

Abstract
A general strategy for the construction of macrocyclic lactones containing conjugated Z,Z-1,3-diene subunits has been is described. The centerpiece of the strategy is a sequential ring-closing metathesis that forms an unsaturated siloxane ring followed by an intramolecular cross-coupling reaction with a pendant alkenyl iodide. A highly modular assembly of the various precursors allowed the preparation of unsaturated macrolactones containing 11-, 12-, 13- and 14-membered rings. Although the ring closing metathesis process proceeded uneventfully, the intramolecular cross-coupling required extensive optimization of palladium source, solvent, fluoride source and particularly fluoride hydration level. Under the optimal conditions (including syringe pump high dilution), the macrolactones were produced in 53-78% yield as single stereoisomers. A benzo fused 12-membered ring macrolactone containing an E,Z-1,3-diene unit was also prepared by the same general strategy. The E-2-styryl iodide was prepared by a novel Heck reaction of an aryl nonaflate with vinyltrimethylsilane followed by iododesilylation with ICl.
Introduction
Highly functionalized macrocycles are considered to be privileged structures because their architectures provide specific disposition of functionalities. Not surprisingly they have found application in supramolecular chemistry1 and in drug discovery.2 In drug development complex macrocycles have been found to bind selectively to proteins without significant loss in entropy upon binding and currently more than 100 marketed drugs are derived from macrocycles that on average contain greater than 16-membered rings.2a The drug-like properties of macrocycles notwithstanding, this class of molecules is still under-explored largely because of concerns about the difficulty of the synthesis.2 Nevertheless, a number of macrocycles are on pre-clinical and clinical development. From a chemical perspective the continuing discovery of macrocycles of natural origin that possess both complex structures and sensitive functional groups in a compact 12-14-membered macrocycle calls for the development of newer more efficient methods.
One such recent discovery of biologically active 12-14-membered macrocycles are the oximidines I3 and III,4 and the resorcylics5 (radicicol6 and monocilin7) that contain a salicylic acid moiety embedded in a 12-membered macrolide ring (Figure 1). These compounds also possess a conjugated diene or triene moiety within the ring that imparts high degree of strain within the macrocycle. Efforts to synthesize these and similar lactones have spurred the development of new synthetic methods capable of efficiently constructing these polyunsaturated macrocycles. In addition, examples of natural products with a conjugated diene system are known that are not salicylic derived such as the 14-membered lactone lactimidimycin.8
Figure 1.

Macrolactone natural products possessing conjugated alkenes.
The construction of medium- (8-to 11-membered) and large-ring (> 12-membered) macrocycles is generally difficult to achieve because of entropy and enthalpic contributions.9,10 To overcome these contributions in macrocyclic synthesis three modes of cyclization are commonly used and they are distinguished by the position of the reacting groups: (1) the end-to-end, (2) the end-to backbone, and (3) the backbone-to-backbone cyclizations.11 To favor cyclization over oligomerization high dilution is effected either by: (1) the reaction is run in a large volume of solvent to make a very dilute solution, or (2) the precursor is added slowly over a long period time into a given volume of solvent to maintain a high dilution of the compound being cyclized. Pioneering kinetic studies done by Galli and Mandolini on effective molarity (EM) now make it possible to determine the optimal concentration required to favor cyclization.12
The methods used to form macrocycles can be broadly classified into two categories: (1) carbon-heteroatom-based cyclization and (2) the carbon-carbon-based cyclization. The carbon-heteroatom-based cyclization to build macrolactams and macrolactones was reported first and since then, many reports of hydroxy acid lactonization or amino acid lactamization that use different activating groups have been developed.13 In addition, lactone formation via a π-allylpalladium intermediates has been reported.14 Cyclization via a carbon-carbon based strategy initially began with nucleophile- and electrophile-promoted methods,9 and later radical and metal-catalyzed cyclizations appeared.15 More recently metal-catalyzed cyclizations have been performed using the ring-closing metathesis (RCM).16 For example, a ruthenium-catalyzed RCM has been applied in a large-scale production of a 15-membered macrocyclic drug candidate.17 In addition a major breakthrough in RCM synthesis of macrolactone has been the ring-closing enyne-yne metathesis18a that in a stereocontrolled process furnishes a 1,3-diene motif within a macrocycle and consequently has been applied in total synthesis.18b,c Another carbon-carbon based macrocyclization technique is the palladium catalyzed cross-coupling reaction such as the Stille, Suzuki, and Hiyama reactions. The earliest examples of cross-coupling reactions in macrocyclization employed organoboranes19 or organostannanes20 as the metallic donor because they were stable and readily prepared. However, the use of organotin has been limited because of tin toxicity and as a result recent reports have focused on using organoboron donors21 or other donors such as silicon. The organosilicon-based partners have emerged as competent donors in the synthesis of medium-ring macrocyclic ethers22a and in the total synthesis of cyclic natural products.22b
Background
The dominant position of palladium catalysis in organic synthesis is largely due to its specificity and functional group tolerance, allowing the planning and execution of complex molecule total synthesis.23 Among the earliest reports of a palladium-catalyzed macrocyclization was the palladium-catalyzed intramolecular alkylation of π-allylpalladium complexes with sulfone-stabilized anions.24 The reaction affords medium- to large-ring macrolactones in good yields and has been applied as the key step in total synthesis of natural products25 Other reports of nucleophilic addition to π-allylpalladium complexes to form macrocycles and macrolactone natural product have appeared.14
Similarly, the assembly of macrocycles by palladium-catalyzed cross-coupling is emerging as a viable alternative. Since the first report19 that used haloalkenylboranes to synthesize the sesquiterpene humulene other palladium-catalyzed cross-coupling reactions of organoboranes have been used to construct complex natural products.21 At the same time that palladium-catalyzed cross-coupling with organoboranes and organostannanes was being developed, examples illustrating the use of organosilicon in the synthesis of macrocycles appeared. Lastly, macrocycles have also synthesized by palladium-catalyzed cross-coupling of alkynes.26
The ring-closing metathesis reaction16 has revolutionized the synthesis of macrocyclic products. Both molybdenum27 (Schrock) and ruthenium28 (Grubbs) complexes have been used to good advantage. Because of their functional group tolerance ruthenium catalyst are preferred for the synthesis of natural and unnatural. However, in substrates containing electron deficient dienes and/or sterically encumbered double bonds, the more reactive molybdenum catalysts are used.
Cleary, both palladium-catalyzed cyclizations and ring-closing metathesis reactions enable synthetic strategies for the total synthesis of macrocyclic natural products. Therefore, the amalgamation of these reactions into a tandem sequence should open new and highly simplifying strategies for the synthesis of macrocycles. Recent disclosures from these laboratories have described a sequential, ring closing metathesis/silicon-assisted intramolecular cross-coupling reaction for the synthesis of medium-sized rings (9-12) containing a cis-cis-diene motif as well as a 9-membered ring ether that culminated in a total synthesis of (+)-brasilenyne.22 The advantages of this approach for macrocycle synthesis are illustrated in Figure 2. To accomplish the strategic disconnection between the double bonds of a generic macrocyclic polyene I with a cross-coupling reaction, the acyclic precursor II must be constructed and the reactive termini bearing X and M but locate each other under catalysis by a transition metal. By constraining one double bond in a smaller ring, the cross-coupling precursor III is less flexible and the distance between the reacting termini is reduced, thus favoring macrocyclization. Moreover, the size and substitution of the smaller ring can be easily adjusted in the preparation of RCM precursor IV.
Figure 2.

Sequential RCM/silicon-assisted cross-coupling strategy.
Although this process provided efficient access to medium-ring ethers, the general applicability and transferability of this chemistry to the synthesis of larger rings as well as in the assembly of the more sensitive macrolactones is still unknown. As an extension of scope, the synthesis of medium to large (11- to 14-membered) ring lactones was targeted as a proving ground. This challenge becomes even more formidable when the flexibility of a macrocycle is constrained by multiple degrees of unsaturation. For example the assembly of the oximidine II core by either ruthenium-catalyzed metathesis29 or Suzuki cross-coupling21d afforded the E,Z,Z-triene macrocycles V and VI in 48% and 42% yields respectively (Figure 3, entries 1 and 2). On the other hand the synthesis of a simpler substrate under Mitsunobu conditions (Figure 2, entry 3) afforded the macrocycle VII in 23% yield.30
Figure 3.

Methods applied to synthesize benzo-fused lactones.
The synthesis of the benzo-fused lactone VII, while simple in appearance, was an especially attractive target for a sequential RCM cross-coupling sequence because it contains the conjugated diene motif within the lactone core. The formation of the constrained macrocycle III under palladium-catalysis was envisioned to proceed via a larger-ring organopalladium-intermediate. Because the carbon-palladium-carbon bonds in the intermediate are longer than carbon-carbon bonds it would have less strain compared to the product being formed. Facile reductive elimination simultaneously affords the desired lactone and regenerates the catalyst. The ultimate end goal now became the efficient assembly of an embellished lactone III analog.
Research Plan
In contrast to the use of the RCM/cross-coupling sequence for the synthesis of medium-sized macrocyclic ethers, the assembly of large-ring lactones such as VII presents additional problems to unite the reacting ends of the now longer acyclic chain. For example, because of the basic nature of fluoride, the TBAF-assisted intramolecular cross-coupling may lead to the cleavage of the lactone. In addition, lactone VII possesses a trans-cis diene system conjugated to a benzene ring that causes the compound to behave as a conjugated triene system that is inherently more rigid and thus more difficult to fuse the reacting termini. Of these problems, the greater ring size and the lability of the ester group in the basic TBAF reaction medium were potentially most challenging and thus led to the investigation of lactones VIII (Figure 4) that are easier to access and modify. These lactones were chosen because they addressed the consequences of placing the carboxyl and hydroxyl groups in different locations with respect to diene moiety. Various ring-sizes of VIII could be accessed from the cyclic siloxanes such as IX. The strategy to use cyclic siloxanes IX was chosen to fix one end of the macrocycle to be formed in a smaller ring to reduce both flexibility and distance between the two reacting termini. The siloxane IX would be derived from ring closing metathesis of silyl ethers X. The silyl ether X could be elaborated with ease from diols XI and the acid XII. The key step in this route is the sequential ring-closing metathesis/intramolecular cross-coupling reaction.
