Vernerova Z, Haluzik M. Physiol Res. 2005;55:233–244. doi: 10.33549/physiolres.930848. [DOI] [PubMed] [Google Scholar]
- 7.Huang L, Li C. Cell Res. 2000;10:81–92. doi: 10.1038/sj.cr.7290038. [DOI] [PubMed] [Google Scholar]
- 8.Rose DP, Komninou D, Stephenson GD. Obes Rev. 2004;5:153–165. doi: 10.1111/j.1467-789X.2004.00142.x. [DOI] [PubMed] [Google Scholar]
- 9.MacDougald OA, Hwang CS, Fan H, Lane MD. Proc Natl Acad Sci U S A. 1995;92:9034–9037. doi: 10.1073/pnas.92.20.9034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ahima RS, Osei SY. Physiol Behav. 2004;81:223–241. doi: 10.1016/j.physbeh.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 11.Bahrenberg G, Behrmann I, Barthel A, Hekerman P, Heinrich PC, Joost HG, Becker W. Mol Endocrinol. 2002;16:859–872. doi: 10.1210/mend.16.4.0800. [DOI] [PubMed] [Google Scholar]
- 12.Bjorbaek C, Uotani S, da Silva B, Flier JS. J Biol Chem. 1997;272:32686–32695. doi: 10.1074/jbc.272.51.32686. [DOI] [PubMed] [Google Scholar]
- 13.Bowman T, Garcia R, Turkson J, Jove R. Oncogene. 2000;19:2474–2488. doi: 10.1038/sj.onc.1203527. [DOI] [PubMed] [Google Scholar]
- 14.Chen H, Lin RJ, Schiltz RL, Chakravarti D, Nash A, Nagy L, Privalsky ML, Nakatani Y, Evans RM. Cell. 1997;90:569–580. doi: 10.1016/s0092-8674(00)80516-4. [DOI] [PubMed] [Google Scholar]
- 15.Ito M, Yuan CX, Malik S, Gu W, Fondell JD, Yamamura S, Fu ZY, Zhang X, Qin J, Roeder RG. Mol Cell. 1999;3:361–370. doi: 10.1016/s1097-2765(00)80463-3. [DOI] [PubMed] [Google Scholar]
- 16.Sharma D, Fondell JD. Proc Natl Acad Sci U S A. 2002;99:7934–7939. doi: 10.1073/pnas.122004799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nakashima K, Yanagisawa M, Arakawa H, Kimura N, Hisatsune T, Kawabata M, Miyazono K, Taga T. Science. 1999;284:479–482. doi: 10.1126/science.284.5413.479. [DOI] [PubMed] [Google Scholar]
- 18.Gertler A. Trends Endocrinol Metab. 2006;17:372–378. doi: 10.1016/j.tem.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 19.Allan AL, Albanese C, Pestell RG, LaMarre J. J Biol Chem. 2001;276:27272–27280. doi: 10.1074/jbc.M103196200. [DOI] [PubMed] [Google Scholar]
- 20.Musgrove EA, Lee CS, Buckley MF, Sutherland RL. Proc Natl Acad Sci U S A. 1994;91:8022–8026. doi: 10.1073/pnas.91.17.8022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sutherland RL, Musgrove EA. J Mammary Gland Biol Neoplasia. 2004;9:95–104. doi: 10.1023/B:JOMG.0000023591.45568.77. [DOI] [PubMed] [Google Scholar]
- 22.Sharma D, Saxena NK, Vertino PM, Anania FA. Endocr-Relat Cancer. 2006;13:1–12. doi: 10.1677/erc.1.01169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee RJ, Albanese C, Stenger RJ, Watanabe G, Inghirami G, Haines GK, Webster M, Muller WJ, Brugge JS, Davis RJ, Pestell RG. J Biol Chem. 1999;274:7341–7350. doi: 10.1074/jbc.274.11.7341. [DOI] [PubMed] [Google Scholar]
- 24.Nakajima K, Yamanaka Y, Nakae K, Kojima H, Ichiba M, Kiuchi N, Kitaoka T, Fukada T, Hibi M, Hirano T. EMBO J. 1996;15:3651–3658. [PMC free article] [PubMed] [Google Scholar]
- 25.Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, Darnell JE. Cell. 1999;98:295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- 26.Shang Y, Brown M. Science. 2002;295:2465–2480. doi: 10.1126/science.1068537. [DOI] [PubMed] [Google Scholar]
- 27.Misra P, Owuor ED, Li W, Yu S, Qi C, Meyer K, Zhu YJ, Rao MS, Kong ANT, Reddy JK. J Biol Chem. 2002;277:48745–48754. doi: 10.1074/jbc.M208829200. [DOI] [PubMed] [Google Scholar]
- 28.Flajollet S, Lefebvre B, Rachez C, Lefebvre P. J Biol Chem. 2006;281:20338–20348. doi: 10.1074/jbc.M603023200. [DOI] [PubMed] [Google Scholar]
- 29.Saxena NK, Titus MA, Ding X, Floyd J, Srinivasan S, Sitaraman SV, Anania FA. FASEB J. 2004;18:1612–1614. doi: 10.1096/fj.04-1847fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sharma D, Saxena NK, Davidson NE, Vertino PM. Cancer Res. 2006;66:6370–6378. doi: 10.1158/0008-5472.CAN-06-0402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fu M, Wang C, Li Z, Sakamaki T, Pestell RG. Endocrinology. 2004;145:5439–5447. doi: 10.1210/en.2004-0959. [DOI] [PubMed] [Google Scholar]
