

Abstract
Genetically modified mice have been used for colon cancer research but findings from these models are confounded by expression of cancer in multiple organs. We sought to create a transgenic mouse with Cre recombinase (Cre) expression limited to the epithelial cells of the large intestine and use this model to study colon cancer driven by adenomatosis polyposis coli (APC) gene inactivation. A promoter/enhancer from the mouse carbonic anhydrase I gene was used to generate a Cre expressing transgenic mouse (CAC). After characterizing transgene expression and distribution, CAC mice were crossed to APC580S mice to generate mice with APC inactivation at one (CAC; APC580S/+) or both alleles (CAC; APC580S/580S). Transgene expression was limited to the epithelial cells of the cecum and colon, extended from the crypt base to the luminal surface, and was expressed in approximately 15% of the crypts. No abnormal gross phenotype was seen in 3 or 6 wk CAC; APC580S/+ mice but CAC; APC580S/580S mice had significant mucosal hyperplasia in the colon at 3 wk that developed into tumors by 6 wk. By 10 wk, 20% of CAC; APC580S/+ mice developed adenomatous lesions in the distal colon (3.0±0.4 mm, 1.1 per mouse). Dextran sulfate sodium treatment increased the incidence and number of tumors and this occurred predominantly in distal colon. Our new model has improved features for colon cancer research i.e. transgene expression is limited to the epithelium of the large bowel with normal cells found next to genetically modified cells.
Keywords: cancer, intestine, APC, Cre recombinase
Introduction
Sporadic colorectal cancer is the second leading cause of cancer-related death in the Western world (1). As a result, many studies have been conducted to understand the pathophysiology of the disease, to evaluate approaches for colorectal cancer prevention, and to test possible treatments.
Animal models for human colon cancer can be useful for studying the mechanism of colon cancer development and for testing cancer prevention and treatment approaches. However, several recent reviews clearly show that the available models are flawed and the cancer that develops in these models is often significantly different from human colon cancer in terms of latency, intestinal location, or molecular signature (2, 3). For example, chemicals that induce colon cancer in animal models affect multiple organs while the incidence of tumors in the colon depends on the carcinogen used, the dosage, route, duration, and frequency of administration (4). In addition, the molecular mutations causing colon cancer in chemically-induced models are often unknown and may not directly relate to the mutations that drive human colorectal cancer. In contrast, the molecular mutations responsible for cancer initiation can be precisely controlled in genetically modified mice, but current transgenic and knockout models for colon cancer research express cancer in other tissues at a higher rate than within the large intestine (e.g. tumors develop predominantly in the small intestine of Apcmin mice (5) and Apcmin mice also develop mammary tumors (6), Msh2 deletion models develop lymphoma and skin tumors (7, 8), and Mlh1 deletion models develop tumors at multiple locations (9)). Another flaw of these models is that the genetic mutations are expressed throughout the colonic epithelium and so they better model familial cancers syndromes like Familial Adenomatous Polyposis (FAP) or Hereditary NonPolyposis Colorectal Cancer (HNPCC) (10) than sporadic colon cancer where normal tissue resides next to diseased tissue. The flaws observed in genetically modified mouse models of colon cancer are an undesired consequence of germline mutation as well as our inability to target gene expression to the large intestine. They confound our ability to evaluate the development of the colon tumor and to test the efficacy of interventions that limit colon cancer.
We believe that several characteristics are important for the next generation of genetically modified animal models of colon cancer. First, the transgene expression should be limited to the colon. Second, expression should be absent or severely limited during embryonic development. These two criteria will avoid unwanted phenotypes associated with extra-colonic expression and fetal lethality. Finally, transgene expression should be limited to the epithelial cell from which most colon tumors originate. In this report we describe our efforts to create a new transgenic mouse model that has improved features for colon cancer research.
Materials and Methods
Bioinformatic analysis of the carbonic anhydrase I (CA1) gene promoter
Genomic DNA sequences from the 30 kb mouse and 36.4 kb human CA1 promoters (www.ensembl.org; human = ENSG00000133742; mouse = ENSMUSG00000027556) were compared using the dottup subroutine in EMBOSS(11) with a 9 bp comparison window. The human CA1 promoter was annotated using data on DNAse I hypersensitive sites (HSS) and transcription factor binding sites previously reported by others (12, 13). Conserved regions were used to generate the transgene construct.
Transgene construction
The 10.6 kb colon-specific promoter and shared 2.5 kb erythroid/colon enhancer fragment of the mouse carbonic anhydrase I (mCA1) gene were PCR amplified and assembled to generate the pUC13kb-mCA-cre plasmid. amplified as 5 fragments by PCR and the sequence of each fragment was confirmed by DNA sequencing. The mCA1 promoter/enhancer fragments were subcloned sequentially into pUC12.4kb-villin (a gift from Dr. Debra Gumucio, University of Michigan Medical School, Ann Arbor, Michigan) after removing the villin promoter sequence using XhoI and Sal I to generate the pUC13kb-mCA plasmid. The Cre recombinase gene from pPGK-cre-bpA (a gift from Drs. Klaus Rajewsky, Harvard Medical School, Boston, MA) was subcloned into the Xho I and Sac II site of pUC13kb-mCA to generate the pUC13kb-mCA-cre plasmid.