Figure 4.

General synthetic sequence for the construction of macrolactones.
The conjugated triene problem will be investigated by the synthesis of siloxane XIII from alcohol XIV. Alcohol XIV in turn would arise from elaboration of 2,6-dihydroxybenzoic acid XV and alcohol XVI. We describe herein, the successful implementation of this plan which culminated in the syntheses of medium- to large-ring macrolactones VIII the fused benzolactone VII.
Results
To initiate the study of the silicon-assisted, cross-coupling macrocyclization, an efficient and versatile synthetic sequence was needed to generate the cycloalkenylsiloxanes 7a-7e (Schemes 1 and 3) and 10 (Scheme 5) in high yield. The cyclic alkenylsiloxanes were prepared in 7 steps from 4-pentyn-1-ol. The synthesis of the styryl siloxane 10 on the other hand required eight steps. The selection of the reaction sequence was guided by the stability of the products along each pathway. Because the silyl ethers and the siloxanes are sensitive compounds, the syntheses of these products were designed such that they are last compounds prepared prior to macrocyclization. The details of each synthetic sequence are described below.
Scheme 1a.

a Conditions: (a) 1. I2, KOH, MeOH; 2. KO2CN=NCO2K, AcOH, THF/i-PrOH, (62%); (b) CrO3/H5IO5, MeCN/H2O (9/1), (97%); (c) 1. COCl2, benzene; 2. 3a, collidine, CH2Cl2, –78 °C, (79%); (d) chlorodimethylvinylsilane, Et3N, CH2Cl2 (93%); (e) 6, benzene, rt (87%). The yields are of analytically pure material.
Scheme 3.

Scheme 5.

1. Preparation of Alkenylsiloxane 7a
Siloxane 7a (the cross-coupling precursor used for optimization) was prepared from 4-pentyn-1-ol as depicted in Scheme 1. Iodination of 4-pentyn-1-ol with iodine in aqueous KOH furnished 5-iodo-4-pentyn-1-ol that was immediately subjected to a selective cis reduction with diimide31 generated in situ from potassium azodicarboxylate,32 to afford (Z)-5-iodo-4-pentenol 133 in 62% yield over 2 steps. Alcohol 1 was then oxidized by a CrO3-catalyzed periodate oxidation34 to form (Z)-5-iodopent-4-enoic acid (2)33 in 97% yield. Hex-5-ene-1,2-diol (3a) was synthesized from glycidol following the established sequence.35
Acid 2 was converted to its acid chloride, and the crude acid chloride solution was added dropwise to a solution of diol 3 and collidine36 in dichloromethane at −78 °C to afford ester 4a in 79% yield over 2 steps. Silylation of ester 4a with chlorodimethylvinylsilane gave the sensitive silyl ether 5a in 93% yield after purification with silica gel buffered with 1% triethylamine. The alkenylsilyl ether 5a was then subjected to ring-closing metathesis using the Schrock molybdenum carbene complex 627 to afford the alkenylsiloxane 7a in 87% yield.
2. Optimization of the Intramolecular Cross-Coupling Reaction
With alkenylsiloxane 7a in hand, the palladium-catalyzed intramolecular cross-coupling was attempted using the reaction conditions previously developed in these laboratories namely, allylpalladium chloride dimer (APC) as the catalyst and tetrabutylammonium fluoride trihydrate (TBAF•3H2O) as the activator.22 The reaction was carried out under high dilution such that a 0.1 M solution of siloxane 7a was slowly added using a syringe-pump into a mixture of APC (0.1 equiv) and a 1 M solution of TBAF•3H2O in THF (10 equiv) over a 40 h period at room temperature. At the 10 h time point, precipitation of palladium black was observed. After 40 h 1H NMR analysis of the reaction mixture showed traces of opened siloxane and hydrolyzed silyl ether products.
The presence of palladium black implied that catalyst death was occurring during the extended reaction time. To determine if catalyst death was caused by oxygen poisoning, the reaction was repeated under the same conditions but in a Schlenk flask instead of a three-necked, round-bottomed flask. In this case the desired lactone 8a was isolated together with an isomeric lactone 8a' as an inseparable 3:1 mixture in 43% yield (Scheme 2). The identity and ratio of 8a/8a' were determined by 1H NMR analysis.37 Further structural evidence was secured by converting the components to their TES-protected ethers followed by exposure of the mixture to CrO3•2py oxidation38 which converted the primary TES-protected 8a' to an aldehyde while the TES-protected secondary alcohol 8a was recovered intact.37 The isomeric lactone 8a' most likely arose from a base-promoted translactonization initiated by the fluoride ion. Accordingly, further optimization was needed to suppress this undesired process and increase the yield of the desired product.
Scheme 2.

2.1. Optimization of TBAF Hydration Level and Solvent
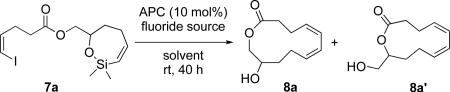
Operating under the hypothesis that the isomeric lactone 8a' arose from 8a because of the basic character of TBAF, measures to lower its basicity while maintaining fluoride anion strength were taken. Detailed studies of the nucleophilicity of quaternary ammonium fluorides have shown that the trihydrated fluoride anion is both very basic and nucleophilic.39 These properties can be attenuated with protic solvents and modulated by varying the hydration level of the quaternary ammonium salt. This strategy was critical for the successful cross-coupling of silanols with aryl triflates40 and alkenyl iodides.41 To address the problem of ester hydrolysis two experiments were performed. The fluoride source (TBAF•3H2O) was further hydrated by adding the appropriate amounts of water to a 1 M solution of TBAF•3H2O in THF to form solutions of TBAF•4H2O and TBAF•6H2O. The palladium-catalyzed macrocyclization reaction using the TBAF•4H2O afforded a higher yield of product (64%) but again as a 3:1 mixture of lactones 8a and 8a', whereas the reaction with TBAF•6H2O produced the macrolactones in a slightly lower 59% yield (Table 1, entries 3-4). Higher hydration levels, TBAF•10H2O and TBAF•15H2O, gave lower yields (46% and 39% respectively) but afforded excellent selectivity, only macrocycle 8a was formed (Table 1, entries 10-11). The hydrate (TBAF•6H2O) was chosen as the fluoride source in the next stage of optimization because of its lower fluoride anion strength.39
Table 1.
Optimization of the Fluoride Source and Solvent for the Macrocyclizationa
 | |||||
|---|---|---|---|---|---|
| entry | fluoride source | equiv | solvent | yield, % 8a+8a' | ratiob8a+8a' |
| 1 | TBAF•3H2O | 10 | THF | 0 | - |
| 2 | TBAF•3H2O | 10 | THF | 43 | 3:1 |
| 3 | TBAF•4H2O | 10 | THF | 64 | 3:1 |
| 4 | TBAF•6H2O | 10 | THF | 59 | 3:1 |
| 5 | TBAF•6H2O | 5 | THF | 72 | 3:1 |
| 6 | TBAF•6H2O | 2.5 | THF | 26 | 3:1 |
| 7 | TBAF•6H2O | 5 | dioxane | 76 | 3:1 |
| 8 | TBAF•6H2O | 5 | DMF | 84 | 2:1 |
| 9 | TBAF•7H2O | 5 | DMF | 79 | 2:1 |
| 10 | TBAF•10H2O | 5 | THF | 46 | 100:0 |
| 11 | TBAF•15H2O | 5 | THF | 39 | 100:0 |
Reaction conditions employed 0.25 mmol of 7a at 0.1 M.
Ratio determined by 1H NMR analysis.
The problem of post-facto isomerization was addressed by adjusting the amount of TBAF•6H2O. Lowering the TBAF•6H2O loading from 10 to 5 equiv improved the yield significantly to 72% but the ratio of the two isomers 8a and 8a' remained the same (Table 1, entry 5). However, when the amount of TBAF•6H2O was reduced to 2.5 equiv the macrolactones 8b and 8b' were isolated in only 26% yield (Table 1, entry 6). For the purpose of further optimization, 5 equiv of TBAF•6H2O was selected.
Because the reaction optimization up to this juncture had been conducted with an ethereal solvent (THF), dioxane was selected next because it is a competent solvent for alkenyl-alkenyl cross-coupling reactions.40 The use of dioxane led to an increase in yield (76%) of the macrocycles (Table 1, entry 7). This result suggested that solvent polarity may have an effect on product yield because dioxane is marginally more polar than THF [dioxane (Aj + Bj) = 0.86 versus THF (Aj + Bj) = 0.84].42 The use of DMF [(Aj + Bj) = (1.23)] led to a still higher yield of the macrocycles 8a and 8a' (84%) but lower selectivity (2:1 ratio) (Table 1, entry 8). Because DMF afforded the macrolactones 8a and 8a' in higher yield compared to THF or dioxane it was selected as the solvent of choice going forward.43
2.2. Survey of Fluoride Sources
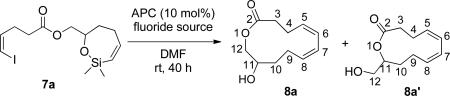
Many fluoride sources are commercially available or are easily accessed in a few synthetic steps. The survey of fluoride sources other than TBAF•6H2O began with the commercially available tetramethylammonium fluoride tetrahydrate (TMAF•4H2O). This salt is superior to TBAF•3H2O in the cross-coupling of aryl triflates and alkenylsilanols.40 Tetramethylammonium (TMAF) is the smallest of all tetralkylammonium cations and has a symmetrical, almost spherical structure.44 By adapting the conditions developed previously, but now with TMAF•4H2O as the fluoride source, the cross-coupling of siloxane 7a afforded macrocycle 8a as a single isomer albeit in only 47% yield (Table 2, entry 1). Increasing the hydration level to TMAF•6H2O afforded the cross-coupled product in 34% yield (Table 2, entry 2). Despite the low yields of 8a obtained with TMAF•6H2O the high selectivity was particularly attractive and encouraged further study.