- 32.Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD. Annu Rev Biochem. 1998;67:227–264. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- 33.Dechow TN, Pedranzini L, Leitch A, Leslie K, Gerald WL, Linkov I, Bromberg JF. Proc Natl Acad Sci U S A. 2004;101:10602–10607. doi: 10.1073/pnas.0404100101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Albanese C, Johnson J, Watanabe G, Eklund N, Vu D, Arnold A, Pestell RG. J Biol Chem. 1995;270:23589–23597. doi: 10.1074/jbc.270.40.23589. [DOI] [PubMed] [Google Scholar]
- 35.Sharma D, Blum J, Yang X, Beaulieu N, Macleod AR, Davidson NE. Mol Endocrinol. 2005;19:1740–1751. doi: 10.1210/me.2004-0011. [DOI] [PubMed] [Google Scholar]
- 36.Palamara KL, Mogul HR, Peterson SJ, Frishman WH. Cardiol Rev. 2006;14:238–258. doi: 10.1097/01.crd.0000233903.57946.fd. [DOI] [PubMed] [Google Scholar]
- 37.Manabe Y, Toda S, Miyazaki K, Sugihara H. J Pathol. 2003;201:221–228. doi: 10.1002/path.1430. [DOI] [PubMed] [Google Scholar]
- 38.Iyengar P, Combs TP, Shah SJ, Gouon-Evans V, Pollard JW, Albanese C, Flanagan L, Tenniswood MP, Guha C, Lisanti MP, Pestell RG, Scherer PE. Oncogene. 2003;22:6408–6423. doi: 10.1038/sj.onc.1206737. [DOI] [PubMed] [Google Scholar]
- 39.Cleary MP, Phillips FC, Getzin SC, Jacobson TL, Jacobson MK, Christensen TA, Juneja SC, Grande JP, Maihle NJ. Breast Cancer Res Treat. 2003;77:205–215. doi: 10.1023/a:1021891825399. [DOI] [PubMed] [Google Scholar]
- 40.Cleary MP, Juneja SC, Phillips FC, Hu X, Grande JP, Maihle NJ. Exp Biol Med (Maywood) 2004;229:182–193. doi: 10.1177/153537020422900207. [DOI] [PubMed] [Google Scholar]
- 41.Ishikawa M, Kitayama J, Nagawa H. Clin Cancer Res. 2004;10:4325–4331. doi: 10.1158/1078-0432.CCR-03-0749. [DOI] [PubMed] [Google Scholar]
- 42.Leslie K, Lang C, Devgan G, Azare J, Berishaj M, Gerald W, Kim YB, Paz K, Darnell JE, Albanese C, Sakamaki T, Pestell R, Bromberg J. Cancer Res. 2006;66:2544–2552. doi: 10.1158/0008-5472.CAN-05-2203. [DOI] [PubMed] [Google Scholar]
- 43.Wang TC, Cardiff RD, Zukerberg L, Lees E, Arnold A, Schmidt EV. Nature. 1994;369:669–671. doi: 10.1038/369669a0. [DOI] [PubMed] [Google Scholar]
- 44.Gillett C, Smith P, Gregory W, Richards M, Millis R, Peters G, Barnes D. Int J Cancer. 1996;69:92–99. doi: 10.1002/(SICI)1097-0215(19960422)69:2<92::AID-IJC4>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 45.McIntosh GG, Anderson JJ, Milton I, Steward M, Parr AH, Thomas MD, Henry JA, Angus B, Lennard TW, Home CH. Oncogene. 1995;11:885–891. [PubMed] [Google Scholar]
- 46.Kenny FS, Hui R, Musgrove EA, Gee JM, Blamey RW, Nicholson RI, Sutherland RL, Robertson JF. Clin Cancer Res. 1999;5:2069–2076. [PubMed] [Google Scholar]
- 47.Han S, Park K, Bae BN, Kim KH, Kim HJ, Kim YD, Kim HY. Oncol Rep. 2003;10:141–144. [PubMed] [Google Scholar]
- 48.Sulkowska M, Golaszewska J, Wincewicz A, Koda M, Baltaziak M, Sulkowski S. Pathol Oncol Res. 2006;12:69–72. doi: 10.1007/BF02893446. [DOI] [PubMed] [Google Scholar]
- 49.Darnell JE. Science. 1997;277:1630–1635. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- 50.Meydan N, Grunberger T, Dadi H, Shahar M, Arpaia E, Lapidot Z, Leeder JS, Freedman M, Cohen A, Gazit A, Levitzki A, Roifman CM. Nature. 1996;379:645–648. doi: 10.1038/379645a0. [DOI] [PubMed] [Google Scholar]
- 51.Burdelya L, Catlett-Falcone R, Levitzki A, Cheng F, Mora LB, Sotomayor E, Coppola D, Sun J, Sebti S, Dalton WS, Jove R, Yu H. Mol Cancer Ther. 2002;1:893–899. [PubMed] [Google Scholar]
- 52.Catlett-Falcone R, Landowski TH, Oshiro MM, Turkson J, Levitzki A, Savino R, Ciliberto G, Moscinski L, Fernandez-Luna JL, Nunez G, Dalton WS, Jove R. Immunity. 1999;10:105–115. doi: 10.1016/s1074-7613(00)80011-4. [DOI] [PubMed] [Google Scholar]
- 53.Grandis JR, Drenning SD, Chakraborty A, Zhou MY, Zeng Q, Pitt AS, Tweardy DJ. J Clin Investig. 1998;102:1385–1392. doi: 10.1172/JCI3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xi S, Gooding WE, Grandis JR. Oncogene. 2005;24:970–979. doi: 10.1038/sj.onc.1208316. [DOI] [PubMed] [Google Scholar]
- 55.Song H, Wang R, Wang S, Lin J. Proc Natl Acad Sci U S A. 2005;102:4700–4705. doi: 10.1073/pnas.0409894102. [DOI] [PMC free article] [PubMed] [Google Scholar]