Generation and identification of transgenic mice
The carbonic anhydrase 1 promoter/enhancer-cre recombinase transgene (CAC) was isolated from pUC13kb-mCA-cre by Pme I restriction endonuclease digestion and transgenic mice were created in the Purdue University Transgenic Mouse Core Facility using standard protocols. PCR analysis of tail DNA was used to detect the CAC transgene (forward: 5’ACCAGCCAGCTATCAACTCG 3’, reverse: 5’TTACATTGGTCCAGCCACC 3’, 199 bp). Transgenic founders were crossed with C57BL6/J mice to perpetuate the transgenic lines. See the Supplemental Information for a detailed description of the genotyping procedure.
Animals
All animal experiments were reviewed and approved by the Purdue Animal Care and Use Committee. Gt(ROSA)26Sor mice (ROSA26R) were from the Jackson Laboratory (Bar Harbor, Maine). APC580S mice carrying an APC gene allele with LoxP sites flanking exon14 (APC580S/580S) were from the Mouse Model of Human Cancers Consortium (NCI-Frederick, MD). PCR genotyping for APC alleles was conducted using conditions and primers described previously (14) to yield the PCR products: 320 bp WT allele, 430 bp floxed allele; 500 bp APC deleted allele. All mice were housed individually and exposed to a 12 h light/12 h dark cycle. Mice were given a standard chow diet and water ad libitum.
Tissue distribution and expression level of the CAC transgene mRNA
Messenger RNA was extracted from kidney, stomach, bone marrow, prostate, lung, thymus, liver, heart, spleen, pancreas and the scraped mucosa of duodenum, jejunum, ileum, cecum, proximal colon, and distal colon using TriReagent (Molecular Research Center Inc., Cincinnati, OH), treated with DNase I to remove contaminating DNA, and reverse-transcribed into cDNA as previously described (15). CAC primers used in the genotyping reaction were used to detect transgene mRNA and GAPDH mRNA was used as a control transcript as previously reported (15).
Assessment of β-galactosidase activity in CAC x ROSA26R mice
CAC5 line were crossed to ROSA26R mice to generate CAC;R26R mice. The gastrointestinal tract from CAC;R26R and wild type (WT) mice were removed, opened longitudinally, and washed with PBS at 4°C. Embryos (at 8.5, 10.5 and 12.5 days post coitum (dpc)) or gastrointestinal tract (at 14.5 dpc) were fixed on ice for 10 min in 4% paraformaldehyde containing 1.25 mM EGTA and 2 mM MgCl2 in PBS. Both whole mount embryos and intestines were incubated with 1 mg/ml 5-bromo-4-chloro-3-indoyl-β-D-galactopyranoside (X-gal, Invitrogen, Carlshad, CA) in PBS containing N,N-dimethylformamide, 5 mM K3Fe(CN)6, 5 mM K4Fe(CN)6-3H2O, and 2 mM MgCl2 for 2–10 h at 37 °C, depending on tissue size and embryo stage. Stained tissues were post-fixed for 10 min at room temperature with 0.2% glutaraldehyde (1.25 mM EGTA, 2 mM MgCl2 in PBS) followed by 4% paraformaldehyde for 4 h at 4 °C.
The cellular pattern of Cre expression from the CAC transgene was examined in intestinal tissue from CAC;R26R and WT mice that was fixed on ice for 10 min in PBS with 4% paraformaldehyde, 1.25 mM EGTA and 2 mM MgCl2, embedded in OCT (TissueTek, Torrance, CA), and snap frozen in liquid nitrogen. Tissue blocks were cut on a Microm HM 500M cryostat (GMI Inc., Ramsey, Minnesota), dried for 5 min at 25 °C, and stained with X-gal and counterstained with eosin as previously described(16). Slides were analyzed, and digital images were captured (Fuji Finepix S2 camera, Olympus BX-50 microscope, Olympus, Nashua, NH).
Experimental design for studies of mice with tissue-specific Apc allele deletion
For experiment I, CAC5 mice were crossed to APC580S/580S mice to generate mice with large-bowel-specific deletion of a single APC gene allele (CAC;APC580S/+). 50 CAC;APC580S/+ and 30 wild type-mice (CAC−/−;APC580S/+, WT) were studied at 10 wk (n=10 per genotype), 15 wk (10 CAC;APC580S/+and 6 WT mice), 20 wk (10 CAC;APC580S/+and 6 WT mice) or 30 wk (20 CAC;APC580S/+and 8 WT mice).
Inflammatory bowel disease (IBD) increases the risk for developing colon cancer and Dextran Sulfate Sodium (DSS) treatment can induce colonic mucosal inflammation leading to promotion of colorectal carcinogenesis in Apcmin mice (17). To investigate whether acute inflammation induced by DSS enhances large intestinal carcinogenesis, CAC;APC580S/+and WT mice were given 2% (w/v) DSS in drinking water (MW = 36,000–50,000 d, MP Biomedicals, LLC, Solon, OH) for either 5 days (8 CAC;APC580S/+) or 7 days (40 CAC;APC580S/+and 20 WT) starting at 10 wks of age. Significant mortality resulted within 4 days of terminating both the 5d (20%) and 7d (55%) DSS treatment regimes; all remaining mice survived until the study termination points. Mice were examined at 15 wk (7 d DSS; 6 CAC;APC580S/+and 4 WT), 17 wk (7 d DSS; 12 CAC;APC580S/+and 5 WT) or 20 wk of age (5 d DSS; 8 CAC;APC580S/+).
For experiment II, Apc580S/580S mice were crossed with CAC;APC580S/+ to generate mice lacking none (CAC−/−;APC580S/+, control), one (CAC;APC580S/+), or both (CAC;APC580S/580S) APC gene alleles. Mice were sacrificed at 3 wk and 6 wk of age (n=6 per genotype per time point).