Table 2.
Survey of the fluoride sources for the macrocyclizationa
 | |||||
|---|---|---|---|---|---|
| entry | fluoride source | solvent | equiv | yield, % 8a+8a' | ratiob8a+8a' |
| 1 | TMAF•4H2O | DMF | 5 | 47 | 100:0 |
| 2 | TMAF•6H2O | DMF | 5 | 34 | 100:0 |
| 3 | BTMAF | DMF | 5 | 39 | 2:1 |
| 4 | BTMAF•6H2O | DMF | 5 | 49 | 6:1 |
| 5 | BTMAF•10H2O | DMF | 5 | 0 | - |
| 6 | TEAF•2H2O | DMF | 5 | 32 | 2:1 |
| 7 | TEAF•6H2O | DMF | 5 | 40 | 2:1 |
| 8 | TBAT | THF | 5 | 29 | 100:0 |
| 9 | TASF | THF | 5 | 22 | 100:0 |
| 10 | CsF | DMF | 5 | 0 | - |
Reaction conditions employed 0.25 mmol of 7 at 0.1 M.
Ratio determined by 1H NMR.
Benzyltrimethylammonium fluoride (BTMAF) is a close analog of TMAF and was easily prepared from benzyltrimethylammonium hydroxide in 73% yield.45 The use of BTMAF (with hydration assumed to be < 0.5 after drying)46 in the macrocyclization reaction of 7a afforded the macrocycles 8a and 8a' in 39% yield in a 2:1 ratio (Table 2, entry 3). To suppress the isomerization, hydrated BTMAF was used (BTMAF•6H2O) and the product was formed in higher yield (49% versus 39%) and improved selectivity (6:1 versus 2:1) (Table 2, entry 4). Unfortunately, increasing the hydration level to BTMAF•10H2O was not productive (Table 2, entry 5).
Next, the commercially available tetraethylammonium fluoride dihydrate (TEAF•2H2O) was investigated. With this activator, the desired macrocycle was obtained in a 32% yield as 2:1 ratio of 8a and 8a' (Table 2, entry 6). Increasing the hydration to level to the TEAF•6H2O gave a modest increase in yield of 8a and 8a' (40%) but the selectivity remained the same (Table 2, entry 7).
The last phase of this survey was the investigation of anhydrous fluoride sources. A number of anhydrous fluoride sources have been used for silicon-based cross-coupling such as tris(diethylamino)sulfonium difluorotrimethysilicate (TASF),47 tetrabutylammonium triphenyldifluorosilicate (TBAT),48 and cesium fluoride (CsF).49 Reaction of siloxane 7a with TBAT afforded the macrocycle 8a in 29% yield as a single isomer (Table 2, entry 8), while the activation with TASF afforded 8a in 22% and with the same selectivity (Table 2, entry 9). Cesium fluoride led only to starting material decomposition and no macrocycle was detected (Table 2, entry 10).
Integrating all of the optimization data, the combination that employed 0.1 equiv of APC, 5 equiv of TBAF•6H2O as the fluoride source, and DMF as the solvent was chosen as the optimal conditions because it afforded the highest yield of 8a. A 1.0-mmol scale macrocyclization reaction of siloxane 7a under these conditions after 48 h furnished 8a and 8a' in 78% yield in a 2:1 ratio. The diagnostic alkene resonances of 8a, namely the HC(6) and HC(7) appeared at 6.08 ppm (dd, 3J = 10.8, 7.0 Hz) and 5.98 ppm (3J = 11.0, 7.0 Hz) respectively. The resonance for H2C(12) appeared as an ABX quartet at 4.19 ppm (dd, 3J = 11.3, 3.0 Hz) and 3.93 ppm (3J = 11.5, 5.0 Hz). On the other hand, compound 8a’ displayed a single, well-resolved alkene resonance for HC(6) and HC(7) at 5.88 ppm (d, 3J = 11.0 Hz). The signal for H2C(12) appeared as an ABX quartet at 3.66 ppm (dd, 3J = 11.9, 5.8, 3.5 Hz) and 3.55 ppm (dd, 3J = 12.0, 10.0 Hz). All attempts to resolve alkene peaks for HC(5) and HC(8) were not successful50 but to the best of our knowledge the macrolactones are formed in a stereospecific manner.
Preliminary molecular modeling calculations51 show that 8a is 0.26 Kcal/mol lower in energy than 8a', which corresponds to a predicted ratio of 1.6:1 at room temperature. Close to the observed ratio of 2:1. Although a good yield of the 12-ring macrolactone could be obtained, it was not possible to suppress the formation of the 11-ring isomer without a substantial decrease in yield. Thus, before further optimization was undertaken it was deemed prudent to establish if isomerization is a general phenomenon or unique to this structure. Accordingly, a selection of other precursors was prepared to evaluate the generality of the cross-coupling macrocyclization.
3. Scope of the Intramolecular Cross-Coupling Reaction
To evaluate the scope of this cross-coupling reaction for the construction of macrocycles the siloxanes 7b-f (Scheme 3) were synthesized in a similar fashion to that shown for synthesis of siloxane 7a. The assembly began with the conversion of acid 2 to the acid chloride that was then added dropwise to cooled solutions of known diols 3b-f52 in collidine to provide esters 4b-f in 72-79% yields (Table 3). The esters were converted to their respective silyl ethers 5b-f and then were subjected to ring closing metathesis with catalyst 6 as before to afford siloxanes 7b-f.
Table 3.
Preparation of Esters 4, Silyl Ether 5, and Siloxanes 7.
| m, n | ester 4 (yield, %)a | silyl ether 5 (yield, %)a | siloxane 7 (yield, %)a |
|---|---|---|---|
| 1, 1 | 4a (79) | 5a (93) | 7b (87) |
| 2, 0 | 4b (72) | 5b (79) | 7b (89) |
| 1, 0 | 4c (76) | 5c (89) | 7c (81)b |
| 2, 1 | 4d (70) | 5d (90) | 7d (84) |
| 3, 0 | 4e (76) | 5e (95) | 7d (83) |
| 3, 1 | 4f (72) | 5f (91)b | 7f (76) |
Yields of analytically pure material.
Yields of chromatographically homogeneous material.
The scope of the intramolecular cross-coupling reaction was investigated on a 1.0 mmol scale under the optimized reaction conditions (0.1 equiv of APC and 5 equiv of TBAF•6H2O in DMF (0.1 M) using syringe pump addition at room temperature). The cyclization of siloxane 7b was carried out first because the product, 12-membered macrocycle 8b, had the same ring size as 8a obtained earlier (Scheme 4). Under the conditions described above siloxane 7b afforded macrocycle 8b in 70% yield as a single isomer. This outcome suggested that the translactonization observed in synthesis of 8a may be substrate dependent.
Scheme 4.

Gratifyingly, siloxane 7c afforded the 11-membered macrolactone 8c in 58% yield again as a single isomer. The diagnostic alkene resonances for HC(6) and HC(7) appeared at 6.04 ppm (dd, 3J = 9.8, 9.8 Hz). The synthesis of 11-membered macrocycles is known to be more difficult than 12-membered rings.10
Next, this process was extended to the synthesis of 13- and 14-membered rings. Thus, cross-coupling of siloxane 7d afforded the 13-membered macrocycle 8d in 61% yield. The closure of this macrolactone required longer reaction time (54 h) compared to the 12-membered lactone (40 h). The synthesis of another 13-membered ring 8e that contained a different substitution pattern proceeded in 53% after 48 h. Unfortunately, the alkene region of the 1H NMR spectra of both 13-membered lactones 8d and 8f could not be fully analyzed.50 Finally, the synthesis of the 14-membered ring 8f was achieved from siloxane 7f in 69% yield after 54 h. Gratifyingly, the alkene resonances for HC(6) and HC(7) were well resolved and showed two doublets of doublets at 6.32 ppm (dd, 3J = 10.5, 10.5 Hz) and 6.23 ppm (dd, 3J = 10.8, 10.8 Hz), respectively.
4. Formation of Benzo-fused Macrocyclic Diene (E,Z)-9
The success of the intramolecular cross-coupling for the synthesis of 11-, 12-, 13-, and 14-membered lactones was gratifying and served to demonstrate that this method could be applied in synthesis of natural products that contain the conjugated diene moiety as a part of a macrolactone. However, a more challenging goal was to apply this approach to synthesize a conjugated triene motif. As an extension of the chemistry described above the synthesis of lactone 9 (Scheme 5) was identified as a model study that could be used to access oximidine III and eventually the triene conjugated oximidine II natural product.
The synthesis of the benzo-fused lactone 9 would result from macrocyclization of the styryl iodide 10 (Scheme 5). The siloxane portion of 10 would arise from elaboration of the silyl ether 11, itself prepared from coupling of styryl iodide 12 with alcohol 13. Synthesis of such styryl iodides can be achieved by Takai olefination of aromatic aldehydes,53 halodecarboxylation of cinnamic acids (Hunsdiecker reaction),54 reduction of dihaloalkenes,55 base promoted homologation of benzyl bromides,56 or ruthenium-catalyzed silylation of styrene.57 Although useful, all of the above methods have various drawbacks. A simpler approach would involve the iododesilylation58 of vinylsilanes themselves obtained either by a Mizoroki-Heck59 reaction of an aryl halide with vinyltrimethylsilane,60 or a hydrosilylation of an aryl alkyne61 obtained from a Sonagashira reaction of an aryl halide. However, in the case at hand these methods could not be used because a suitable aryl halide precursor to compound 14 is not readily available. An alternative solution was to find conditions that could effect a Mizoroki-Heck59 reaction between aryl triflate 1562 and vinyldimethylsilane.
4.1. Optimization of the Mizoroki-Heck Reaction of Vinyltrimethysilane
To the best of our knowledge, the Mizoroki-Heck reaction of an aryl fluoroalkylsulfonate with trimethylvinylsilane is unprecedented and this example represented an even more daunting challenge because of the ortho-substituent on aryl triflate 15.63 The initial survey employed the phosphine-free “Jeffrey conditions”64 (Table 4, entry 1). Unfortunately, when run at ambient temperature the reaction stalled at 43% conversion, but increasing the temperature to 80 °C improved the conversion to 73% (Table 4, entry 2). The change of solvent to MeCN did not offer any significant improvement (Table 4, entry 3).
Table 4.
Optimization of the Heck reaction of Aryl Triflate 15a
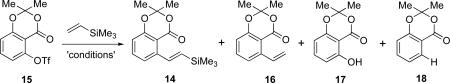 | ||||||
|---|---|---|---|---|---|---|
| entrya | Pd source (5 mol % Pd) | 15, %b | 14, %b | 16, %b | 17, %b | 18, %b |
| 1c | Pd(OAc)2, n-Bu4NOAc, DMF, rt | 57 | 43 | |||
| 2 | Pd(OAc)2, nBu4NOAc, DMF | 27 | 73 | |||
| 3 | Pd(OAc)2, nBu4NOAc, MeCN | 30 | 70 | |||
| 4 | 19,d DMF | 58 | 27 | 15 | ||
| 5 | 20,d DMF | 71 | 22 | 7 | ||
| 6 | Pd(OAc)2, Ph3P, DMF | 46 | 54 | |||
| 7 | Pd(OAc)2, dppf, DMF | 27 | 34 | 17 | 22 | |
| 8 | Pd(OAc)2, BIPHEP, DMF | 19 | 10 | 71 | ||
| 9 | Pd(OAc)2, t-Bu3P•HBF4, DMF | 60 | 30 | |||
| 10 | Pd(OAc)2, t-Bu3P•HBF4, DMSO | 74 | 18 | 8 | ||
| 11 | Pd(OAc)2, t-Bu3P•HBF4, NMP | 88 | 12 | |||
| 12 | Pd(OAc)2, BPTBP,e DMF | 74 | 16 | |||
| 13 | Pd(OAc)2, BPTBP,e DMSO | 87 | 3 | 10 | ||
| 14 | Pd(OAc)2, BPTBP,e NMP | 42 | 48 | 19 | ||
| 15 | Pd(OAc)2, BPTBP,e MeCN | 85 | 10 | |||
Reaction conditions employed 0.25 mmol of 15, 3 equiv of trimethylvinylsilane, 2 equiv of Et3N, 80 °C.
Yields of 14, 16, 17, and 18 were determined by GC analysis without an internal standard.
No Et3N was used as base.
Ligands: VII and VIII.
2-(di-tert-butylphosphino)biphenyl (BPTBP).
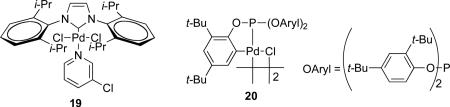
A survey of Pd-sources that represent the three most widely used ligand classes, namely N-heterocyclic carbene (NHC) 19,65,66,67 phosphite 20,65,68 and phosphines65,67 (Table 4, entries 4-6) was carried out. Two important observations were made: (1) triphenylphosphine (Table 4, entry 6) provided a significantly increased yield compared to 19 and 20 (Table 4, entries 4 and 5), and (2) palladium acetate (Table 4, entry 6) was the best palladium source affording the desired arylvinylsilane 14 in 54% yield together with a 46% yield of unreacted starting material.69 Most importantly, this combination did not lead to the formation of byproducts. Moreover, the reactivity and selectivity of Pd(OAc)2 can be tuned with different phosphine ligands.
Catalysis of the Heck reaction with palladium complexes bearing electron-rich arylphosphines such as tri-o-tolylphosphine [(o-Tol)3P] and 1,2-bis(diphenylphosphino)ferrocene (dppf) often have higher reaction rates and turnovers compared to Ph3P.70 In addition, tri-(tert-butyl)phosphine (t-Bu3P) and di(tertbutylphosphino)ferrocene are effective ligands for Heck reactions.67,71
Thus, a survey of electron-rich, sterically hindered monodentate and bidentate phosphine ligands was carried out with Pd(OAc)2. The first reactions were run using the bidentate ligands dppf and BIPHEP (Table 4, entry 7 and 8). The use of dppf (Table 4, entry 7) gave the expected silyl ether 14 in 34% yield together with 27% yield of unreacted starting material 15. In addition, the desilylated compound 16 and the hydrolysis byproduct 17 were observed in 17% and 22% yields, respectively. The ligand BIPHEP on the other hand gave the desilylated product 16 in 71% yield and only 10% yield of the desired material 14. Cleary, bidentate ligands were inferior to Ph3P, so monodentate ligands were surveyed next.
To evaluate t-Bu3P, the commercially available tri-tert-butylphosphine tetrafluoroborate salt (t-Bu3P•HBF4) was employed and complete consumption of the starting material occurred to afford 14 in 60% yield together with reduction product 18 in 30% yield, however no other byproducts were detected by GC analysis (Table 4, entry 9). The formation of reduced products is known in cross-coupling reactions particularly in DMF with tertiary amines.72 To remedy this problem, both DMSO and NMP were tested (Table 4, entries 10 and 11). The use of DMSO increased the yield of 14 to 74% yield together with 11% of 8 but also led to formation of the hydrolyzed product 17 in 18% yield. The use of NMP led to an increase in yield of 14 (88%) albeit with 12% yield of 18.
On the other hand, the use of 2-(di-tert-butylphosphino)biphenyl (BPTBP) in DMF gave 14 in 74% yield together with 18 in 12% yield, a significantly better result than the use of t-Bu3P and DMF (Table 4, entry 12). Moreover, DMSO led to the formation of 14 in 87% yield together with 18 (10%) and 17 (3%) (Table 4, entry 13). In NMP, the reaction stalled and 18 was obtained in 48% yield accompanied by a 10% yield of 18 and a 42% recovery of the starting material 15 (Table 4, entry 14). However, upon using MeCN, 14 was obtained in 85% yield together with a 10% yield of the reduced product 10.
From the ligand and solvent survey conducted above, the reaction conditions that employed t-Bu3P•HBF4 in NMP and BPTBP in MeCN were identified as the best ligand/solvent combinations that gave the highest yields of 14 without creating more than one byproduct. However, of the two ligands employed, BPTBP was chosen for scale-up because the reaction appeared to be cleaner by GC analysis. On a 1.3-mmol scale this reaction afforded the desired arylvinylsilane 14 in 66% yield after chromatographic purification.
Among the optimization studies, entry 6 in Table 4, was intriguing because, aside from the Jeffery conditions, this was the only reaction that did not give byproducts. This result indicated that a more reactive electrophile might be a better substrate. Accordingly, an aryl nonaflate, known to be 1.4 times more reactive73 than the corresponding aryl triflate, was tested. The desired aryl nonaflate 21 (Scheme 6) was efficiently prepared in 88% yield from the corresponding phenol.74 Subjecting 19 to the reaction conditions in Table 4, entry 6, afforded a single product by GC analysis. On a 10-mmol scale this reaction yielded the analytically pure arylvinylsilane 14 in 84% yield after chromatography and Kugelrohr distillation (Scheme 6).
Scheme 6.

a Conditions: (a) Pd(OAc)2 (5 mol%), Ph3P (10 mol%), chlorodimethylvinylsilane, Et3N, MeCN, 80 °C (84%); (b) ICl, MeCN, 0 °C (83%); (c) 13, Otera cat., toluene, reflux (91%); (d) CH3I, Cs2CO3, THF (96%); (e) CAN, MeCN/H2O (86%); (f) chlorodimethylvinylsilane, Et3N, CH2Cl2 (91%); (g) 6, benzene, rt (89%); (h) [allylPdCl]2 (10 mol %), TBAF•6H2O, DMF (74%).
Now with a robust and easily scaled process of synthesizing arylvinylsilane 14 in hand the elaboration to the macrocyclization precursor siloxane 10 began in earnest.
4.2. Synthesis of the Benzo-fused Macrolactone (E,Z)-9
Iododesilylation of 14 with ICl58 afforded arylvinyl iodide 12 in 83% yield with complete retention of the double bond configuration (Scheme 6). Attempts to open lactone 12 with the sodium or potassium alkoxide of 1374 (Scheme 5) failed and led either to vinyl iodide decomposition and/or inconsistent conversion to the expected ester 22. Similarly, attempts to effect acylation with titanium(IV) isopropoxide75 met with failure. However, acylation using 1-hydroxy-3-(isothiocyanato)tetrabutyldistannoxane (Otera's catalyst)76 in toluene at reflux furnished the analytically pure ester 22 in 91% yield after chromatography. Methylation of the hydroxyl group of 22 with methyl iodide and Cs2CO3 afforded 23 in 96% yield. Removal of the PMB group was then effected using ceric ammonium nitrate (CAN) to afford alcohol 24 in 86% yield. Alcohol 24 was then silylated with chlorodimethylvinylsilane to afford the sensitive silyl ether 11 in 95% yield. The silyl ether 11 was then subjected to ring closing metathesis with catalyst 6 to furnish siloxane 10 in 89% yield. Slow addition of 0.1 M solution of siloxane 10 in DMF into a solution of APC and TBAF•6H2O by syringe pump over 48 h furnished macrocycle 9 in a gratifying 74% yield as an oil whose molecular composition was confirmed by high-resolution mass spectrometry. The benzo-fused lactone 9 was formed stereospecifically as indicated by analysis of its 1H NMR spectrum. The diagnostic alkene resonance for HC(8) appeared at 6.53 ppm (d, 3J = 15.5 Hz) that correlated well with the coupling constant found for a simpler compound.77 The other alkenyl protons HC(9), HC(10) and HC(11) appeared at 6.73 ppm (dd, 3J = 15.5, 9.0 Hz), 6.13 ppm (dd, 3J = 10.0 Hz), and 5.72 ppm (dt, 3J = 10.5, 7.5 Hz), respectively, that also matched well with known analogs.77
Discussion
The primary goals of this study were to develop an intramolecular cross-coupling reaction of siloxanes that affords macrolactones in good yields, and to prepare a benzo-fused lactone that could serve as the model system for the synthesis of oximidine II. The preparation of macrolactones was enabled by the discovery that fluoride activation of the cross-coupling was effective. The generality of this finding was demonstrated by the synthesis of various macrolactones. The second aim of this study was facilitated by the development of a Mizoroki-Heck reaction of aryl trifluoralkylsulfonates and a vinylsilane. The elaboration of the Mizoroki-Heck product led to assembly of a benzo-fused macrolactone possessing the core structure of oximidine II.