Histological and immunohistological analysis
Mice were killed and the large bowel was removed, split lengthwise, and a digital image was collected. Visible tumors were dissected and the remaining portion of the large intestine was prepared using the Swiss-roll technique. Both tumors and Swiss rolls were fixed in 10% buffered formalin phosphate (Fisher Scientific, Fair lawn, NJ), processed, and embedded in paraffin using standard procedures. 5 μm sections were deparafinized in xylene followed by alcohol rehydration and then stained using hematoxylin and eosin Y (Surgipath®, Richmond, IL) or used for immunohistochemistry.
Immunohistochemistry
For antigen retrieval, slides were submerged in 0.01 M sodium citrate (pH 6.0) and heated for 20–30 minutes in a steamer. Immunohistochemistry was performed on a Dako Autostainer (Dakocytomation, Carpinteria, CA). Slides were incubated with 0.03% hydrogen peroxide (5 min) and serum-free protein block (10 min) (Dakocytomation, Carpinteria, CA). After 60 min incubation with primary antibodies (1: 200 rabbit monoclonal anti-β-catenin; Abcam, Cambridge, MA and 1:50 rat-anti-mouse Ki-67; Dakocytomation, Carpinteria, CA) antigen binding was detected by an anti-rabbit horseradish peroxidase (HRP) labelled polymer (Envision+ System-HRP, Dakocytomation, Carpinteria, CA) or biotinylated anti-rat immunoglobulin and streptavidin-HRP (Dakocytomation, Carpinteria, CA) with 3,3′-diaminobenzidine (DAB) as the chromogen. Slides were counterstained with Harris hematoxylin (EK Industries, Joliet, IL) and digital images were collected (SPOT Insight™ Firewire Color Mosaic digital camera (Diagnostic Instruments, Sterling Heights, MI); Nikon Eclipse 4000 light microscope (Nikon Instruments, Melville, NY)).
Image analysis
Mucosal thickening and lesion area in the large intestine of 3 wk old Apc−/−, Apc+/−, and Apc+/+ mice was assessed using Image J (http://rsbweb.nih.gov/ij/). Mucosal thickening was measured in each mouse from luminal surface to lamina muscularis in two representative regions from distal colon or proximal colon within the Swiss-rolled large intestine.
Results
mCA promoter/enhancer limits cre recombinase gene expression
The human carbonic anhydrase I (hCA1) gene has three alternate exon 1s in its promoter and the distance between the most proximal (E1a) and distal (E1c) versions span 36.4 kb (12). DNase I hypersensitivity assays have identified four hypersensitive sites (HSS) that are exclusive to erythroid tissue (−1e, −2e, +3e and +4e), two distal sites (+5c and +6c) that are exclusive to colon, and two HSS present in tissues (+1ec and +2ec) (Figure 1A, a) (12, 18). Others have reported that the colon uses E1c while erythroid tissue uses E1a (12). Regions conserved between the mouse and human CA1 promoters included all of the human HSS except +4e. Using this information, we developed the mCA1 promoter/enhancer-cre recombinase (CAC) transgene construct (Figure 1A, b). Mice expressing the CAC transgene were generated and two lines were selected for further characterization: CAC5 (40 copies) and CAC6 (20 copies). PCR analysis showed that the transgene mRNA level was 10-fold higher in CAC5 compared to CAC6 mice (Figure 1B, a). In both lines, the transgene mRNA was detectable within the intestine only in the large intestine from cecum to distal colon. There was a low level of transgene mRNA expression in the liver but no other tissue we examined expressed the transgene (Figure 1B, b).
Figure 1. Cre expression mediated by the mCA1 promoter/enhancer is restricted within the intestine to the large bowel.

(A) (a) Schematic of the mouse carbonic anhydrase I (mCA) locus with regions of DNA conserved between mouse and human (grey bars) and hypersensitive sites (numbers under the gene) identified. E= exon; e = erthyroid cell; c = colon; ec = both erythroid cell and colon. (b) Schematic of mCA Cre (CAC) transgene construct.. BspEI (B), NsiI (N), Sw (SwaI), X (XhoI), Sa (SacII) and S (SaII) restriction enzyme sites are indicated. (B) (a) Cre mRNA expression from the transgene measured in the proximal colon (CoP) of two transgenic lines (CAC5 and CAC6). (b) Survey of Cre mRNA expression in tissues from the CAC5 line. (C) (a) Whole mount staining for β-galactosidase (β-gal) in the intestine of 6 wk-old wild-type (WT) and CAC;R26R mice. Bar = 1 cm. (b) β-gal expression in colon (arrow) of E14.5 WT and CAC;R26R embryos, Bar = 1 cm. (D) (a) High magnification of β-gal staining in whole mount of proximal colon from CAC;R26R mice, Bar = 2 mm. β-gal expression in cryostat section of proximal colon: (b) Bar = 500 μm. (c) Bar = 100 μm.
In whole-mount preparations from 6 wk old CAC;R26R mice, β-galactosidase activity was strongly expressed in large intestine from cecum to rectum (Figure 1C,a) while there was also faint staining for β-galactosidase activity in the liver (data not shown). No other gross expression was observed in any other tissue and no expression was seen in the intestine of wild-type mice (Figure 1C, a). Expression was not seen in 8.5, 10.5, or 12.5 dpc CAC;R26R embryos (data not shown) but by 14.5 dpc β-galactosidase was detected only in the large intestine (Figure 1C, b). In the cecum and colon from 6-week-old CAC;R26R mice, expression of β-galactosidase was mosaic (Figure 1D, a). Histologic examination confirmed the presence of strong β-galactosidase activity in the epithelial cells of the lower bowel (Figure 1D, b) and faint activity in the liver (Supplemental Figure 2S). In the lower bowel approximately 15% of the epithelial cells in colon tissue stained strongly for β-galactosidase activity (Figure 1D, b); staining was observed in individual crypts and reached from the crypt base to the luminal surface (Figure 1D, c).