1. Preparation of Siloxanes 7a-f
The synthesis of the siloxanes used in this study was accomplished in a multi-step sequence whereby each reaction step was a modification of a closely related report. The syntheses of siloxanes 7a-f began from 4-pentyn-1-ol and in a seven-step sequence proceeded uneventfully to yield analytically pure materials in good to excellent yields for all the steps and in all the cases. However, the unexpected sensitivity of the silyl ethers 5a-f warrants further comment. Initial attempts to purify the silyl ethers by silica gel chromatography led to extensive desilylation and inconsistent results. Although the use of neutral alumina remedied the desilylation problem the product contained an impurity that led to death of the molybdenum catalyst in the following ring closing metathesis step. However, adding 1% of Et3N to the eluting solvent for the silica gel purification eliminated the problem of desilylation. Silyl ethers 5a-f could be isolated in analytically pure form in excellent yields.
2. Optimization of the Intramolecular Cross-Coupling Reaction
In planning for the key intramolecular macrocyclization reaction of siloxanes 7a-f it was expected that only minor adjustments of the conditions developed earlier would be required. Orienting experiments with 7a quickly revealed the failure of that assumption, but repeating the experiment with rigorous exclusion of oxygen afforded the desired macrocycle 8a in 46% yield together with the unexpected isomer 8a' as an inseparable mixture. Isomer 8a' most likely arose from a rapid, base-catalyzed translactonization after formation of 8a. This assumption was confirmed by analyzing the reaction every 10 h which showed that the ratio of 8a:8a' remained constant. Furthermore, when pure 8a (obtained from subsequent, selective cyclizations) was exposed to TBAF/THF within 1 h the ratio of 8a:8a' was 5.5:1 and after 40 h the ratio was essentially 3:1 (by 1H NMR analysis). The ratio obtained experimentally (2-3:1) correlates well with molecular modeling calculations that predict a 1.6:1 ratio of 8a:8a' from the 0.26 kcal/mol difference between 8a and 8a'. Thus, to suppress the formation of 8a' and improve the yield of 8a, the basicity of TBAF was attenuated. However, conclusive evidence that the fluoride was the culprit could not be inferred since TBAF had been used successfully before with macrolactones.78
Landini and Maia found that the basicity and nucleophilicity of the fluoride ion in TBAF could be modulated by adjusting the hydration levels and this strategy succeeded here as well.39,79,80 For example, TBAF•4H2O or TBAF•6H2O afforded desired macrolactones 8a and 8a' in higher yields (64% and 59%) compared to TBAF•3H2O. In the more highly hydrated ammonium salts, the fluoride anion forms H-bonds with the water leading to decrease in the basicity of TBAF without greatly affecting the nucleophilicity.39,81 Thus, at these higher hydration levels the fluoride anion is nucleophilic enough to form the pentacoordinate fluorosiliconate intermediate necessary for transmetalation.82 The hydrated TBAF•6H2O was selected over TBAF•4H2O because at this hydration level the fluoride anion basicity is negligible.39 Investigation of higher hydration levels (TBAF•10H2O and TBAF•15H2O) gave lower yields because at these levels, free water is not bound within TBAF matrix which affects the nucleophilicity of the fluoride anion.83
Decreasing the loading of TBAF from 10 to 5 equiv increased the yield of lactones 8a and 8a'. Although this outcome appears contradictory, it can be understood because the overall reaction concentration has increased. The reaction solvent is a stock solution of 1 M TBAF in THF so reducing the number of equivalents of TBAF also constitutes a reduction in the reaction volume. The resulting higher concentration of palladium catalyst leads to a faster rate and ultimately a higher yield. Further increasing the concentration of palladium by using 2.5 equiv of TBAF•6H2O unfortunately led to low yields of isolated macrolactone presumably because at this loading fluoride activation is not sufficient to promote transmetalation.
2.1 Survey of Fluoride Promoters for the Intramolecular Cross-Coupling Reaction
A systematic survey of anhydrous and hydrated fluoride promoters revealed that the hydrated fluoride source afforded the highest yields of macrolactones 8a and 8a' while the anhydrous fluoride sources produced the best selectivity. The higher selectivity may be because TBAT and TASF are fluorosilicates and are less basic and nucleophilic compared to TBAF.84 The hydrated tetraalkylammonium salts gave variable yields and selectivities that were largely influenced by the structure of the alkyl groups comprising the ammonium salt. For example, TMAF•4H2O and BTMAF•6H2O gave similar yields of macrocycle 8a essentially as a single isomer (Table 3, entries 1-4). The similar reactivity exhibited by these two tetraalkylammonium fluoride ions probably arises from the similarity of their structures and therefore comparable electrostatic interactions of the two fluoride sources. The structure of the tetraalkylammonium salt strongly affects the reactivity of the fluoride ion. Whereas tetramethylammonium is a Td symmetrical ion, tetraethylammonium is found crystallographically in a “Nordic cross” conformation (S4)85 and tetrabutylammonium exists in both a flattened cross conformation of D2d symmetry and a more tetrahedral-like conformation of S4 symmetry.86 These structures influence the reactivity of the attendant fluoride ion in two ways, first through the accessibility of the positive charge for Coulombic stabilization87 and second, through the availability of CH•••F– hydrogen bonding interactions.88 These two features combine to most effectively attenuate the reactivity of the fluoride ion in the tetramethylammonium salt. With only 4.0 equiv of water, TMAF does not induce the base-promoted isomerization of 8a to 8a’ whereas 10.0 equiv of water are required to suppress this isomerization with TBAF (cf. Table 1 entry 10 vs. Table 2 entry 1).44
The highest yields of the macrolactones 8a and 8a' were achieved with TBAF but with variable selectivities. The solvent and the hydration level of the TBAF greatly influenced the outcome. For example, ethereal solvents (THF and dioxane) afforded lower yields of the macrocycles 8a and 8a' compared with the more polar DMF but led to higher selectivity. The higher yield in DMF may be due to increased catalytic efficiency of palladium brought about by weak complexation with DMF.65 Another likely contribution by DMF might be its assistance in ionic palladium-ligand exchange reactions.89 In silicon-based cross-coupling reactions the critical, anionic hypervalent siliconate intermediate needed to effect transmetalation is formed in a preequilibrium step from TBAF. The position of this equilibrium is very likely solvent dependent.82,90
3. Scope of the Intramolecular Cross-Coupling Reaction
The intramolecular cross-coupling reaction displayed good generality for the construction of 11-, 12-, 13-, and 14-membered macrolactones from siloxanes 8a-f. Comparison of the results shows that even-numbered rings are formed in slightly higher yields compared to the odd-numbered rings (Scheme 4). This trend has been observed previously in the synthesis of macrocycles.11 It is worthy to note that the size of the siloxane ring has a moderate effect on the yields of the macrolactones. For example, in the synthesis of the 12-membered macrolactones, the seven-membered siloxane 7a afforded 8a and 8a' in 78% yield compared to the six-membered siloxane 7b that gave 8b in 70% yield. Similarly, in the synthesis of the 13-membered macrolactone, the seven-membered siloxane 7d afforded 8d in 61% yield while the six-membered siloxane 7e furnished 8e in 53% yield. The seven-membered siloxane has a higher strain compared to the six-membered siloxanes and thus upon fluoride activation will undergo a more facile transmetalation compared to the six-membered lactone in order to relieve the strain. The less strained siloxane on the other hand may undergo other destructive pathways prior to transmetalation. While this indirect correlation cannot be ascertained with the data at hand a similar trend was observed in one case in the synthesis of medium-ring cyclic ethers.22
4. The Mizoroki-Heck Reaction of Vinyltrimethylsilane
The Mizoroki-Heck reaction of aryl triflates with vinyltrimethylsilane is challenging because aryl sulfonates possess moderate reactivity.73,91 The Heck reaction of aryl triflate 21 under Jeffrey conditions at both ambient temperature and heating at 80 °C (Table 4, entry 1 and 2) stalled at 43% and 73% conversions, respectively. The inability of the reactions to go to full completion is presumably because oxidative addition could not occur efficiently without ligands.
A survey of ligands known to be useful in Heck reactions identified monodentate phosphines as superior additives. The higher selectivity and yields exhibited by monodentate phosphines compared to bidentate ligands arises because monodentate ligands do not form the unreactive bisphosphine complexes formed by bidentate ligands because of steric crowding.65 The success of the bulky monophosphines arises from their ability to effect a fast oxidative addition. In addition, they are able to generate the active T-shaped, three-coordinate palladium complexes.71 Because of the open coordinate site, these complexes are capable of undergoing facile olefin insertion leading to the Heck product. The polar, aprotic solvents acetonitrile and NMP gave the best results and are known to increase the rate of Heck reactions.89,92
The successful reaction of aryl nonaflate 21 with trimethylvinylsilane under condition wherein 15 stalled is not unexpected because aryl nonaflates display higher reactivity than aryl triflates.73 The enhanced electrophilicity of aryl nonaflates renders them better electrophiles for rapid oxidative addition onto the palladium center and thereupon renders the palladium atom more electrophilic for the carbo metalation of the vinylsilane.