CAC;APC580S/+ and CAC;APC580S/580S mice develop colon tumors
In CAC;APC580S/+ mice, cre recombinase deleted exon 14 in the APC gene in the large intestine (Figure 2). Visible, gross tumors were apparent in CAC;APC580S/+ mice by 10 wks of age and the incidence did not increase with time up to 30 wks. Microscopic examination revealed two morphologic adenomatous variants of the lesions (i.e. microadenomas and adenomas, Figure 3C) with an over-representation of both lesions in the distal colon (Figure 3A, B and data not shown). Ten of the 50 CAC;APC580S/+ mice studied (20%) had grossly identifiable adenomatous polyps characterized microscopically as sessile or pedunculated (Figure 3C, 3D, Table 1). In addition, 7 microadenomas in the distal colon and 3 microadenomas in the proximal colon were identified upon microscopic examination of a single cross section of each Swiss roll (Figure 3C, a, Table 1). Lesions consistent with carcinoma were not observed and no gross abnormalities were present in other organs examined. Six of the 11 CAC;APC580S/+ mice with tumors had bloody stool or rectal prolapse and mice with tumors were mildly anemic (hematocrit = 37% for Apc+/− vs. 48% for wild-type mice). There were no differences in body weight between CAC;APC580S/+ mice and control mice even when CAC;APC580S/+ mice had tumors.
Figure 2. Deletion of the floxed APC allele in 6 wk-old CAC;APC580S/+ mice.
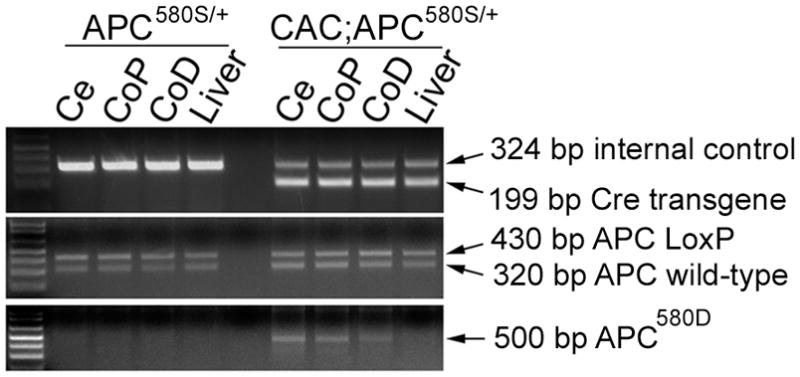
Genomic DNA from cecum (Ce), CoP (proximal colon), CoD (distal colon) and liver was examined for the presence of the Cre transgene and an internal control gene, as well as the wild-type APC allele, the floxed APC allele (APC LoxP), and the Cre-deleted APC allele (APC580D). Because genotyping was conducted on whole tissue containing epithelial and other cell types containing the Cre transgene, the floxed Apc allele is still visible even as the Cre-recombined Apc allele appears.
Figure 3. Tumorigenesis in CAC;APC580S/+ mice.

(A) Representative images of colorectal tumors (arrow) in CAC;APC580S/+ mice without DSS (a) or after 2% DSS for 7d (b) or 5d (c). Bar =1 cm. (B) Distribution and size of tumors in the lower bowel of CAC;APC580S/+ mice without DSS (a) or after 2% DSS for 5d or 7d (b). (C) Examples of H&E stained sections representing different colonic lesions seen in CAC;APC580S/+ mice. (a) microadenoma (arrow), Bar = 100 μm, (b, c) adenomas, Bar = 1 mm. N, normal-looking tissue. A, adenomatous tissue. (D) Examples of H&E stained sections representing simple adenomas from CAC;APC580S/+ mice without DSS treatment (a) and complex, multiple adenomas from DSS treated CAC;APC580S/+ mice. Bar = 1 mm.
Table 1.
Tumor Characteristics in CAC;APC580S/+ mice
| Group | # mice | # with gross tumors | Incidence (%) | # gross tumors | Tumor size (mm) | Tumor location# |
|---|---|---|---|---|---|---|
| No DSS | 50 | 10 | 20 | 12 | 3.0±0.4 | 2.4±0.5 |
| 2% DSS 7d | 18 | 12 | 66.7 | 70 | 3.2±0.2 | 3.1±0.3 |
| 2% DSS 5d | 8 | 4 | 50 | 12 | 2.3±0.4 | 2.0±0.3 |
| Micro adenomas* | # mice | # with micro lesions | Incidence (%) | # micro lesions | ||
| No DSS | 50 | 5 | 10 | 10 | ||
| 2% DSS 7d | 18 | 11 | 61.1 | 41 | ||
| 2% DSS 5d | 8 | 3 | 37.5 | 5 | ||
| Total Lesions | # mice | # mice with lesion | Incidence (%) | # lesions | ||
| No DSS | 50 | 13 | 26 | 22 | ||
| 2% DSS 7d | 18 | 13 | 72.2 | 111 | ||
| 2% DSS 5d | 8 | 4 | 50 | 17 | ||
From a single section of a Swiss Roll
cm from rectum
We identified grossly visible adenomatous lesions in 12 of the 18 CAC;APC580S/+ mice receiving DSS treatment for 7d (66.7% incidence, 5.8 tumors per affected mouse, average size = 3.2±0.2 mm; Table 1 and Figure 3B, b). As with the non-DSS treated group, most of the tumors were observed in the distal colon (73%). In the 5d DSS treatment group, 50% of the CAC;APC580S/+ mice had grossly visible tumors at 20 wks (average 3 tumors per affected mouse, average size = 2.3±0.4 mm, Table 1) and all were located in the distal colon. Upon examination of a single cross section from each Swiss roll we identified 26 microadenomas in the distal colon, 15 microadenomas in the proximal colon, and 5 microadenomas in the cecum of DSS treated CAC;APC580S/+ mice (Table 1).
Because transgene expression was limited to the colon, CAC;APC580S/580S mice lacking both APC alleles were viable and were able to survive up to 6 wks of age. All CAC;APC580S/580S mice developed rectal prolapse by 3 wks and the body weights of CAC;APC580S/580S mice were reduced by 12.8% and 20% at 3 and 6 wks, respectively (Figure 4A). The hematocrit of CAC;APC580S/580S mice was 41.8% lower than CAC;APC580S/+ mice at 6 wk (27.6±1.5 vs 42.5±1.0 %, respectively). At 3 and 6 wks, raised lesions were observed grossly in the large intestine of CAC;APC580S/580S (Figure 4B, b, c). Examination of these areas revealed mucosal thickening, with the distal colon being more severely affected than the proximal colon (Table 2). Histologically, the affected areas were characterized by dysplastic epithelial cells that exhibited increased nuclear to cytoplasmic ratio, loss of nuclear polarity, and increased number of mitotic figures characteristic of microadenomas (Figure 4C, a). At 6 wks of age, adenomas and microadenomas were multifocally distributed throughout distal colon and proximal colon of CAC;APC580S/580S mice (Figure 4B, c and 4C, b).
Figure 4. Analysis of the phenotype of wild-type (APC+/+) CAC;APC580S/+ (APC+/−), and CAC;APC580S/580S (APC−/−) mice.

(A) Growth curves. * significantly less than APC+/+ mice p<0.05. (B) Representative images of colorectal appearance in (a) APC+/−, (b) 3 wk-old APC−/−, and (c) 6 wk-old APC−/− mice (arrows = raised mucosal lesions). Bar = 1 cm. (C) Representative H&E stained sections of distal colon in (a) 3 wk-old and (b) 6 wk-old APC−/− mice. Bar = 500 μm; N = normal epithelium, A = adenoma. (D) Immunuhistochemical labeling of step sections from the distal colon of 3 wk-old APC−/− mice for (a) β-catenin and (b) Ki-67.
Arrow = a microadenoma. Bar = 100μm.
Table 2.
Effect of APC allele deletion on mucosal thickening and lesion size.
| Mucoal Thickness (mm) | Lesion area (% total area) | |
|---|---|---|
| APC+/+ | ||
| CoP | 0.23±0.01 | 0 |
| CoD | 0.22±0.03 | 0 |
| APC+/− | ||
| CoP | 0.20±0.02 | 0 |
| CoD | 0.26±0.03 | 0 |
| APC+/− | ||
| CoP | 0.39±0.07* | 16.0+3.3* |
| CoD | 0.99±0.08* | 48.7+3.7* |
p < 0.05 vs APC+/+
In normal mouse colon, labeling for the proliferation marker Ki67 is limited to the cells at the crypt base and staining for the transcriptional activator β-catenin is concentrated at the cell membrane and diffusely present in the cytoplasm (Supplemental Figure 3S).
Immunohistochemical analysis of the adenomatous lesions from CAC;APC580S/580S mice showed significant cytoplasmic and nuclear accumulation of β-catenin in dysplastic epithelial cells as early as 3 wks of age (Figure 4D, a). This is consistent with the loss of APC-mediated effects onβ-catenin degradation and localization(19). In the crypts whose cells have nuclear accumulation of β-catenin, Ki-67 labeling was increased and extended beyond the crypt base. (Figure 4D, b). In CAC;APC580S/+ mice, the accumulation of β-catenin and the increase in Ki-67-stained cells outside the crypt base was apparent only after obvious changes in epithelial cell morphology were present, i.e. in microadenomas and adenomas (Supplemental Figure 3S). This suggests that the development of these phenotypes occurs only after the loss of heterozygosity at the APC alleles, a situation seen in other mouse models (6, 20–23).
Discussion
Many animal models have been developed and characterized for colon cancer research (see recent reviews (2, 3)). In this report we describe a new transgenic mouse model that meets all three of the criterion we identified as important for an improved animal model of human colon cancer: the transgene expression in our CAC mouse is restricted primarily to the colon, transgene expression is limited to the epithelial cell from which most colon tumors originate, and expression during embryonic development is limited to the large intestine of late stage embryos. We confirmed the utility of the CAC mouse for colon cancer research by crossing them to mice with a floxed APC allele and demonstrating the development of intestinal lesions that morphologically resemble those seen in humans. Like the early stage lesions described in the Vogelstein model of colon cancer (24), the lesions in CAC-APC580S/+ mice are found predominantly in the distal colon and they are pedunculated, well-differentiated, epithelial-rich lesions with hyperplasia that is associated with increased cellular and nuclear β-catenin accumulation. We also demonstrate that, similar to the situation in humans (25), colonic inflammation increases the formation of colon lesions in CAC;APC580S/+ mice.