6. Synthesis of the Benzo-fused Lactone 9
The synthesis of 9 followed the established route except for the opening of lactone 12 with alcohol 13, by employing the Otera catalyst 22 was then elaborated in 4 steps to siloxane 10. Under the optimized cross-coupling conditions, siloxane 10 furnished the benzo-fused macrocycle (E,Z)-9 in excellent yield (74%) as a single isomer. The yield is comparable to the other two 12-membered lactones 8a and 8a' and 8b that were obtained in 78% and 70% yield respectively.
The high yield of this silicon-assisted cross-coupling reaction over the Mitsunobu protocol used to synthesize a similar analog30 can be ascribed to: (1) the added rigidity provided by the benzene ring reduces the entropic penalty of cyclization, (2) the slow addition of the siloxane into the reaction mixture to maintain a low concentration of the reactive intermediates to avoid polymerization, and, (3) the palladium macrocycle intermediate formed after oxidative addition and transmetalation has reduced strain (compared to the lactonization transition structure) because the two palladium-carbon bonds are longer compared to carbon-carbon bonds. Furthermore, the energetically favored reductive elimination step should overcome the barrier imposed by ring formation more easily than the Mitsunobu reaction that relies on the carboxylate to effect ring closure. This result brings to the fore the power of palladium-catalyzed reactions in facile formation of macrocycles21,22-24,93 that take advantage of the reductive elimination as the key step.
Conclusion
The intramolecular cross-coupling reaction of olefinic iodides and cycloalkenylsiloxanes under fluoride activation effectively produced 11-, 12-, 13-, and 14-membered macrolactones containing a conjugated 4,6-Z,Z- or 1,3-E,Z-diene unit. The fluoride hydration level was critical for controlling the yield and selectivity of macrolactone formation. Key features of this process include: (1) high stereospecificity, (2) modular construction of cyclization precursors, (3) flexibility in locating the hydroxyl group at multiple positions in a macrocycle, and (4) generality of ring size. The insights garnered from these studies are being applied in the total syntheses of complex macrocyclic natural products. The results of those studies will be disclosed in due course.
Supplementary Material
Acknowledgement
We are grateful to the National Institutes of Health for generous financial support (GM63167). J.M.M. acknowledges the NIH for a postdoctoral fellowship.
Footnotes
Supporting Information Available: Detailed experimental procedures, optimization studies, full characterization of all products. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Wessjohann LA, Rivera DG, Vercillo OE. Chem. Rev. 2009;109:796–814. doi: 10.1021/cr8003407. [DOI] [PubMed] [Google Scholar]
- 2.a Driggers EM, Hale SP, Lee J, Terrett NK. Nat. Rev. Drug Discovery. 2008;7:608–624. doi: 10.1038/nrd2590. [DOI] [PubMed] [Google Scholar]; b Harvey AL. Drug Discov. Today. 2008;13:894–901. doi: 10.1016/j.drudis.2008.07.004. [DOI] [PubMed] [Google Scholar]; c Butler MS. Nat. Prod. Rep. 2008;25:475–516. doi: 10.1039/b514294f. [DOI] [PubMed] [Google Scholar]; d Butler MS. Nat. Prod. Rep. 2005;22:162–195. doi: 10.1039/b402985m. [DOI] [PubMed] [Google Scholar]
- 3.Kim JW, Shin-Ya K, Furihata K, Hayakawa Y, Seto H. J. Org. Chem. 1999;64:153–155. doi: 10.1021/jo9814997. [DOI] [PubMed] [Google Scholar]
- 4.Hayakawa Y, Tomikawa T, Shin-ya K, Arao N, Nagai K, Suzuki K, Furihata K. J. Antibiot. 2003;56:905–908. doi: 10.7164/antibiotics.56.905. [DOI] [PubMed] [Google Scholar]
- 5.Winssinger N, Barluenga S. Chem. Commun. 2007:22–36. doi: 10.1039/b610344h. [DOI] [PubMed] [Google Scholar]
- 6.Delmotte P, Delmotte-Plaquee J. Nature. 1953;171:344. doi: 10.1038/171344a0. [DOI] [PubMed] [Google Scholar]
- 7.Ayer WA, Lee SP, Tsuneda A, Hiratsuka Y. Can. J. Microbiol. 1980;26:766–773. [Google Scholar]
- 8.Sugawara K, Nishiyama Y, Toda S, Komiyama N, Hatori M, Moriyama T, Sawada Y, Kamei H, Konishi M, Oki T. J. Antibiot. 1992;45:1433–1441. doi: 10.7164/antibiotics.45.1433. [DOI] [PubMed] [Google Scholar]
- 9.a Langer P, Freiberg W. Chem. Rev. 2004;104:4125–4149. doi: 10.1021/cr010203l. [DOI] [PubMed] [Google Scholar]; b Rousseau G. Tetrahedron. 1995;51:2777–2849. [Google Scholar]
- 10.a Mandolini L. Adv. Phys. Org. Chem. 1986;22:1–111. [Google Scholar]; b Illuminati G, Mandolini L. Acc. Chem. Res. 1981;14:95–102. [Google Scholar]
- 11.Winnik MA. Chem. Rev. 1981;81:491–524. [Google Scholar]
- 12.Galli C, Mandolini L. Eur. J. Org. Chem. 2000:3117–3125. and references cited therein. [Google Scholar]
- 13.Parenty A, Moreau X, Campagne J-M. Chem. Rev. 2006;106:911–939. doi: 10.1021/cr0301402. Hamada Y, Shioiri T. Chem. Rev. 2005;105:4441–4482. doi: 10.1021/cr0406312. Wessjohann LA, Ruijter E. Top. Curr. Chem. 2005;243:137–184. Yet L. Chem. Rev. 2003;103:4283–4306. doi: 10.1021/cr030035s. Roxburgh CJ. Tetrahedron. 1995;51:9767–9822. (f) for an example of macrocyclization by oxidative allylic transposition see: Jung HH, Seiders JR, Floreancig PE. Angew. Chem. Int. Ed. 2007;46:8464–8467. doi: 10.1002/anie.200702999.
- 14.a Rousseau G, Homsi F. Chem. Soc. Rev. 1997;26:453–461. [Google Scholar]; b Stang EM, White MC. Nature Chem. 2009;1:547–551. doi: 10.1038/nchem.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Edmonds DJ, Johnston D, Procter DP. Chem. Rev. 2004;104:3371–3403. doi: 10.1021/cr030017a. [DOI] [PubMed] [Google Scholar]
- 16.a Monfette S, Fogg DE. Chem. Rev. 2009;109:3783–3816. doi: 10.1021/cr800541y. [DOI] [PubMed] [Google Scholar]; b Deiters A, Martin SF. Chem. Rev. 2004;104:2199–2238. doi: 10.1021/cr0200872. [DOI] [PubMed] [Google Scholar]; c Hoveyda AH, Zhugralin AR. Nature. 2007;450:243–251. doi: 10.1038/nature06351. [DOI] [PubMed] [Google Scholar]; d Majumdar KC, Rahaman H, Roy B. Curr. Org. Chem. 2007;11:1339–1365. [Google Scholar]; e Nicolaou KC, Bulger PG, Sarlah D. Angew. Chem. Int. Ed. 2005;44:4490–4527. doi: 10.1002/anie.200500369. [DOI] [PubMed] [Google Scholar]; f Diver ST, Giessert AJ. Chem. Rev. 2004;104:1317–1382. doi: 10.1021/cr020009e. [DOI] [PubMed] [Google Scholar]
- 17.Nicola T, Brenner M, Donsbach K, Kreye P. Org. Process Res. Dev. 2005;9:513–515. [Google Scholar]
- 18.a Lacombe F, Radkowski K, Seidel G, Fürstner A. Tetrahedron. 2004;60:7315–7324. [Google Scholar]; b Fürstner A, Turet L. Angew. Chem. Int. Ed. 2005;44:3462–3466. doi: 10.1002/anie.200500390. [DOI] [PubMed] [Google Scholar]; c Fürstner A, Bonnekessel M, Blank JT, Radkowski K, Seidel G, Lacombe F, Gabor B, Mynott R. Chem. Eur. J. 2007;13:8762–8783. doi: 10.1002/chem.200700926. [DOI] [PubMed] [Google Scholar]
- 19.Miyaura N, Suginome H, Suzuki A. Tetrahedron Lett. 1984;25:761–764. [Google Scholar]
- 20.a Stille JK, Tanaka M. J. Am. Chem. Soc. 1987;109:3785–3786. [Google Scholar]; b Pattenden G, Sinclair DJ. J. Organomet. Chem. 2002;653:261–268. [Google Scholar]; c Dunctun MAJ, Pattenden G. J. Chem. Soc., Perkin. Trans. 1. 1999:1235–1246. [Google Scholar]; d Smith AB, III, Condon SM, Leazer JL, Jr., Leahy JW, Maleczka RE. J. Am. Chem. Soc. 1995;117:5407–5408. [Google Scholar]
- 21.For selected examples of alkene-alkene Suzuki macrocyclizations, see: Ghidu VP, Wang JQ, Wu B, Liu QS, Jacobs A, Marnett LJ, Sulikowski GA. J. Org. Chem. 2008;73:4949–4955. doi: 10.1021/jo800545r. Tortosa M, Yakelis NA, Roush WR. J. Am. Chem. Soc. 2008;130:2722–2723. doi: 10.1021/ja710238h. Nicolaou KC, Nold AL, Milburn RR, Schindler CS, Cole KP, Yamaguchi J. J. Am. Chem. Soc. 2007;129:1760–1768. doi: 10.1021/ja068053p. Molander GA, Dehmel F. J. Am. Chem. Soc. 2004;126:10313–10318. doi: 10.1021/ja047190o. Wu B, Liu Q, Sulikowski GA. Angew. Chem., Int. Ed. 2004;43:6673–6675. doi: 10.1002/anie.200461469. For selected examples of ary-aryl Suzuki macrocyclizations, see: Roberts TC, Smith PA, Cirz RT, Romesberg FE. J. Am. Chem. Soc. 2007;129:15830–15838. doi: 10.1021/ja073340u. Lepine R, Zhu JP. Org. Lett. 2005;7:2981–2984. doi: 10.1021/ol050949w. Kaiser M, Siciliano C, Assfalg-Machleidt I, Groll M, Milbradt AG, Moroder L. Org. Lett. 2003;5:3435–3437. doi: 10.1021/ol035178f. Carbonnelle AC, Zhu JP. Org. Lett. 2000;2:3477–3480. doi: 10.1021/ol006520g.