Several groups have previously attempted to improve the animal models available for colon cancer research. For example, several APC mutant mice were designed to increase the incidence of colon tumors (e.g. APCΔ716, APC1638N, APCΔ14, APCmin-FCCC) but all of these models still develop polyps primarily in the small intestine (6, 26–28). In addition, mice with germline mutations often form tumors outside of the gastrointestinal tract, e.g. mammary gland tumors in APCmin, APCΔ14 and APCmin-FCCC mice (6, 28), a wide spectrum of tumor development in mice with mutant mismatch repair enzyme genes (2). In contrast, our model had weak transgene expression in the liver but we saw no evidence of liver pathology in CAC;APC580S/+. While further studies are needed to reveal whether the low level transgene expression in liver causes cancer after deletion of other floxed alleles, our CAC mouse has the most favorable restriction of transgene expression reported to date for a colon cancer model. Other groups have attempted to develop promoters that restrict the expression of transgenes to specific intestinal locations but with moderate success in the context of colon cancer. Transgene expression from the 12.4 kb villin promoter/enhancer-Cre construct is limited to the epithelial cells of the intestine (29). However, villin-cre; APC580D/+ transgenic mice have an intestinal distribution of tumors that is similar to the APCmin mouse (small intestine≫colon) (30). Tuveson et al. (31) restricted expression of an activated K-Ras mutant (k-rasG12D) transgene to intestinal epithelium using the fatty acid binding protein (FaBP) promoter. However, others have shown that the FaBP promoter drives transgene expression in all segments of intestine (ileum > cecum > colon > duodenum = jejunum) as well as causing very high transgene expression in the bladder (32). Gum et al. used the intestinal trefoil factor gene promoter to drive expression of a SV40 large T antigen transgene to the intestine (33). While this resulted in colon tumor formation, the cancer originated in the enteroendocrine cell and more closely resembled small cell carcinoma, a rare form of colorectal cancer in humans.
Shibata et al. (34) and Hung et al., (35) have attempted to bypass the problem of tissue-specific transgene expression by introducing a Cre-expressing adenovirus directly into the colon. The intrarectal administration of the adenovirus used by Shibata et al. (34) was only effective in Apc580S/580S mice and not Apc580S/+, an outcome that reflects the poor ability of the approach to influence the proliferating cells at the crypt base (i.e. the deletion is not induced in self-renewing cells) (35). Hung et al. used surgical clips to limit adenovirus effects to the distal colon and found that this surgical approach was effective at inducing adenocarcinoma in Apc580S/580S mice as well as inducing colon carcinoma and liver metastasis in Apc580S/580S mice with one activated K-Ras allele. While infection with Cre expressing adenovirus is clearly an effective way to induce colon cancer in mice, the requirement for surgical manipulation severely limits this model for larger studies on chemoprevention or testing of therapeutics.
A more promising model is the one developed recently by Hinoi T et al. (20) using a 9.5 kb fragment of the CDX2 gene promoter to drive Cre-recombinase expression (CPC). In adult mice, expression of this transgene is limited to the ileum, cecum, and colon. Compound CPC; APCfloxed/+ mice develop intestinal lesions that show a clear adenoma-carcinoma progression with 61% of the tumors occurring in the colon. Unfortunately, there is significant embryonic expression of this transgene in the skin and muscle of the lower limbs as well as in kidney, thymus, adrenal glands, spleen, and testis. As such, it is not clear whether the tumor phenotype of these mice will be limited to the large intestine of CPC;APCfloxed/+ mice as this data was not reported.
Another unique feature of our CAC mouse is that only 15% of the large intestinal crypts express the transgene. Uniform expression of genetic mutations throughout the epithelium mimics the tissue and cellular environment of heritable cancers like FAP and HNPCC (10). However, while our transgene expression pattern was unexpected, it reflects a distribution of mutant cells that is more similar to that seen in sporadic cancer (normal tissue next to abnormal cells). Other groups have recently reported methods to create mouse models that reflect the sporadic nature of human cancer (30, 36). These methods use Cre alleles modified to contain long mononucleotide repeat tracts that disrupt the reading frame of Cre. Random mutations in the nucleotide repeat within a subset of somatic stem cells revert the Cre allele to a translatable reading frame and permit deletion of floxed alleles. By using the 9.5 kb CDX2 promoter to drive a disrupted Cre allele, Akyol et al. (30) got Cre mediated deletion of alleles that was limited to the cecum and proximal colon (and some tail expression) and that caused a mosaic transgene expression pattern similar to what we see throughout the large intestine of our CAC mouse. Although the Akyol model will be useful for restricting genetic modifications to the proximal colon, our CAC mouse compares favorably in all other respects.
There are some potential weaknesses of our new model. First, the embryonic expression may lead to early colon cancer development when multiple floxed alleles are combined and this may limit the ability of researchers to test therapeutic or chemopreventative agents. Use of the CA1 promoter/enhancer to drive an inducible Cre-ERT2 transgene (37) may overcome this limitation. Second, the incidence of lesion formation in the large intestine of CAC;APC580S/+ mice without inflammation is low (20% with gross lesions) and will require increased animal numbers for colon cancer studies. However, this issue will be unavoidable in models that are more representative of human sporadic colon cancer. Finally, we created the mouse on the C57BL/6J background that is relatively resistant to colon cancer (38). This choice was driven by the large availability of genetically modified lines with floxed alleles on the C57BL/6 background but could be overcome by backcrossing the transgene into other genetic backgrounds.
Supplementary Material
Acknowledgments
This work was supported by NCI award R21CA124527 to JCF.
The authors would like to thank: Purdue University: Ms. Rebecca McCreedy (Dept. of Foods and Nutrition, organ harvests). University of Michigan: Dr. Debra Gumucio for the pUC12.4kb-villin plasmid. The CBR Institute for Biomedical Research, Harvard Medical School: Drs. Klaus Rajewsky and Marc Schmidt-Supprian for the pPGK-cre-bpA plasmid.