- 22.a Denmark SE, Yang SM. J. Am. Chem. Soc. 2002;124:2102–2103. doi: 10.1021/ja0178158. [DOI] [PubMed] [Google Scholar]; b Denmark SE, Yang SM. J. Am. Chem. Soc. 2002;124:15196–15197. doi: 10.1021/ja028936q. [DOI] [PubMed] [Google Scholar]; c Denmark SE, Yang SM. J. Am. Chem. Soc. 2004;126:12432–12440. doi: 10.1021/ja0466863. [DOI] [PubMed] [Google Scholar]; d Denmark SE, Yang S-M. In: Strategies and Tactics in Organic Synthesis. Harmata MA, editor. Vol. 6. Elsevier; Amsterdam: 2005. Chapter. 4. [Google Scholar]
- 23.Nicolaou KC, Bulger PG, Sarlah D. Angew. Chem. Int. Ed. 2005;44:4442–4489. doi: 10.1002/anie.200500368. [DOI] [PubMed] [Google Scholar]
- 24.a Trost BM, Verhoeven TR. J. Am. Chem. Soc. 1977;99:3867–3868. [Google Scholar]; b Trost BM, Verhoeven TR. J. Am. Chem. Soc. 1979;101:1595–1597. [Google Scholar]; c Trost BM, Verhoeven TR. J. Am. Chem. Soc. 1980;102:4743–4763. [Google Scholar]
- 25.Vosburg DA, Vanderwal CD, Sorensen EJ. J. Am. Chem. Soc. 2002;124:4552–4553. doi: 10.1021/ja025885o. Trost BM, Ohmori M, Boyd SA, Okawara H, Brickner SJ. J. Am. Chem. Soc. 1989;111:8281–8284. Fürstner A, Weintritt H. J. Am. Chem. Soc. 1998;120:2817–2825. Marshall JA, Andrews RC, Lebioda L. J. Org. Chem. 1987;52:2378–2388. For reviews see: Heumann A, Reglier M. Tetrahedron. 1995;51:975–1015. Trost BM. Angew. Chem., Int. Ed. 1989;28:1173–1192.
- 26.Trost BM, Dong G. Nature. 2008;456:485–488. doi: 10.1038/nature07543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.a Bazan GC, Khosravi E, Schrock RR, Feast WJ, Gibson VC, Oregan MB, Thomas JK, Davis WM. J. Am. Chem. Soc. 1990;112:8378–8387. [Google Scholar]; b Bazan GC, Oskam JH, Cho HN, Park LY, Schrock RR. J. Am. Chem. Soc. 1991;113:6899–6907. [Google Scholar]; c Schrock RR, Murdzek JS, Bazan GC, Robbins J, Dimare M, Oregan M. J. Am. Chem. Soc. 1990;112:3875–3886. [Google Scholar]
- 28.a Schwab P, France MB, Ziller JW, Grubbs RH. Angew. Chem., Int. Ed. 1995;34:2039–2041. [Google Scholar]; b Schwab P, Grubbs RH, Ziller JW. J. Am. Chem. Soc. 1996;118:100–110. [Google Scholar]; c Scholl M, Ding S, Lee CW, Grubbs RH. Org. Lett. 1999;1:953–956. doi: 10.1021/ol990909q. [DOI] [PubMed] [Google Scholar]; d Trnka TM, Morgan JP, Sanford MS, Wilhelm TE, Scholl M, Choi TL, Ding S, Day MW, Grubbs RH. J. Am. Chem. Soc. 2003;125:2546–2558. doi: 10.1021/ja021146w. [DOI] [PubMed] [Google Scholar]
- 29.Wang X, Porco JA. J. Am. Chem. Soc. 2003;125:6040–6041. doi: 10.1021/ja034030o. [DOI] [PubMed] [Google Scholar]
- 30.Coleman RS, Garg R. Org. Lett. 2001;3:3487–3490. doi: 10.1021/ol016744e. [DOI] [PubMed] [Google Scholar]
- 31.Luthy C, Konstantin P, Untch KG. J. Am. Chem. Soc. 1978;100:6211–6217. [Google Scholar]
- 32.Groves JT, Ma KW. J. Am. Chem. Soc. 1977;99:4076–4082. [Google Scholar]
- 33.Wu Y, Gao J. Org. Lett. 2008;10:1533–1536. doi: 10.1021/ol800137f. [DOI] [PubMed] [Google Scholar]
- 34.Zhao MZ, Li J, Song ZG, Desmond R, Tschaen DM, Grabowski EJJ, Reider PJ. Tetrahedron Lett. 1998;39:5323–5326. [Google Scholar]
- 35.Pospisil J, Marko IE. Tetrahedron Lett. 2006;47:5933–5937. [Google Scholar]
- 36.Ishihara K, Kurihara H, Yamamoto H. J. Org. Chem. 1993;58:3791–3793. [Google Scholar]
- 37.See Supporting Information for details.
- 38.Smith AB, III, Lupo AT, Ohba M, Chen K. J. Am. Chem. Soc. 1989;111:6648–6656. [Google Scholar]
- 39.Landini D, Maia A, Rampoldi A. J. Org. Chem. 1989;54:328–332. [Google Scholar]
- 40.Denmark SE, Sweis RF. Org. Lett. 2002;4:3771–3774. doi: 10.1021/ol026900x. [DOI] [PubMed] [Google Scholar]
- 41.Denmark SD, Liu JH-C, Muhuhi MJ. J. Am. Chem. Soc. 2009;131:14188–14189. doi: 10.1021/ja9063475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reichardt C. In Solvents and Solvents Effects in Org. Chem. 3rd. Wiley-VCH; Weinheim: 2003. updated and enl. Ed., Chap 7. Aj represents acidity, a solvent anion-solvating tendency while Bj represents basicity, a solvent cation-solvating tendency. The sum of acidity and basicity is a solvent solvation capability or its polarity.
- 43.The DMF used in these investigations was dried by percolation through an activity 1 alumina bed. The water content was determined by triplicate Karl Fischer titration to be 3.3 µg/100 µL or 33 ppm.
- 44.Harmon KM, Gennick I. Inorg. Chem. 1975;14:1840–1845. [Google Scholar]
- 45.Kuwajima I, Nakamura E, Shimizu M. J. Am. Chem. Soc. 1982;104:1025–1030. [Google Scholar]
- 46.a Cox DF, Terpinski J, Lawrynowicz W. J. Org. Chem. 1984;49:3216–3219. [Google Scholar]; b Albanese D, landini D, Penso M. J. Org. Chem. 1998;63:9587–9589. [Google Scholar]
- 47.Hatanaka Y, Hiyama T. J. Org. Chem. 1988;53:918–920. [Google Scholar]
- 48.a DeShong P, Handy CJ, Mowery ME. Pure Appl. Chem. 2000;72:1655–1658. [Google Scholar]; b Mowery ME, DeShong P. J. Org. Chem. 1999;64:3266–3270. doi: 10.1021/jo990072c. [DOI] [PubMed] [Google Scholar]
- 49.Denmark SE, Kobayashi T. J. Org. Chem. 2003;68:5153–5159. doi: 10.1021/jo034064e. [DOI] [PubMed] [Google Scholar]
- 50.All attempts to resolve the olefinic region by recording the 1H NMR spectra with various deuterated solvents (CDCl3, CD2Cl2, C6D6, CD3OD, toluene-d, DMSO-d, pyridine-d, tetrahydrofuran-d, 1,4-dioxane-d) and with lanthanide shift reagents (EuFOD and YbFOD) were unsuccessful.
- 51.Spartan 08, DFT, B3LYP, 6-31*G.
- 52.a Takaoka LR, Buckmelter AJ, LaCruz TE, Rychnovsky SD. J. Am. Chem. Soc. 2005;127:528–529. doi: 10.1021/ja044642o. [DOI] [PubMed] [Google Scholar]; b Crawford RJ, Lutener SB, Cockcroft RD. Can. J. Chem. 1976;54:3364–3376. [Google Scholar]; c Sugawara M, Yoshida J. Tetrahedron Lett. 1999;40:1717–1720. [Google Scholar]; d Nishikawa T, Shinokubo H, Oshima K. Tetrahedron. 2003;59:9661–9668. [Google Scholar]; e Klos AM, Heintzelman GR, Weinreb SM. J. Org. Chem. 1997;62:3758–3761. [Google Scholar]
- 53.a Takai K, Nitta K, Utimoto K. J. Am. Chem. Soc. 1986;108:7408–7410. doi: 10.1021/ja00279a068. [DOI] [PubMed] [Google Scholar]; b Takai K, Ichiguchi T, Hikasa S. Synlett. 1999:1268–1270. [Google Scholar]; c Haack T, Kurtkaya S, Snyder JP, Georg GI. Org. Lett. 2003;5:5019–5022. doi: 10.1021/ol036007d. [DOI] [PubMed] [Google Scholar]
- 54.a Kuang CX, Yang Q, Senboku H, Tokuda M. Synthesis. 2005:1319–1325. [Google Scholar]; b Das JP, Roy S. J. Org. Chem. 2002;67:7861–7864. doi: 10.1021/jo025868h. [DOI] [PubMed] [Google Scholar]; c You H-W, Lee K-J. Synlett. 2001:105–107. [Google Scholar]; d Naskar D, Roy S. Tetrahedron. 2000;56:1369–1377. [Google Scholar]
- 55.Horibe H, Kondo K, Okuno H, Aoyama T. Synthesis. 2004:986–988. [Google Scholar]
- 56.Bull JA, Mousseau JJ, Charette AB. Org. Lett. 2008;10:5485–5488. doi: 10.1021/ol802315k. [DOI] [PubMed] [Google Scholar]
- 57.Pawluc P, Hreczycho G, Szudkowska J, Kubicki M, Marciniec B. Org. Lett. 2009;11:3390–3393. doi: 10.1021/ol901233j. [DOI] [PubMed] [Google Scholar]
- 58.a Stamos DP, Taylor AG, Kishi Y. Tetrahedron Lett. 1996;37:8647–8650. [Google Scholar]; b Tamao K, Akita M, Maeda K, Kumada M. J. Org. Chem. 1987;52:1100–1106. [Google Scholar]
- 59.a Mizoroki T, Mori K, Ozaki A. Bull. Chem. Soc. Jpn. 1971;44:581. [Google Scholar]; b Heck RF, Nolley JP. J. Org. Chem. 1972;37:2320–2322. [Google Scholar]
- 60.a Karabelas K, Hallberg A. J. Org. Chem. 1986;51:5286–5290. [Google Scholar]; b Garst ME, McBride BJ. J. Org. Chem. 1989;54:249–250. [Google Scholar]; c Battace A, Zair T, Doucet H, Santelli M. J. Organomet. Chem. 2005;690:3790–3802. [Google Scholar]
- 61.Hamze A, Provot O, Brion JD, Alami M. J. Organomet. Chem. 2008;693:2789–2797. [Google Scholar]
- 62.Fürstner A, Dierkes T, Thiel OR, Blanda G. Chem. Eur. J. 2001;7:5286–5298. doi: 10.1002/1521-3765(20011217)7:24<5286::aid-chem5286>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 63.Neuville L, Bigot A, Dau METH, Zhu JP. J. Org. Chem. 1999;64:7638–7642. [Google Scholar]
- 64.a Jeffery T, Ferber B. Tetrahedron Lett. 2003;44:193–197. [Google Scholar]; b Jeffery T. Tetrahedron Lett. 1999;40:1673–1676. [Google Scholar]; c Jeffery T. Tetrahedron. 1996;52:10113–10130. [Google Scholar]
- 65.Beletskaya IP, Cheprakov AV. Chem. Rev. 2000;100:3009–3066. doi: 10.1021/cr9903048. [DOI] [PubMed] [Google Scholar]
- 66.Herrmann WA. Angew. Chem., Int. Ed. 2002;41:1290–1309. doi: 10.1002/1521-3773(20020415)41:8<1290::aid-anie1290>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 67.Littke AF, Fu GC. Angew. Chem., Int. Ed. 2002;41:4176–4211. doi: 10.1002/1521-3773(20021115)41:22<4176::AID-ANIE4176>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 68.Beletskaya IP, Cheprakov AV. J. Organomet. Chem. 2004;689:4055–4082. [Google Scholar]
- 69.(PPh3)2PdCl2 afforded 14 (49%), 16 (38%) along with 15% of recovered 15.
- 70.Qadir M, Mochel T, Hii KK. Tetrahedron. 2000;56:7975–7979. [Google Scholar]
- 71.a Shaughnessy KH, Kim P, Hartwig JF. J. Am. Chem. Soc. 1999;121:2123–2132. [Google Scholar]; b Barrios-Landeros F, Carrow BP, Hartwig JF. J. Am. Chem. Soc. 2008;130:5842–5843. doi: 10.1021/ja711159y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.a Ghosez L, Franc C, Denonne F, Cuisinier C, Touillaux R. Can. J. Chem. 2001;79:1827–1839. [Google Scholar]; b Handy ST, Bregman H, Lewis J, Zhang XL, Zhang YN. Tetrahedron Lett. 2003;44:427–430. [Google Scholar]
- 73.Rottlander M, Knochel P. J. Org. Chem. 1998;63:203–208. doi: 10.1021/jo971636k. Lipshutz BH, Buzard DJ, Yun CS. Tetrahedron Lett. 1999;40:201–204. (c) for review of nonafluorobutane sulfonate chemistry see: Högermeier J, Reissig HU. Adv. Synth. Catal. 2009;351:2747–2763. Stang PJ, Hanack M, Subramanian LR. Synthesis. 1982:85–126.
- 74.Fürstner A, Nagano T, Muller C, Seidel G, Muller O. Chem. Eur. J. 2007;13:1452–1462. doi: 10.1002/chem.200601370. [DOI] [PubMed] [Google Scholar]
- 75.Rehwinkel H, Steglich W. Synthesis. 1982:826–827. [Google Scholar]
- 76.Otera J, Danoh N, Nozaki H. J. Org. Chem. 1991;56:5307–5311. [Google Scholar]
- 77.For a less functionalized analog of the same ring system: HC(8), d, J = 15.5 Hz.; HC(9), dd, J = 15.5, 11.1 Hz.; HC(10) app t, J = 11.1 Hz.; HC(11), dt, J = 10.6, 4.9 Hz. Coleman RS, Garg R. Org. Lett. 2001;3:3487–3490. doi: 10.1021/ol016744e.
- 78.a Anquetin G, Rawe SL, McMahon K, Murphy EP, Murphy PV. Chem. Eur. J. 2008;14:1592–1600. doi: 10.1002/chem.200701033. [DOI] [PubMed] [Google Scholar]; b Williams DR, Plummer SV, Patnaik S. Angew. Chem. Int. Ed. 2003;42:3934–3938. doi: 10.1002/anie.200351817. [DOI] [PubMed] [Google Scholar]; c Fettes A, Carreira EM. J. Org. Chem. 2003;68:9274–9283. doi: 10.1021/jo034964v. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2002;41:4098–4101. doi: 10.1002/1521-3773(20021104)41:21<4098::AID-ANIE4098>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]; d Evans DA, Hu E, Burch JD, Jaeschke G. J. Am. Chem. Soc. 2002;124:5654–5655. doi: 10.1021/ja026235n. [DOI] [PubMed] [Google Scholar]
- 79.The structures of hydrated tetraalkylammonium ions have been the subject of careful investigation for many years. A continuum of structures has been identified (depending upon ion structure and hydration level) that ranges from pure anion clathrates through framework structures through hydrogen bonded networks and finally to discrete polyhedral cluster anions, see: Jeffrey GA, Mak TC. Science. 1965;149:178–179. doi: 10.1126/science.149.3680.178. McLean WJ, Jeffrey GA. J. Chem. Phys. 1967;47:414–417. Gennick I, Harmon KM, Hartwig J. Inorg. Chem. 1977;16:2241–2248.
- 80.Clark JH. Chem. Rev. 1980;80:429–452. [Google Scholar]
- 81.It has been suggested that a stable geometry exists of six water molecules around the fluoride ion of TBAF in an aqueous solution. See Craig JDC, Booker MH. J. Solution Chem. 2000;29:879.
- 82.Denmark SE, Sweis RF, Wehrli D. J. Am. Chem. Soc. 2004;126:4865–4875. doi: 10.1021/ja037234d. [DOI] [PubMed] [Google Scholar]
- 83.Rozhkov IN, Kuleshova ND. Bull. Acad. Sci. USSR Div. Chem. Sci. 1976;25:1919–1923. [Google Scholar]; Izv. Akad. Nauk SSSR, Ser. Khim. 1976;9:2048–2052. [Google Scholar]
- 84.Pilcher AS, DeShong P. J. Org. Chem. 1996;61:6901–6905. doi: 10.1021/jo960922d. Pilcher AS, Ammon HL, DeShong P. J. Am. Chem. Soc. 1995;117:5166–5167. (c) for an example of reduced TASF basicity over TBAF in a total synthesis see: Fürstner A, Nagano T. J. Am. Chem. Soc. 2007;129:1906–1907. doi: 10.1021/ja068901g.
- 85.Wait E, Powell HM. J. Chem. Soc. 1958:1872–1875. [Google Scholar]
- 86.a Snow MR, Ibers JA. Inorg. Chem. 1973;12:249–254. [Google Scholar]; b Andrenini BP, Dell'Amico DB, Calderazzo F, Venturi MG. J. Organomet. Chem. 1988;354:369–380. and references cited therein. [Google Scholar]
- 87.Halpern M, Sasson Y, Rabinovitz M. Tetrahedron. 1982;38:3183–3187. [Google Scholar]
- 88.Harmon KM, Gennick I, Madeira SL. J. Phys. Chem. 1974;78:2585–2591. [Google Scholar]
- 89.Spencer A. J. Organomet. Chem. 1983;258:101–108. [Google Scholar]
- 90.Sugiyama A, Ohnishi YY, Nakaoka M, Nakao Y, Sato H, Sakaki S, Nakao Y, Hiyama T. J. Am. Chem. Soc. 2008;130:12975–12985. doi: 10.1021/ja801362e. [DOI] [PubMed] [Google Scholar]
- 91.Ritter K. Synthesis. 1993:735–762. [Google Scholar]
- 92.Scott WJ, Stille JK. J. Am. Chem. Soc. 1986;108:3033–3040. [Google Scholar]
- 93.a Jägel J, Maier ME. Synthesis. 2009:2881–2892. [Google Scholar]; b Evans DA, Hu E, Burch JD, Jaeschke G. J. Am. Chem. Soc. 2002;124:5654–5655. doi: 10.1021/ja026235n. [DOI] [PubMed] [Google Scholar]; c Stille JK, Su H, Hill DH, Schnelder P, Tanaka M, Morrison DL, Hegedus LS. Organometallics. 1991;10:1993. [Google Scholar]; d Stille JK, Tanaka M. J. Am. Chem. Soc. 1987;109:3785–3786. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.