Abbreviations
- APC
adenomatosis polyposis coli
- CA1
carbonic anhydrase I
- CDX2
caudal type homeobox 2
- DSS
dextran sulfate sodium
- Mlh1
mutL homolog 1
- Msh2
mutS homolog 2
- WT
wild-type
Footnotes
Conflicts: The authors have no conflicts to report.
Reference List
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–49. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 2.Taketo MM, Edelmann W. Mouse models of colon cancer. Gastroenterology. 2009;136:780–98. doi: 10.1053/j.gastro.2008.12.049. [DOI] [PubMed] [Google Scholar]
- 3.Rosenberg DW, Giardina C, Tanaka T. Mouse models for the study of colon carcinogenesis. Carcinogenesis. 2009;30:183–96. doi: 10.1093/carcin/bgn267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heijstek MW, Kranenburg O, Borel RI. Mouse models of colorectal cancer and liver metastases. Dig Surg. 2005;22:16–25. doi: 10.1159/000085342. [DOI] [PubMed] [Google Scholar]
- 5.Moser AR, Pitot HC, Dove WF. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science. 1990;247:322–4. doi: 10.1126/science.2296722. [DOI] [PubMed] [Google Scholar]
- 6.Colnot S, Niwa-Kawakita M, Hamard G, Godard C, Le Plenier S, Houbron C, et al. Colorectal cancers in a new mouse model of familial adenomatous polyposis: influence of genetic and environmental modifiers. Laboratory Investigation. 2004;84:1619–30. doi: 10.1038/labinvest.3700180. [DOI] [PubMed] [Google Scholar]
- 7.de Wind N, Dekker M, Berns A, Radman M, te RH. Inactivation of the mouse Msh2 gene results in mismatch repair deficiency, methylation tolerance, hyperrecombination, and predisposition to cancer. Cell. 1995;82:321–30. doi: 10.1016/0092-8674(95)90319-4. [DOI] [PubMed] [Google Scholar]
- 8.Reitmair AH, Cai JC, Bjerknes M, Redston M, Cheng H, Pind MT, et al. MSH2 deficiency contributes to accelerated APC-mediated intestinal tumorigenesis. Cancer Res. 1996;56:2922–6. [PubMed] [Google Scholar]
- 9.Prolla TA, Baker SM, Harris AC, Tsao JL, Yao X, Bronner CE, et al. Tumour susceptibility and spontaneous mutation in mice deficient in Mlh1, Pms1 and Pms2 DNA mismatch repair. Nat Genet. 1998;18:276–9. doi: 10.1038/ng0398-276. [DOI] [PubMed] [Google Scholar]
- 10.Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159–70. doi: 10.1016/s0092-8674(00)81333-1. [DOI] [PubMed] [Google Scholar]
- 11.Rice P, Longden I, Bleasby A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 2000;16:276–7. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 12.Sowden J, Edwards M, Morrison K, Butterworth PH, Edwards YH. Erythroid expression and DNAaseI-hypersensitive sites of the carbonic anhydrase 1 gene. Biochem J. 1992;288:545–51. doi: 10.1042/bj2880545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Drummond F, Sowden J, Morrison K, Edwards YH. The caudal-type homeobox protein Cdx-2 binds to the colon promoter of the carbonic anhydrase 1 gene. European Journal of Biochemistry. 1996;236:670–81. doi: 10.1111/j.1432-1033.1996.t01-1-00670.x. [DOI] [PubMed] [Google Scholar]
- 14.Kuraguchi M, Wang XP, Bronson RT, Rothenberg R, Ohene-Baah NY, Lund JJ, et al. Adenomatous polyposis coli (APC) is required for normal development of skin and thymus. Plos Genetics. 2006;2:1362–74. doi: 10.1371/journal.pgen.0020146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fleet JC, Wood RJ. Specific 1,25(OH)2 D3-mediated regulation of transcellular calcium transport in Caco-2 cells. Am J Physiol. 1999;276:G958–G964. doi: 10.1152/ajpgi.1999.276.4.G958. [DOI] [PubMed] [Google Scholar]
- 16.Braunstein EM, Qiao XTT, Madison B, Pinson K, Dunbar L, Gumucio DL. Villin: A marker for development of the epithelial pyloric border. Developmental Dynamics. 2002;224:90–102. doi: 10.1002/dvdy.10091. [DOI] [PubMed] [Google Scholar]
- 17.Tanaka T, Kohno H, Suzuki R, Hata K, Sugie S, Niho N, et al. Dextran sodium sulfate strongly promotes colorectal carcinogenesis in Apc(Min/+) mice: inflammatory stimuli by dextran sodium sulfate results in development of multiple colonic neoplasms. Int J Cancer. 2006;118:25–34. doi: 10.1002/ijc.21282. [DOI] [PubMed] [Google Scholar]
- 18.Sowden J, Leigh S, Talbot I, Delhanty J, Edwards Y. Expression from the proximal promoter of the carbonic anhydrase 1 gene as a marker for differentiation in colon epithelia. Differentiation. 1993;53:67–74. doi: 10.1111/j.1432-0436.1993.tb00647.x. [DOI] [PubMed] [Google Scholar]
- 19.Voutsadakis IA. The ubiquitin-proteasome system in colorectal cancer. Biochim Biophys Acta. 2008;1782:800–8. doi: 10.1016/j.bbadis.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 20.Hinoi T, Akyol A, Theisen BK, Ferguson DO, Greenson JK, Williams BO, et al. Mouse model of colonic adenoma-carcinoma progression based on somatic Apc inactivation. Cancer Res. 2007;67:9721–30. doi: 10.1158/0008-5472.CAN-07-2735. [DOI] [PubMed] [Google Scholar]
- 21.Levy DB, Smith KJ, Beazerbarclay Y, Hamilton SR, Vogelstein B, Kinzler KW. Inactivation of Both Apc Alleles in Human and Mouse-Tumors. Cancer Research. 1994;54:5953–8. [PubMed] [Google Scholar]
- 22.Luongo C, Moser AR, Gledhill S, Dove WF. Loss of Apc(+) in Intestinal Adenomas from Min Mice. Cancer Research. 1994;54:5947–52. [PubMed] [Google Scholar]
- 23.Smits R, Kartheuser A, JagmohanChangur S, Leblanc V, Breukel C, deVries A, et al. Loss of Apc and the entire chromosome 18 but absence of mutations at the Ras and Tp53 genes in intestinal tumors from Apc1638N, a mouse model for Apc-driven carcinogenesis. Carcinogenesis. 1997;18:321–7. doi: 10.1093/carcin/18.2.321. [DOI] [PubMed] [Google Scholar]
- 24.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–67. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 25.Xie J, Itzkowitz SH. Cancer in inflammatory bowel disease. World J Gastroenterol. 2008;14:378–89. doi: 10.3748/wjg.14.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fodde R, Edelmann W, Yang K, van LC, Carlson C, Renault B, et al. A targeted chain-termination mutation in the mouse Apc gene results in multiple intestinal tumors. Proc Natl Acad Sci U S A. 1994;91:8969–73. doi: 10.1073/pnas.91.19.8969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oshima M, Oshima H, Kitagawa K, Kobayashi M, Itakura C, Taketo M. Loss of Apc heterozygosity and abnormal tissue building in nascent intestinal polyps in mice carrying a truncated Apc gene. Proc Natl Acad Sci U S A. 1995;92:4482–6. doi: 10.1073/pnas.92.10.4482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cooper HS, Chang WC, Coudry R, Gary MA, Everley L, Spittle CS, et al. Generation of a unique strain of multiple intestinal neoplasia (Apc(+/Min-FCCC)) mice with significantly increased numbers of colorectal adenomas. Mol Carcinog. 2005;44:31–41. doi: 10.1002/mc.20114. [DOI] [PubMed] [Google Scholar]
- 29.Madison BB, Dunbar L, Qiao XT, Braunstein K, Braunstein E, Gumucio DL. Cis elements of the villin gene control expression in restricted domains of the vertical (crypt) and horizontal (duodenum, cecum) axes of the intestine. J Biol Chem. 2002;277:33275–83. doi: 10.1074/jbc.M204935200. [DOI] [PubMed] [Google Scholar]
- 30.Akyol A, Hinoi T, Feng Y, Bommer GT, Glaser TM, Fearon ER. Generating somatic mosaicism with a Cre recombinase-microsatellite sequence transgene. Nat Methods. 2008;5:231–3. doi: 10.1038/NMETH.1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tuveson DA, Shaw AT, Willis NA, Silver DP, Jackson EL, Chang S, et al. Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell. 2004;5:375–87. doi: 10.1016/s1535-6108(04)00085-6. [DOI] [PubMed] [Google Scholar]
- 32.Saam JR, Gordon JI. Inducible gene knockouts in the small intestinal and colonic epithelium. J Biol Chem. 1999;274:38071–82. doi: 10.1074/jbc.274.53.38071. [DOI] [PubMed] [Google Scholar]
- 33.Gum JR, Jr, Hicks JW, Crawley SC, Yang SC, Borowsky AD, Dahl CM, et al. Mice expressing SV40 T antigen directed by the intestinal trefoil factor promoter develop tumors resembling human small cell carcinoma of the colon. Mol Cancer Res. 2004;2:504–13. [PubMed] [Google Scholar]
- 34.Shibata H, Toyama K, Shioya H, Ito M, Hirota M, Hasegawa S, et al. Rapid colorectal adenoma formation initiated by conditional targeting of the Apc gene. Science. 1997;278:120–3. doi: 10.1126/science.278.5335.120. [DOI] [PubMed] [Google Scholar]
- 35.Hung KE, Maricevich MA, Richard LG, Chen WY, Richardson MP, Kunin A, et al. Development of a mouse model for sporadic and metastatic colon tumors and its use in assessing drug treatment. Proc Natl Acad Sci U S A. 2010;107:1565–70. doi: 10.1073/pnas.0908682107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miller AJ, Dudley SD, Tsao JL, Shibata D, Liskay RM. Tractable Cre-lox system for stochastic alteration of genes in mice. Nat Methods. 2008;5:227–9. doi: 10.1038/NMETH.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Indra AK, Warot X, Brocard J, Bornert JM, Xiao JH, Chambon P, et al. Temporally-controlled site-specific mutagenesis in the basal layer of the epidermis: comparison of the recombinase activity of the tamoxifen-inducible Cre-ER(T) and Cre-ER(T2) recombinases. Nucleic Acids Res. 1999;27:4324–7. doi: 10.1093/nar/27.22.4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deschner EE, Long FC, Hakissian M, Herrmann SL. Differential susceptibility of AKR, C57BL/6J, and CF1 mice to 1,2-dimethylhydrazine-induced colonic tumor formation predicted by proliferative characteristics of colonic epithelial cells. J Natl Cancer Inst. 1983;70:279–82. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.